
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting PLA melt strength by controlling the chirality of co-monomer incorporation†
An Sofie
Narmon
 a,
Annelies
Dewaele
a,
Kevin
Bruyninckx
a,
Bert F.
Sels
a,
Peter
Van Puyvelde
*b and
Michiel
Dusselier
*a
a,
Annelies
Dewaele
a,
Kevin
Bruyninckx
a,
Bert F.
Sels
a,
Peter
Van Puyvelde
*b and
Michiel
Dusselier
*a
aDepartment of Microbial and Molecular Systems, Centre for Sustainable Catalysis and Engineering, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: michiel.dusselier@kuleuven.be; Web: https://dusselier-lab.org/
bDepartment of Chemical Engineering, Soft Matter, Rheology and Technology, KU Leuven, Celestijnenlaan 200F, 3001 Leuven, Belgium. E-mail: peter.vanpuyvelde@kuleuven.be
First published on 10th March 2021
Abstract
Bio-based and degradable polymers such as poly(lactic acid) (PLA) have become prominent. In spite of encouraging features, PLA has a low melt strength and melt elasticity, resulting in processing and application limitations that diminish its substitution potential vis-a-vis classic plastics. Here, we demonstrate a large increase in zero shear viscosity, melt elasticity, elongational viscosity and melt strength by random co-polymerization of lactide with small amounts, viz. 0.4–10 mol%, of diethylglycolide of opposite chiral nature. These enantiomerically pure monomers can be synthesized using one-step zeolite catalysis. Screening of the ester linkages in the final PLA chains by the ethyl side groups is suggested to create an expanding effect on the polymer coils in molten state by weakening of chain–chain interactions. This effect is suspected to increase the radius of gyration, enabling more chain entanglements and consequently increasing the melt strength. A stronger melt could enable access to more cost-competitive and sustainable PLA-based biomaterials with a broader application window. Amongst others, blow molding of bottles, film blowing, fiber spinning and foaming could be facilitated by PLA materials exhibiting a higher melt strength.
Introduction
Biobased and biodegradable polymers are appealing alternatives for petroleum-based plastics, as the latter are associated with environmental pollution, triggering worldwide public concern.1–3 Poly(lactic acid) (PLA), an aliphatic polyester made up of carbohydrate-sourced lactic acid (LA) monomers, is nowadays one of the most promising bioplastics on the market.4,5 PLA is biodegradable under industrial composting conditions and exhibits adequate mechanical and physical properties, comparable with those of commodity plastics such as polystyrene and poly(ethylene terephthalate).6–8Certain factors restrain the adoption of PLA. At present, high-molecular weight PLA is synthesized by ring-opening polymerization (ROP) of the dilactone, lactide (LD), which is made by back-biting depolymerization of polycondensed LA.4,9 While dilactones and ROP are indispensable, making LD is a two-step, time and energy consuming process that requires additional purification, driving up the cost of production. Moreover, LD yields are mediocre and the process is confronted with racemization, losing control over the enantiopurity of (L,L)-lactide (L-LD), and thus of PLA.10,11 Recently, some of us developed a zeolite-based catalytic process, enabling the single-step conversion of LA to LD. The highly selective route outperforms the two-step procedure, potentially in cost and certainly in atom efficiency (few side-products) and in preserving stereochemistry (no racemization).12 Other routes to lactide are also being developed and gear toward solving the cost issue.13–15 A more intrinsic drawback of PLA is associated with its processing performance and consequently its applicability. PLA has a poor melt strength and melt elasticity, which are crucial properties for elongational flow dominated processes such as film blowing, blow molding, fiber spinning, foaming, etc.16–18 Different strategies for improving the melt strength properties of PLA have been described. Some efforts focus on the use of chain extenders to create long chain, branched, cross-linked or star-shaped structures of PLA, to enhance chain entanglements.19–25 Although chain extenders can boost numerous properties of PLA, final polymer structures are often difficult to control, and seeking the optimal extender concentration remains challenging. In addition, chain extended polymer structures can exhibit reduced degradability,26–28 whereas extending agents themselves are often not biodegradable or biocompatible and in some cases are even toxic (diisocyanates). A second strategy consists of blending PLA with other polymers, as well as compounding it with different micro- or nanosized fillers (e.g. 5 wt%) to enhance its melt behavior.29 However, homogeneous blends or equal particle dispersion are not readily obtained. Moreover, blending can result in recycling or (bio)degradation limitations.30
Finally, stereocomplex crystallites, induced by mixing PLAs of opposite chiral nature (L-PLA with 5 wt% D-PLA), can act as crosslinking points, resulting in a melt behavior similar to that of chain-extended PLA structures.31–33 However, synthesis of D-PLA requires large scale availability of pure D-LA, which is currently not trivial.34 In contrast to these strategies, studies linking the composition of the backbone (linear lactide-based co-polymers) to rheological properties are scarce, despite the obvious value of microstructure – viscoelasticity relations for these polyesters. One group performed a few rheological studies on syndio- and heterotactically enriched PLAs.35 The effect of incorporating D-LD in L-PLA has been a topic of investigation but without unambiguously clarifying the impact of the D/L ratio or showing a negligible change in polymer melt viscosity.36,37
Here, we describe the unprecedented improvement of melt strength behavior of PLA by ring-opening co-polymerization of LD in the presence of (small amounts of) diethylglycolide (EG) of opposite chiral nature (Fig. 1A). Although diethylglycolide has been polymerized before, its traditional synthesis is homogeneously catalyzed, inefficient and laborious, ending up with a mixture of diastereomers.38–40 Zeolite-based cyclization of α-hydroxy acids here not only generates efficiency and high yields, but also essentially maintains the full stereochemistry of the substrate, which is of utmost importance for this research. Access to enantiomerically pure cyclic ester monomers enables the exploration of a wide range of high-molecular weight (co)-polymers with possibly attractive new properties.
 | ||
Fig. 1 (A) Catalytic one-step synthesis of diethylglycolide (EG) and random co-polymerization with lactide (LD) toward high melt strength PLA. (B) 1H-NMR (400 MHz, dmso-d6) results from catalytic cyclization of L-α-HBA and rac-α-HBA, resulting in an EG yield of 94.0% (e.e. = 99.2%) and 56.0% (e.e. = 2.6%), respectively. (C) Chiral GC results from catalytic cyclization of L-α-HBA and rac-α-HBA, showing the absence of racemization and pure (L,L)-EG and a statistical mixture of (L,L), (D,D) and meso-EG, respectively. (D) Monomer conversion (%) of (D,D)-LD and 1 mol% of (L,L)-EG over 60 minutes of polymerization (bulk, 170 °C, Sn(Oct)2 (monomer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :catalyst = 489), 1-dodecanol (initiator:catalyst = 0.7)). :catalyst = 489), 1-dodecanol (initiator:catalyst = 0.7)). | ||
In this research, rheological studies on co-polymers of enantiomerically pure LD and EG revealed a strong increase in melt elasticity and melt strength compared to those of conventional PLA, with clearly observed differences associated with the chiral nature of the monomers. While improvements in melt properties are usually obtained through chain extension, blending or compounding, a simple co-polymerization here suffices, even for small EG incorporation. A promising strategy for large scale implementation thus arises, given the ease of dropping-in 1% of EG into the lactide batch for classic L-PLA production. The gains in melt strength could translate into lower weight of plastic needed per functional item (e.g. blow molding a thinner bottle) and expand the application horizon for PLA.
Experimental
Synthesis of diethylglycolide
In a typical reaction, 0.01 mol of (L)- or (D)-α-hydroxybutyric acid and 0.5 g of H-beta zeolite (SiO2/Al2O3 = 25 or 150, (Zeolyst and Südchemie)) are added to a round bottom flask with 20 mL of toluene or o-xylene. On top of the round bottom flask, a custom made Dean–Stark trap, filled with solvent, is installed. The setup is connected to a condenser, and heated in an oil bath at 130 or 170 °C. The mixture is stirred for 3 hours. After the reaction, the mixture is homogenized by the addition of 15 mL of acetonitrile. After homogenization, the zeolite is removed by filtration over a glass frit filter under vacuum, and rinsed with another 10 mL of acetonitrile.Ring-opening polymerization
Solvent-free ring-opening polymerization is carried out in a dry, custom-made round-bottom flask. In a typical experiment, a desired amount of monomer is added to a flask in an oxygen and moisture free environment. A solution of stannous octoate in toluene as a catalyst (monomer:catalyst = 2500:1) and 1-dodecanol as an initiator (70 mol% of the catalyst) are added to the monomer. The solvent is removed in vacuo and the flask is filled with argon and immersed in an oil bath at 170 °C for 70 minutes. After polymerization, the flask is cooled, and the polymer is dissolved in chloroform. The synthesized polymers are separated from the remaining monomers and oligomers via precipitation in methanol, filtered, and dried under reduced pressure.
Small amplitude oscillatory shear
Small amplitude oscillatory shear (SAOS) measurements are carried out on an AresMelts rheometer, using a parallel plate set-up (8 mm diameter) surrounded by a convection oven, purged with N2 gas. Prior to rheology measurements, the polymer samples are compression molded into discs of 8 mm diameter and a thickness of 1 mm. The polymer discs are vacuum dried overnight at 80 °C prior to SAOS measurements. First, dynamic time sweep measurements are performed to verify the thermal stability of the polymer samples under the applied test method. The changes in G′ and G′′ are determined over time at an angular frequency of 10 rad s−1, a strain amplitude of 1% and a temperature of 185 °C during 300 s. Secondly, strain sweep tests are performed by varying the strain amplitude between 0.1 and 10% to determine the linear viscoelastic regime of the materials. Frequency sweep measurements are carried out at 185 °C with dynamic frequencies ranging from 0.1 to 100 rad s−1 at a strain amplitude of 1–10%. Consecutively, rate sweep tests are performed applying shear rates between 0.01 and 1 s−1. At each shear rate a waiting time of 30 s is installed to guarantee steady state, while a measuring time of 10 s is applied.Extensional viscosity fixture
Extensional flow properties of the polymers are determined on an AresMelts rheometer, using an Extensional Viscosity Fixture (EVF) set-up. Extensional viscosities are measured in strain-controlled stretch experiments with a Hencky strain of 3.4 at 185 °C in a N2 atmosphere. The polymer samples are compression molded into rectangular plates of 18.0 mm long, 10.0 mm (±0.10 mm) wide and 0.80 mm (±0.05 mm) thick. Prior to stretch measurements, the plates are dried at 80 °C overnight. The experimental protocol consists of three steps. During the first step a pre-stretch with a stretch rate of 0.0075 s−1 is performed on the polymers to compensate for thermal expansion during heating. Before pre-stretch, a delay time of 50.0 s is applied to ensure that the polymer samples are completely molten. The pre-stretch is followed by a relaxation step of 5.0 s to remove the residual stress in the sample. Finally, the stretch measurement was performed at a constant Hencky strain rate (0.1, 0.5, 1, and 3.5 s−1).Haul-off
To determine the extensional properties of the polymer melts, a Göttfert 2002 capillary heometer is used in combination with a Haul-off apparatus. The polymer material is added to the barrel of the capillary rheometer at 185 °C. To create molten polymer strands, the melt is pushed out of the barrel by the piston with a diameter of 12 mm and a die of 2 mm at a piston speed of 0.05 mm s−1. The molten strands are attached to the Haul-off apparatus which spins the molten strands on a wheel, rotating at a pull-off speed of 100 mm s−1. The speed is linearly increased at an acceleration of 0.12 mm s−1 till the polymer melt breaks.Results
Catalytic monomer synthesis and co-polymerization
The scope of the one-step catalytic conversion of lactic acid (LA) to lactide (LD) in the presence of Brønsted acidic zeolite beta (H-BEA) is extended towards the cyclization of α-hydroxybutyric acid (α-HBA) to form diethylglycolide (EG). A complete overview of all catalytic syntheses of symmetric and asymmetric cyclic esters can be found in the ESI (Tables S1–S2 and Fig. S1†). Applying the same reaction conditions to L-α-HBA as those applied to L-LA by Dusselier et al.12 (130 °C, 3 h, toluene, H-BEA (SiO2/Al2O3 = 25)) only yielded 5% L-EG, compared to 78% for L-LD. This could be overcome by increasing the reaction temperature to 170 °C (o-xylene, yield = 76%), indicating a reactivity difference between LA and α-HBA. The catalyst-free reactions revealed – next to the expected low cyclic ester selectivity - a conversion decline with increasing steric hindrance of the alkyl chains at the α-position (see Fig. S2† for more details comparing the four different substrates). This confirms a slower spontaneous condensation (reactivity) of α-HBA compared to LA. In this regard, one could expect that α-HBA would tend to form smaller products and perhaps favor ring-closure. Nonetheless, a reaction with a soluble acid catalyst (sulphuric acid) at full monomer conversion yielded less than 5% EG, indicating the need for confining the acid function in a shape-selective catalyst such as H-BEA. Under optimized conditions, the zeolite reaction (with H-BEA, SiO2/Al2O3 = 150) proceeded even faster and was more selective towards (L,L)-diethylglycolide (L-EG) (yield = 88%). Distillation of the α-HBA substrate before reaction yielded 94% L-EG in a single step. In addition, 1H-NMR showed an excellent enantiomeric purity of 99.6% L-EG (e.e. = 99.2%) with formation of only 0.4% meso-EG (= (L,D)-EG). In contrast to enantiopure α-HBA, racemic α-HBA (rac-α-HBA) conversion is slower and less selective (yield = 56%) for cyclic esters, but still a statistical mixture of the L,L; D,D- and L,D-diastereomers is achieved confirming the absence of racemization via the direct zeolite-catalyzed route (Fig. 1(B) and (C)). After the reaction, the heterogeneous catalyst is easily removed and cyclic esters are separated from the linear oligomers and unreacted monomers by phase extraction. Subsequent crystallization leads to an overall EG purity of 99.5% (e.e. ≈ 100%) and an overall isolated yield of 48% (Fig. S3†). Obtaining higher isolated yields should be possible, but this was not the scope here as purity for ring-opening polymerization (ROP) was the main concern.ROP, based on the work by Fisher et al.,41 was performed with the stannous octanoate (Sn(Oct)2) catalyst and n-dodecanol initiator. This catalytic system ensures preservation of the stereochemistry of the monomers within the polymer chains, high molecular weight and low polydispersity.42,43 Diverse lactide-based polyesters were synthesized with different incorporations of EG, varying between 0.3 and 10 mol%, i.e. n/(m + n) in Fig. 1(A). The polymers vary in stereochemistry of the co-monomers (L-EG or D-EG with L-LD or D-LD, Fig. S4†). 1H-NMR proved successful incorporation of the co-monomers in polylactide (Fig. S5†), and weight-average molecular weights (Mws) varied between 101 and 149 kg mol−1 (Tables 1 and S3† have a full dataset). Various benchmarks were synthesized, i.e. a co-polymer of L-LD with 10 mol% D-LD and pure poly(L-lactide) (P(L-LD)) and poly(D-lactide) (P(D-LD)), or purchased. The latter, a commercial PLA (Ingeo 7001D, NatureWorks) has specifications targeting use in injection stretch blow molded bottle applications.
| Build-ina % | GPC | DSC | SAOS | |||||
|---|---|---|---|---|---|---|---|---|
| Polymer | Schematic chain structure | Co-monomer | M w (kg mol−1) | D | T g (°C) | T m (°C) | η 0 (kPa s) | ω c (rad s−1) |
| a Average build-in % of methyl and methine protons (when both visible), determined by 1H-NMR (400 MHz, CDCl3). b Polystyrene standards were used to calibrate weight-average molecular weights (Mw). The experimental molecular weight was corrected considering the Mark–Houwink parameters for PLA, and the same parameters are used as an estimation for the different co-polymers.6 c Commercial grade PLA from NatureWorks. d Large scale polymerization reactions (50–100 g). e Added amounts before reaction; L- and D-LD cannot be distinguished from each other in 1H-NMR. | ||||||||

|

|
— | 135 | 1.7 | 58 | 177 | 9.7 | 62.9 |

|

|
— | 117 | 1.7 | 57 | 179 | 12.1 | 84.5 |

|
— | 83.3 | 1.5 | 58 | 149 | 5.9 | >100 | |

|

|
10e | 128 | 1.7 | 54 | — | 4.8 | >100 |

|

|
9.0 | 146 | 1.6 | 43 | 148 | 8.3 | >100 |

|
0.7 | 133 | 1.4 | 54 | 177 | 23.1 | 46.7 | |

|

|
10.3 | 101 | 1.4 | 46 | 127 | 15.2 | 80.4 |

|
1.2 | 115 | 1.7 | 56 | 169 | 143.3 | 7.83 | |

|
0.7 | 138 | 1.6 | 54 | 170 | 33.9 | 32.1 | |

|
0.4 | 128 | 1.8 | 57 | 165 | 54.8 | 24.3 | |
|

|
0.6 | 142 | 1.6 | 56 | 170 | 27.8 | 32.7 |

|
|
10.5 | 131 | 1.7 | 45 | 118 | 71.8 | 19.1 |

|
4.1 | 129 | 1.7 | 54 | 154 | 108.2 | 8.17 | |

|
2.0 | 149 | 1.3 | 56 | 160 | 17.3 | 51.0 | |

|
1.2 | 118 | 1.7 | 56 | 171 | 33.3 | 24.3 | |

|
0.7 | 128 | 1.5 | 58 | 172 | 191.1 | 6.64 | |

|
0.3 | 129 | 1.6 | 56 | 177 | 22.9 | 76.6 | |
|
||||||||
| Large scale reactions | ||||||||

|
— | 106 | 1.6 | 52 | 177 | 7.1 | >100 | |

|
1.2 | 119 | 1.9 | 59 | 170 | 14.8 | 55.9 | |
Chain structures of co-polymers are well-known to be dependent on the relative polymerization rates of the different monomers. If propagation rates are comparable, random chain structures are obtained, whereas a significant rate difference leads to block co-polymers. Various researchers have described the decrease in the polymerization rate of alkyl-substituted lactides with increasing steric hindrance of the substituents38,39,44–46 This has been attributed to a more difficult nucleophilic attack of the initiator.38,46 In addition, a decreased polymerisability is observed with increased intramolecular steric repulsion in the polymer chains (less present in the cyclic di-esters) which decreases the polymerization enthalpy.44 Nevertheless, the difference in the polymerization rate between rac-LD and rac-EG has been described by Baker et al. as rather small when comparing the reactivity ratios, indicating a high degree of randomness within their co-polymer chains.38,39 Here, the randomness of incorporation was quasi confirmed by determining the time evolution of the co-monomers during ROP (Fig. 1(D); Table S4 and Fig. S6†). Only insubstantial differences in the relative progression of the conversions are detected. L-EG polymerization progresses (relative to its starting amount of 1%) only slightly slower than D-LD polymerization (99%), most probably giving rise to strongly randomized co-polymers (note that the degree of randomness can vary with the co-monomer ratio).
Differences in the thermal properties of the various co-polymers are observed. In general, a downward trend in glass transition temperature (Tg), melt temperature (Tm) and degree of crystallinity (χc) is obtained with increasing amounts of EG co-monomer (see Tables 1 and S3, and Fig. S7†). Tm and χc show a stronger decrease when co-monomers with opposite stereochemistry are used. Baker et al.47 explained the decrease in Tg of poly(alkyl glycolide) polymers with alkyl side chains of increasing length (2–8 carbons) compared to that of polylactide based on weaker dipole–dipole interactions between polyester chains as due to a stronger screening of the ester groups by the alkyl side chains and thus less chain–chain interactions. The decrease in Tm and χc is explained by the introduction of defects in the regular structure of P(L- or D-LD), preventing chains from folding into close crystalline structures. This effect is expected to be stronger when co-monomers with opposite stereochemistry are used.4
Melt rheology
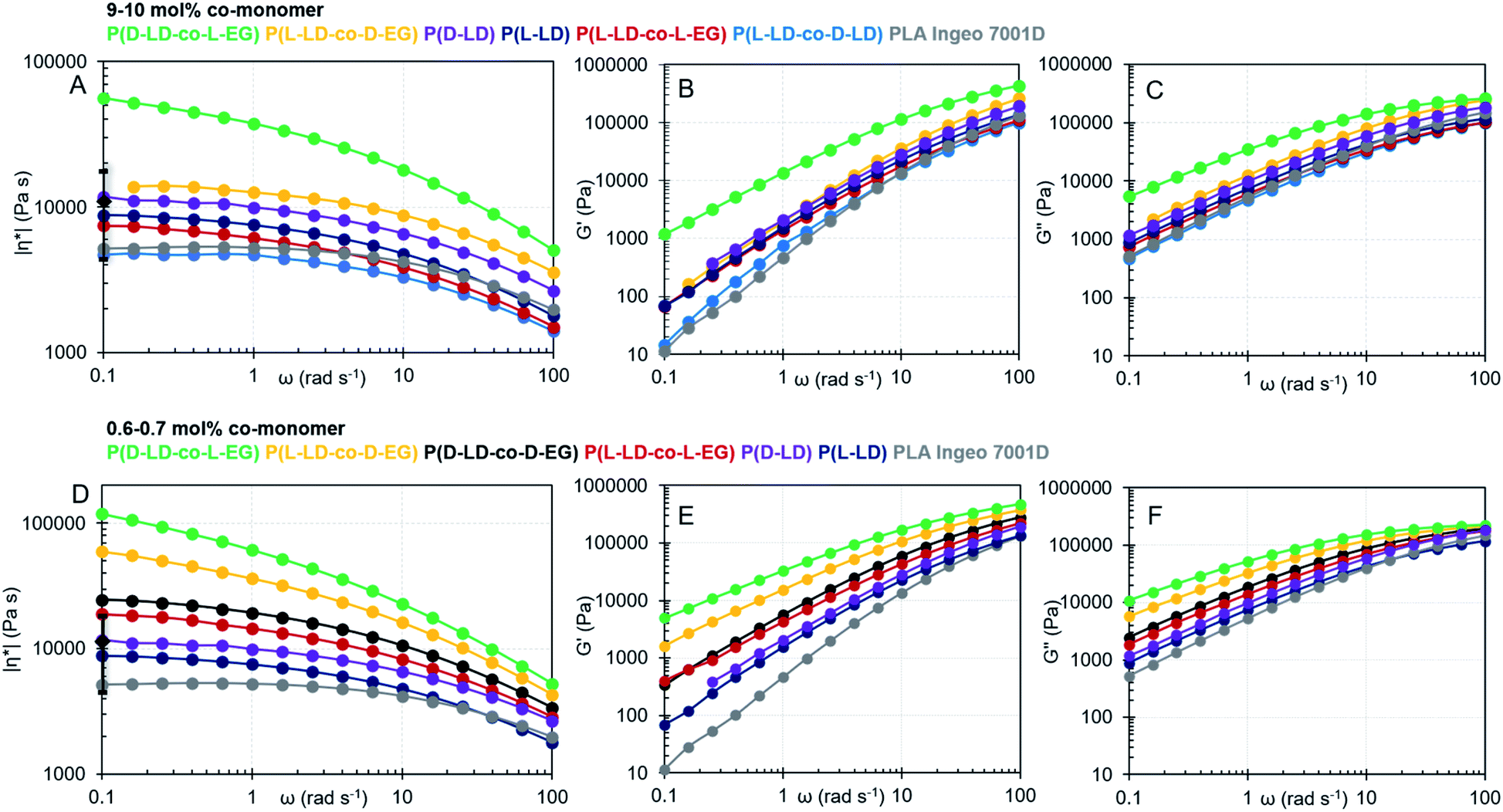 | ||
| Fig. 2 (A) Complex shear viscosity (|η*|), (B) elastic modulus (G′) and (C) viscous modulus (G′′) as a function of angular frequency (ω) for co-polymers with 9–10 mol% of co-monomer. (D) Complex shear viscosity (|η*|), (E) elastic modulus (G′) and (F) viscous modulus (G′′) as a function of angular frequency (ω) for co-polymers with 0.6–0.7 mol% of co-monomer. ♦ and error bars at 0.1 rad s−1 in (A) and (D) indicate the average value and standard deviation as calculated from different synthesized homopolymers of pure L or D-LD (see the ESI† for more details). The colors are also traceable in Table 1. | ||
All polymer melts tend to a Newtonian plateau at low frequencies, while shear-thinning occurs at higher frequencies (Fig. 1(A) and (D)). Several observations emerge from the data by comparing the viscoelastic properties of the co-polymers with those of P(L-LD), a self-made control with a similar Mw, and the commercial grade PLA (Ingeo) designed for injection stretch blow molding. Adding 10 mol% of D-LD to P(L-LD) seems to decrease the viscosity and elasticity of pure P(L-LD) to a small extent, in line with earlier studies noting only subtle differences in the viscoelastic properties of polylactides with various D-LD contents.36,37,49 While the viscoelastic properties do not seem to significantly change with incorporation of 10 mol% L-EG in P(L-LD), co-polymers with 10 mol% EG and LD with opposite stereochemistry exhibit remarkable increases in both complex viscosity and elasticity of the polymer melts (Fig. 2(A–C) and Table 1). While the average η0 of P(L-LD) and P(D-LD) (Table S6†) is calculated to be 12324 Pa s (standard deviation of 7585 Pa s) and the average ωc lies around 72 rad s−1, for an average Mw of 122.5 kg mol−1, the values of P(L-LD-co-D-EG) and P(D-LD-co-L-EG) are estimated to be 15212 (ωc = 80.4 rad s−1) and 71783 Pa s (ωc = 19.1 rad s−1), respectively (Carreau-Yasuda model, see S7.1† for more details). Since the melt viscosity and elasticity are strongly dependent on the chain length,19,50 one could argue that the increase in P(L-LD-co-D-EG) is limited due to its rather low Mw (101 kg mol−1). Nevertheless, P(D-LD-co-L-EG) exceeds the zero shear viscosity of P(L-LD) with a factor 6, exhibiting a unique beneficial impact on the viscoelastic properties of polylactide. Interestingly, comparable results were obtained when much lower amounts, viz. 0.6–0.7 mol%, of EG were incorporated in polylactide (Fig. 2(D–F) and Table 1). Notably, co-polymers in which EG and LD have the same stereochemistry, (P(L-LD-co-L-EG) and P(D-LD-co-D-EG)) exhibited a limited increase in viscosity and elasticity, while the increase is most significant for co-polymers of L-LD and D-EG or D-LD and L-EG. To make sure effects are not largely attributable to variations in Mw, a Mark–Houwink correction was applied to the calculated zero shear viscosities shown in Table 1, which can be consulted in Fig. S16.† Note that as an estimate the same parameters were used for all different co-polymers. This correction further strengthens the possible melt-strength improving effects of EG.
To study the effect of the co-monomer ratio (EG:LD) on the viscoelastic properties of the materials, various co-polymers (P(L-LD-co-D-EG) and P(D-LD-co-L-EG)) with different percentages of EG were analyzed (Tables 1 and S6, and Fig. S12–S15†). It can be noticed that for both P(L-LD-co-D-EG) and P(D-LD-co-L-EG) no clear correlation exists between viscosity or elasticity and the co-monomer ratio. For P(D-LD-co-L-EG) rather low η0s and high ωcs are observed when 0.3 or 2.0 mol% of EG is incorporated, while incorporation of 0.7, 4.1 or 10.5% exhibits a remarkable improvement in viscoelastic properties. For P(L-LD-co-D-EG) very high viscosity and elasticity are seen when 1.2 or 0.4% of D-EG is used. Nevertheless, the co-polymer with 10.3 mol% D-EG shows a much smaller increase, which is likely attributed to its rather low Mw compared to the other polymers. The non-directional trends potentially derive from changes in the mode of incorporation with the percentages (e.g. random vs. block) or other polymer microstructure effects. Different reactivity ratios have been reported when changing co-monomer percentages.54 Although Mws vary to some extent, the variation is limited and unlikely to cause such large effects (see Mw correction Fig. S16†). In addition, these effects seem to be reproducible in different co-polymers of LD and EG. Although the data described here cannot be compared to the literature data directly (due to differences in molecular weight, (D,D)-lactide content, additive mixing techniques, etc.), one can conclude that the zero shear viscosity values described here are in the same range as values obtained in the literature when mixing PLA with 1–3 wt% of Joncryl, a reactive additive.51–53
Extensional viscosity fixture measurements were performed at different Hencky strain rates (0.1, 0.5, 1 and 3.5 s−1) to determine the extensional viscosity (ηe) as a function of extension time (timee) (Fig. 3(A): Hencky strain rate = 3.5 s−1, data at other rates can be found in the ESI (S7.2†)). Extensional viscosity is related to η0 by a factor of three as explained using the Trouton ratio (ηe = 3η0) (see S7.2† for more details).
 | ||
| Fig. 3 Large scale extensional viscosity measurements on P(L-LD), P(L-LD-co-D-EG) (1.2 mol% D-EG) and PLA Ingeo 7001D (A) extensional viscosity (ηe) as a function of extension time (timee) at a Hencky strain rate of 3.5 s−1 determined with an extensional viscosity fixture rheometer. (B) Average extensional force exerted by the polymer materials determined by using a Haul-off extensional rheometer. | ||
A comparable extensional viscosity is observed for the commercial PLA grade and the synthesized P(L-LD) sample, while P(L-LD-co-D-EG) confirms its higher viscosity. Although a strong difference in viscosity is noticed, no strain hardening behavior is observed at these Hencky strain rates, in agreement with the literature on extensional viscosity of linear polylactide.55,56
Finally, to directly measure the melt strength, vertical polymer melt strands (Fig. S18†) were spun on a wheel at a linearly accelerating velocity (100–5000 mm s−1) and the extensional force exerted by the materials was determined using a Haul-off extensional rheometer (details in S7.3†). While strong variations were seen in the rotation speed at which the polymer materials broke, forces seemed to be quite constant. P(L-LD-co-D-EG) exhibited an average force of about 24.4 N (Fig. 3(B)), which is about two times higher than the force of P(L-LD) (12.4 N). P(L-LD) and the commercial PLA showed very comparable values. This large scale – from a polymer chemist point of view – measurement consolidates the presumption of a severe rise in the melt strength of polylactide with incorporation of small amounts of EG with opposite stereochemistry.
Discussion
Co-polymerization of LD and EG with a reverse stereo-configuration seems to uniquely impact the viscoelastic properties of PLA. While improvements in melt properties are mostly the results of chain extension, stereocomplexation, blending, compounding, etc., obtaining these effects here through co-polymerization is unexpected and may open new ways to more functional PLA. A higher melt strength and melt elasticity could facilitate certain elongational dominated processes (extrusion blow molding, thermoforming, film blowing, foaming, etc.), broadening the application window of PLA bioplastics, simply by adding a small amount of co-monomer to the existing ROP procedure. Moreover, if a thinner stretch can be achieved without breakage of the stronger melt, less polymer is required for a given application, substantially reducing the ecological footprint of the plastic application. Since the ethyl side groups, here incorporated into the polylactide backbone, are too short to induce extra side-chain entanglements,19,36 other explanations should be invoked and investigated for this spectacular increase in melt strength and melt elasticity.Yamane et al.32 obtained indicative, but less strong, viscoelastic effects by blending small amounts of low molecular weight P(D-LD) into P(L-LD). This was explained by P(D-LD) acting as branching points on the P(L-LD) chains, forming small stereocomplex crystallites at the contact points of P(L-LD) and P(D-LD) chains. These crystallites stay solid above the Tm of P(L-LD), counteracting disentanglement under shear deformation. Similar interactions may occur between the P(L-LD-co-D-EG) or P(D-LD-co-L-EG) chains in our co-polymers. Stereocomplexation between blends of P(L-α-HBA) and P(D-LA)57 or P(L-α-HBA) and P(D-α-HBA)58 as well as stereocomplexation between stereoblock co-polymers of P(D-α-HBA) and P(L-LA) (note that these are polycondensation-derived) have been described in the literature.59 However, the ring-opening-polymerization-derived co-polymers in our work only contain very small amounts of co-monomers, randomly distributed throughout the PLA chains. Moreover, no proof of stereocomplexation was observed in the DSC measurements, which should be manifested as a melting peak at higher temperatures, i.e. above the melting peaks of both homopolymers of LD and EG (Fig. S7†), rendering the stereocomplex hypothesis unlikely. To further ensure the absence of stereocomplex crystallites, G′ and G′′ values of some of the prepared co-polymers were measured at increasing temperatures up to 230 °C to detect sudden drops in elasticity or viscosity of the materials. Since no remarkable effects were observed (Fig. S17†), presence of such crystallites with higher Tm can be discarded.
Even though the ethyl side groups are unable to foresee strong entanglements among polymer chains, their presence will likely affect the expansion of the polymer coils in molten state due to decreased inter- and intramolecular chain interactions. This effect might increase the radius of gyration (Rg) of the polymer coils (Fig. 4), rendering them more amenable to entanglements, resulting in higher elasticity and melt strength. This hypothesis agrees with the weaker chain–chain interactions between polydiethylglycolide chains compared to those in polylactide chains, as discussed above. Baker et al.47 described the screening effect of alkyl side chains with increasing length (2–8 carbons) on the ester groups in poly(alkyl glycolide) polymers and decreasing Tg as a result of reduced dipole–dipole interactions between polymer chains. One can hypothesize here that such screening effects are stronger when LD and EG are of opposite chirality, as the possibility to stack the chains is reduced. Interchain interactions are expected to dominate in polymer melts as models and theories describing polymer melt associations neglect the effects of intrachain interactions.60–62 However, a recent study by Carrillo et al.63 demonstrates the importance of the often under-investigated intrachain associations or intrachain loops in determining the structure of associating polymers in the melt state and their influence on the viscoelastic properties of these materials. On the other hand, the exact relation between viscoelastic properties and radius of gyration is still under discussion.64 This relation is described as dependent on both Mw and the density of the polymer coils in molten state. While the effect of Mw is already discussed above, the density of polymer coils is expected to be limited when dealing with very comparable polymer structures.65
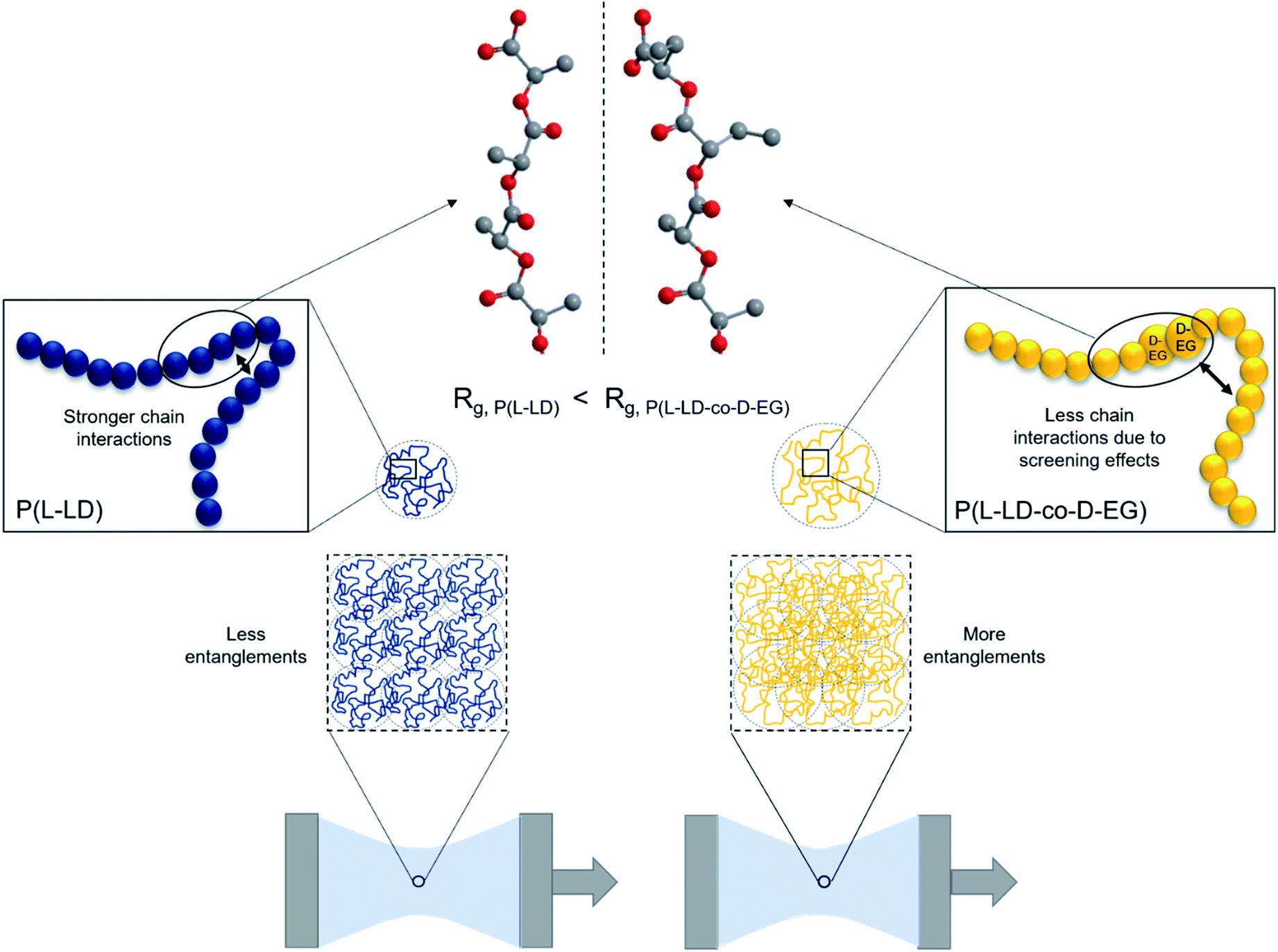 | ||
| Fig. 4 Hypothesis for the observed increases in melt strength based on Rg and screening effects, and the molecular structure in the inset showing the location of the inverse enantiomer of the incorporated ethyl side group in P(L-LD-co-D-EG). | ||
Conclusions
The one-step conversion of α-hydroxybutyric acid in the presence of zeolite H-BEA to diethylglycolide in high yields and enantiomeric purity has been shown. Access to the enantiomerically pure cyclic ester allowed us to investigate a range of unexplored high-molecular weight, random co-polymers with lactide, exhibiting spectacular melt properties. α-Hydroxybutyric acid, the precursor to diethylglycolide, is naturally produced in mammalian hepatic tissues.66 (Bio)chemical syntheses of this hydroxy acid are known, starting from naturally occurring compounds such as α-ketobutyric acid,67L-threonine,68 alpha-aminobutyric acid69 or methyl vinyl glycolate.70 Although chemical syntheses by reacting butyraldehyde with NaClO or by heating α-bromobutyric acid with formamide71 are probably the main routes at the moment, biotechnological routes to obtain α-hydroxybutyric acid seem feasible. PLA co-polymers containing randomly distributed, small amounts (up to 10 mol%) of diethylglycolide, importantly of opposite chiral nature compared to lactide, result in unique polymer materials with high melt elasticity. The effect was successfully reproduced at large scale, while additional extensional viscosity measurements on the large-scale co-polymer samples confirmed the presumption of the melt strength improving effect of EG. A large scale co-polymer of (L,L)-lactide with (D,D)-diethylglycolide (1.2 mol%) exhibited an extensional viscosity and extensional force both substantially higher than those of common L-PLA. Interestingly, these values were also significantly better than those of the commercial grade PLA (Ingeo 7001D) reference, that is deliberately designed for use in injection stretch blow molding applications. It is hypothesized that the beneficial melt elasticity effects are related to the screening role of the ethyl side groups, lowering interchain interactions and the presence of intrachain loops, and foreseeing polymer coils with a larger radius of gyration that are prone to chain entanglements. Given that this work is the first of its kind that demonstrates such effects in PLA, discarding the use of chain extenders, blending, compounding or stereocomplexation, it may lay a new foundation for unexplored pathways towards high melt strength materials via co-polymerization. A stronger melt can translate into less plastic usage per item, for the same functionality, thus lowering the overall ecological and economic footprint of bioplastic items, where strong extensional forces are needed in the processing (e.g. during blow molding of bottles, fibre spinning, foaming, etc.).Author contributions
A.D., P.V.P and M.D. initiated the research and discovered the first proofs of concept. A.S.N confirmed the proofs, elaborated most experimental findings and did large scale rheology testing. K.B. assisted with and established polymerization protocols. All authors analyzed the data. A.S.N. and M.D. wrote the manuscript with critical input by P.V.P, K.B. and B.S.Conflicts of interest
There are no conflicts to declare. A.S.N, A.D., B.S., P.V.P and M.D. filed a European patent application on this invention.Acknowledgements
A.D., P.V.P and M.D. acknowledge KU Leuven for a BOF-PDM grant to A.D. M.D. acknowledges KU Leuven for startup funding as BOFZAP. The authors acknowledge Corbion for kindly providing (L,L) and (D,D)-lactide.Notes and references
- J. Hammer, M. H. S. Kraak and J. R. Parsons, in Reviews of Environmental Contamination and Toxicology, Springer, New York, 2012, pp. 1–44 Search PubMed.
- C. Wilcox, E. Van Sebille and B. D. Hardesty, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 11899–11904 CrossRef CAS PubMed.
- M. Xiong, D. K. Schneiderman, F. S. Bates, M. A. Hillmyer and K. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8357–8362 CrossRef CAS PubMed.
- R. Auras, L.-T. Lim, S. E. M. Selke and H. Tsuji, Poly(lactic acid): Synthesis, Structures, Properties, Processing, and Applications, John Wiley & Sons, Hoboken, New Jersey, 2010 Search PubMed.
- M. Jamshidian, E. A. Tehrany, M. Imran, M. Jacquot and S. Desobry, Compr. Rev. Food Sci. Food Saf., 2010, 9, 552–571 CrossRef CAS PubMed.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- S. Farah, D. G. Anderson and R. Langer, Adv. Drug Delivery Rev., 2016, 107, 367–392 CrossRef CAS PubMed.
- S. Inkinen, M. Hakkarainen, A.-C. Albertsson and A. Södergård, Biomacromolecules, 2011, 12, 523–532 CrossRef CAS PubMed.
- P. Gruber, et al., US Pat.5142023, 1992 Search PubMed.
- D. K. Yoo and D. Kim, Macromol. Res., 2006, 14, 510–516 CrossRef CAS.
- M. Dusselier, P. Van Wouwe, A. Dewaele, P. A. Jacobs and B. F. Sels, Science, 2015, 349, 79–81 CrossRef PubMed.
- P. P. Upare, J. W. Yoon, D. W. Hwang, U. H. Lee, Y. K. Hwang, D. Y. Hong, J. C. Kim, J. H. Lee, S. K. Kwak, H. Shin, H. Kim and J. S. Chang, Green Chem., 2016, 18, 5978–5983 RSC.
- R. De Clercq, M. Dusselier, E. Makshina and B. F. Sels, Angew. Chem., 2018, 57, 3074–3078 CrossRef CAS.
- S. Heo, H. W. Park, J. H. Lee and Y. K. Chang, ACS Sustainable Chem. Eng., 2019, 7, 6178–6184 CrossRef CAS.
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820–852 CrossRef CAS.
- J. Dorgan, in Poly(lactic acid): Synthesis, Structure, Properties, Processing and Applications, ed. H. T. Rafael Auras, Loong-Tak Lim and Susan E. M. Selke, John Wiley & Sons, New York, 2010, pp. 125–139 Search PubMed.
- M. Nofar and C. B. Park, Prog. Polym. Sci., 2014, 39, 1721–1741 CrossRef CAS.
- J. R. Dorgan, J. S. Williams and D. N. Lewis, Rheol. Bull., 1999, 43, 1141–1155 CrossRef CAS.
- A. Michalski, M. Brzezinski, G. Lapienis and T. Biela, Prog. Polym. Sci., 2019, 89, 159–212 CrossRef CAS.
- J. Liu, L. Lou, W. Yu, R. Liao, R. Li and C. Zhou, Polymer, 2010, 51, 5186–5197 CrossRef CAS.
- Y. M. Corre, J. Duchet, J. Reignier and A. Maazouz, Rheol. Acta, 2011, 50, 613–629 CrossRef CAS.
- L. Wang, X. Jing, H. Cheng, X. Hu, L. Yang and Y. Huang, Ind. Eng. Chem. Res., 2012, 51, 10731–10741 CrossRef CAS.
- S. Nouri, C. Dubois and P. G. Lafleur, J. Polym. Sci., Part B: Polym. Phys., 2015, 53, 522–531 CrossRef CAS.
- M. Mihai, M. A. Huneault and B. D. Favis, Polym. Eng. Sci., 2010, 50, 629–642 CrossRef CAS.
- M. A. Ghalia and Y. Dahman, Int. J. Biol. Macromol., 2017, 95, 494–504 CrossRef PubMed.
- F. Iñiguez-Franco, R. Auras, J. Ahmed, S. Selke, M. Rubino, K. Dolan and H. Soto-Valdez, Polym. Test., 2018, 67, 190–196 CrossRef.
- W. Limsukon, R. Auras and S. Selke, Polym. Test., 2019, 80, 106108 CrossRef CAS.
- M. Nofar, R. Salehiyan and S. Sinha Ray, Polym. Rev., 2019, 59, 465–509 CrossRef CAS.
- M. Murariu and P. Dubois, Adv. Drug Delivery Rev., 2016, 107, 17–46 CrossRef CAS PubMed.
- Y. Ikada, K. Jamshidi, H. Tsuji and S. H. Hyon, Macromolecules, 1987, 20, 904–906 CrossRef CAS.
- H. Yamane, K. Sasai, M. Takano and M. Takahashi, Rheol. Bull., 2004, 48, 599–609 CrossRef CAS.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, J. Appl. Polym. Sci., 2014, 41073, 1–8 Search PubMed.
- P. Van Wouwe, M. Dusselier, E. Vanleeuw and B. Sels, ChemSusChem, 2015, 9, 907–921 CrossRef PubMed.
- L.-E. Chile, P. Mehrkhodavandi and S. G. Hatzikiriakos, Macromolecules, 2016, 49, 909–919 CrossRef CAS.
- J. R. Dorgan, J. Janzen, M. P. Clayton, S. B. Hait and D. M. Knauss, Rheol. Bull., 2005, 49, 607–619 CrossRef CAS.
- N. Othman, A. Acosta-Ramírez, P. Mehrkhodavandi, J. R. Dorgan and S. G. Hatzikiriakos, Rheol. Bull., 2011, 55, 987–1005 CrossRef CAS.
- M. Yin and G. L. Baker, Macromolecules, 1999, 32, 7711–7718 CrossRef CAS.
- E. Vogel, M. R. Smith III and G. L. Baker, in Proceedings of the American Chemical Society, 2007, p. 2 Search PubMed.
- K. D. Jandt and U. S. Schubert, Polym. Chem., 2019, 10, 5440–5451 RSC.
- S. Kaihara, S. Matsumura, A. G. Mikos and J. P. Fisher, Nat. Protoc., 2007, 2, 2767–2771 CrossRef CAS PubMed.
- H. R. Kricheldorf, I. Kreiser-Saunders and C. Boettcher, Polymer, 1995, 36, 1253–1259 CrossRef CAS.
- M. Ryner, K. Stridsberg, A. C. Albertsson, H. Von Schenck and M. Svensson, Macromolecules, 2001, 34, 3877–3881 CrossRef CAS.
- W. Saiyasombat, R. Molloy, T. M. Nicholson, A. F. Johnson, I. M. Ward and S. Poshyachinda, Polymer, 1998, 39, 5581–5585 CrossRef CAS.
- F. Jing, M. R. Smith and G. L. Baker, Macromolecules, 2007, 40, 9304–9312 CrossRef CAS.
- T. Trimaille, M. Möller and R. Gurny, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4379–4391 CrossRef CAS.
- G. L. Baker, E. B. Vogel and M. R. Smith III, Polym. Rev., 2008, 48, 64–84 CrossRef CAS.
- F. T. Trouton, Proc. R. Soc. London, Ser. A, 1906, A77, 426–440 Search PubMed.
- L. I. Palade, H. J. Lehermeier and J. R. Dorgan, Macromolecules, 2001, 34, 1384–1390 CrossRef CAS.
- J. J. Cooper-white and M. E. Mackay, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 1803–1814 CrossRef CAS.
- N. Najafi, M. C. Heuzey, P. J. Carreau, D. Therriault and C. B. Park, Rheol. Acta, 2014, 53, 779–790 CrossRef CAS.
- R. Al-itry, K. Lamnawar and A. Maazouz, Eur. Polym. J., 2014, 58, 90–102 CrossRef CAS.
- Y.-M. Corre, A. Maazouz, J. Duchet and J. Reignier, J. Supercrit. Fluids, 2011, 58, 177–188 CrossRef CAS.
- M. Gagliardi and A. Bifone, PLoS One, 2018, 13, 1–15 Search PubMed.
- A. T. Hedegaard, L. Gu and C. W. Macosko, J. Rheol., 2015, 59, 1397–1417 CrossRef CAS.
- L. Gu, Y. Xu, G. W. Fahnhorst and C. W. Macosko, J. Rheol., 2017, 61, 785–796 CrossRef CAS.
- H. Tsuji, S. Yamamoto, A. Okumura and Y. Sugiura, Biomacromolecules, 2010, 11, 252–258 CrossRef CAS PubMed.
- H. Tsuji and A. Okumura, Macromolecules, 2009, 42, 7263–7266 CrossRef CAS.
- H. Tsuji, K. Shimizu, Y. Sakamoto and A. Okumura, Polymer, 2011, 52, 1318–1325 CrossRef CAS.
- M. Rubinstein and A. N. Semenov, Macromolecules, 2001, 34, 1058–1068 CrossRef CAS.
- W. C. Forsman, Macromolecules, 1982, 15, 1032–1040 CrossRef CAS.
- L. G. Baxandall, Macromolecules, 1989, 22, 1982–1988 CrossRef CAS.
- J. Y. Carrillo, W. Chen, Z. Wang, B. G. Sumpter and Y. Wang, J. Phys. Commun., 2019, 3, 1–6 Search PubMed.
- D. E. Dunstan, Sci. Rep., 2019, 9, 1–9 CAS.
- L. J. Fetters, D. J. Lohse, D. Richter, T. A. Witten and A. Zirkel, Macromolecules, 1994, 27, 4639–4647 CrossRef CAS.
- W. E. Gall, K. Beebe, K. A. Lawton, K.-P. Adam, M. W. Mitchell, P. J. Nakhle, J. A. Ryals, M. V. Milburn, M. Nannipieri, S. Camastra, A. Natali and E. Ferrannini, PLoS One, 2010, 5, 1–11 CrossRef PubMed.
- M.-J. Kim and G. M. Whitesides, J. Am. Chem. Soc., 1988, 110, 2959–2964 CrossRef CAS.
- P. Yao, Y. Cui, S. Yu, Y. Du, J. Feng, Q. Wu and D. Zhu, Adv. Synth. Catal., 2016, 358, 2923–2928 CrossRef CAS.
- H. Tsuji, K. Nakayama and Y. Arakawa, RSC Adv., 2020, 10, 39000–39007 RSC.
- A. Sølvhøj, E. Taarning and R. Madsen, Green Chem., 2016, 18, 5448–5455 RSC.
- K. Miltenberger, in Ullmann's Encyclopedia of Industrial Chemistry, 2012, pp. 481–491 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Extended information about materials and methods (S1–S2), extended reading (S3–S7), and extended data in Tables S1–S6 and in Fig. S1–S18. See DOI: 10.1039/d1sc00040c |
| This journal is © The Royal Society of Chemistry 2021 |