
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe dome of gold nanolized for catalysis†
Yao
Peng
,
Cheng
Shang
 * and
Zhi-Pan
Liu
*
* and
Zhi-Pan
Liu
*
Collaborative Innovation Center of Chemistry for Energy Material, Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Key Laboratory of Computational Physical Science, Department of Chemistry, Fudan University, Shanghai 200433, China. E-mail: cshang@fudan.edu.cn; zpliu@fudan.edu.cn
First published on 8th March 2021
Abstract
Gold is noble in bulk but turns out to be a superior catalyst at the nanoscale when supported on oxides, in particular titania. The critical thickness for activity, namely two-layer gold particles on titania, observed two decades ago represents one of the most influential mysteries in the recent history of heterogeneous catalysis. By developing a Bayesian optimization controlled global potential energy surface exploration tool with machine learning potential, here we determine the atomic structures of gold particles within ∼2 nm on a TiO2 surface. We show that the smallest stable Au nanoparticle is Au24 which is pinned on the oxygen-rich TiO2 and exhibits an unprecedented dome architecture made by a single-layer Au sheet but with an apparent two-atomic-layer height. Importantly, this has the highest activity for CO oxidation at room temperature. The physical origin of the high activity is the outstanding electron storage ability of the nano-dome, which activates the lattice oxygen of the oxide. The determined CO oxidation mechanism, the simulated rate and the fitted apparent energy barrier are consistent with known experimental facts, providing key evidence for the presence of both the high-activity Au dome and the low-activity close-packed Au particles in real catalysts. The future direction for the preparation of active and stable Au-based catalysts is thus outlined.
1. Introduction
Metal and metal oxide composites represent a classical type of heterogeneous catalyst, renowned for complex multi-element nanostructures at interfaces.1–4 Knowledge about the active sites of these catalysts, although of great significance to industry, is however largely speculative due to the lack of efficient tools to map out atoms at interfaces.5–7 Experimental studies face great challenges in achieving high local resolution of both geometry and electronic structures,8 whilst theory generally fails to give either high speed or high accuracy in exploring the potential energy surface (PES) of interfaces.9 Here we show that the latest machine learning (ML)-based atomic simulation can resolve the atomic structure of gold nanoparticles on TiO2 in low temperature CO oxidation, a well-known but puzzling example used to illustrate the phenomenological synergetic effect of composites in catalysis.10–12 By meticulously scanning gold particles with subnano to ∼2 nm in dimension, we identify a unique gold nano-dome of 0.62 nm in height anchored on oxygen-rich TiO2, which is not only the smallest stable particle but also the most active for low temperature CO oxidation.Oxide-supported Au catalysts exhibit attractive low temperature (down to 200 K) CO oxidation activity.13,14 Since the first report of Au/TiO2 by Haruta et al. in the 1980s,14,15 a large volume of literature, encompassing both experimental and theoretical studies, has been devoted to understanding its physical properties, focusing on the active site and the mechanism.16–22 Goodman's group laid a milestone for understanding the Au/TiO2 composite system,10 using surface science techniques to establish that the most active Au particles are nanosized and have a two-layer height (0.7 nm). However, Emmanuel et al. recently used a model catalyst experiment and only found an exponential activity increase with a reduction of particle diameter from 6 to 1.5 nm; there was no abrupt activity jump at a certain particle size.23 Indeed, because supported particles generally have a wide size distribution, it has not been possible to either confirm the critical thickness for activity or identify the true active site in supported gold catalysts.
Apart from the structural uncertainty, the CO oxidation mechanism is highly controversial, with uncertainty over whether or not the reaction follows the Mars–van Krevelen (M–vK) mechanism24 in which the lattice O of oxide takes part in CO oxidation. A recent experiment by Behm and co-workers showed the participation of atomic O (stable up to 353 K) in CO oxidation,25,26 indicating the dissociative adsorption of molecular O2. However, the atomic O from O2 may well become the interfacial O between Au and TiO2 (an interface Langmuir–Hinshelwood (LH) mechanism), rather than forming the lattice O, and thus there is no conclusive evidence for the occurrence of the M–vK mechanism. On the other hand, the past two decades have witnessed a large volume of theoretical studies using different models for Au/TiO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27–31 and the computed data for activity appears diverse. The CO oxidation barrier is reported to be 0.3 eV on Au18/TiO2 (110) with an LH mechanism,27,28 but it jumps to 0.8 eV for a supported Au20 particle with a M–vK mechanism.29,30 In contrast, using periodic Au stripe models on TiO2, the barrier turns out to be ∼0.5 eV where O2 dissociation is the rate determining factor.32,33 Since there is no quantitative consensus for the activity reported from different hypothetical models, the key to these puzzles boils down to the “true” atomic structure of the gold nanoparticles on TiO2 under the reaction conditions.
27–31 and the computed data for activity appears diverse. The CO oxidation barrier is reported to be 0.3 eV on Au18/TiO2 (110) with an LH mechanism,27,28 but it jumps to 0.8 eV for a supported Au20 particle with a M–vK mechanism.29,30 In contrast, using periodic Au stripe models on TiO2, the barrier turns out to be ∼0.5 eV where O2 dissociation is the rate determining factor.32,33 Since there is no quantitative consensus for the activity reported from different hypothetical models, the key to these puzzles boils down to the “true” atomic structure of the gold nanoparticles on TiO2 under the reaction conditions.
Recent years have seen an increasing number of atomic simulation applications with ML potential for structure determination.34–37 The applications are, however, generally limited to relatively simple systems (e.g. bulk with few elements) due to the great difficulty in generating reliable multiple-element potentials for structure prediction, which requires the good description of the global PES.38 Here a Bayesian-guided global optimization tool is developed to advance the simulation of complex catalytic reactions under realistic conditions, with the aim of efficient structure determination of a multi-element composite. The method utilizes a Bayesian optimization algorithm39 to guide the ML-based global PES search,40–43 in which the stochastic surface walking (SSW)40,41 global optimization with global neural network (G-NN) potential acts as the key tool for ML potential construction and global PES exploration.44–47 Using this Bayesian-guided SSW-NN search, we managed to reveal the global minima (GM) of gold particles up to Au52 on reduced, stoichiometric and oxidized TiO2 (110) surfaces. The smallest stable Au cluster, i.e. an Au24O4 dome made by a single-layer Au sheet but with an apparent two-atomic-layer height, is identified as the key component for catalyzing low temperature CO oxidation.
2. Methods and calculation details
2.1. Bayesian-guided SSW-NN simulation
The GM of all gold particles was obtained using our recently-developed SSW-NN method as implemented in LASP code (http://www.lasphub.com),48 in which the SSW-NN achieves fast global PES exploration. Fig. 1a illustrates two key steps for Bayesian-guided SSW-NN simulation. First the ML potential (the G-NN potential) is generated by the standard SSW-NN process via the iterative self-learning of the plane-wave density functional theory (DFT) global PES dataset generated from SSW exploration. There are three steps: (i) global dataset generation based on DFT calculations using selected structures from SSW simulation from random bulk and slab structures, (ii) G-NN potential fitting and (iii) SSW global optimization using the G-NN potential. These three steps are iteratively performed until the G-NN potential is transferable and robust enough to describe the global PES. | ||
| Fig. 1 AunOm/TiO2 structure determination using Au–Ti–O G-NN potential-based global optimization. (a) Illustration of the Bayesian-SSW-NN methodology for identifying the GM of AunOm/TiO2. (b) Example of AunOm/TiO2 utilized in the Bayesian-SSW-NN search. TiO2 is represented by a p(9 × 3) TiO2(110) surface (the side view is inset). (c) Global PES contour plot of AunOm/TiO2 systems containing 3 × 105 low energy minima from SSW-NN. The x axis is a distance-weighted Steinhart order parameter (OP), the y axis is the free formation energy (Gf) of AunOm and the color indicates the density of states for the minima. The GM of representative clusters are labelled in the map as black and red dots. (d) Gf of the GM of AunOm/TiO2 (n = 6, 12, 18, 24, 30, 35, 37 and 52). | ||
Second, Bayesian optimization is utilized to drive the iterative acquisition-surrogate procedure, i.e. predicting new GM structures by SSW-NN searches and updating the G-NN potential by learning thse GM structures. Bayesian optimization is terminated when the GM predicted from SSW-NN is confirmed after more than 5 Bayesian cycles. The procedure is standardized in LASP as described recently by Ma et al.49
We should emphasize that the structures that were iteratively added into the dataset during the G-NN potential generation, were always selected from SSW global optimization trajectories according to the structure and energy criteria. These structures either have distinct local structure as reflected by structure descriptors, or have a large energy deviation between NN and DFT results. To generate the AuTiO potential, we obtained 44096 structures after the standard SSW-NN procedure, and we further added 418 AuOx/TiO2 structures, including 187 Au24Ox/TiO2 structures, into the final stage of Bayesian optimization to search for AuOx particles on TiO2. The AuTiCO global dataset consisted of 54127 structures, including 53582 from the standard SSW-NN procedure, and 545 from Bayesian optimization. All structures in the dataset were calculated by plane–wave DFT calculation with average setups (also see Section 2.2 for details). The input files of these calculations to reproduce the training set can be found on the lasphub website.50 The AuTiO G-NN potential has a four-layer (198-50-50-1) feed-forward NN structure, with a total of 37753 fitting parameters. The root-mean-square (RMS) errors for the energy and the force of the G-NN are 6.3 meV per atom and 0.155 eV Å−1, respectively (see ESI† part 3 for more details).
2.2. DFT calculations
All DFT calculations were performed by using plane-wave DFT code, VASP,51,52 in which the electron–ion interaction is represented by the projector augmented wave (PAW) pseudopotential.51 The kinetic energy cutoff was 450 eV. The exchange functional GGA-PBE53 was used for training dataset generation. The CO oxidation mechanism exploration and the analyses of the projected density of states (p-DOS) and Bader charge were carried out using the spin-polarized GGA-PBE with the local Hubbard term U correction (PBE + U) (U = 5 eV) for the Ti 3d orbital.54 The dispersion effect on reactions was examined by using Grimme's D3 correction (PBE-D3)55 (see ESI, Table S4†), which shows that the inclusion of dispersion does not change the CO oxidation kinetics. The first Brillouin zone k-point sampling utilized the Monkhorst–Pack scheme with an automated mesh determined by 25 times the reciprocal lattice vectors for bulk, and only the Γ point for surface in the slab model [a large unit cell of 26.7 Å × 19.7 Å, corresponding to the p(9 × 3) rutile (110) surface]. The energy and force criteria for convergence of the electron density and structure optimization were set at 10−5 eV and 0.01 eV Å−1, respectively. All the transition states (TS) were located using the double-ended surface walking method (DESW).563. Results and discussions
3.1. Structure of supported Au particles
To map out the active site responsible for CO oxidation, we investigated a series of gold particles on titania, namely AunOm/TiO2 (illustrated in Fig. 1b) with Bayesian-guided SSW-NN, with each composition (by varying n and m) being visited by more than 105 minima. The numbers of Au atoms (“n” of AunOm) studied in this work were 6, 12, 17, 18, 20, 22, 24, 30, 31, 32, 35, 36, 37 and 52, and here only the most representative examples are reported in Fig. 1b for clarity. TiO2 was modelled as rutile TiO2 (110) with up to p(9 × 3) surface area, as illustrated in Fig. 1b. This corresponds to Au exposure from 8% to 71% ML (1 ML is 1.387 × 1015 Au atoms per cm2 corresponding to Au (111)). The shortest distance between adjacent Au particles was at least 9.38 Å (occurred in the largest Au52O4 particle), which is large enough to avoid lateral interactions between particles. The formal oxidation state of Au in AunOm particles was subject to the O2 chemical potential (see ESI Table S1†) and thus the amount of O (m value) in the simulation varied from −1 to 5 (−1 indicates an O vacancy near Au). The free formation energy of AunOm, Gf (eV per Au atom), could be determined by reference to the TiO2 stoichiometric surface, Au (111) surface and O2 at ambient conditions (eqn (S1) and (S2) in ESI†). In computing the free energy, we utilized the DFT total energy to approximate the free energy for solid states (AuOx/TiO2, TiO2) because the vibration entropy and the pV term contributions of solid phases are negligibly small; and we corrected the zero-point-energy, thermal and entropy contributions for gas phase O2 molecule at finite temperatures.57 While all structure candidates were obtained from SSW-NN, the low energy structures were always checked by DFT to achieve high accuracy, and all key energetics reported in this work are DFT results.In Fig. 1c, we show the global PES contour map of AunOm/TiO2 by plotting Gf against a distance-weighted Steinhart order parameter58,59 (OP) of all low energy minima. The smaller the OP value is, the more close-packed the Au particles will be. Fig. 1c shows clearly that the larger particles have lower Gf, indicating the sintering tendency of very small-sized particles (<20 atoms), as also observed in experiments.10,11 We note that the bare Au particles on the stoichiometric surface and the reduced surface (with oxygen vacancies) are the least stable. As shown in ESI Fig. S1,† the GMs of AunO−1/TiO2 and Aun/TiO2 (n = 12, 18 and 24) adopt the close-packed stick configuration. The stick grows in the groove between two protruding rows of bridge O (Obr) on TiO2(110) along [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0], interacting with the surface via eight Au–Obr bonds. For AunO−1/TiO2, the Au particles prefer to sit away from the surface Obr vacancies, which is consistent with the previous experimental observation.11 Importantly, the presence of additional O, i.e. forming O-containing AunOm particles (m > 1), not only stabilizes the particles, but also largely smoothens the Gf difference between particles. From Au18 to Au24, Gf drops by 0.05 eV, and it changes by only 0.007 eV from Au18O2 to Au24O2. In the bottom of the global PES, we identify Au24O4 and Au52O4, which are the two most stable particles with Gf values of −0.04 and −0.05 eV per Au, respectively. Our results indicate that subnano and nano Au particles can resist sintering (Gf < 0) as long as the TiO2 surface is O-rich, which is indeed possible under oxidation conditions.11 The thermodynamic stability of these particles is summarized in Fig. 1d for each size. We can see that with increasing particle size, more O atoms are required to stablize the particle. In all cases, these extra O near to Au (Oad) reside on the exposed five-coordinated Ti site (Ti5c, see Fig. 1b) at the Au/TiO2 interface, acting as the anchor to stabilize Au particles. In the following, we describe the GMs for three representative sizes.
0], interacting with the surface via eight Au–Obr bonds. For AunO−1/TiO2, the Au particles prefer to sit away from the surface Obr vacancies, which is consistent with the previous experimental observation.11 Importantly, the presence of additional O, i.e. forming O-containing AunOm particles (m > 1), not only stabilizes the particles, but also largely smoothens the Gf difference between particles. From Au18 to Au24, Gf drops by 0.05 eV, and it changes by only 0.007 eV from Au18O2 to Au24O2. In the bottom of the global PES, we identify Au24O4 and Au52O4, which are the two most stable particles with Gf values of −0.04 and −0.05 eV per Au, respectively. Our results indicate that subnano and nano Au particles can resist sintering (Gf < 0) as long as the TiO2 surface is O-rich, which is indeed possible under oxidation conditions.11 The thermodynamic stability of these particles is summarized in Fig. 1d for each size. We can see that with increasing particle size, more O atoms are required to stablize the particle. In all cases, these extra O near to Au (Oad) reside on the exposed five-coordinated Ti site (Ti5c, see Fig. 1b) at the Au/TiO2 interface, acting as the anchor to stabilize Au particles. In the following, we describe the GMs for three representative sizes.
Au6O in Fig. 2 has a structure typical of Au particles below 20 atoms, and is highly unstable with a Gf value of +0.3 eV. It grows in the groove between two protruding rows of bridge O (Obr) on TiO2 (110) along [10], anchored to the surface via Au–Obr bonds.10,60 The particle is a single-layer thick, but the Au atoms not linked with Obr are higher in position. The Au6O particle is 0.30 nm high and 0.44 nm in diameter. The Au coordination number (CN, Au–Au coordination shell) is very low, being only 4.25 on average (c.f. CN = 9 for Au in the Au (111) surface).
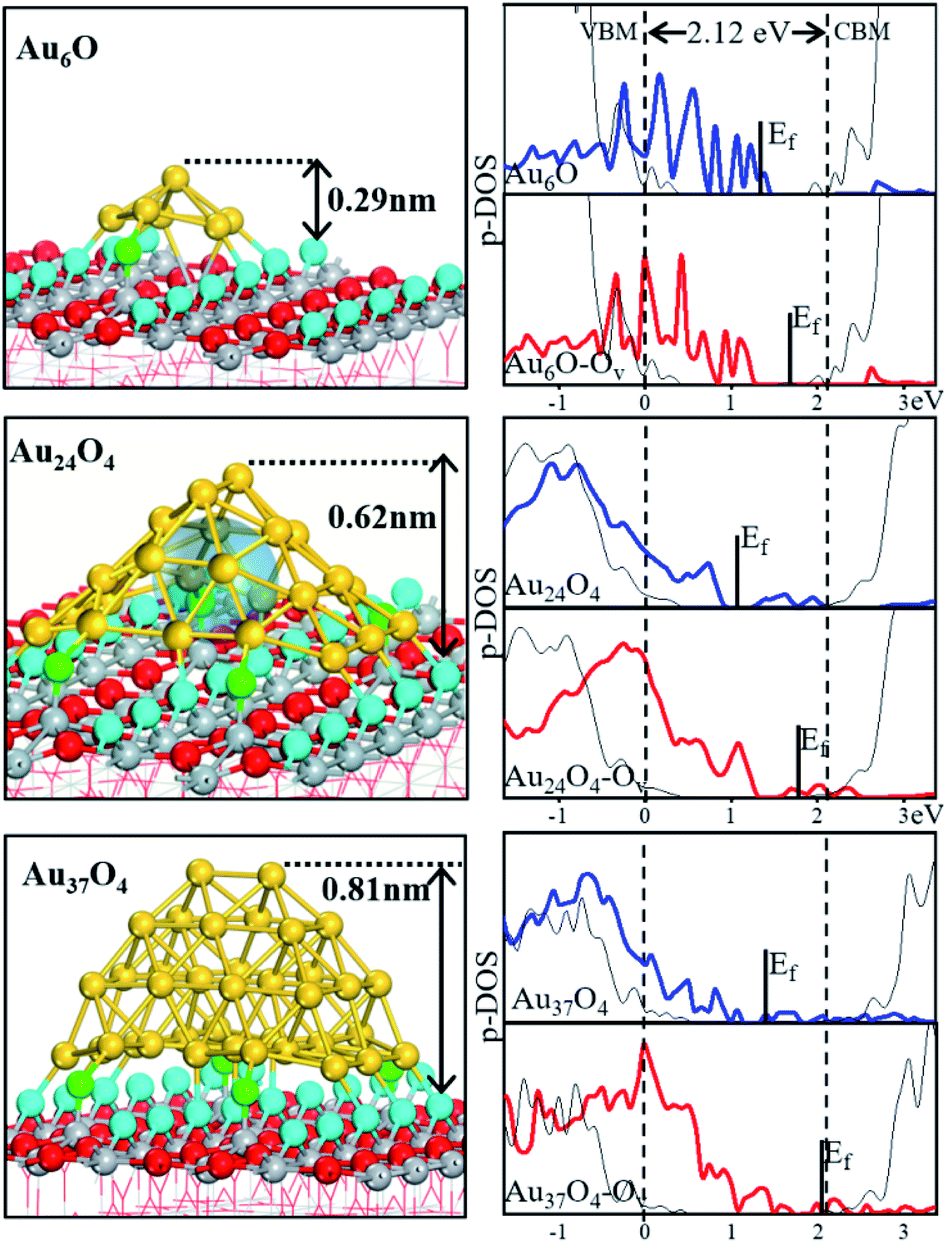 | ||
| Fig. 2 Geometrical structures and p-DOS of AunOm/TiO2. Geometrical structures of Au6O, Au24O4 and Au37O4 on TiO2 (110) (left panel) and the corresponding p-DOS for the 5d (Au) before (blue) and after (red) the formation of a surface O vacancy (right panel). All the DOSs are aligned via the Ti 3d states of the bulk TiO2. Ef indicates the Fermi level; the black lines represent the total DOS of bulk TiO2; the vertical dot lines indicate the valence band minimum (VBM) and CBM of bulk TiO2, respectively. The colour scheme of atoms is the same as that used in Fig. 1b, and the transparent ball represents the void space. | ||
Turning to Au24, the particle can hold four additional Oad atoms at the perimeter sites, i.e. Au24O4, and is thus much stabilized. The particle expands to cover three Obr rows, and the Au atoms form one highly curved two-dimensional sheet. Interestingly, the particle has a dome architecture with C2v symmetry and a large hollow void in between the Au sheet and TiO2 (see the transparent ball in Fig. 2). It reaches 0.62 nm in height and spans 1.16 nm in diameter. Due to its single-layer nature, the Au atoms still have low coordination numbers (CN = 4.7 on average) and thus locate at the bottom-right corner in the Fig. 1c global PES. We note that structures in this region (i.e. OP > 0.41) all have the dome structure, clearly different from close-packing configurations (OP < 0.2).
Further increasing the Au coverage gives Au particles that are not only thicker but also more close-packed. At Au37, the GM is Au37O4, which is 0.81 nm high but still 1.16 nm in diameter. Compared to Au24O4, the additional Au atoms fill in the void of the dome by first sealing the bottom to form a close-packed Au layer base (Au35O4, ESI Fig. S2†) and then occupying the middle cavity until Au37O4 forms (Fig. 2). By further increasing Au atoms, Au52O4 is eventually formed (ESI Fig. S2†), which is 1.3 nm in diameter and 1.01 nm in height, having a similar stability to Au24O4. Both Au37O4 and Au52O4 are fcc close-packed Au pyramids in which the average CN of Au in Au52O4 is 6.9, close to that for Au in the Au (111) surface (CN = 9).
3.2. Electronic structure of supported Au particles
The O-rich TiO2 surface not only helps to stabilize Au particles but also influences the nearby lattice O stability. We have computed the energy required to create one O vacancy (EOv) by removing the Obr nearest to the Au particles (ESI Table S2†). For small particles, e.g. Au6O to Au18O2, EOv remains similar to or is even higher than that for bare TiO2 (110) (2.36 eV). However, for the larger particles, i.e. Au24O4 and Au35O4, EOv reduces significantly to 1.66 eV and then it increases to 2.08 eV for Au37O4 and 2.21 eV for Au52O4. These results indicate that this Obr atom is significantly activated only when the Au particle is larger than 1 nm, especially when the void is present. Because the lattice Obr is a negatively charged anion, the creation of Ov on the TiO2 surface leads to the emergence of high energy electrons near Ov.11 The low-lying unoccupied states in Au, if present, should help to stabilize the Ov, through electron transfer from the surface to Au. Indeed, our Bader charge analyses confirm a significant charge transfer for Au24 and Au35 systems, much more than is found for Au6: the entire particle gains 0.02, 0.65 and 0.54e after Ov formation for Au6O, Au24O4 and Au37O4, respectively. To provide deeper insights, we therefore considered the electronic structures. In Fig. 2 (right panel), we have plotted the p-DOSs for the Au 5d states in three representative systems (Au6O, Au24O4 and Au37O4 on TiO2), both before and after the formation of a surface O vacancy, namely AunOm and AunOm–Ov. These p-DOSs (5d) reflect the different responses of the Au particles in creating Ov, as described below.In Au6O/TiO2, most Au 5d states are occupied and the remaining antibonding 5d states are far above the conduction band minimum (CBM) of TiO2. The Fermi level states are related to the delocalized bonding between Au and Obr. After the Obr removal, these Au 5d states diminish, indicating that the creation of Ov is at the cost of the Au–Obr interaction.
In Au24O4/TiO2 and Au37O4/TiO2, a significant number of low-lying unoccupied Au 5d states are present, i.e. below the CBM, before Ov creation, suggesting their capacity to store extra electrons. After Obr removal, there is an up-shift of the Fermi level that crosses the continuous Au 5d states in both systems. Obviously, this is the consequence of electron transfer from the oxide surface to Au, leading to electron accumulation on the Au 5d states. For Au37O4, the highest occupied molecular orbital (HOMO) of Au is relatively high, which is due to the fact that the close-packed morphology is similar to that of the bulk structure, and thus reduces the ability of the Au particles to receive electrons. Such results are consistent with the experimental observation that Au particles start to exhibit metallicity when the height is 0.6–1 nm.61 We note that these unoccupied 5d states of Au24O4 have aromatic bonding characteristics, exhibiting a highly delocalized nature throughout the whole particle (ESI Fig. S3†). They are similar to those in the stand-alone Au32 hollow cage, which also has aromatic 5d states (ESI Fig. S4†). Apparently, the delocalized 5d bonding is the key to stabilizing the dome structure, which is favored over the close-packed configuration in small Au particles (Au24, Au30 and Au37 investigated in this work).
3.3. CO oxidation reaction
With the elucidation of the atomic structure of supported Au particles, we are now at the position to explore CO oxidation activity. We constructed a four-element G-NN potential, Au–Ti–C–O, based on the dataset of the Au–Ti–O potential, to which a number of structure configurations for CO adsorption and CO oxidation in the AuTiO system were added from the self-learning SSW reaction sampling data. Because we limited the reaction to the CO oxidation at the Au–TiO2 interface, the training of the four-element potential was relatively facile, but it is tremendously helpful for exploring the possible CO oxidation channels. The detail of the construction of the AuTiCO potential can be found in the ESI.† Considering that the supported Au particles need to be sufficiently thermodynamically stable during catalysis, we focused on reactions on Au24O4 and Au37O4. Au24O4 is the smallest stable particle with void morphology while Au37O4 is the smallest close-packed structure representative of the larger fcc particles in general. The dynamic stability of these particles at finite temperatures was examined and is discussed later. Since CO oxidation occurs under an O2 atmosphere and thus the supply of interfacial O is guaranteed, the surface O vacancies should be filled readily during the steady state conversion.Utilizing the G-NN potential, we were able to explore all the likely reaction pathways on the supported catalysts, and the lowest energy pathways identified from the G-NN simulation were then confirmed by DFT calculations. The reaction mechanisms on the two supported Au particle types were found to be similar, both following the M–vK mechanism in which the lattice oxygen at the surface takes part in reacting with CO molecules to form CO2. Overall, the reaction can be divided into four key elementary steps:
(i) CO* + Obr → CO2 + Ov. An adsorbed CO (CO*) on interfacial Au reacts with the nearby lattice Obr.
(ii)  An O2 adsorbs at the Obr vacancy.
An O2 adsorbs at the Obr vacancy.
(iii)  The adsorbed O2 reacts with the second adsorbed CO, forming an OCOO intermediate.
The adsorbed O2 reacts with the second adsorbed CO, forming an OCOO intermediate.
(iv) OCOO → CO2 + Obr. The OCOO intermediate dissociates into CO2 and regenerates the catalyst.
We show in Fig. 3 the reaction free energy profiles for the best CO oxidation channel at 200 K and at 300 K, which occur at different Au particles, i.e. on Au37O4 at 200 K, and on Au24O4 at 300 K (both under 1 atm with CO 1% and O2 21%). The reaction profiles at the two temperatures thus highlight the fact that the particle size matters for the activity: Au24O4 is more active at ambient temperature, while the close-packed particles as represented by Au37O4 are active only at extremely low temperatures (i.e. ∼200 K). This is mainly because CO adsorption on Au37O4 is much weaker than that on Au24O4, leading to the temperature-dependence of the best reaction channel, as elaborated below. The gas phase free energy correction was made for CO and O2 and the zero-point energy for all the surface states were taken into account in deriving the free energy profiles.
 | ||
| Fig. 3 Minimum free energy profiles for CO oxidation on Au24O4/TiO2 at 300 K (red) and Au37O4/TiO2 at 200 K (blue), both under 1 atm (CO 1%, O2 21%). The snapshots of the three transition states (TS1, TS2 and TS3) on Au24O4 are shown in the lower panel, in which the key distances of the reactants are labelled with dotted lines. Au: yellow; C: gray; O in CO and O2: red; Oad: green; Obr: cyan. | ||
Taking CO oxidation on Au24O4 as an example, we illustrate how the reaction occurs. The most stable CO at the interfacial Au is at the Au site (Au* in Fig. 3) that is bonded with the lattice Obr ( in Fig. 3). Upon this adsorption, the Au atom moves outside along TiO2 [001] by ∼1 Å to maximally stabilize CO. The CO adsorption at this CO–Au–Obr configuration gains 1.10 eV in energy, and 0.46 eV in free energy (at 300 K and p(CO) = 0.01 atm). These results are consistent with the general knowledge that CO prefers to adsorb at the defective sites of Au and can reconstruct small Au particles. This adsorbed CO can react with its nearby Obr (next to the
in Fig. 3). Upon this adsorption, the Au atom moves outside along TiO2 [001] by ∼1 Å to maximally stabilize CO. The CO adsorption at this CO–Au–Obr configuration gains 1.10 eV in energy, and 0.46 eV in free energy (at 300 K and p(CO) = 0.01 atm). These results are consistent with the general knowledge that CO prefers to adsorb at the defective sites of Au and can reconstruct small Au particles. This adsorbed CO can react with its nearby Obr (next to the  ) of the oxide to produce the first CO2 with a barrier (Eb) of 0.57 eV (Fig. 3, TS1), in which the Obr–CO distance at the TS is 2.02 Å, which is quite typical for CO oxidation on metals. After the reaction, an Ov is created that is then filled by a gas-phase O2 with the free adsorption energy of 0.42 eV. The adsorbed O2 can react with a nearby adsorbed CO to form an OCOO complex by overcoming a low barrier of 0.31 eV (Fig. 3, TS2) and the reaction is exothermic by 0.32 eV. The dissociation of OCOO is also facile with a barrier of 0.34 eV (Fig. 3, TS3), after which a second CO2 is released and the catalyst is recovered.
) of the oxide to produce the first CO2 with a barrier (Eb) of 0.57 eV (Fig. 3, TS1), in which the Obr–CO distance at the TS is 2.02 Å, which is quite typical for CO oxidation on metals. After the reaction, an Ov is created that is then filled by a gas-phase O2 with the free adsorption energy of 0.42 eV. The adsorbed O2 can react with a nearby adsorbed CO to form an OCOO complex by overcoming a low barrier of 0.31 eV (Fig. 3, TS2) and the reaction is exothermic by 0.32 eV. The dissociation of OCOO is also facile with a barrier of 0.34 eV (Fig. 3, TS3), after which a second CO2 is released and the catalyst is recovered.
Compared to Au24O4, CO adsorption on Au37O4 is much weaker, which is due to the fact that the interfacial Au in Au37O4, (being close-packed) has a higher CN than that in Au24O4. Even at 200 K, CO adsorption on Au37O4 at the Au site analogous to that in Au24O4 is already endothermic by 0.12 eV for free energy (exothermic by 0.28 eV for energy). The weakly adsorbed CO can react with the nearby Obr with a very low barrier of only 0.13 eV. The subsequent OCOO formation becomes the rate-determining step with an overall free energy barrier of 0.52 eV (see Fig. 3).
To properly address the kinetics at different temperatures, we performed a microkinetic simulation for CO oxidation on the two catalysts using the free energetics derived from DFT and the results are shown in Fig. 4a. For the dome Au24O4 particle (red curve in Fig. 4a), the CO2 production rate increases sharply from 0.29 mmol gAu−1 s−1 at 220 K to a maximum of 45 mmol gAu−1 s−1 at 300 K, then reduces to 28 mmol gAu−1 s−1 at 350 K. For the Au37O4 catalyst (green in Fig. 4a), the activity grows slowly from 0.55 mmol gAu−1 s−1 at 220 K to 1.1 mmol gAu−1 s−1 at 350 K. Therefore, Au24O4 has a higher activity than Au37O4 above 230 K. Supported Au catalysts generally have a wide size distribution of Au particles, and so for simplicity, we considered a model catalyst that contains 10% Au24O4, 10% Au37O4 and 80% 2 nm Au particles and we evaluated its overall activity, as shown in the blue curve in Fig. 4a. Since Au particles larger than Au37 are close-packed and their activity at the interface is similar to that of Au37, the activity of 2 nm Au can be deduced from Au37O4 as r37/d2 (where r37 is the rate of Au37O4 and d is the diameter of the particle, i.e. 2 nm in this model). The overall rate as shown by the blue curve follows the same trend as that of Au24O4, reaching a maximum of 4.8 mmol gAu−1 s−1 at 300 K and slightly reducing to 3.1 mmol gAu−1 s−1 at 350 K.
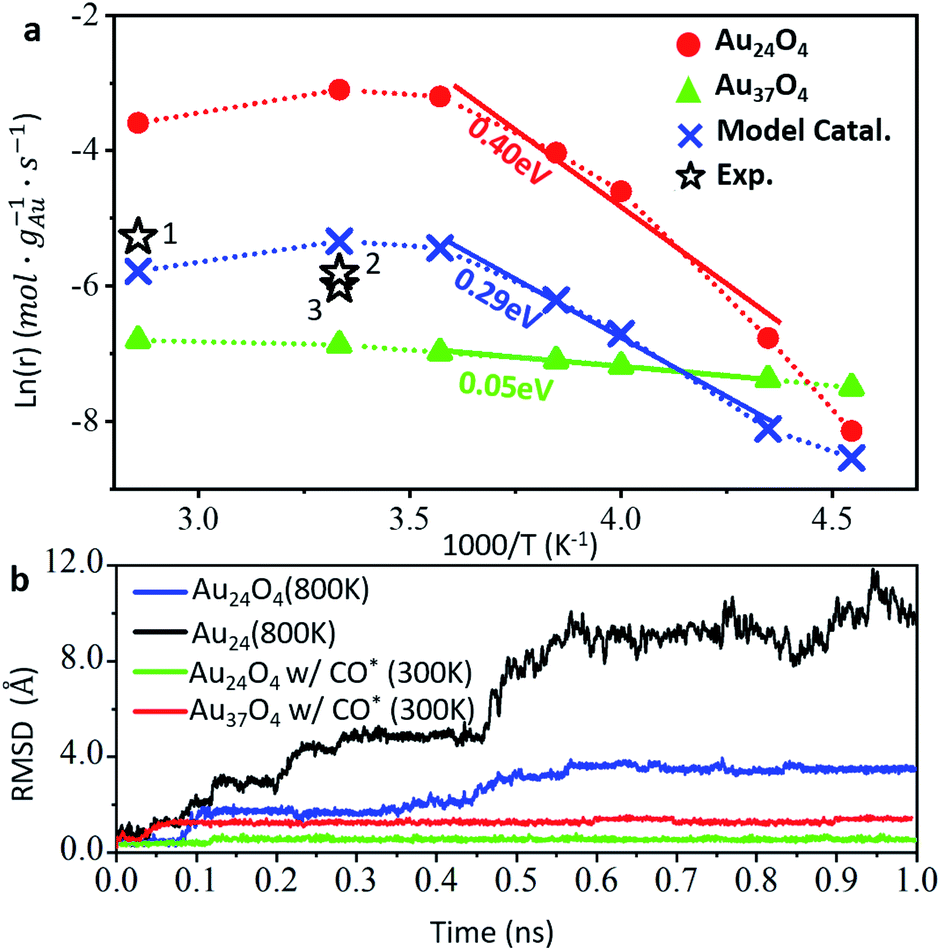 | ||
| Fig. 4 Activity and stability of Au particles in CO oxidation. (a). Logarithm of CO2 production rates versus the reciprocal of the temperatures. Red line: Au24O4/TiO2; green line: Au37O4/TiO2; blue line: model catalyst containing 10% Au24, 10% Au37 and 80% Au2nm particles. The hollow pentastars represent experimental results ((1) Wang et al.,62 (2) Yan et al.,63 (3) Shao et al.64). Ea denotes the linearly fitted theoretical apparent barrier at 230–280 K. (b). The plot of the RMSD of Au atoms for different Au24Om and Au37O4 particles with (w/) or without CO adsorption taken from MD simulations over time at different temperatures. | ||
It is of significance to compare our theoretical prediction with the available experiments that reported high CO oxidation activity. Although supported Au catalysts can be synthesized by different methods to control the size of small Au particles, we found that the agreement for the overall rate is satisfactory between our model catalyst and different experimental data (star symbols numbered from 1 to 3 in Fig. 4a). For example, a 1% Au/TiO2 catalyst synthesized from a tetranuclear gold complex by Goodman and co-workers was reported to have a CO oxidation rate of 3 mmol gAu−1 s−1 at 300 K.63 By carefully controlling the size of the Au particles, Shao et al. synthesized a 2.2 nm Au/TiO2 catalyst, which could catalyze CO oxidation at a rate of 2.5 mmol gAu−1 s−1 at 300 K.64 These data compare well with 4.8 mmol gAu−1 s−1 for our model catalyst. At 353 K, Behm and co-workers reported a CO oxidation rate of 5 mmol gAu−1 s−1using a commercial Au/TiO2 catalyst.,62 which is only slightly larger than 3.1 mmol gAu−1 s−1 predicted from theory.
In addition to the rate, it is possible to deduce the apparent barrier (Ea) from Arrhenius plots. As shown in Fig. 4a, the fitted Ea values for the temperature range 230 to 280 K are 0.40, 0.05 and 0.29 eV for Au24O4, Au37O4 and our model catalyst, respectively. In contrast to Au24O4 which has a high activity and high apparent barrier, Au37O4 has a low apparent barrier and low activity, which can be attributed to the low barrier to surface reactions on Au37O4 (temperature-independent) and the overall high free energy barrier due to the low CO adsorption ability. The experimental apparent barrier is generally ∼0.3 eV below 300 K as measured at low conversion conditions, being consistent with our data.63–66 Interestingly, by synthesizing a series of Au/TiO2 catalysts with different methods and gold precursors, Goodman and co-workers observed that the supported Au catalyst with higher activity exhibited higher Ea.63 The best catalyst with an average diameter of 3 nm had the apparent Ea of 0.38 eV; the worst catalyst with a diameter of 7.7 nm had an apparent Ea of 0.19 eV. Our explanation for this intriguing phenomenon is now straightforward: it is related to the concentration of very small gold particles that can adsorb CO strongly, such as Au24O4, for which the surface reaction of adsorbed CO reacting with lattice O contributes to the apparent barrier.
3.4. Stability of Au particles under reaction conditions
Finally, we would like to provide insights into the thermodynamic stability of Au particles under reaction conditions, which is one of the key factors for supported Au catalysts. Molecular dynamics (MD) calculations were carried out to measure directly the thermal stability of supported Au particles in the presence of CO and O2. Firstly, we took Au24O4 systems as the example and performed 1 ns MD simulations at 800 K as shown in Fig. 4b and also in ESI Fig. S5.† We found that the Au24O4 dome is basically unchanged with the root mean square displacement (RMSD, eqn (S5) in ESI†) of Au, converging to 0.35 nm per atom, where Au atoms only locally exchange between neighbours. For comparison, in the absence of Oad, the bare Au24 on TiO2 undergoes a dramatic structure deformation at 800 K with a RMSD that is three times larger than that of Au24O4. It transforms eventually from the dome to a close-packed stick-like structure with a Gf value of +0.21 eV (also see Fig. 1c), which suggests the sintering tendency of Au particles on TiO2 in the absence of an O2 atmosphere.Then, we considered the effect of CO adsorption on the particle stability. For 3 CO molecules adsorbed on Au24O4/TiO2, the 1 ns MD simulation at 300 K shows that the structure is immobile, with an RMSD of only 0.080 nm. A similar result was also obtained for Au37O4/TiO2 with 5 CO adsorbed at the interfacial Au, where the RMSD is 0.16 nm. These results indicate that due to the presence of the anchoring Oad at Au periphery site, which is less reactive than lattice O, the Au particles are stable under reaction condition.
Our MD simulations confirm the high stability of Au particles on O-rich TiO2 surfaces during CO oxidation, demonstrating the key anchoring role of the Oad at the interface. These Oad are not active during CO oxidation, but act as structural stabilizers to immobilize the Au particles. These results explain the previous experimental findings. For example, scanning tunneling microscopy experiments have shown that small Au particles prefer to attach to Obr rows.10,60 In an O2 atmosphere the particles can be more stable, e.g. Au7 survives up to 341 K.11 At elevated temperatures, CO oxidation with Oad (the barrier is ∼1.0 eV from our calculations) occurs, which leads to the formation of bare Au particles, and the catalyst soon undergoes sintering due to the high mobility of bare Au particles on TiO2, as shown in the black curve in Fig. 4b.
4. Conclusion
The extreme size sensitivity of supported Au catalysts revealed theoretically not only explains the critical thickness of Au particles on TiO2 for CO oxidation, but also reveals the potential to boost the catalytic activity by finely controlling the Au particle size. For titania-supported Au catalysts, the smallest stable structure, the Au24 dome, is in fact the key component for catalyzing low temperature CO oxidation. The presence of oxidation conditions which introduce interfacial O atoms is the prerequisite for stabilizing such small Au particles. With the advent of ML-based global PES exploration, we believe that many more catalyst active sites can be resolved technically under reaction conditions, which should not only bring us new chemistry, but also pave the way towards the rational design of catalysts through the optimization of the active site structure and composition.Author contributions
C. S. and Z.-P. L. conceived the project and contributed to the design of the calculations and analyses of the data. Y. P. carried out most of the calculations and wrote the draft of the paper. All the authors discussed the results and commented on the manuscripts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2018YFA0208600) and the National Science Foundation of China (91945301, 92061112, 21533001, 22033003, 91645201 and 91745201).References
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- Q. Fu, F. Yang and X. Bao, Acc. Chem. Res., 2013, 46, 1692–1701 CrossRef CAS PubMed.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- T. W. van Deelen, C. Hernández Mejía and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- A. T. Bell, Science, 2003, 299, 1688–1691 CrossRef CAS PubMed.
- R. Ciriminna, E. Falletta, C. Della Pina, J. H. Teles and M. Pagliaro, Angew. Chem., Int. Ed., 2016, 55, 14210–14217 CrossRef CAS.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS.
- A. Bruix, J. T. Margraf, M. Andersen and K. Reuter, Nat. Catal., 2019, 2, 659–670 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647–1650 CrossRef CAS PubMed.
- D. Matthey, J. G. Wang, S. Wendt, J. Matthiesen, R. Schaub, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2007, 315, 1692 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Chen, Y. Cai, Z. Yan and D. W. Goodman, J. Am. Chem. Soc., 2006, 128, 6341–6346 CrossRef CAS.
- I. N. Remediakis, N. Lopez and J. K. Norskov, Angew. Chem., Int. Ed., 2005, 44, 1824–1826 CrossRef CAS PubMed.
- N. Lopez and J. K. Norskov, J. Am. Chem. Soc., 2002, 124, 11262–11263 CrossRef CAS PubMed.
- M. Haruta, Gold Bull., 2004, 37, 27–36 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Gold Bull., 2000, 33, 41–51 CrossRef CAS.
- G. J. Hutchings, M. S. Hall, A. F. Carley, P. Landon, B. E. Solsona, C. J. Kiely, A. Herzing, M. Makkee, J. A. Moulijn, A. Overweg, J. C. Fierro-Gonzalez, J. Guzman and B. C. Gates, J. Catal., 2006, 242, 71–81 CrossRef CAS.
- J. Emmanuel, B. E. Hayden and J. Saleh-Subaie, J. Catal., 2019, 369, 175–180 CrossRef CAS.
- P. Mars and D. W. van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- P. Schlexer, D. Widmann, R. J. Behm and G. Pacchioni, ACS Catal., 2018, 8, 6513–6525 CrossRef CAS.
- D. Widmann and R. J. Behm, J. Catal., 2018, 357, 263–273 CrossRef.
- L. Li and X. C. Zeng, J. Am. Chem. Soc., 2014, 136, 15857–15860 CrossRef CAS PubMed.
- L. Li, Y. Gao, H. Li, Y. Zhao, Y. Pei, Z. Chen and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 19336–19346 CrossRef CAS PubMed.
- Y.-G. Wang, D. C. Cantu, M.-S. Lee, J. Li, V.-A. Glezakou and R. Rousseau, J. Am. Chem. Soc., 2016, 138, 10467–10476 CrossRef CAS PubMed.
- Y.-G. Wang, Y. Yoon, V.-A. Glezakou, J. Li and R. Rousseau, J. Am. Chem. Soc., 2013, 135, 10673–10683 CrossRef CAS PubMed.
- L. B. Vilhelmsen and B. Hammer, ACS Catal., 2014, 4, 1626–1631 CrossRef CAS.
- Z.-P. Liu, X.-Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef PubMed.
- Z. Duan and G. Henkelman, ACS Catal., 2018, 8, 1376–1383 CrossRef CAS.
- V. L. Deringer, M. A. Caro and G. Csányi, Adv. Mater., 2019, 31, 1902765 CrossRef CAS PubMed.
- K. T. Butler, D. W. Davies, H. Cartwright, O. Isayev and A. Walsh, Nature, 2018, 559, 547–555 CrossRef CAS PubMed.
- H. Wang, Y. Ji and Y. Li, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1421 CAS.
- M. L. Paleico and J. Behler, J. Chem. Phys., 2020, 153, 054704 CrossRef CAS.
- J. Behler, Angew. Chem., Int. Ed., 2017, 56, 12828–12840 CrossRef CAS PubMed.
- J. Močkus, in Optimization Techniques IFIP Technical Conference: Novosibirsk, July 1–7, 1974, ed. G. I. Marchuk, Springer Berlin Heidelberg, Berlin, Heidelberg, 1975, pp. 400–404, DOI:10.1007/978-3-662-38527-2_55.
- C. Shang, X.-J. Zhang and Z.-P. Liu, Phys. Chem. Chem. Phys., 2014, 16, 17845–17856 RSC.
- C. Shang and Z.-P. Liu, J. Chem. Theory Comput., 2013, 9, 1838–1845 CrossRef CAS PubMed.
- S.-D. Huang, C. Shang, P.-L. Kang and Z.-P. Liu, Chem. Sci., 2018, 9, 8644–8655 RSC.
- S.-D. Huang, C. Shang, X.-J. Zhang and Z.-P. Liu, Chem. Sci., 2017, 8, 6327–6337 RSC.
- S. Ma, C. Shang, C.-M. Wang and Z.-P. Liu, Chem. Sci., 2020, 11, 10113–10118 RSC.
- S. Ma, S.-D. Huang and Z.-P. Liu, Nat. Catal., 2019, 2, 671–677 CrossRef CAS.
- P.-L. Kang, C. Shang and Z.-P. Liu, J. Am. Chem. Soc., 2019, 141, 20525–20536 CrossRef CAS PubMed.
- P.-L. Kang, C. Shang and Z.-P. Liu, Acc. Chem. Res., 2020, 53, 2119–2129 CrossRef CAS PubMed.
- S.-D. Huang, C. Shang, P.-L. Kang, X.-J. Zhang and Z.-P. Liu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2019, 9, e1415 CAS.
- S.-C. Ma, C. Shang and Z.-P. Liu, J. Chem. Phys., 2019, 151, 050901 CrossRef.
- www.lasphub.com/supportings/AuTiCHO.pdf, accessed Feb. 22nd, 2021.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. J. Morgan and G. W. Watson, Surf. Sci., 2007, 601, 5034–5041 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132 Search PubMed.
- X.-J. Zhang, C. Shang and Z.-P. Liu, J. Chem. Theory Comput., 2013, 9, 5745–5753 CrossRef CAS PubMed.
- R. D. Lide, CRC Handbook of Chemistry and Physics, CRC press, 84th edn, 2003 Search PubMed.
- P. J. Steinhardt, D. R. Nelson and M. Ronchetti, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 28, 784–805 CrossRef CAS.
- X.-J. Zhang, C. Shang and Z.-P. Liu, Phys. Chem. Chem. Phys., 2017, 19, 4725–4733 RSC.
- F. Cosandey and T. E. Madey, Surf. Rev. Lett., 2001, 8, 73–93 CrossRef CAS.
- Y. Maeda, M. Okumura, S. Tsubota, M. Kohyama and M. Haruta, Appl. Surf. Sci., 2004, 222, 409–414 CrossRef CAS.
- Y. Wang, D. Widmann and R. J. Behm, ACS Catal., 2017, 7, 2339–2345 CrossRef CAS.
- Z. Yan, S. Chinta, A. A. Mohamed, J. P. Fackler and D. W. Goodman, Catal. Lett., 2006, 111, 15–18 CrossRef CAS.
- B. Shao, J. Zhang, J. Huang, B. Qiao, Y. Su, S. Miao, Y. Zhou, D. Li, W. Huang and W. Shen, Small Methods, 2018, 2, 1800273 CrossRef.
- G. R. Bamwenda, S. K. Tsubota, T. Nakamura and M. M. Haruta, Catal. Lett., 1997, 44, 83–87 CrossRef CAS.
- Y. Tai, W. Yamaguchi, K. Tajiri and H. Kageyama, Appl. Catal., A, 2009, 364, 143–149 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods; the global minima of other AunOm/TiO2 particles; training dataset and benchmark of the G-NN potential. See DOI: 10.1039/d0sc06502a |
| This journal is © The Royal Society of Chemistry 2021 |