
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA single point mutation converts a proton-pumping rhodopsin into a red-shifted, turn-on fluorescent sensor for chloride†
Jasmine N.
Tutol
 a,
Jessica
Lee
ab,
Hsichuan
Chi
ab,
Farah N.
Faizuddin
ab,
Sameera S.
Abeyrathna
a,
Qin
Zhou
b,
Faruck
Morcos
bc,
Gabriele
Meloni
a and
Sheel C.
Dodani
*a
a,
Jessica
Lee
ab,
Hsichuan
Chi
ab,
Farah N.
Faizuddin
ab,
Sameera S.
Abeyrathna
a,
Qin
Zhou
b,
Faruck
Morcos
bc,
Gabriele
Meloni
a and
Sheel C.
Dodani
*a
aDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson, TX 75080, USA. E-mail: sheel.dodani@utdallas.edu
bDepartment of Biological Sciences, The University of Texas at Dallas, Richardson, TX 75080, USA
cDepartment of Bioengineering, The University of Texas at Dallas, Richardson, TX 75080, USA
First published on 17th March 2021
Abstract
The visualization of chloride in living cells with fluorescent sensors is linked to our ability to design hosts that can overcome the energetic penalty of desolvation to bind chloride in water. Fluorescent proteins can be used as biological supramolecular hosts to address this fundamental challenge. Here, we showcase the power of protein engineering to convert the fluorescent proton-pumping rhodopsin GR from Gloeobacter violaceus into GR1, a red-shifted, turn-on fluorescent sensor for chloride in detergent micelles and in live Escherichia coli. This non-natural function was unlocked by mutating D121, which serves as the counterion to the protonated retinylidene Schiff base chromophore. Substitution from aspartate to valine at this position (D121V) creates a binding site for chloride. The binding of chloride tunes the pKa of the chromophore towards the protonated, fluorescent state to generate a pH-dependent response. Moreover, ion pumping assays combined with bulk fluorescence and single-cell fluorescence microscopy experiments with E. coli, expressing a GR1 fusion with a cyan fluorescent protein, show that GR1 does not pump ions nor sense membrane potential but instead provides a reversible, ratiometric readout of changes in extracellular chloride at the membrane. This discovery sets the stage to use natural and laboratory-guided evolution to build a family of rhodopsin-based fluorescent chloride sensors with improved properties for cellular applications and learn how proteins can evolve and adapt to bind anions in water.
Introduction
Chloride is an essential inorganic ion for life.1,2 Beyond its role as an electrolyte, chloride mobilization across cellular membranes is known to be involved in a wide range of homeostatic processes including, but not limited to, pH regulation, fluid excretion, and electrical activity.2–4 The growing importance of chloride in normal physiology and disease is further evident by recent discoveries of new transporters and biological roles, which have been accelerated through the application of fluorescent sensors for chloride.5–9 The fluorescence imaging of chloride in living cells is linked to our ability to build hosts that can recognize chloride in water, a fundamental challenge in the field of supramolecular chemistry.10–12 Advances along these lines could contribute to our general understanding of aqueous anion recognition and to the design of new fluorescent sensors for chloride.A key limitation for the recognition of chloride in water is the inherent energetic penalty for desolvation.10,12–15 To overcome this, successful examples of fluorescent molecular and polymer hosts integrate Nature's design principles and combine cooperative, non-covalent interactions, including hydrogen bonding, ion-pairing, and Van der Waals with the hydrophobic effect.4,11,16–20 However, synthetic hosts that allow for the fluorescence imaging of chloride in living cells remain rare and have been limited to quinolinium and acridinium-based fluorophores. The positively charged pyridinium cation directly interacts with chloride, bromide, and even iodide resulting in collisional fluorescence quenching.11,18,21,22 These molecular fluorescent sensors are pH-independent, and additional bioconjugation strategies have enabled improved cellular uptake, organelle targeting, and ratiometric fluorescent outputs.4,23–28 It is important to note that the lack of selectivity has not been a point of improvement for cellular applications since chloride is the most abundant halide.4
As a complementary approach, we use and evolve Nature to build protein-based hosts for chloride. Even though there are only twenty proteinogenic amino acids, protein sequence space is vast and can be further enriched through protein engineering methods.29 By selecting for a desired function or property, laboratory-guided evolution allows for this sequence space to be quickly sampled and filtered.29 For our goal, this approach affords a diverse pool of hosts with new and improved properties for the fluorescence imaging of chloride in living cells. Along these lines, the green fluorescent protein family (GFP, Pfam: PF01353) has been widely explored and engineered to build fluorescent sensors for chloride.4,30,31 Indeed, pioneering work from Wachter and Remington established that variants of the green fluorescent protein (avGFP) from the jellyfish Aequorea victoria are sensitive to chloride.32–34 Of these, the yellow fluorescent protein avYFP-H148Q is well characterized. As shown in the crystal structure bound to iodide (PDB ID: 1F09), the halide binding pocket in avYFP-H148Q is lined with hydrophobic residues and four polar amino acids, including two glutamines, arginine, and tyrosine, that can hydrogen bond with the anion (Fig. 1A and B).34 The latter tyrosine residue connects the binding of chloride through a π–π interaction with the 4-(p-hydroxybenzylidene)imidazolidin-5-one chromophore.34 Because of this close proximity, chloride binding increases the pKa of the chromophore and shifts the equilibrium from the fluorescent phenolate state towards the non-fluorescent phenol state, generating a turn-off fluorescence response.34,35 The E2GFP variant of avGFP is also sensitive to chloride but operates via static fluorescence quenching with no change in the chromophore equilibrium.36 This sensing mechanism could be attributed to a different chloride binding pocket; the chloride ion forms four hydrogen bonds: one with a tyrosine residue, two with the peptide backbone connecting the chromophore to valine and glutamine residues, and one with a water molecule (Fig. 1C).36 Both avYFP-H148Q and E2GFP, and variants thereof, have been applied for the fluorescence imaging of chloride despite being pH-dependent, turn-off, and not selective over bromide, iodide, or nitrate.31,35,37–41 However, fusion with other fluorescent proteins have enabled ratiometric fluorescent outputs that account for variability in protein expression or pH dependence in living cells but still require rigorous experimental controls.42–48
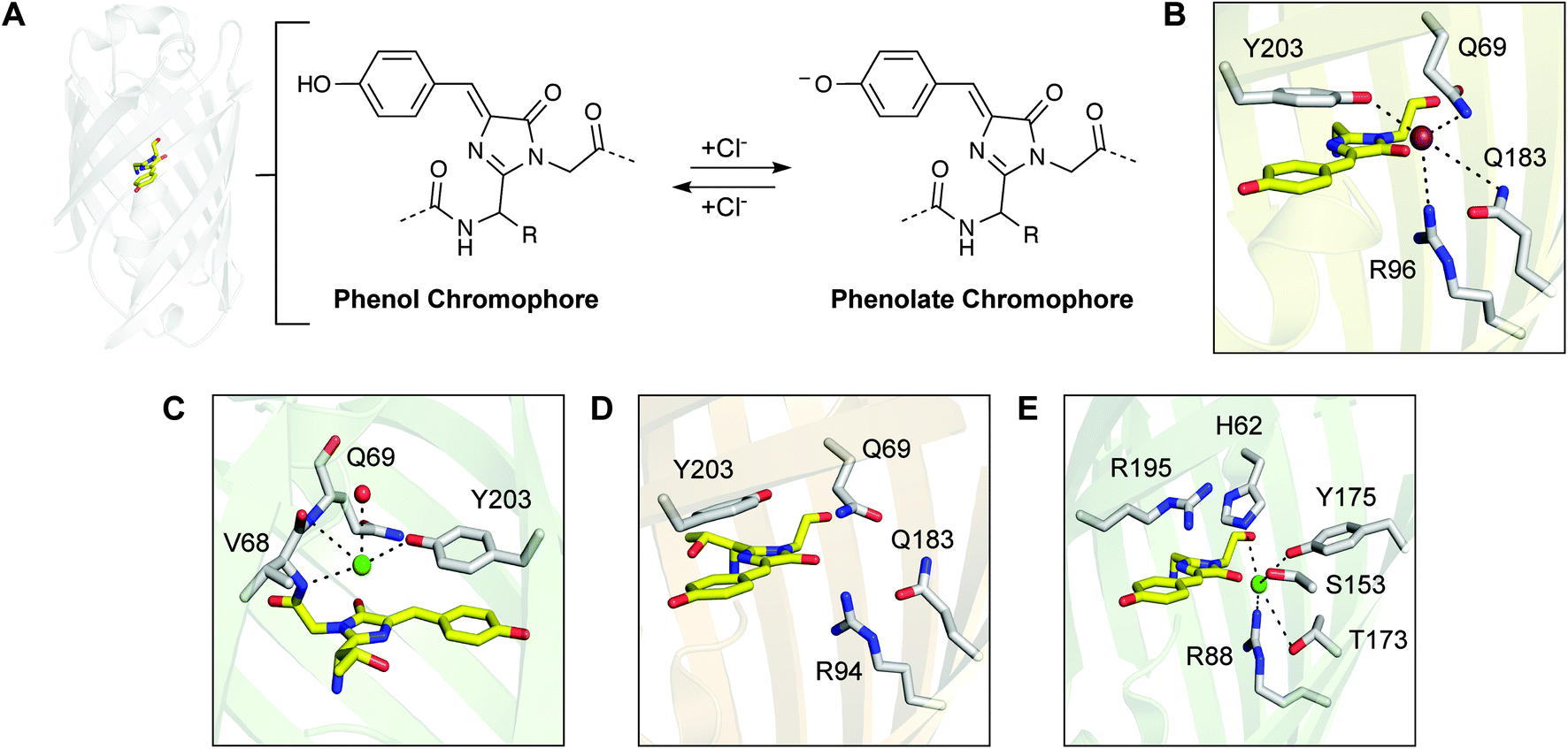 | ||
| Fig. 1 (A) The overall β-barrel structure of a representative chloride-sensitive fluorescent protein with the chromophore shown as sticks in yellow (left, PDB ID: 1F09), and a general schematic of the chloride-dependent conversion of the two chromophore states (right). Comparison of the anion binding sites in the crystal structures of (B) avYFP-H148Q with iodide (PDB ID: 1F09), (C) E2GFP with chloride (PDB ID: 2O24), (D) a variant of phiYFP (PDB ID: 4HE4), and (E) mNeonGreen with chloride (PDB ID: 5LTP). Note: the structure in panel C is rotated by 180° in the counter-clockwise direction. For each protein, the chromophore is shown as yellow sticks, and the residues corresponding to the anion binding pocket are shown as light gray sticks with the oxygen (red) and nitrogen (blue) atoms. Residues are labeled with the position number and the single letter amino acid code. Hydrogen bonds are shown as black dashed lines. The iodide ion, chloride ions, and water molecule are shown as maroon, green, and red spheres, respectively. | ||
Relative to these literature precedents, we have identified low sequence identity homologues that are also sensitive to chloride but with different properties.49,50 The chloride binding pocket in the naturally occurring yellow fluorescent protein from the jellyfish Phialidium sp. (phiYFP) is identical to that of avYFP-H148Q (Fig. 1D).34,51 Similarly, chloride binding increases the pKa of the chromophore but generates an excitation ratiometric, turn-on fluorescence response, likely through an excited state proton transfer process.49 In addition, through a structure-guided search, we identified that the yellow fluorescent protein lanYFP from the cephalochordate Branchiostoma lanceolatum and its engineered monomer mNeonGreen also bind chloride.52 In mNeonGreen, chloride forms five hydrogen bonds with histidine, arginine, serine, threonine, and tyrosine residues near the chromophore (Fig. 1E).52 Based on this, we recently reported that chloride binding decreases the pKa of the chromophore and shifts the equilibrium from the non-fluorescent phenol state to the highly fluorescent phenolate state, generating a turn-on fluorescence response.50 Furthermore, we attributed this unique sensing mechanism to a non-coordinating arginine residue, instead of a tyrosine residue, above the chromophore. Together, these homologues highlight how differences in the amino acid residues, within and outside of the chloride binding pocket and even in the chromophore, can influence sensor properties. This is a key advantage of sampling and enriching the vast protein sequence space to build hosts for chloride.
To date, protein-based fluorescent sensors for chloride have only been based on the GFP family. Beyond these advances, we envisioned that microbial rhodopsins (Bac_rhodopsin, Pfam: PF01036) could serve as new scaffolds to engineer fluorescent sensors for chloride with the added advantages of red-shifted spectral properties and membrane localization. The latter could expand current biological applications to monitor the uptake and recycling of chloride at the plasma or organelle membranes.53 Microbial rhodopsins are light-activated, transmembrane ion pumps with seven α-helices and a covalently bound retinylidene Schiff base chromophore (SBC) – generated from the condensation of all-trans-retinal and a lysine residue (Fig. 2A).54–58 The SBC and its protonation state are stabilized by a key counterion position, which plays a role in the ion pumping pathway and selectivity.59 In chloride-pumping rhodopsins (CPRs), this counterion position corresponds to a threonine or asparagine, such that the chloride ion serves as the counterion to the protonated SBC.54,56,59–65 This is observed in the X-ray crystal structures of representative CPRs from Halobacterium salinarum (HR) and Nonlabens marinus S1-08T (ClR) (Fig. 2B and C).60,63,64 The chloride binding pocket also consists of polar amino acids, including serine and threonine, and highly ordered water molecules that can form hydrogen bonding interactions with the chloride ion.63,64 However, the homologous counterion position in proton-pumping rhodopsins (PPRs) corresponds to a negatively charged aspartate, which accepts the proton from the protonated SBC (Fig. 2D).56,62,66 Interestingly, the well-studied PPR bacteriorhodopsin (BR) from Halobacterium salinarum can be converted into a CPR by substituting the aspartate counterion position (D85) with serine, threonine, or asparagine.67 Absorption spectroscopy indicates that these variants bind chloride and other anions, including bromide, iodide, and nitrate, but have varying pumping activities.67–70 Of the three variants, BR D85S has been crystallized with bromide (Fig. 2E).71 Similar to CPRs, the serine residue at the counterion position directly interacts with the bromide ion, which forms a hydrogen bond with the protonated SBC.71 The residues R82, mediated by a highly ordered water molecule, and D212 form two hydrogen bonding interactions with bromide, thus adapting to accommodate this non-natural function.71 Outside of these contexts, when illuminated with near-infrared light, PPRs can also access fluorescent states in the photocycle.72–75 Because of this property, engineered PPRs have been repurposed as fluorescent proton sensors to provide a readout of the membrane potential at plasma or mitochondrial membranes in cells and in vivo.53,73,74,76–78 On a molecular level, the protonation state of the SBC controls the on and off fluorescent states, which can be influenced by the counterion position.73,74
 | ||
| Fig. 2 (A) Microbial rhodopsins are light-activated, transmembrane ion pumps with a covalently bound retinylidene Schiff base chromophore (SBC), formed from all-trans-retinal (yellow sticks) and a conserved lysine residue (Lys, black sticks). The counterion position (black sticks) stabilizes the protonated SBC (PSBC) and controls ion selectivity (PDB ID: 6NWD). Comparison of (B) the chloride-pumping rhodopsin (CPR) from Halobacterium salinarum (HR) with chloride (PDB ID: 5AHY), (C) the CPR from Nonlabens marinus S1-08T (ClR) with chloride (PDB ID: 5G28), (D) the proton-pumping rhodopsin (PPR) from Halobacterium salinarum (BR, PDB ID: 1KGB), (E) the engineered PPR BR D85S with bromide (PDB ID: 1MGY), and (F) the fluorescent PPR from Gloeobacter violaceus (wtGR, PDB ID: 6NWD) selected for this study. For each structure, the residues corresponding to the anion binding pocket in the CPRs and BR D85S are shown as light gray sticks with the oxygen (red) and nitrogen (blue) atoms. Hydrogen bonds are shown as black dashed lines. Each residue is labeled with the position number and the single letter amino acid code. The retinal (RET) is shown as sticks in yellow covalently bound to a conserved lysine residue, generating the SBC. The counterion position to the SBC is labeled in bold. The water molecules, chloride ions, and bromide ion are shown as spheres in red, green, and purple, respectively. | ||
Motivated by these studies, we hypothesized that chloride could replace the aspartate as the counterion in a fluorescent PPR, form an electrostatic interaction with the protonated SBC, and tune the optical properties to generate a fluorescent sensor for chloride. To test this, we selected the fluorescent PPR from the cyanobacterium Gloeobacter violaceus (GR) because it has been spectroscopically and structurally characterized, can readily be expressed in Escherichia coli, and remains fluorescent even after the introduction of multiple mutations (Fig. 2F).79–91 We have developed a workflow to rapidly evolve, screen, and discover chloride-sensitive variants of GR. Based on this screen, we highlight how a single point mutation at the SB counterion position from aspartate to valine, a non-natural substitution, transforms wild-type (wt) GR from a proton pump into GR1, a red-shifted, turn-on fluorescent sensor for chloride. In vitro data indicates that chloride binding to GR1, but not wtGR, increases the pKa of the SBC, shifting the equilibrium towards the protonated, fluorescent state, generating a pH-dependent, turn-on response. Ion pumping assays combined with bulk fluorescence measurements and single-cell fluorescence imaging of live E. coli demonstrate that GR1 does not pump ions nor sense membrane potential but instead is a reversible sensor for changes in extracellular chloride at the membrane.
Results and discussion
Fluorescence library screening and identification of GR sensors for chloride
The fluorescence of wtGR was evaluated in live E. coli at pH 5, 6, and 7 in the absence and presence of 400 mM sodium chloride. This screening method relies on the fact that the bacterial periplasmic space can readily equilibrate with the conditions of the extracellular environment.92 Since wtGR is localized in the inner-membrane and is exposed to the periplasm, it can provide a readout for changes in extracellular chloride levels at the membrane. To account for any variability in protein expression, wtGR was fused to a chloride-insensitive cyan fluorescent protein (CFP) to generate the wtGR–CFP construct as previously described (Fig. S1–S3†).90 The soluble CFP tag is localized to the cytosol and normalization to this signal generates a ratiometric output. Upon excitation at 530 nm, the rhodopsin-based emission of wtGR–CFP is centered at 710 nm and does not change in the presence of chloride (Fig. 3 and S3†). Next, site-saturation mutagenesis (SSM) was carried out at position 121 to sample all possible amino acid substitutions that could give rise to a chloride-sensitive variant. To our surprise, mutation from aspartate to valine (GR1–CFP) results in a ca. 2.2-fold turn-on fluorescence response with 400 mM sodium chloride at pH 5 (Fig. 3 and S4, S5†). In addition, substitution with leucine (ca. 1.2-fold), threonine (ca. 1.3-fold), asparagine (ca. 1.3-fold), or isoleucine (ca. 1.5-fold) results in a turn-on fluorescence response, albeit to a lesser extent (Fig. S4 and S6–S9†). No chloride-sensitive variants were identified in the library screen at pH 6 or pH 7. However, upon rescreening, GR1–CFP shows a ca. 1.2-fold turn-on fluorescence response at pH 6 (Fig. 3). Given these results, the CFP tag was removed from the wtGR and GR1 constructs, and comparable results were observed in live E. coli (Fig. S10†). Interestingly, asparagine and threonine are enriched at this counterion position in a subset of CPRs, whereas, the hydrophobic substitutions, such as leucine, isoleucine, and valine, have yet to be identified at this position in the rhodopsin family (sequence identity > 23%, Fig. S4C†). | ||
| Fig. 3 Site-saturation mutagenesis at the D121 counterion position converts wtGR–CFP into the chloride-sensitive variant GR1–CFP. Bars represent the integrated emission response of the rhodopsin (FGR) normalized by the integrated emission of CFP (FCFP) in the absence (black bars) and presence of 400 mM sodium chloride (gray bars). Spectra were acquired with live Escherichia coli expressing wtGR–CFP or GR1–CFP in 50 mM sodium acetate buffer at pH 5, 50 mM sodium phosphate buffer at pH 6, and 50 mM sodium phosphate buffer at pH 7. Excitation for the rhodopsin was provided at 530 nm, and the emission was collected and integrated from 600–800 nm (FGR). Excitation for CFP was provided at 390 nm, and the emission was collected and integrated from 425–560 nm (FCFP). The average of three biological replicates with standard error of the mean is reported. | ||
Spectroscopic characterization of purified wtGR and GR1
Next, the in vitro properties of purified wtGR and GR1 in detergent micelles were compared in sodium acetate buffer at pH 5, supplemented with sodium gluconate for protein stability (Fig. S11 and S12†).69,93,94 Consistent with previously reported absorbance spectra, wtGR has one absorption maximum at 545 nm that corresponds to the covalently bound SBC (Fig. S13†).80,81,86,87,89,91 Upon excitation at 530 nm, wtGR has a broad emission band with a maximum centered at 705 nm (Φapo = 2.0 × 10−3, Fig. 3A and S14†).84 The wtGR absorption and emission maxima do not shift upon the addition of 400 mM sodium chloride (Fig. S13†). Instead, the emission intensity increases ca. 1.2-fold with no clear indication of binding (ΦCl = 2.4 × 10−3, Fig. 4A and S14†). As expected, the absorption maximum of GR1 is red-shifted to 560 nm and does not change upon the addition of sodium chloride (Fig. S15†).90 However, with excitation at 530 nm, GR1 displays a ca. 1.6-fold turn-on fluorescence response that saturates with 400 mM sodium chloride at 705 nm (Φapo = 3.0 × 10−3, ΦCl = 3.9 × 10−3) with no shift in the emission maximum, suggesting an excited state process (Fig. 4B and S14, S15†). This titration data can be fitted to a single site binding model for chloride resulting in an apparent dissociation constant (Kd) of 203 ± 41 mM (Fig. S15†). Of note, GR1 can detect as low as 12.5 mM sodium chloride with a ca. 1.1 turn-on fluorescence response, whereas wtGR does not show a response. | ||
| Fig. 4 Characterization of purified wtGR and GR1 in detergent micelles shows that GR1 has a chloride binding site and is also sensitive to bromide, iodide, and nitrate. Fluorescence spectra of ∼3 μM (A) wtGR and (B) GR1 in the presence of 0 (bold), 12.5, 25, 50, 100, 200, 300, and 400 mM (red) sodium chloride. Insets: integrated emission response (y-axis) to sodium chloride. (C) Integrated emission response of ∼3 μM GR1 to 0 mM (Fi, Ctrl) or 400 mM (Ff) sodium chloride, bromide, iodide, nitrate, or dihydrogen phosphate. All spectra were acquired in 50 mM sodium acetate buffer containing 600 mM sodium gluconate at pH 5. Excitation for the rhodopsin was provided at 530 nm, and the emission was collected and integrated from 600–800 nm. The average of three technical replicates with standard error of the mean is reported. | ||
Moreover, a turn-on fluorescence response is also observed with 400 mM of sodium bromide (ca. 1.6-fold, Kd = 194 ± 14 mM) and iodide (ca. 1.8-fold, Kd = 170 ± 34 mM), but quenching is observed with sodium nitrate (∼20%) and no change is observed with sodium dihydrogen phosphate (Fig. 4C and S16–S18†). As described above, these anion preferences are not only found in GFP-based sensors but also naturally occurring CPRs and engineered variants of the PPR BR suggesting that the anion binding site in GR1 could be adjacent to the SBC. To determine how the binding of chloride contributes to the turn-on fluorescence response, we next measured the pKa of the SBC with fluorescence spectroscopy. The pKa for apo wtGR is 4.9 ± 0.1 and does not significantly change upon the addition of 400 mM sodium chloride (pKa = 4.6 ± 0.1, p-value > 0.05, Fig. S19†). These values are consistent with that previously reported for wtGR (pKa = 4.5) in the presence of 300 mM sodium chloride using absorption spectroscopy.87 Interestingly, the pKa for GR1 increases from 3.1 ± 0.1 to 4.8 ± 0.1 with 400 mM sodium chloride (Fig. S20†). These data provide evidence that the binding of chloride to GR1 shifts the equilibrium towards the protonated, fluorescent SBC resulting in the observed turn-on fluorescence response. Although it has been demonstrated that the fluorescent state of the SBC is linked to the SBC protonation state, we cannot rule out other factors that could be contributing to the chloride sensing mechanism.73,74 We do note that below pH 5, wtGR, like GR1, has a turn-on fluorescence response to 400 mM sodium chloride, but comparatively, the binding with chloride cannot be saturated (for wtGR: Kd = 944 ± 123 mM at pH 4, Fig. S21;† for GR1: Kd = 97 ± 6 mM at pH 4, Fig. S22†). Indeed, at pH values below the pKa, protonation of D121 and other residues could give rise to a weak binding site for chloride in wtGR. Nonetheless, together our data demonstrates that mutation of the SBC counterion residue creates a chloride binding site, generating a turn-on fluorescent sensor. We speculate that, like BR D85S, GR1 adapts to this new function. In line with our hypothesis, substitution of the negatively charged aspartate with a hydrophobic valine allows for the chloride ion to bind instead and interact with the protonated SBC along with nearby hydrogen-bond donating residues or water molecules (Fig. 2E and F). These additional interactions combined with the desolvation of the chloride ion could contribute favorably to the overall driving force for chloride binding. Moreover, the gain from the desolvation of GR1 would likely be minimal since it is a hydrophobic, transmembrane protein.13–15,95
Application and validation of GR1–CFP in live E. coli
With in vitro characterization in hand, we turned our attention to validating GR1–CFP as a ratiometric fluorescent sensor for chloride in living cells using bulk and single-cell fluorescence measurements. For this proof-of-concept, E. coli cells were selected due to their tolerance in pH 5 buffer supplemented with chloride.92,96 Consistent with the library screening, cells expressing wtGR–CFP show a measurable level of fluorescence that does not change with increasing concentrations of sodium chloride (Fig. 5A and S23†). In contrast, the fluorescence of cells expressing GR1–CFP increases, in a dose-dependent manner, by ca. 2.5-fold with 400 mM sodium chloride and no interference from sodium acetate (Fig. 5B and S24, S25†). A fluorescence response (ca. 1.3-fold) is detected with as little as 12.5 mM sodium chloride.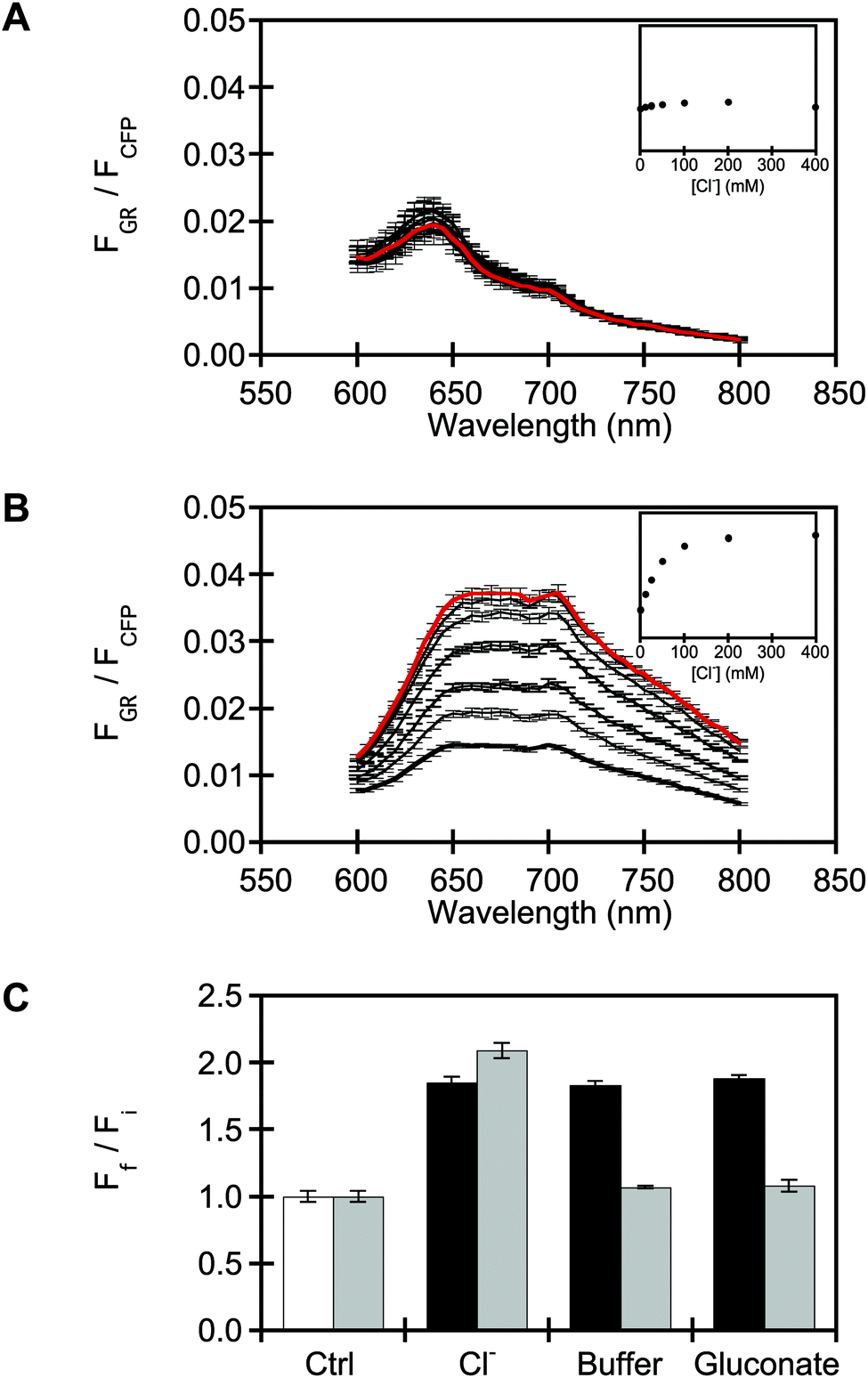 | ||
| Fig. 5 Bulk fluorescence measurements show that GR1–CFP is a ratiometric, turn-on fluorescent sensor that dynamically responds to changes in sodium chloride concentrations in live E. coli cells. Normalized fluorescence spectra of (A) wtGR–CFP and (B) GR1–CFP in the presence of 0 (bold), 12.5 25, 50, 100, 200, and 400 mM (red) sodium chloride. Inset: integrated emission response of the rhodopsin (FGR) over the integrated emission of CFP (FCFP) (y-axis) to sodium chloride. (C) Normalized integrated emission response (FGR/FCFP) of GR1–CFP in the presence of buffer (Ctrl, Fi, white bar) and 400 mM (Ff) sodium chloride (black bars). Buffer-treated cells were re-equilibrated with buffer only (Ctrl, Fi, gray bar), and sodium chloride-treated cells were re-equilibrated with 400 mM sodium chloride, sodium acetate buffer, or sodium gluconate (Ff, gray bars). All spectra were acquired with E. coli expressing wtGR–CFP or GR1–CFP in 50 mM sodium acetate buffer at pH 5. Excitation for the rhodopsin was provided at 530 nm, and the emission was collected and integrated from 600–800 nm (FGR). Excitation for CFP was provided at 390 nm, and the emission was collected and integrated from 425–560 nm (FCFP). The average of three biological replicates with standard error of the mean is reported. | ||
Similar to the in vitro measurements, a single site binding model can also be used to fit this titration data, but an improved affinity for chloride (Kd = 42 ± 1 mM) is observed in cells (Fig. S24†). This result and the larger dynamic range are in line with the fact that GR1–CFP maintains proper folding when expressed in live and intact membranes compared to purified GR1. To test the reversibility of the turn-on response for GR1–CFP, cells were equilibrated in buffer or 400 mM sodium chloride followed by re-equilibration in sodium chloride, sodium acetate buffer, or sodium gluconate (Fig. 5C). With this washout experiment, the fluorescence response was restored to baseline levels, indicating that GR1–CFP can sense dynamic changes in extracellular chloride that can readily equilibrate with the periplasmic space.92 Finally, GR1–CFP was applied at the single cell level using confocal fluorescence microscopy. As can be seen in Fig. 6, GR1 localizes to the cell membrane, whereas CFP is diffusely localized in the cytosol. Cells expressing GR1–CFP immobilized on agarose pads supplemented with 400 mM sodium chloride display a ca. 1.8-fold turn-on response. Under the same conditions, wtGR–CFP does not show any response to chloride (Fig. 6 and S26†).
 | ||
| Fig. 6 Molecular imaging shows that GR1–CFP is a fluorescent chloride sensor at the single cell level in live E. coli. Representative confocal fluorescence microscopy images of E. coli expressing (A) wtGR–CFP and (C) GR1–CFP immobilized on 1.5% agarose pads containing 0 mM and 400 mM sodium chloride in 50 mM sodium acetate buffer at pH 5. Images are shown for the rhodopsin in red (left) and an overlay of the CFP in cyan with the transmitted light image (right). Scale bar = 2 μm. Single-cell analysis of the normalized emission response to 400 mM sodium chloride for (B) wtGR–CFP (for 0 mM chloride n = 2829 regions of interest (ROIs); for 400 mM chloride n = 3818 ROIs) and (D) GR1–CFP (for 0 mM chloride n = 1890 ROIs; for 400 mM chloride n = 2913 ROIs). The median fluorescence intensity of the rhodopsin (FGR) was normalized to the median fluorescence intensity of CFP (FCFP). Boxplots represent the analysis for all cells in the fields of view with the lower and upper quartile data enclosed by the gray box. The median values are indicated by the red line, and the minimum and maximum values for each data set are indicated by the lines extending below and above the gray box. Data points that fall outside of these parameters are considered outliers and are shown as open circles. At least four different fields were analyzed for each biological replicate (n = 3). | ||
Finally, since wtGR is a light-activated outward proton pump, ion pumping assays were conducted in 400 mM sodium chloride. White light LED illumination of E. coli expressing wtGR–CFP results in a time-dependent decrease in the extracellular pH (Fig. S27†). Upon removal of LED illumination, the extracellular pH is restored to baseline levels. Moreover, treatment with the protonophore carbonyl cyanide 3-chlorophenylhydrazone (CCCP, 30 μM) serves as a control to abolish the proton gradient and equalize the membrane potential, thus validating our experimental setup.97 Under the same conditions, GR1–CFP does not pump protons; these observations are in line with dye-based proton-pumping assays.90 We can also conclude that GR1–CFP is not a light-activated chloride pump because the extracellular pH does not change as observed in other CPRs and engineered PPRs.67,97–100 Furthermore, since PPRs can also be used as fluorescent indicators of membrane potential, chloride titrations were carried out at pH 5 in the presence of CCCP.73 Even though CCCP treatment uncouples the membrane potential, as independently confirmed with a commercially available dye, the fluorescence response of both wtGR–CFP and GR1–CFP to chloride are not affected (Fig. S28 and S29†).101 Taken together, our data show that wtGR–CFP can be converted into a non-pumping, ratiometric fluorescent sensor for chloride that is red-shifted and independent of membrane potential; thus, allowing for the reversible detection of chloride in exogenously supplemented E. coli. We envision that by improving the dynamic range, affinity, and operational pH of GR1 the imaging of endogenous chloride dynamics at the membranes of bacteria and eukaryotic cells could be possible. Moreover, future generations of GR1 will enable multiplexing with GFP-based sensors for chloride or other analytes to provide a more complete picture for the roles of chloride in cell biology.
Conclusion
From a molecular perspective, naturally occurring CPRs combine the hydrophobic effect with multivalent, cooperative interactions to achieve anion recognition in water. Drawing on these design principles, we showcase how protein engineering can be used to create a chloride binding site in Gloeobacter rhodopsin (wtGR), resulting in a non-pumping, red-shifted, and turn-on fluorescent sensor for chloride (GR1) in detergent micelles and live E. coli. This non-natural function was unlocked by substituting the aspartate counterion to the protonated SBC with valine at position 121, which to date has not been identified in the rhodopsin family. In vitro characterization shows that chloride binding tunes the pKa of the SBC towards the protonated, fluorescent state to generate a pH-dependent response to not only chloride but also bromide, iodide, and nitrate. Based on these spectroscopic data, it is likely that chloride forms an electrostatic interaction with the protonated SBC in the excited state. Future investigations with time-resolved spectroscopy will be used to confirm this mechanism.74,75 Moreover, bulk and single-cell fluorescence measurements of E. coli expressing the GR1–CFP fusion show that GR1 does not sense membrane potential but instead provides a reversible, ratiometric readout of changes in extracellular chloride at the membrane. Directed evolution efforts are currently in progress to improve and alter the properties of GR1 for expanded cellular applications in bacteria and eukaryotic cell types. Our discovery establishes that rhodopsins can be a new platform to build fluorescent sensors for studying chloride in living cells and provides an opportunity at the intersection of protein engineering and molecular recognition to create biological supramolecular hosts for chloride.Author contributions
J. N. T., J. L., C. H., F. F., and S. C. D. designed and performed research and analyzed data. S. S. A. and G. M. contributed technical expertise for protein purification. Q. Z. and F. M. generated and analyzed sequence data for the rhodopsin family. J. N. T. and S. C. D. wrote the manuscript with input from all co-authors.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank members of the Dodani and Meloni labs, Dr Alexander R. Lippert, Dr John W. Sibert, and Dr Ronald A. Smaldone for helpful discussions. We also acknowledge Dr Jacob Henderson (Flow Cytometry Core at UT Dallas) and Dr Ved Prakash (Olympus Discovery Center at UT Dallas) for expert technical assistance with method development and data analysis. F. M. acknowledges support from the National Science Foundation (MCB-1943442) and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM133631). G. M. acknowledges support from the Welch Foundation (AT-1935-20170325) and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128704). S. C. D. acknowledges support from The University of Texas at Dallas start-up funds, the Welch Foundation (AT-1918-20170325), and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128923). This work is the sole responsibility of the authors and does not represent the views of the funding agencies.References
- J. A. Raven and C. Raines, J. Exp. Bot., 2017, 68, 359–367 CAS
.
-
U. Lindh, in Essentials of Medical Geology, ed. O. Selinus, Springer, Dordrecht, 2nd edn, 2013, ch. 7, pp. 129–177 Search PubMed
- T. J. Jentsch and M. Pusch, Physiol. Rev., 2018, 98, 1493–1590 CrossRef CAS PubMed
- M. Zajac, K. Chakraborty, S. Saha, V. Mahadevan, D. T. Infield, A. Accardi, Z. Qiu and Y. Krishnan, J. Cell Sci., 2020, 133, 1–13 CrossRef PubMed
- M. Heubl, J. Zhang, J. C. Pressey, S. Al Awabdh, M. Renner, F. Gomez-Castro, I. Moutkine, E. Eugène, M. Russeau, K. T. Kahle, J. C. Poncer and S. Lévi, Nat. Commun., 2017, 8, 1–17 CrossRef CAS PubMed
- T. Wasiluk, M. Roueinfar, K. Hiryak, M. Torsiello, A. Miner, J. Lee, M. Venditto, W. Terzaghi, D. Lucent and A. L. VanWert, Biol. Open, 2019, 8, 1–8 Search PubMed
- J. K. Roebber, S. D. Roper and X. N. Chaudhari, J. Neurosci., 2019, 39, 6224–6232 CrossRef CAS PubMed
- J. Yang, J. Chen, M. Del Carmen Vitery, J. Osei-Owusu, J. Chu, H. Yu, S. Sun and Z. Qiu, Science, 2019, 364, 395–399 CrossRef CAS PubMed
- F. Ullrich, S. Blin, K. Lazarow, T. Daubitz, J. Peter Von Kries and T. J. Jentsch, eLife, 2019, 8, 1–25 CrossRef PubMed
- S. Kubik, Chem. Soc. Rev., 2010, 39, 3648–3663 RSC
- T. Ashton, K. A. Jolliffe and F. M. Pfeffer, Chem. Soc. Rev., 2014, 44, 4547–4595 RSC
- M. J. Langton, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 1974–1987 CrossRef CAS PubMed
-
J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, 2nd edn, 2009 Search PubMed
- H. I. Okur, J. Hladílková, K. B. Rembert, Y. Cho, J. Heyda, J. Dzubiella, P. S. Cremer and P. Jungwirth, J. Phys. Chem. B, 2017, 121, 1997–2014 CrossRef CAS PubMed
- P. S. Cremer, A. H. Flood, B. C. Gibb and D. L. Mobley, Nat. Chem., 2018, 10, 8–16 CrossRef CAS PubMed
- J. J. Gassensmith, S. Matthys, J.-J. Lee, A. Wojcik, P. V Kamat and B. D. Smith, Chem.–Eur. J., 2010, 16, 2916–2921 CrossRef CAS
- S. Amatori, G. Ambrosi, E. Borgogelli, M. Fanelli, M. Formica, V. Fusi, L. Giorgi, E. Macedi, M. Micheloni, P. Paoli, P. Rossi and A. Tassoni, Inorg. Chem., 2014, 53, 4560–4569 CrossRef CAS
- J. P. Kim, Z. Xie, M. Creer, Z. Liu and J. Yang, Chem. Sci., 2017, 8, 550–558 RSC
- M. M. Watt, J. M. Engle, K. C. Fairley, T. E. Robitshek, M. M. Haley and D. W. Johnson, Org. Biomol. Chem., 2015, 13, 4266–4270 RSC
- S. Vallejos, E. Hernando, M. Trigo, F. García, M. García-Valverde, D. Iturbe, M. J. Cabero, R. Quesada and J. M. García, J. Mater. Chem. B, 2018, 6, 3735–3741 RSC
- A. S. Verkman, Am. J. Physiol., 1990, 259, C375–C388 CrossRef CAS PubMed
- C. D. Geddes, Meas. Sci. Technol., 2001, 12, R53–R88 CrossRef CAS
- N. D. Sonawane, J. R. Thiagarajah and A. S. Verkman, J. Biol. Chem., 2002, 277, 5506–5513 CrossRef CAS
- A. Graefe, S. E. Stanca, S. Nietzsche, L. Kubicova, R. Beckert, C. Biskup and G. J. Mohr, Anal. Chem., 2008, 80, 6526–6531 CrossRef CAS PubMed
- S. Saha, V. Prakash, S. Halder, K. Chakraborty and Y. Krishnan, Nat. Nanotechnol., 2015, 10, 645–652 CrossRef CAS PubMed
- V. Prakash, S. Saha, K. Chakraborty and Y. Krishnan, Chem. Sci., 2016, 7, 1946–1953 RSC
- K. Chakraborty, K. Leung and Y. Krishnan, eLife, 2017, 6, 1–21 CrossRef PubMed
- S. H. Park, J. Y. Hyun and I. Shin, Chem. Sci., 2019, 10, 56–66 RSC
- P. A. Romero and F. H. Arnold, Nat. Rev. Mol. Cell Biol., 2009, 10, 866–876 CrossRef CAS PubMed
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS
- D. Arosio and G. M. Ratto, Front. Cell. Neurosci., 2014, 8, 1–12 Search PubMed
- R. M. Wachter, M.-A. Elsliger, K. Kallio, G. T. Hanson and J. Remington, Structure, 1998, 6, 1267–1277 CrossRef CAS PubMed
- R. M. Wachter and S. James Remington, Curr. Biol., 1999, 9, R628–R629 CrossRef CAS PubMed
- R. M. Wachter, D. Yarbrough, K. Kallio and S. J. Remington, J. Mol. Biol., 2000, 301, 157–171 CrossRef CAS PubMed
- S. Jayaraman, P. Haggie, R. M. Wachter, S. J. Remington and A. S. Verkman, J. Biol. Chem., 2000, 275, 6047–6050 CrossRef CAS PubMed
- D. Arosio, G. Garau, F. Ricci, L. Marchetti, R. Bizzarri, R. Nifosì and F. Beltram, Biophys. J., 2007, 93, 232–244 CrossRef CAS PubMed
- L. J. V Galietta, P. M. Haggie and A. S. Verkman, FEBS Lett., 2001, 499, 220–224 CrossRef
- T. Nagai, K. Ibata, E. S. Park, M. Kubota, K. Mikoshiba and A. Miyawaki, Nat. Biotechnol., 2002, 20, 87–90 CrossRef CAS PubMed
- O. Markova, M. Mukhtarov, E. Real, Y. Jacob and P. Bregestovski, J. Neurosci. Methods, 2008, 170, 67–76 CrossRef CAS PubMed
- S. D. Watts, K. L. Suchland, S. G. Amara and S. L. Ingram, PLoS One, 2012, 7, 1–9 CrossRef PubMed
- S. Zhong, D. Navaratnam and J. Santos-Sacchi, PLoS One, 2014, 9, 1–9 Search PubMed
- T. Kuner and G. J. Augustine, Neuron, 2000, 27, 447–459 CrossRef CAS PubMed
- T. Waseem, M. Mukhtarov, S. Buldakova, I. Medina and P. Bregestovski, J.
Neurosci. Methods, 2010, 193, 14–23 CrossRef CAS PubMed
- D. Arosio, F. Ricci, L. Marchetti, R. Gualdani, L. Albertazzi and F. Beltram, Nat. Methods, 2010, 7, 516–518 CrossRef CAS PubMed
- M. Mukhtarov, L. Liguori, T. Waseem, F. Rocca, S. Buldakova, D. Arosio and P. Bregestovski, Front. Mol. Neurosci., 2013, 6, 1–12 Search PubMed
- J. M. Paredes, A. I. Idilli, L. Mariotti, G. Losi, L. R. Arslanbaeva, S. S. Sato, P. Artoni, J. Szczurkowska, L. Cancedda, G. M. Ratto, G. Carmignoto and D. Arosio, ACS Chem. Biol., 2016, 11, 1652–1660 CrossRef CAS PubMed
- S. S. Sato, P. Artoni, S. Landi, O. Cozzolino, R. Parra, E. Pracucci, F. Trovato, J. Szczurkowska, S. Luin, D. Arosio, F. Beltram, L. Cancedda, K. Kaila and G. M. Ratto, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E8770–E8779 CrossRef CAS PubMed
- E. Demes, L. Besse, P. Cubero-Font, B. Satiat-Jeunemaitre, S. Thomine and A. De Angeli, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 15343–15353 CrossRef PubMed
- J. N. Tutol, W. Peng and S. C. Dodani, Biochemistry, 2019, 58, 31–35 CrossRef CAS PubMed
- J. N. Tutol, H. C. Kam and S. C. Dodani, ChemBioChem, 2019, 20, 1759–1765 CAS
- N. V Pletneva, V. Z. Pletnev, E. Souslova, D. M. Chudakov, S. Lukyanov, V. I. Martynov, S. Arhipova, I. Artemyev, A. Wlodawer, Z. Dauter and S. Pletnev, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1005–1012 CrossRef PubMed
- D. Clavel, G. Gotthard, D. Von Stetten, D. De Sanctis, H. Pasquier, G. G. Lambert, N. C. Shaner and A. Royant, Acta Crystallogr., 2016, D72, 1298–1307 Search PubMed
- P. Ernst, N. Xu, J. Qu, H. Chen, M. S. Goldberg, V. Darley-Usmar, J. J. Zhang, B. O’rourke, X. Liu and L. Zhou, Biophys. J., 2019, 117, 631–645 CrossRef CAS PubMed
- J. P. Klare, I. Chizhov and M. Engelhard, Results Probl. Cell Differ., 2007, 45, 73–122 Search PubMed
- O. Yizhar, L. Fenno, F. Zhang, P. Hegemann and K. Diesseroth, Cold Spring Harb. Protoc., 2011, 3, 273–282 Search PubMed
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed
- M. Kurihara and Y. Sudo, Biophys. Physicobiol., 2015, 12, 121–129 CrossRef CAS PubMed
- E. G. Govorunova, O. A. Sineshchekov, H. Li and J. L. Spudich, Annu. Rev. Biochem., 2017, 86, 845–872 CrossRef CAS PubMed
- K. Tsutsui and Y. Shichida, Photochem. Photobiol. Sci., 2010, 9, 1426–1434 CrossRef CAS PubMed
- M. Kolbe, H. Besir, L.-O. Essen and D. Oesterhelt, Science, 2000, 288, 1390–1396 CrossRef CAS PubMed
- T. Kouyama, S. Kanada, Y. Takeguchi, A. Narusawa, M. Murakami and K. Ihara, J. Mol. Biol., 2010, 396, 564–579 CrossRef CAS PubMed
- K. Inoue, Y. Kato and H. Kandori, Trends Microbiol., 2015, 23, 91–98 CrossRef CAS PubMed
- M. Schreiner, R. Schlesinger, J. Heberle and H. H. Niemann, J. Struct. Biol., 2015, 190, 373–378 CrossRef CAS PubMed
- K. Kim, S.-K. Kwon, S.-H. Jun, J. S. Cha, H. Kim, W. Lee, J. F. Kim and H.-S. Cho, Nat. Commun., 2016, 7, 1–10 Search PubMed
- J. E. Besaw, W.-L. Ou, T. Morizumi, B. T. Eger, J. D. Sanchez Vasquez, J. H. Y. Chu, A. Harris, L. S. Brown, R. J. Dwayne Miller and O. P. Ernst, J. Biol. Chem., 2020, 295, 14793–14804 CrossRef CAS PubMed
- M. T. Facciotti, S. Rouhani, F. T. Burkard, F. M. Betancourt, K. H. Downing, R. B. Rose, G. McDermott and R. M. Glaeser, Biophys. J., 2001, 81, 3442–3455 CrossRef CAS PubMed
- J. Sasaki, L. S. Brown, Y.-S. Chon, H. Kandori, A. Maeda, R. Needleman and J. K. Lanyi, Science, 1995, 269, 73–75 CrossRef CAS PubMed
- L. S. Brown, R. Needleman and J. K. Lanyi, Biochemistry, 1996, 35, 16048–16054 CrossRef CAS PubMed
- J. Tittor, U. Haupts, C. Haupts, D. Oesterhelt, A. Becker and E. Bamberg, J. Mol. Biol., 1997, 271, 405–416 CrossRef CAS PubMed
- C. Ganea, J. Tittor, E. Bamberg and D. Oesterhelt, Biochim. Biophys. Acta, 1998, 1368, 84–96 CrossRef CAS
- M. T. Facciotti, V. S. Cheung, D. Nguyen, S. Rouhani and R. M. Glaeser, Biophys. J., 2003, 85, 451–458 CrossRef CAS
- R. A. Bogomolni, L. Stubbs and J. K. Lanyi, Biochemistry, 1978, 17, 1037–1041 CrossRef CAS PubMed
- J. M. Kralj, D. R. Hochbaum, A. D. Douglass and A. E. Cohen, Science, 2011, 333, 345–348 CrossRef CAS PubMed
- D. Maclaurin, V. Venkatachalam, H. Lee and A. E. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 5939–5944 CrossRef CAS PubMed
- J. K. Yu, R. Liang, F. Liu and T. J. Martínez, J. Am. Chem. Soc., 2019, 141, 18193–18203 CrossRef CAS PubMed
- J. M. Kralj, A. D. Douglass, D. R. Hochbaum, D. Maclaurin and A. E. Cohen, Nat. Methods, 2012, 9, 90–95 CrossRef CAS PubMed
- Y. Bando, C. Grimm, V. H. Cornejo and R. Yuste, BMC Biol., 2019, 17, 1–12 CrossRef CAS
- K. Kojima, A. Shibukawa and Y. Sudo, Biochemistry, 2020, 59, 218–229 CrossRef CAS PubMed
- Y. Nakamura, T. Kaneko, S. Sato, M. Mimuro, H. Miyashita, T. Tsuchiya, S. Sasamoto, A. Watanabe, K. Kawashima, Y. Kishida, C. Kiyokawa, M. Kohara, M. Matsumoto, A. Matsuno, N. Nakazaki, S. Shimpo, C. Takeuchi, M. Yamada and S. Tabata, DNA Res., 2003, 10, 137–145 CrossRef CAS PubMed
- M. R. M. Miranda, A. R. Choi, L. Shi, A. G. Bezerra, K.-H. Jung and L. S. Brown, Biophys. J., 2009, 96, 1471–1481 CrossRef CAS PubMed
- S. Ganapathy, H. Venselaar, Q. Chen, H. J. M. De Groot, K. J. Hellingwerf and W. J. De Grip, J. Am. Chem. Soc., 2017, 139, 2338–2344 CrossRef CAS PubMed
- T. Morizumi, W.-L. Ou, N. Van Eps, K. Inoue, H. Kandori, L. S. Brown and O. P. Ernst, Sci. Rep., 2019, 9, 1–14 CAS
- S. Jana, K. H. Jung and M. Sheves, Sci. Rep., 2020, 10, 1–13 CrossRef PubMed
- E. S. Imasheva, S. P. Balashov, A. R. Choi, K.-H. Jung and J. K. Lanyi, Biochemistry, 2009, 48, 10948–10955 CrossRef CAS
- K. Hashimoto, A. R. Choi, Y. Furutani, K.-H. Jung and H. Kandori, Biochemistry, 2010, 49, 3343–3350 CrossRef CAS PubMed
- A. Vogt, J. Wietek and P. Hegemann, Biophys. J., 2013, 105, 2055–2063 CrossRef CAS PubMed
- T. Tsukamoto, T. Kikukawa, T. Kurata, K.-H. Jung, N. Kamo and M. Demura, FEBS Lett., 2013, 587, 322–327 CrossRef CAS PubMed
- T. Tsukamoto, K. Inoue, H. Kandori and Y. Sudo, J. Biol. Chem., 2013, 288, 21581–21592 CrossRef CAS PubMed
- A. R. Choi, L. Shi, L. S. Brown and K.-H. Jung, PLoS One, 2014, 9, 1–10 Search PubMed
- M. K. M. Engqvist, R. S. McIsaac, P. Dollinger, N. C. Flytzanis, M. Abrams, S. Schor and F. H. Arnold, J. Mol. Biol., 2015, 427, 205–220 CrossRef CAS PubMed
- S. Jana, T. Eliash, K.-H. Jung and M. Sheves, J. Phys. Chem. B, 2017, 121, 10759–10769 CrossRef CAS PubMed
- J. C. Wilks and J. L. Slonczewski, J. Bacteriol., 2007, 189, 5601–5607 CrossRef CAS PubMed
- J. K. Kaushik and R. Bhat, Protein Sci., 1999, 8, 222–223 CrossRef CAS
- M. T. Facciotti, V. S. Cheung, C. S. Lunde, S. Rouhani, N. S. Baliga and R. M. Glaeser, Biochemistry, 2004, 43, 4934–4943 CrossRef CAS
-
S. Mangani and M. Ferraroni, in Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, John Wiley & Sons, Inc., New York City, 1997, ch. 3, pp. 63–78 Search PubMed
- M. Ghoul, M. Pommepuy, A. Moillo-Batt and M. Cormier, Appl. Environ. Microbiol., 1989, 55, 1040–1043 CrossRef CAS PubMed
- T. Hasemi, T. Kikukawa, N. Kamo and M. Demura, J. Biol. Chem., 2016, 291, 355–362 CrossRef CAS PubMed
- P. Saeedi, J. M. Moosaabadi, S. S. Sebtahmadi, J. F. Mehrabadi, M. Behmanesh, H. R. Nejad and A. Nazaktabar, Bioengineered, 2012, 3, 275–279 CrossRef CAS PubMed
- S. Yoshizawa, Y. Kumagai, H. Kim, Y. Ogura, T. Hayashi, W. Iwasaki, E. F. DeLong and K. Kogure, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6732–6737 CrossRef CAS
- K. Inoue, S. Ito, Y. Kato, Y. Nomura, M. Shibata, T. Uchihashi, S. P. Tsunoda and H. Kandori, Nat. Commun., 2016, 7, 1–10 Search PubMed
- D. Novo, N. G. Perlmutter, R. H. Hunt and H. M. Shapiro, Cytometry, 1999, 35, 55–63 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06061e |
| This journal is © The Royal Society of Chemistry 2021 |