
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of rare sulfated N-unsubstituted glucosamine based heparan sulfate analogs selectively activating chemokines†
Prashant
Jain‡
b,
Chethan D.
Shanthamurthy‡
b,
Shani
Leviatan Ben-Arye
a,
Robert J.
Woods
c,
Raghavendra
Kikkeri
*b and
Vered
Padler-Karavani
 *a
*a
aDepartment of Cell Research and Immunology, The Shmunis School of Biomedicine and Cancer Research, The George S. Wise Faculty of Life Sciences, Tel Aviv University, Tel Aviv, 69978, Israel. E-mail: vkaravani@tauex.tau.ac.il
bDepartment of Chemistry, Indian Institute of Science Education and Research, Pune-411008, India. E-mail: rkikkeri@iiserpune.ac.in
cComplex Carbohydrate Research Center, University of Georgia, Athens 30606, GA, USA
First published on 28th January 2021
Abstract
Achieving selective inhibition of chemokines with structurally well-defined heparan sulfate (HS) oligosaccharides can provide important insights into cancer cell migration and metastasis. However, HS is highly heterogeneous in chemical composition, which limits its therapeutic use. Here, we report the rational design and synthesis of N-unsubstituted (NU) and N-acetylated (NA) heparan sulfate tetrasaccharides that selectively inhibit structurally homologous chemokines. HS analogs were produced by divergent synthesis, where fully protected HS tetrasaccharide precursor was subjected to selective deprotection and regioselectively O-sulfated, and O-phosphorylated to obtain 13 novel HS tetrasaccharides. HS microarray and SPR analysis with a wide range of chemokines revealed the structural significance of sulfation patterns and NU domain in chemokine activities for the first time. Particularly, HT-3,6S-NH revealed selective recognition by CCL2 chemokine. Further systematic interrogation of the role of HT-3,6S-NH in cancer demonstrated an effective blockade of CCL2 and its receptor CCR2 interactions, thereby impairing cancer cell proliferation, migration and invasion, a step towards designing novel drug molecules.
Introduction
Chemokines are endogenous signaling peptides essential for immuno-surveillance, homeostasis, inflammation, infection and tissue repair.1 Chemokines and their receptor activities depend on how they bind and oligomerize in the presence of glycosaminoglycans (GAGs).2 Humans express 47 chemokines and 20 receptors, and most of the chemokines are highly basic proteins. It has been therefore hypothesized elsewhere that chemokine–GAG binding is non-specific. However, after the discovery of acidic CCL3 and CCL4 chemokines binding to GAGs, it had become clearer that their interaction proceeds in a sequence-dependent manner.3 Moreover, chemokines have shown different interaction strengths with various GAGs, including heparan sulfate (HS), chondroitin sulfate and dermatan sulfate, illustrating that microheterogeneity in GAG structures can modulate binding patterns, for example through uronic acid composition, sulfation patterns and oligosaccharide length.4This has prompted the synthesis of well-described, homogeneous GAG structures, and more specifically HS oligosaccharides, to regulate chemokine activity. For example, Gallagher et al. reported that the CXCL4 chemokine requires HS 2-O-sulfated iduronic acid (IdoA) for tetramerization and binding to its cell surface receptors.5 Elsewhere, Lindahl et al. had shown that interleukin-8 (CXCL8 or IL-8) prefers the IdoA(2-OSO3−)-GlcNSO3−(6OSO3−) repeating unit to activate neutrophil trafficking,6 while Gardiner et al. reported the elegant role of 6-O-sulfation in switching the binding between CXCL12 and IL-8.7 Hesieh-Wilson et al. demonstrated that the trisulfated IdoA(2-OSO3−)-GlcNSO3−(6-OSO3−)-conjugated polymer strongly inhibited RANTES (CCL5)-CCR3-receptor-mediated cell migration.8 In addition, Seeberger et al. showed that CCL21 strongly binds to a hexasaccharide containing the GlcNSO3−(6-OSO3−)-IdoA(2-OSO3−) repeating unit as compared to CXCL12, while CCL19 does not bind to it at all.9 Boons et al. discovered that CCL2 binds to highly sulfated HS compounds and exhibits no preference for the uronic acid component, while both CCL2 and CCL13 displayed promiscuous binding with most of the HS glycans.10 These data suggest that well-defined HS oligosaccharides can provide structural details to target chemokine–GAG interaction to modulate its activities. However, HS is highly heterogeneous in its structure and the majority of the HS libraries that have been used for chemokine studies are composed of N-sulfated and N-acetylated (NA) glucosamine domains.8–10 Native HS also expresses an N-unsubstituted (NU) domains, but this region has not been fully investigated in existing studies.11 To address this gap, and to decipher the sulfation code of chemokine heparin binding, here we report the divergent synthesis of a limited number of NU- and NA domains HS tetrasaccharides (Fig. 1).
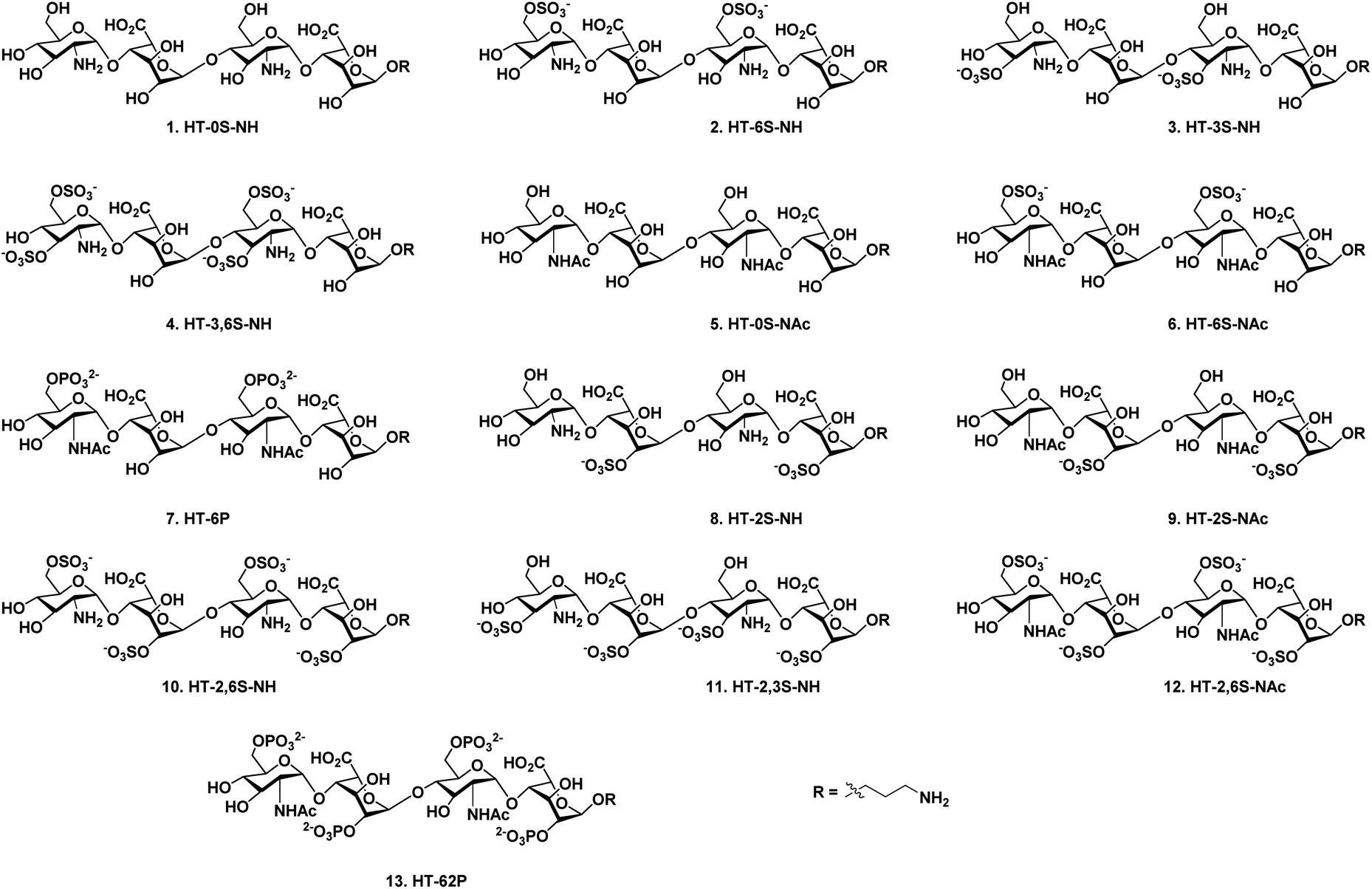 | ||
| Fig. 1 Structures of heparan sulfate tetrasaccharide analogs (1–13). | ||
High-throughput screening of these synthetic HS ligands with a wide range of chemokines revealed selective chemokine binder, which can be used to block chemokine activity to target cancer biology. However thus far, only few heparin binding proteins have been reported to bind NU-domain-containing HS ligands,12 and here we provide the first such example where chemokines activity was interrogated systematically with NU domain ligands.
Results and discussion
N-unsubstituted and N-acetylated HS analog library with regioselective sulfate modifications at 2-O (IdoA), 3-O (GlcN), and 6-O (GlcN) were obtained by single tetrasaccharide 27 and its N-acetate counterpart 28 (Scheme 1) using the divergent synthetic approach. Synthesis of 27 required a carefully designed technique that allowed us to do selective site modifications along the tetrasaccharide backbone in a controlled manner. Efforts led by various research groups revolutionized heparin sulfate synthesis in the past decade.13 Using a similar strategy as Hung13het al. with slight modifications in the protecting group, synthesis of 27 was carried out by disaccharide building blocks 21 and 18. Notably, the use of 4-O-chloroacetate (ClA) at non reducing GlcN residue as a temporary protecting group in 17 was found to be vital for chain elongation than other previously reported 4-O protecting groups such as Lev13f/Fmoc13m/TCA.13r Disaccharides (21 and 18) and monosaccharide precursors were synthesized from glucosamine and iduronic acid building blocks 14, 16, 19 & 20, as previously described.13h,14 Next, we adopted [2 + 2] glycosylation approach with 21 (glycosyl donor) and 18 (glycosyl acceptor) to obtain 1,6 anhydrous tetrasaccharide 22 in excellent yield. Acetolysis of the reducing end IdoA residue of 22 with the aid of acetic anhydride and copper trifluoromethanesulfonate as catalyst followed by phenyl trimethylsulfide and ZnI2 treatment afforded 24 as a thiophenol glycosyl donor. Linker glycosylation of 24, followed by sequential deacetylation and TEMPO mediated oxidation of 25, yielded 27 (72% for three steps). Finally, C-2 azide of glucosamine moieties in 27 was converted into acetate in the presence of Zn/AcOH/Ac2O to develop tetrasaccharide 28, which was further used to synthesize HS analogs with N-acetate backbone (Scheme 1). | ||
Scheme 1 (a) (i) PTSA, CH2Cl2/MeOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), rt, 6 h; (ii) TBDPSCl, imidazole, DMAP, CH2Cl2, 0 °C, 12 h; (iii) (ClAc)2O, CH2Cl2/py (4:1), 0 °C, 20 min. (b) (i) BH3·THF, TMSOTf, CH2Cl2, 0 °C, 6 h; (ii) TBDPSCl, imidazole, DMAP, CH2Cl2, 0 °C, 12 h. (c) NIS, TMSOTf, 4 Å MS, −78 °C to −20 °C, CH2Cl2, 30 min. (d) Thiourea, MeOH/py (1:1), 80 °C, 1 h. (e) NIS, TMSOTf, 4 Å MS -10 °C, CH2Cl2, 30 min. (f) Ac2O, Cu(OTf)2, rt, 12 h. (g) TMSSPh, ZnI2, CH2Cl2, rt, 2 h. (h) Benzyl (3-hydroxypropyl)carbamate, NIS, TfOH, 4 Å MS, rt, CH2Cl2, 30 min. (i) NaOMe, CH2Cl2/MeOH (1:1), rt, 12 h. (j) TEMPO, CH2Cl2/MeOH (1:1), rt, 12 h. (k) Zn, THF/AcOH/Ac2O (3:2:2), rt, 12 h. :2), rt, 6 h; (ii) TBDPSCl, imidazole, DMAP, CH2Cl2, 0 °C, 12 h; (iii) (ClAc)2O, CH2Cl2/py (4:1), 0 °C, 20 min. (b) (i) BH3·THF, TMSOTf, CH2Cl2, 0 °C, 6 h; (ii) TBDPSCl, imidazole, DMAP, CH2Cl2, 0 °C, 12 h. (c) NIS, TMSOTf, 4 Å MS, −78 °C to −20 °C, CH2Cl2, 30 min. (d) Thiourea, MeOH/py (1:1), 80 °C, 1 h. (e) NIS, TMSOTf, 4 Å MS -10 °C, CH2Cl2, 30 min. (f) Ac2O, Cu(OTf)2, rt, 12 h. (g) TMSSPh, ZnI2, CH2Cl2, rt, 2 h. (h) Benzyl (3-hydroxypropyl)carbamate, NIS, TfOH, 4 Å MS, rt, CH2Cl2, 30 min. (i) NaOMe, CH2Cl2/MeOH (1:1), rt, 12 h. (j) TEMPO, CH2Cl2/MeOH (1:1), rt, 12 h. (k) Zn, THF/AcOH/Ac2O (3:2:2), rt, 12 h. | ||
A divergent synthetic approach was followed to develop a combinatorial library of HS oligosaccharides. For instance, silyl protecting group TBDPS was deprotected selectively using 70% HF·py for 6-O-sulfate derivatives precursors (29 & 36). Similarly, NAP deprotection with the help of DDQ yielded 3-O-sulfated precursors (31 & 34). The lactone ring was first opened for the derivatives carrying 2-O-sulfated IdoA, followed by the benzyl esterification to yield 39 & 42 in moderate yield (Scheme 2). Subsequent, selective deprotection was carried out in a similar manner for oligosaccharides having multiple O-2,6 or O-2,3 sulfate modifications (45, 47 & 50). SO3·NEt3 was used for introducing sulfate group in the backbone (30, 32, 35, 37, 40, 43, 46, 48 & 51), whereas for the phosphate derivative (38 & 52) diphenylphsphoryl chloride (DPPC) was utilized. Finally, global deprotection of all the sulfated derivatives, including non-sulfated analogs (29 & 36) yielded desired HS tetrasaccharide 1–13 with the amine linker at the reducing for the generation of HS microarray. Final HS oligosaccharides and intermediates were characterized by 1H, 13C, DEPT and 31P NMR. Additionally, molecular weights for all the compounds were confirmed by high-resolution mass spectroscopy.
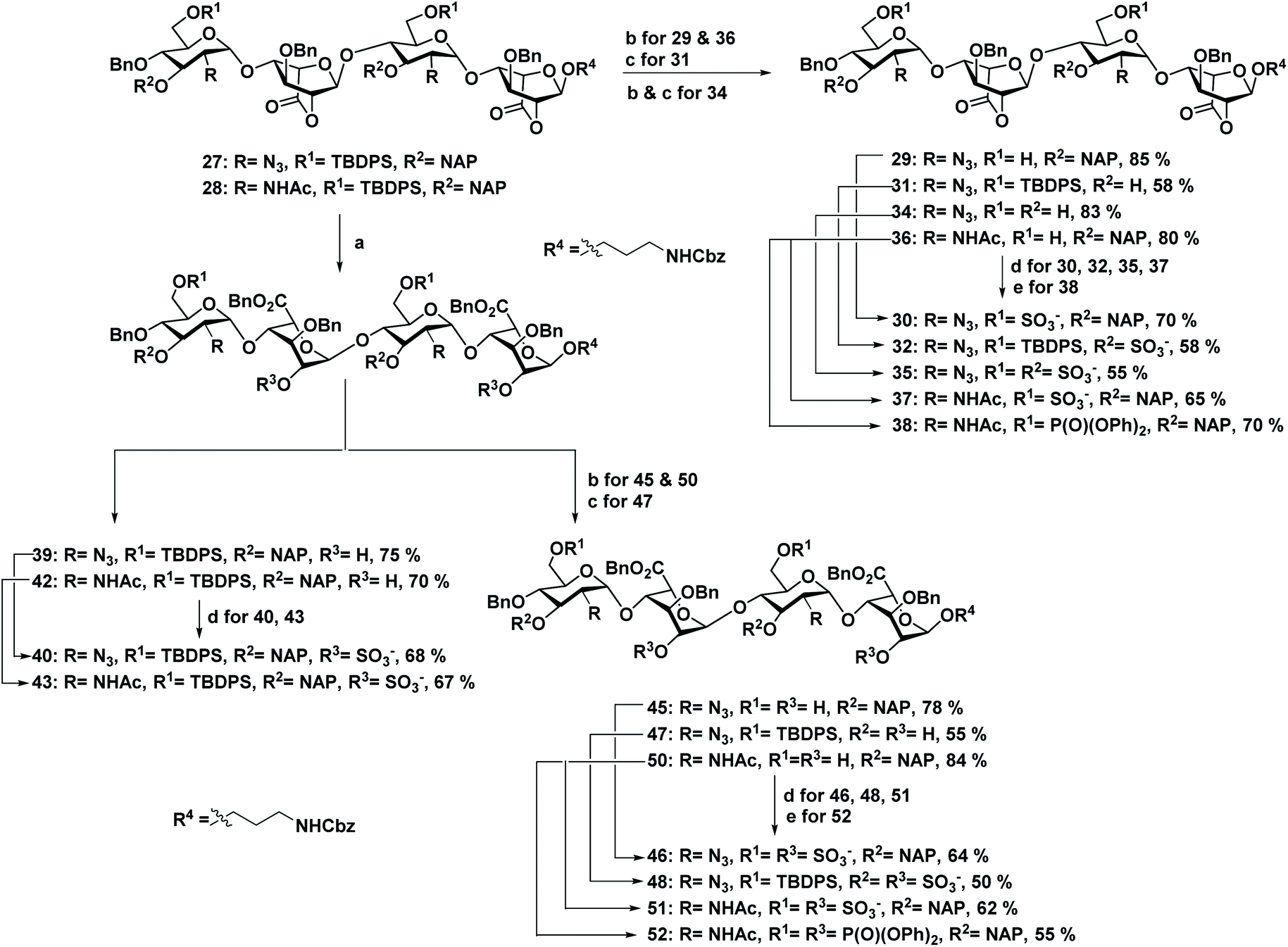 | ||
| Scheme 2 (a) (i) LiOH·H2O, THF/H2O (1:1), rt, 2 h; (ii) BnBr, TBAI, NaHCO3, DMF, 60 °C, 2 h. (b) 70% HF·py, py, 0 °C, 12 h. (c) DDQ, CH2Cl2/H2O (18:1), rt, 1 h. (d) SO3·NEt3, DMF, 60 °C, 72 h. (e) DPPC, DMAP, NEt3, CH2Cl2/py (1:1), 0 °C, 12 h. | ||
To unravel HS–chemokines binding patterns, heparin microarray was fabricated and examined with various biotinylated chemokines, at three different concentrations, followed by detection with Cy3-tagged streptavidin. We interrogated the binding of HS analogs on three homeostatic chemokines (CCL28, CXCL12 and CCL21) and six inflammatory chemokines [CXCL8 (IL-8), CXCL10 (IP-10), CCL2 (MCP-1) CCL7 (MCP-3), CCL13 (MCP-4) and CCL5 (RANTES)]. To rationalize the binding patterns of oligosaccharides, each HS–chemokines interaction was ranked according to percentage of maximal binding. Based on ranking and chemokine binding patterns, they were segregated into four groups (A, B, C and D) (Fig. 2a).
 | ||
| Fig. 2 Chemokine glycan microarray binding assay. (a) Arrays were fabricated on epoxide-activated slides as described.16 Binding was tested at 3 serial dilutions, then detected with the relevant biotinylated secondary antibody (1 μg ml−1) followed by Cy3-streptavidin (1.5 μg ml−1) (Table S1†). Arrays were scanned, relative fluorescent units (RFU) obtained, and maximum RFU determined and set as 100% binding. Then rank binding (per printed glycan per concentration, per each chemokine dilution, per printed block) was determined. Since each glycans was printed at 2 concentration, 100% binding was set separately for each concentration. Then, binding to all the other glycans at the same concentration was ranked in comparison to the maximal binding, and the average rank binding and SEM for each glycan across the two glycan concentrations and three examined dilutions of each chemokine was calculated (n = 6; 2 glycan concentrations, across 3 chemokine dilutions). This analysis allowed to compare the glycan binding profiles of the different chemokines and dissect their binding preferences. The mean rank is shown as a heat map of all the examined binding assays together (red highest, blue lowest and white 50th percentile of ranking). (b) Sulfate groups were added or deleted from the ligand in the crystal structure of the co-complex of a heparin trisaccharide with CCL5 (PDB ID: 5DNF, Chain I) using Chimera (UCSF Chimera – a visualization system for exploratory research and analysis).17 No other changes to the orientation of the ligand or protein were made. Based on glycan microarray screening, binding to CCL5 was high for GAG fragments HT-2,6-NH and HT-3,6S-NH (red), but low to HT-2,3S-NH (blue), consistent with expectations based on the co-complex crystal structure models. | ||
In this analysis, CCL13 (an inflammatory chemokine), CXCL12 and CCL21 (homeostatic chemokine) shared several of the conserved binding patterns, which suggest that these chemokines share several homologous binding pockets. For instance, group A chemokines bind to non-sulfated HT-0S-NH and phosphorylated ligands (HT-6,2P and HT-6P) (Fig. 2a), suggesting that the HS-based structure–activity relationship of these chemokines do not solely depend on the sulfate group. Moreover, group A chemokines showed strong binding with di-sulfated analogs such as HT-2S-NH and HT-6S-NAc. However, its respective NU and NA counterpart (HT-6S-NH and HT-2S-NAc) displayed weak binding (Fig. 2a), illustrating that NU and NA domains display switchable binding patterns via sulfation codes. It is noted that highly sulfated HS ligands displayed moderate to strong binding regardless of sulfation pattern or NU/NA domains. These trends clearly demonstrate that group A chemokines are sensitive to di-sulfation codes, while the highly sulfated HS ligands may improve the binding strength, but with poor selectivity.
In group B, CCL28 chemokine displayed weak binding with non-sulfated analogs (ranked 36% for HT-0S-NH and 22% HT-0S-NAc) and moderate to strong binding with sulfated ligands. Unlike, group A chemokines, CCL28 displayed poor binding with 2-O-sulfated NU ligand (ranked 43% for HT-2S-NH). Whereas, HT-6S-NAc (ranked 79%) and HT-3S-NH (ranked 62%) di-sulfated ligands, H-2,6S-NH (ranked 79%), HT-2,6-NAc (ranked 66%), HT-3,6S-NH (ranked 54%) tetra-sulfated ligands and HT-2,6P (ranked 78%) phosphate ligand displayed moderate to strong binding (Fig. 2a). These results suggest that group A and group B chemokine binding patterns may require a more complex HS library to establish selectivity.
In group C, CCL7 and CXCL10 displayed a sulfation pattern-based binding. Unlike group A and group B chemokines, CCL7 and CXCL10 displayed weak binding with non-sulfated and phosphate HS ligands (ranked between 2–56%). Among six di-sulfated ligands, only HT-3S-NH (ranked 78% for CCL7 and 65% for CXCL10) and HT-6S-NAc (ranked 73% for CCL7) displayed strong binding as compared to the other analogs. Similarly, among four tetra-sulfated HS ligand, HT-2,3S-NH (ranked 35% for CCL7 and 27% for CXCL10) displayed weak binding, whereas HT-2,6S-NH (ranked 91% for CCL7 and 89% for CXCL10), HT-2,6S-NAc (ranked 86% for CCL7 and 64% for CXCL10) and HT-3,6S-NH (ranked 89% for CCL7 and 96% for CXCL10) ligands displayed strong binding (Fig. 2a). These results illustrate that group C chemokines have some selectivity to di-sulfated ligands. Larger HS-disulfated library could further allow to fine-tune their binding patterns characteristics.
Finally, all members of D group chemokines (CXCL8, CCL5 and CCL2) displayed exclusive strong binding to high-sulfated ligands and weak binding with di-sulfated ligands, nonsulfated or phosphorylated ligands. Among high-sulfate ligands, group D chemokines displayed high selectivity to NU domain over NA domain. Notably, CXCL8 and CCL2 displayed strong binding preference to HT-3,6S-NH (ranked 94% for CXCL8 and 95% for CCL2) ligand, whereas, CCL5 showed strong binding to HT-2,6S-NH (ranked 95%) ligand (Fig. 2a), suggesting that NU domain is highly significant in modulating these chemokines activities, particularly of group D chemokines. It had previously been shown that the interaction of heparin tetrasaccharides with CCL5 is modulated by sulfation pattern and pH, however these studies also emphasized the dynamic and often non-specific nature of the ionic GAG-protein contacts.15 Nevertheless, to provide further insights into the interactions between CCL5 and HT compounds of varying sulfation patterns, we used a co-crystal structure of CCL5 co-complexed with a heparin trisaccharide (PDB ID: 5DNF, Chain I), in which sulfate groups were added or deleted from the ligand (Fig. 2b). This analysis revealed that sulfation at the 6 position in GlcNH is preferred over sulfation at the 3 position, because the 6S group can interact with both R59 and K55, whereas the 3S only interacts with R59. For this reason, both GlcNH[6S]-IdoA[2S]-GlcNH[6S] and GlcNH[3S,6S]-IdoA-GlcNH[3S,6S] (related to HT-2,6S-NH and HT-3,6S-NH, ranked 95% and 82% by glycan microarrays, respectively) are stronger binders than GlcNH[3S]-IdoA[2S]-GlcNH[3S] (related to HT-2,3S-NH, ranked 4%) (Fig. 2a).
To quantitatively evaluate the binding patterns between HS ligands and chemokines, SPR experiment was performed with H-3,6S-NH and chemokines (CXCL10, CXCL8, CCL5 and CCL2), which showed strong selective and sensitive binding in microarray experiments. The equilibrium binding constants (KD) measured from steady state fits are listed in Table S2.†HT-3,6S-NH displayed strong binding with inflammatory chemokine CCL2 (1.89 μM) (Fig. 3a–d). This strong binding is attributed to the fast association (Kon) as compared to chemokine–CCL2 interaction. In contrast, HT-2,6SNH displayed strong binding to CCL5 (2.34 μM) (Fig. 3a–d). Interestingly, CXCL8, CCL7 and CXCL10 showed weak binding constants (10–15 μM) for both NU domain ligands (Fig. S2†). Furthermore, SPR analysis of CCL5 and CCL2 displayed 3-fold stronger binding with H-3,6S-NH and HT-2,6S-NH ligands, respectively. Thus, for the first time, we were able to identify key NU domain sulfation pattern that can modulate chemokines activity.
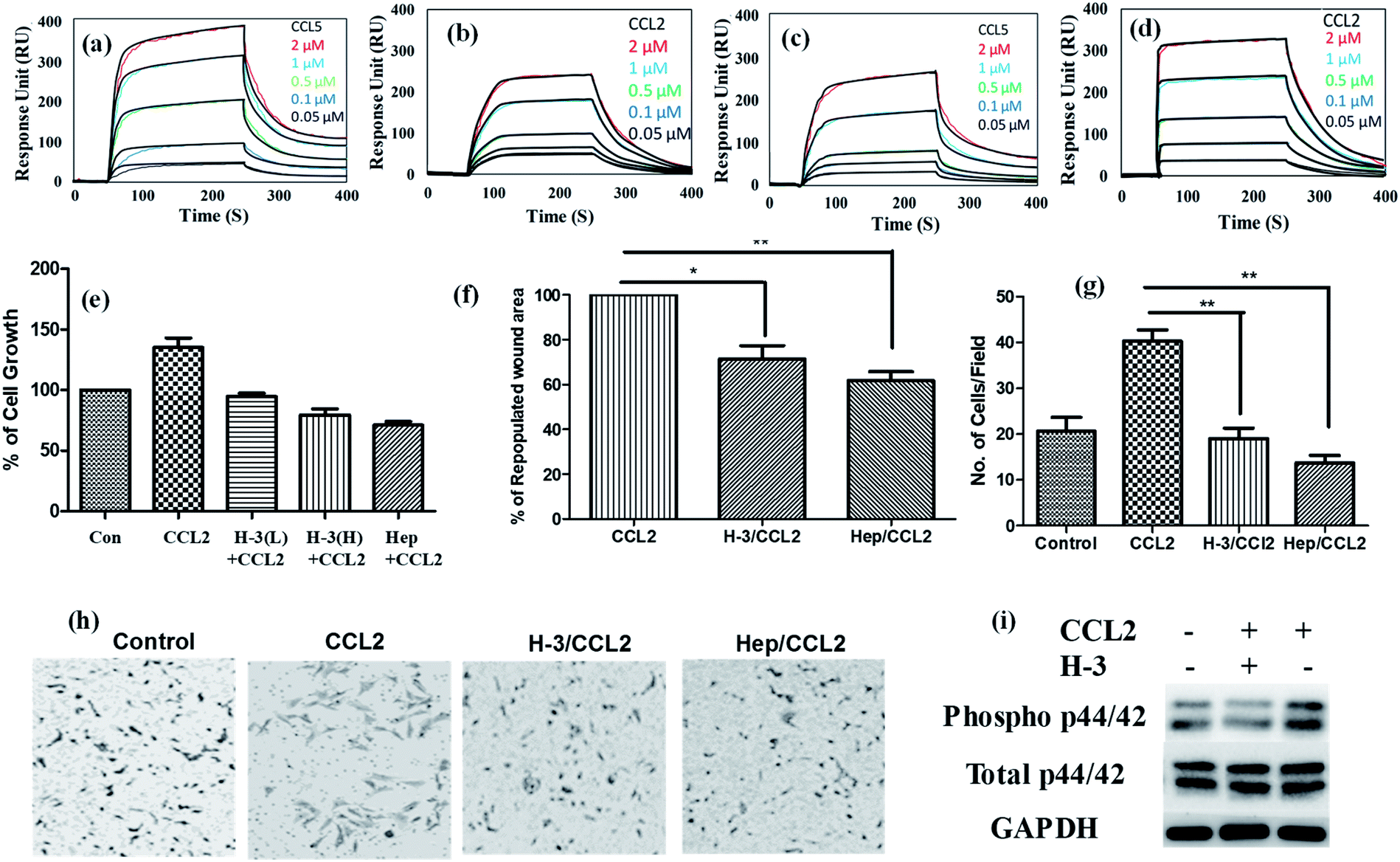 | ||
| Fig. 3 SPR analysis of chemokines binding profile on sensor chip having HT ligands: (a & b) SPR binding analysis of the interaction between HT-2,6S-NH with CCL5 and CCL2 respectively; (c & d) SPR binding analysis of the interaction between HT-3,6S-NH with CCL5 and CCL2, respectively. Concentrations of chemokines were 0.05–2 μM. A global fit according to a 1:1 binding model was applied (black curves); (e) MCF-7 cell proliferation was quantified by WST assay after 72 h treatment with HT-3,6S-NH (H-3) at different concentration with CCL2 chemokine. The bar graphs indicated percentage of cell growth. L corresponds to 10 μg ml−1 concentration; H corresponds to 50 μg ml−1 concentration of ligands ((H-3) and Heparin (Hep)). CCL2 chemokines (50 ng); (f) cell migration assay: area repopulated in 8 h with CCL2 chemokine is considered as 100% wound heal and data expressed as mean ± SD (n = 3; *P < 0.05, *P < 0.01); (g) Boyden chamber assay was performed in presence of HT-3,6S-NH and Hep (50 μg ml−1) with or without CCL2 (50 ng); (h) bright field images of Boyden chamber assay; (i) MAPK pathway analysis: MCF-7 cells were treated with CCL2 (50 ng) with or without HT-3,6S-NH (H-3) ligands (50 μg ml−1) and cell lysate was prepared at 30 min time points and P-p44/42 and total p44/42 was imaged. | ||
Given the high affinity binding of HT-3,6S-NH to CCL2 chemokines, and the link between CCL2 and cancer metastasis, investigating inhibitor effect on CCL2 cancer cell signaling is considered a novel approach to demonstrate the therapeutic potential of HS mimetics.18 To this end, we first studied cancer cells proliferation in the presence of HT-3,6S-NH (H-3) ligand and CCL2. Native heparin was used as a positive control. Cell proliferation was analyzed by WST assay using MCF-7 cell line, as they express high level of the CCL2 specific chemokine receptor (CCR2).19 It was observed that high concentration of HT-3,6S-NH ligand moderately inhibited cell proliferation (Fig. 3e). To understand the mechanism of inhibition, we performed cell-cycle analysis in the presence and absence of HS ligand and CCL2 chemokine, then quantified the DNA content of each cell state by flow-cytometry. The cell cycle analysis clearly revealed that addition of CCL2 induced S and G2/M phase cell-cycles, while high concentration of HT-3,6S-NH and heparin reduced G2/M state from 15% to 10%, indicating that HT-3,6S-NH moderately to poorly activate the cell cycle. Further studies with HT-3,6S-NH multivalent probes are ideal for modulating cancer cell proliferation.
We next examined cell migration by wound healing assay (Fig. 3f) and by Boyden-chamber assay (Fig. 3g and h). Addition of HT-3,6S-NH ligand reduced chemokine activity, where a 24% reduction in cell migration rate and 41% reduction in the wound healing was observed. In addition, a substantial reduction in cell migration was observed in the Boyden chamber assay. Finally, the mechanism of invasiveness was examined further by analyzing the level of phosphorylation of MAP kinase pathway. Western blot analysis of p42/44 showed that MCF-7 cells treated with HT-3,6S-NH/CCL2 expressed low level of MAPK compared to CCL2 treated cells (Fig. 3i). Overall, these results suggest that HT-3,6S-NH is a potential ligand that can modulate CCL2 chemokine activities.
Conclusions
Here, we describe the divergent synthesis of 13 new HS ligands, displaying different sulfate/phosphate patterns with NU/NA glucosamine residues. The binding interactions between HS ligands and chemokines on nano-printed microarray platform displayed several cryptic binding pockets for sulfation patterns with NU domain, which was not identified with previous HS synthetic ligands. Among them, HT-3,6S-NH ligand displayed a marked selectivity and sensitivity to CCL2 chemokine. The biological relevance of such structural binding studies was illustrated by incubating HT-3,6S-NH ligand with cancer cells showing the HS ligand inhibited cancer cells proliferation, migration and invasion. Thus, NU domain is important to regulate specific chemokine biological activities, thereby demonstrating potential novel therapeutic applications of HS ligands. To the best of our knowledge, we have identified CCL2 and CCL5 chemokines as only the fourth and fifth proteins currently known to recognize NU-domain HS ligands with different sulfation patterns.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Financial support from the IISER, Pune, DST (Grant No. SR/NM/NS-1113/2016), DBT (Grant No. BT/PR21934/NNT/28/1242/2017, STARS/APR2019/CS/426/FS, and SERB/F/9228/2019-2020) are gratefully acknowledged (to R. K.). This work was supported by the Israeli Science Foundation (ISF; to V. P.-K.). We would also like to thank Prof. John Gallagher for his contribution to heparin chemistry. This work is dedicated to Prof. J. D. Esko for his contribution to heparan sulfate glycobiology.Notes and references
- (a) A. Mantovani, R. Bonecchi and M. Locati, Nat. Rev. Immunol., 2006, 6, 907–918 CrossRef CAS PubMed; (b) C. R. Mackay, Nat. Immunol., 2001, 2, 95–101 CrossRef CAS PubMed; (c) R. J. Nibbs and G. J. Graham, Nat. Rev. Immunol., 2013, 13, 815–829 CrossRef PubMed; (d) B. Moser and P. Loetscher, Nat. Immunol., 2001, 2, 123–128 CrossRef CAS PubMed; (e) A. Mortier, J. Van Damme and P. Proost, Immunol. Lett., 2012, 145, 2–9 CrossRef CAS PubMed.
- (a) K. Jöhrer, L. Pleyer, A. Olivier, E. Maizner, C. Zelle-Rieser and R. Greil, Expert Opin. Biol. Ther., 2008, 8, 269–290 CrossRef PubMed; (b) W. G. Liang, C. G. Triandafillou, T.-Y. Huang, M. M. L. Zulueta, S. Banerjee, A. R. Dinner, S.-C. Hung and W.-J. Tang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 5000–5005 CrossRef CAS PubMed; (c) D. P. Dyer, C. L. Salanga, B. F. Volkman, T. Kawamura and T. M. Handel, Glycobiology, 2016, 26, 312–326 CAS; (d) L. Wang, M. Fuster, P. Sriramarao and J. D. Esko, Nat. Immunol., 2005, 6, 902–910 CrossRef CAS PubMed; (e) C. R. Parish, Nat. Rev. Immunol., 2006, 6, 633–643 CrossRef CAS PubMed.
- (a) A. E. Proudfoot, T. M. Handel, Z. Johnson, E. K. Lau, P. LiWang, I. Clark-Lewis, F. Borlat, T. N. Wells and M. H. Kosco-Vilbois, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1885–1890 CrossRef CAS PubMed; (b) Z. Johnson, A. Proudfoot and T. Handel, Cytokine Growth Factor Rev., 2005, 16, 625–636 CrossRef CAS PubMed.
- K. M. Sepuru and K. Rajarathnam, J. Biol. Chem., 2019, 294, 15650–15661 CrossRef CAS PubMed.
- S. E. Stringer and J. T. Gallagher, J. Biol. Chem., 1997, 272, 20508–20514 CrossRef CAS PubMed.
- D. Spillmann, D. Witt and U. Lindahl, J. Biol. Chem., 1998, 273, 15487–15493 CrossRef CAS PubMed.
- G. C. Jayson, S. U. Hansen, G. J. Miller, C. L. Cole, G. Rushton, E. Avizienyte and J. M. Gardiner, Chem. Commun., 2015, 51, 13846–13849 RSC.
- G. J. Sheng, Y. I. Oh, S.-K. Chang and L. C. Hsieh-Wilson, J. Am. Chem. Soc., 2013, 135, 10898–10901 CrossRef CAS PubMed.
- J. L. de Paz, E. A. Moseman, C. Noti, L. Polito, U. H. von Andrian and P. H. Seeberger, ACS Chem. Biol., 2007, 2, 735–744 CrossRef CAS PubMed.
- C. Zong, A. Venot, X. Li, W. Lu, W. Xiao, J.-S. L. Wilkes, C. L. Salanga, T. M. Handel, L. Wang, M. A. Wolfert and G. J. Boons, J. Am. Chem. Soc., 2017, 139, 9534–9543 CrossRef CAS PubMed.
- (a) T. Toida, H. Yoshida, H. Toyoda, I. Koshiishi, T. Imanari, R. E. Hileman, J. R. Fromm and R. J. Linhardt, Biochem. J., 1997, 322, 499–506 CrossRef CAS PubMed; (b) C. Westling and U. Lindahl, J. Biol. Chem., 2002, 277, 49247–49255 CrossRef CAS PubMed.
- (a) C. Vanpouille, A. Deligny, M. Delehedde, A. Denys, A. Melchior, X. Liénard, M. Lyon, J. Mazurier, D. G. Fernig and F. Allain, J. Biol. Chem., 2007, 282, 24416–24429 CrossRef CAS PubMed; (b) Z. Wei, M. Lyon and J. T. Gallagher, J. Biol. Chem., 2005, 280, 15742–15748 CrossRef CAS PubMed; (c) Z. Wei, J. A. Deakin, B. S. Blaum, D. Uhrín, J. T. Gallagher and M. Lyon, Glycoconjugate J., 2011, 28, 525–535 CrossRef CAS PubMed; (d) A. Koenig, K. Norgard-Sumnicht, R. Linhardt and A. Varki, J. Clin. Invest., 1998, 101, 877–889 CrossRef CAS PubMed; (e) K. Norgard-Sumnicht and A. Varki, J. Biol. Chem., 1995, 270, 12012–12024 CrossRef CAS PubMed.
- (a) M. Mende, C. Bednarek, M. Wawryszyn, P. Sauter, M. B. Biskup, U. Schepers and S. Bräse, Chem. Rev., 2016, 116, 8193–8255 CrossRef CAS PubMed; (b) W. Lu, C. Zong, P. Chopra, L. E. Pepi, Y. Xu, I. J. Amster, J. Liu and G. J. Boons, Angew. Chem., Int. Ed., 2018, 57, 5340–5344 CrossRef CAS PubMed; (c) S. U. Hansen, G. J. Miller, M. J. Cliff, G. C. Jayson and J. M. Gardiner, Chem. Sci., 2015, 6, 6158–6164 RSC; (d) Y. P. Hu, S. Y. Lin, C. Y. Huang, M. M. L. Zulueta, J. Y. Liu, W. Chang and S. C. Hung, Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed; (e) X. Zhang, V. Pagadala, H. M. Jester, A. M. Lim, T. Q. Pham, A. M. P. Goulas, J. Liu and R. J. Linhardt, Chem. Sci., 2017, 8, 7932–7940 RSC; (f) C. Noti, J. L. de Paz, L. Polito and P. H. Seeberger, Chem. - Eur. J., 2006, 12, 8664–8686 CrossRef CAS PubMed; (g) J. L. de Paz, C. Noti and P. H. Seeberger, J. Am. Chem. Soc., 2006, 128, 2766–2767 CrossRef CAS PubMed; (h) Y.-P. Hu, Y.-Q. Zhong, Z.-G. Chen, C.-Y. Chen, Z. Shi, M. M. L. Zulueta, C.-C. Ku, P.-Y. Lee, C.-C. Wang and S.-C. Hung, J. Am. Chem. Soc., 2012, 134, 20722–20727 CrossRef CAS PubMed; (i) N. J. Pawar, L. Wang, T. Higo, C. Bhattacharya, P. K. Kancharla, F. Zhang, K. Baryal, C. X. Huo, J. L. Jian, R. J. Linhardt, X. H. Xuefei and L. C. Hsieh-Wilson, Angew. Chem., Int. Ed., 2019, 58, 18577–18583 CrossRef CAS PubMed; (j) R. S. Boothello, A. Sarkar, V. M. Tran, T. K. Nguyen, N. V. Sankaranarayanan, A. Y. Mehta, A. Alabbas, S. Brown, A. Rossi, A. C. Joice, C. P. Mencio, M. V. Quintero, B. Kuberan and U. R. Desai, ACS Chem. Biol., 2015, 10, 1485–1494 CrossRef CAS PubMed; (k) N. V. Sankarayanarayanan, T. R. Strebel, R. S. Boothello, K. Sheerin, A. Raghuraman, F. Sallas, P. D. Mosier, N. D. Watermeyer, S. Oscarson and U. R. Desai, Angew. Chem., Int. Ed., 2017, 56, 2312–2317 CrossRef CAS PubMed; (l) L. Sun, P. Chopra and G.-J. Boons, J. Org. Chem., 2020, 85(24), 16082–16098 CrossRef CAS PubMed; (m) S. Arungundram, K. Al-Mafraji, J. Asong, F. E. Leach III, I. J. Amster, A. Venot, J. E. Turnbull and G.-J. Boons, J. Am. Chem. Soc., 2009, 131, 17394–17405 CrossRef CAS PubMed; (n) N. V. Sankaranarayanan, T. R. Strebel, R. S. Boothello, K. Sheerin, A. Raghuraman, F. Sallas, P. D. Mosier, N. D. Watermeyer, S. Oscarson and U. R. Desai, Angew. Chem., Int. Ed., 2017, 56, 2312–2317 CrossRef PubMed; (o) C. H. Chang, L. S. Lico, T. Y. Huang, S. Y. Lin, C. L. Chang, S. D. Arco and S. C. Hung, Angew. Chem., Int. Ed., 2014, 126, 10034–10037 CrossRef; (p) T. N. Laremore, F. Zhang, J. S. Dordick, J. Liu and R. J. Linhardt, Curr. Opin. Chem. Biol., 2009, 13, 633–640 CrossRef CAS PubMed; (q) S. B. Dulaney and X. Huang, Adv. Carbohydr. Chem. Biochem., 2012, 67, 95–136 CrossRef CAS PubMed; (r) S. U. Hansen, G. J. Miller, G. C. Jayson and J. M. Gardiner, Org. Lett., 2013, 15, 88–91 CrossRef CAS PubMed.
- (a) C. D. Shanthamurthy and R. Kikkeri, Eur. J. Org. Chem., 2019, 2019, 2950–2953 CrossRef CAS; (b) S. Anand, S. Mardhekar, R. Raigawali, N. Mohanta, P. Jain, C. D. Shanthamurthy, B. Gnanaprakasam and R. Kikkeri, Org. Lett., 2020, 22, 3402–3406 CrossRef CAS PubMed.
- A. Singh, W. C. Kett, I. C. Severin, I. Agyekum, J. Duan, I. J. Amster, A. E. Proudfoot, D. R. Coombe and R. J. Woods, J. Biol. Chem., 2015, 290, 15421–15436 CrossRef CAS PubMed.
- (a) V. Padler-Karavani, X. Song, H. Yu, N. Hurtado-Ziola, S. Huang, S. Muthana, H. A. Chokhawala, J. Cheng, A. Verhagen and M. A. Langereis, J. Biol. Chem., 2012, 287, 22593–22608 CrossRef CAS PubMed; (b) S. L. Ben-Arye, H. Yu, X. Chen and V. Padler-Karavani, J. Visualized Exp., 2017, e56094 Search PubMed; (c) M. Gade, C. Alex, S. L. Ben-Arye, J. T. Monteiro, S. Yehuda, B. Lepenies, V. Padler-Karavani and R. Kikkeri, Chembiochem, 2018, 19, 1170–1177 CrossRef CAS PubMed; (d) C. D. Shanthamurthy, P. Jain, S. Yehuda, J. T. Monteiro, S. L. Ben-Arye, B. Subramani, B. Lepenies, V. Padler-Karavani and R. Kikkeri, Sci. Rep., 2018, 8, 1–7 CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS.
- Y. Lu, Z. Cai, G. Xiao, Y. Liu, E. T. Keller, Z. Yao and J. Zhang, J. Cell. Biochem., 2007, 101, 676–685 CrossRef CAS.
- P. Dutta, M. Sarkissyan, K. Paico, Y. Wu and J. V. Vadgama, Breast Cancer Res. Treat., 2018, 170, 477–486 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05862a |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2021 |