
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrifluoromethanesulfonyl azide as a bifunctional reagent for metal-free azidotrifluoromethylation of unactivated alkenes†
Hong-Gui
Huang
,
Weishuang
Li
,
Dayou
Zhong
,
Hu-Chong
Wang
,
Jing
Zhao
and
Wen-Bo
Liu
 *
*
Sauvage Center for Molecular Sciences, Engineering Research Center of Organosilicon Compounds & Materials (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, 299 Bayi Road, Wuhan 430072, Hubei, China. E-mail: wenboliu@whu.edu.cn
First published on 7th January 2021
Abstract
Vicinal trifluoromethyl azides have widespread applications in organic synthesis and drug development. However, their preparation is generally limited to transition-metal-catalyzed three-component reactions. We report here a simple and metal-free method that rapidly provides these building blocks from abundant alkenes and trifluoromethanesulfonyl azide (N3SO2CF3). This unprecedented two-component reaction employs readily available N3SO2CF3 as a bifunctional reagent to concurrently incorporate both CF3 and N3 groups, which avoids the use of their expensive and low atom economic precursors. A wide range of functional groups, including bio-relevant heterocycles and amino acids, were tolerated. Application of this method was further demonstrated by scale-up synthesis (5 mmol), product derivatization to CF3-containing medicinal chemistry motifs, as well as late-stage modification of natural product and drug derivatives.
Introduction
The development of efficient synthetic methods, ideally those that convert chemical feedstocks into biologically important and complex structures, has significantly advanced drug discovery and development.1 Direct difunctionalization of abundant alkenes is of fundamental importance to the synthesis of high-value molecules as evidenced by increasing research interests of the synthetic community in recent years.2 Selective azidotrifluoromethylation of the carbon–carbon double bond enables to concomitantly assembly of two vicinal functional groups (N3 and CF3) in a single step. These structures have high biological significance due to the unique physical and electronic properties of the trifluoromethyl group.3 On the other hand, the azide also serves as a handle for downstream derivatization to trifluoromethylated amines and heterocycles.4 In particular, the well-established click chemistry of azides has empowered chemists to rapidly generate large libraries of CF3-containing compounds for screening in drug discovery.5To date, catalytic methods for efficient access to vicinal trifluoromethyl azides from alkenes are still limited. In 2014, Liu and co-workers pioneered the direct azidotrifluoromethylation of alkenes using copper catalysis (Scheme 1a).6 In their report, Togni's reagent II and TMSN3 were employed as the CF3 and N3 sources, respectively. Following this pattern, iron-catalyzed processes with extraordinary substrate scope were later reported by Hartwig7 and Xu8 (Scheme 1a). Other methods including oxidative radical relay9 and photo–chemical reactions10 were also successfully developed by a prudent choice of the precursors of CF3 radical and azide. While these advances are invaluable, from a practical point of view, the requirement of transition-metal catalysts and expensive CF3 sources (e.g., Togni's reagents, Umemoto's reagent) has impeded their application especially for scale-up syntheses. Additionally, state-of-the-art approaches rely on the same three-component framework: addition of a CF3 radical onto an alkene to form an alkyl radical or cation11 and then trapped by a separate azide reagent, which generally constitutes a low level of mass efficiency by producing stoichiometric side-products (e.g., 2-iodobenzoic acid).12 Therefore, the development of more environmentally benign and atom-economic alternatives is practically appealing and conceptually novel.
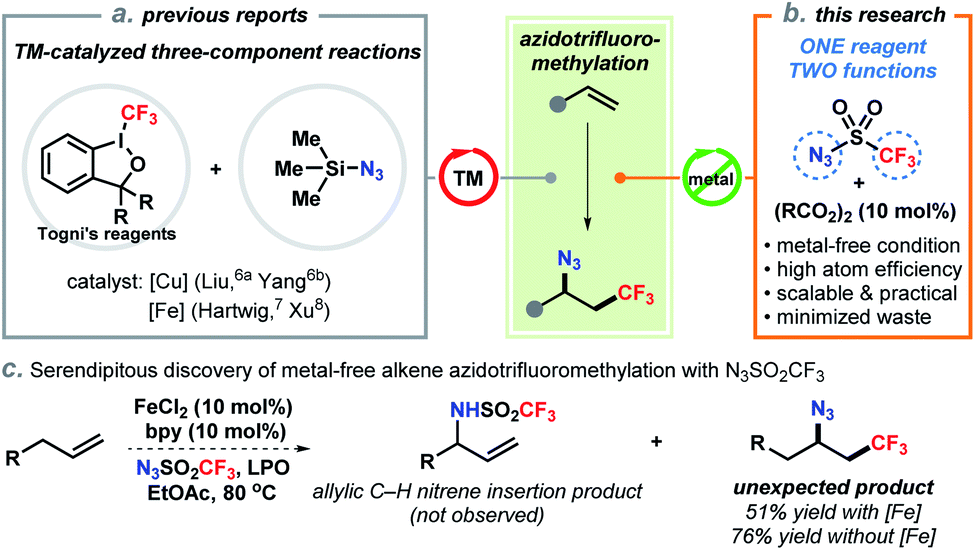 | ||
| Scheme 1 Azidotrifluoromethylation of alkenes. | ||
Trifluoromethanesulfonyl azide (N3SO2CF3) is readily synthesized in situ by reacting trifluoromethanesulfonic anhydride with sodium azide in hexane (see ESI†). Although N3SO2CF3 has been commonly used as a diazo- or trifluoromethanesulfonylamino-transfer reagent,13 its ability to act as a radical precursor received little attention. The homolytic decomposition of N3SO2CF3 to form a trifluoromethyl radical was first observed by Kamigata et al. in 1984.14 In 2018, a significant work by the Studer group employed N3SO2CF3 as an azide source for remote radical C–H azidation of amides by a fragmentation/1,5-HAT process.15 Very recently, a notable radical 1,3-difunctionalization of allylboronic esters using N3SO2CF3 was also reported by the same group.16 These remarkable studies exemplified the potential utilization of N3SO2CF3 as a bifunctional reagent.17 Encouraged by this precedent and our serendipitous discovery (vide infra), we have developed a two-component azidotrifluoromethylation reaction of unactivated alkenes and N3SO2CF3 (Scheme 1b). In this unprecedented strategy, N3SO2CF3 serves as the sole precursor for both CF3 and N3 by releasing SO2 gas, which therefore minimizes waste generation. This radical chain reaction is initiated by an acyl peroxide, avoids the use of transition-metal catalysts, tolerates a wide range of functional groups, and is amenable to late-stage modification of natural products and drug derivatives.
Results and discussion
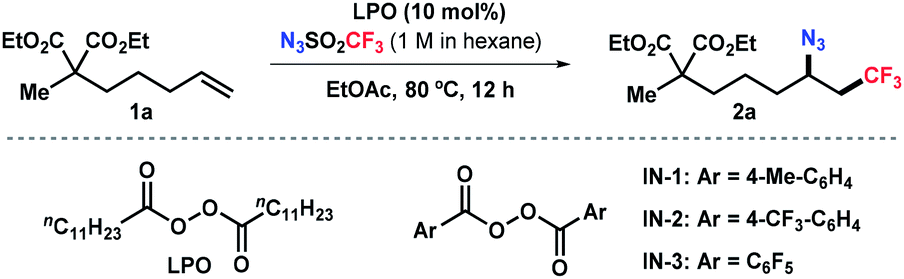
Our group has been interested in developing environmentally benign direct C–H amination methodologies.18 In our efforts toward using trifluoromethanesulfonyl azide as a nitrene precursor for iron-catalyzed intermolecular allylic C–H amination, we unexpectedly observed azidotrifluoromethylation of the alkene (Scheme 1c). A control experiment disclosed that the reaction occurred without iron catalysts. Encouraged by this serendipity, we commenced the reaction optimization utilizing alkene 1a as a model substrate as outlined in Table 1 (see Tables S1–S3 in ESI† for details). The desired product 2a was obtained in 48% yield using 10 mol% of lauroyl peroxide (LPO) as a radical initiator and EtOAc as solvent (entry 1). Reactions in other solvents (e.g., DCM, DMF, MeCN, entry 2 and Table S1†) were less effective. Varying the concentration of the reaction did not further improve the productivity. After surveying commonly used thermal radical initiators, such as organic peroxides and aliphatic azo compounds (entries 3–7), pentafluorobenzoyl peroxide (IN-3) emerged as the choice resulting in 65% yield of 2a (entry 7). To our delight, an increased yield (87%) was obtained by using 2.5 equivalent of N3SO2CF3 (entry 8). Unsurprisingly, radical initiators were indispensable and no desired product was detected in their absence (entry 9). It should be noted that N3SO2CF3 should be handled with cautions, although its stock solution in hexane is fairly stable (see ESI†).|
|
||
|---|---|---|
| Entrya | Deviation | 2a (%) |
| a Reaction conducted with 1a (0.2 mmol), LPO (10 mol%), and N3SO2CF3 (1.5 equiv., 1 M in hexane) in EtOAc (2 mL) at 80 °C for 12 h. b Determined by 19F NMR of the crude reaction mixture using an internal standard. c Isolated yield. TBHP, tert-butyl hydroperoxide. DTBP, di-tert-butyl peroxide. AIBN, 2,2′-azobis(2-methylpropionitrile). N.R., no reaction. | ||
| 1 | None | 48 |
| 2 | Dioxane, MeCN, DMF, or DCE instead of EtOAc | 17–33 |
| 3 | TBHP or DTBP instead of LPO | Trace |
| 4 | AIBN instead of LPO | 20 |
| 5 | IN-1 instead of LPO | 52 |
| 6 | IN-2 instead of LPO | 50 |
| 7 | IN-3 instead of LPO | 65 |
| 8 | IN-3 instead of LPO and with 2.5 equiv of N3SO2CF3 (standard conditions) | 86 (87)c |
| 9 | w/o initiator | N.R. |
Next, we set out to explore the substrate scope of this reaction (Scheme 2). Aliphatic alkenes bearing a broad array of functional groups, such as ester, ketone, tosylate, phosphate, and phthalimide, were viable substrates under the standard conditions, delivering the corresponding products in moderate to good yields (2a–2k). Heterocycles relevant to medicinal chemistry, including furan, thiophene, and pyridine, were tolerated as well, albeit in moderate yields (2l–2n). The mild, base- and transition-metal-free reaction conditions also allowed the compatibility of chloride- and bromide-containing substrates with the halides unscathed (2o–2q). Moreover, abundant unsaturated hydrocarbons were efficiently converted into high-value bifunctionalized products in 58–62% yields (2r–2t). This simple process also pertains to sterically hindered alkenes. 1,1-Disubstituted alkenes were selectively transformed into the tertiary azides 2u–2w in 71–81% yields. The reaction with 1,1,2-trisubstituted alkenes delivered the corresponding bifunctionalized product 2x in moderate yield. This method also enabled the efficient installation of perfluoroalkyl groups (e.g., C4F9, eqn (1)), given the readily synthesis of the corresponding sulfonyl azides. Surprisingly, further expanding the scope to cyclic olefin 1y and styrene 1z unexpectively delivering imine 2y and aldehyde 2z in 92% and 45% yield, respectively (eqn (2) and (3)), which probably formed through a [3 + 2] cycloaddition pathway.19 The reaction with vinyl ether resulted in a complex mixture.19b Attempts to employ acrylate or vinyl amide under the standard conditions failed to give any desired azidotrifluoromethylation product but led to polymerization of the substrates (Scheme S1 in ESI†).
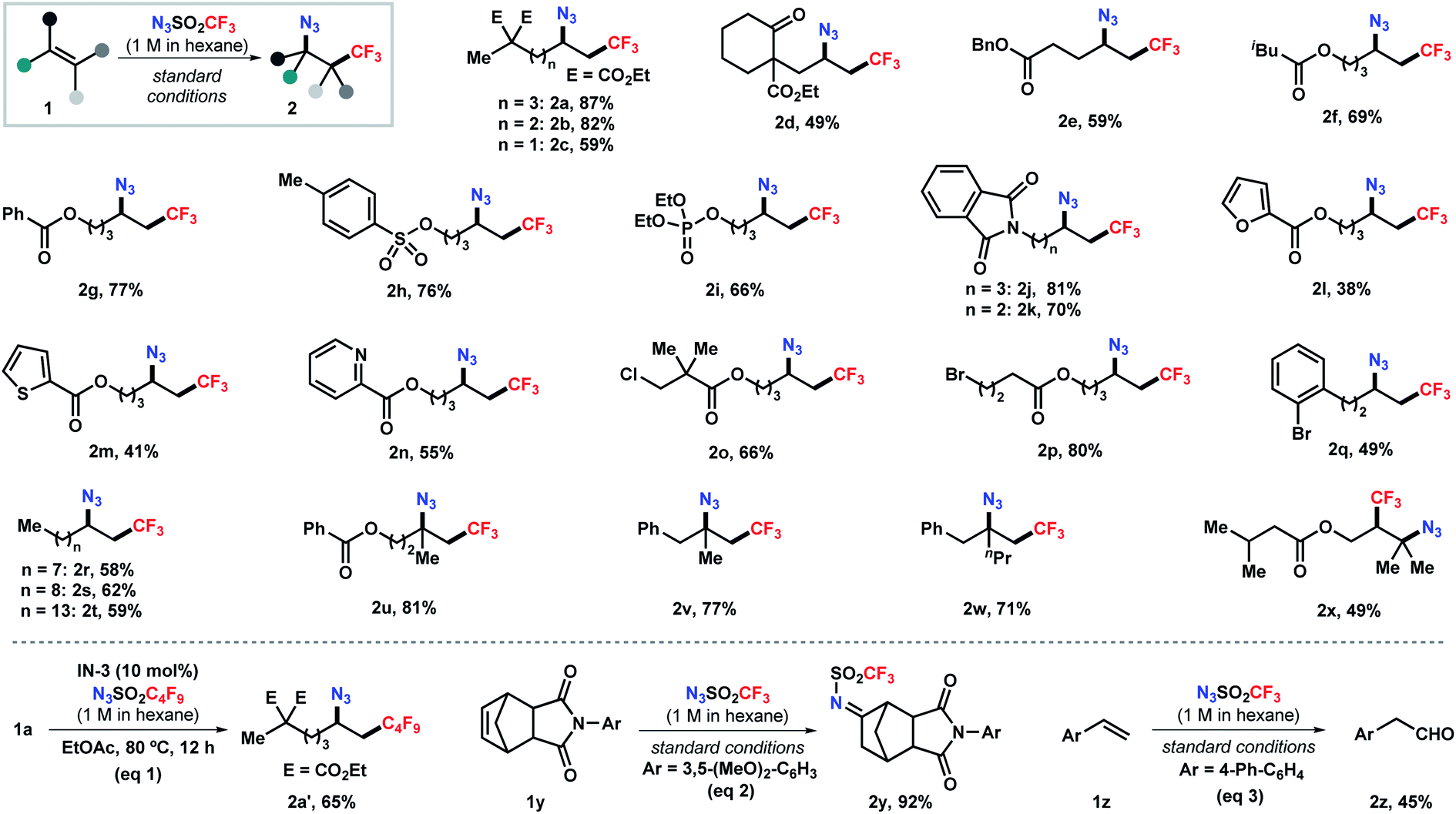 | ||
| Scheme 2 Scope of the alkene azidotrifluoromethylation. Reactions conducted on a 0.2 mmol scale (except 0.4 mmol for 2e) under the standard conditions (entry 8, Table 1). | ||
The absence of transition metals and operationally simple procedure of this reaction offers a practical tool in late-stage modification of densely functionalized biologically relevant molecules(Scheme 3). Functionalization of a series of complex structures (3a–3g) derived from pharmaceuticals were successfully achieved in good yields (4a–4g). The alkenes synthesized from terpenes, norneol and menthol, reacted smoothly to the corresponding azidotrifluoromethylated products (4j, 4k) in 71% and 72% yields, respectively. Remarkably, derivatives of amino acid including glycine (4l), tyrosine (4m), valine (4n), phenylalanine (4o), and aspartic acid (4p), were amenable as well to the reaction yielding the corresponding difunctionalized products smoothly. Moreover, a steroid example and a carbohydrate substrate were also included (4q, 4r). Overall, these results showcased the broad applicability and usefulness of our chemistry.
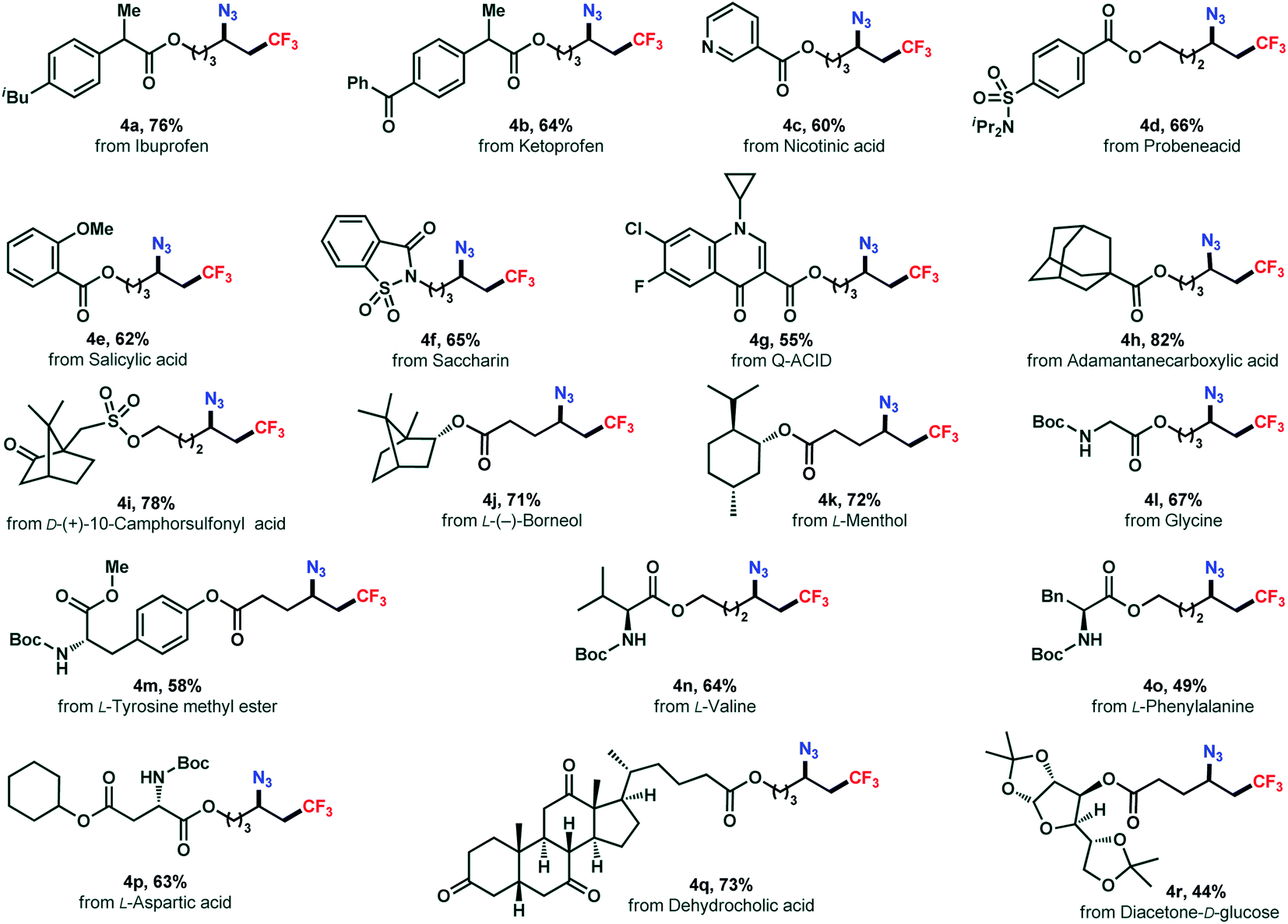 | ||
| Scheme 3 Late-stage modification of drug and natural product derivatives. Reactions conducted on a 0.2 mmol scale under the standard conditions (entry 8, Table 1). | ||
As shown in Scheme 4, the robustness of the reaction was demonstrated by a gram–scale reaction (5 mmol) under the standard conditions providing azidotrifluoromethylation product 2j in 88% yield. The azide unit of the products accessed above can be conveniently converted into privileged medicinal chemistry motifs such as amines and heterocycles. Reduction of azide 2j with In/NH4Cl produced a vicinal trifluoromethylated primary amine 5 in 69% yield. Notably, treatment of 2j with triphenylphosphine formed tetrahydro-1,3-diazepine 6via a Staudinger reaction/aza-Wittig sequence. Moreover, [3 + 2] annulation of 2j with ethynylbenzene and benzoyl cyanide resulted in the formation of CF3-containing triazole 7 and tetrazole 8, respectively. Additionally, selective reduction of an azide 2e bearing a pendant ester group proceeded with concomitant aminolysis to provide γ-butyrolactam 9 in 92% yield. In a two-step protocol, reduction of azide 2q to amine followed by subsequent cross-coupling delivered tetrahydroquinoline 10 in 57% overall yield.
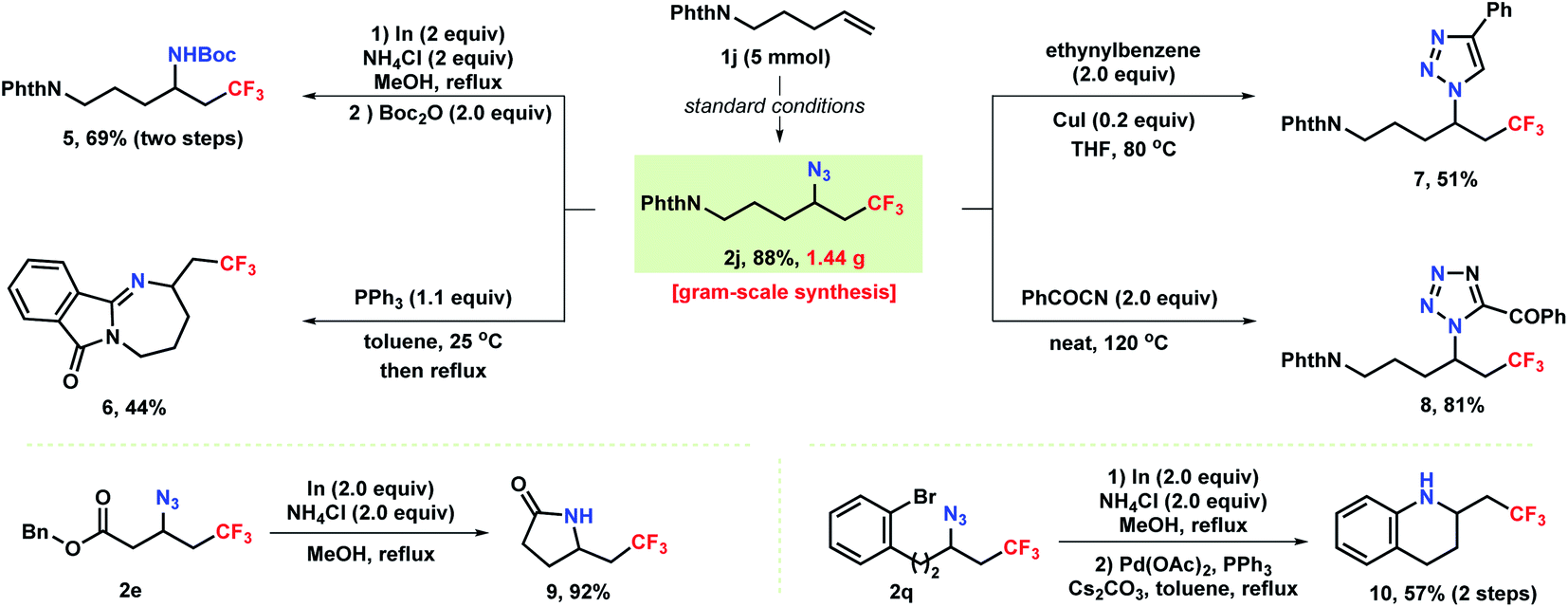 | ||
| Scheme 4 Synthetic application. | ||
Notably, all the trifluoromethyl-containing compounds accessed through these derivatizations are highly prevalent and commonly used in pharmaceuticals or as biological probes, demonstrating the ease of our chemistry in incorporating of trifluoromethyl group into these reminiscent motifs.
To gain mechanistic insight into this reaction, control experiments were carried out (Scheme 5). Addition of a radical scavenger TEMPO completely inhibited the reaction (Scheme 5a). Similarly, butylated hydroxytoluene (BHT) as an additive prevented the formation of the azidotrifluoromethylation product. The detection of trifluoromethyl adduct 11 by GC-MS (MS = 288.1) indicates that the trifluoromethyl radical is likely involved in this transformation (Scheme 5b). It is known that 5-hexenyl radical analogues undergo 5-exo-cyclization to the cyclopentylcarbinyl radical and are commonly employed as radical clocks.20 The reaction with substituted 1,6-diene 12 delivered a cyclization product 13 which suggests that the addition of trifluoromethyl radical to the alkene forms an alkyl radical followed by 5-exo-cyclization (Scheme 5c). On the basis of the above observations and literature reports,21 a putative radical chain pathway was illustrated as shown in Scheme 5d. First, thermal decomposition of the radical initiator IN-3 produces an aryl carboxylic radical I or an aryl radical I′,22 which reacts with trifluoromethanesulfonyl azide generating trifluoromethanesulfonyl radical II, following by SO2 release to form the trifluoromethyl radical III.23 Subsequent addition of III onto the alkene provides an alkyl radical IV. Finally, azidation of IV with N3SO2CF3 yields the desired product 2 along with regeneration of the chain carrying radical II.
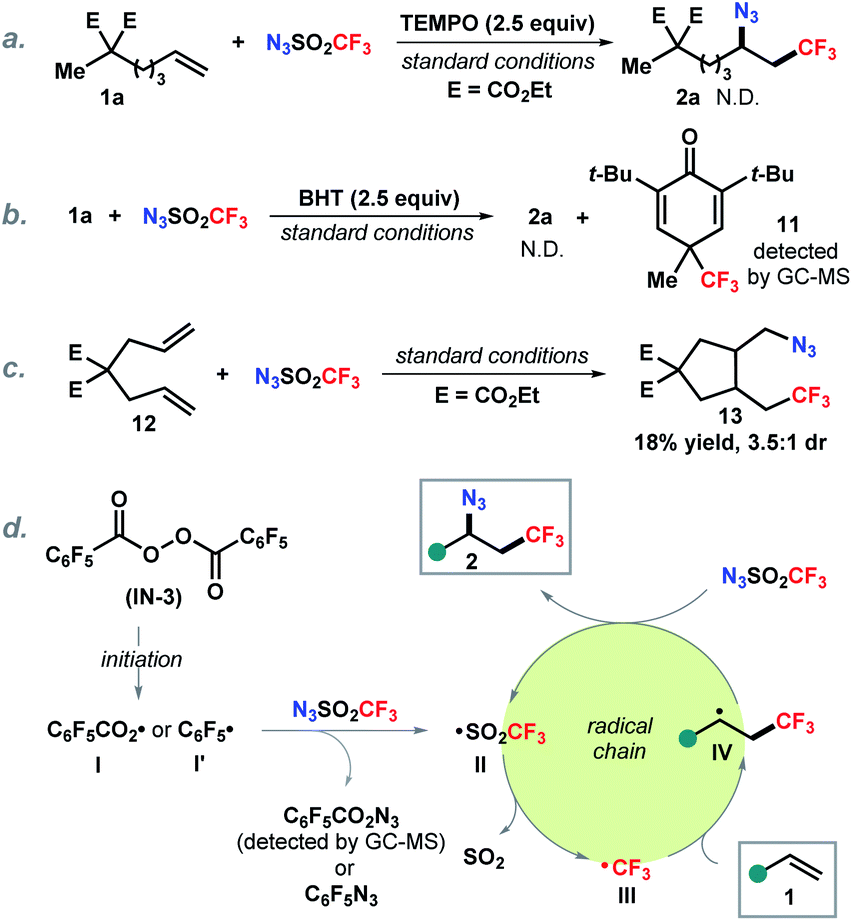 | ||
| Scheme 5 Mechanistic investigation. | ||
Conclusions
In summary, we have developed a transition-metal free azidotrifluoromethylation reaction of unactivated alkenes for the synthesis of vicinal trifluoromethyl azides. This unprecedented difunctionalization process employs trifluoromethanesulfonyl azide as the sole precursor for both CF3 and N3. The application was demonstrated by late-stage functionalization of complex bio-important molecules and converting the products into the corresponding CF3-containing amine, lactam, and other privileged heterocycles. This study provides an atom economic, cost efficient, and operationally simple method for the synthesis of valuable vicinal difunctionalized structures from chemical feedstocks.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from NSFC (21971198 and 21772148), the Fundamental Research Funds for Central Universities (2042019kf0208), Large-scale Instrument and Equipment Sharing Foundation of Wuhan University and the Natural Science Foundation of Hubei Province (Grant No. 2020CFA036). We also thank Dr Yiyang Liu (Pfizer) for helpful discussion.Notes and references
- K. R. Campos, P. J. Coleman, J. C. Alvarez, S. D. Dreher, R. M. Garbaccio, N. K. Terrett, R. D. Tillyer, M. D. Truppo and E. R. Parmee, Science, 2019, 363, eaat0805 CrossRef CAS
.
- Selected examples:
(a) T. Koike and M. Akita, Chem, 2018, 4, 409 CrossRef CAS
-
(a) K. Müller, C. Faeh and F. Diederich, Science, 2017, 317, 1881 CrossRef
-
(a) S. Chiba, Synlett, 2012, 23, 21 CrossRef CAS
-
(a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS
-
(a) F. Wang, X. Qi, Z. Liang, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2014, 53, 1881 CrossRef CAS
- R. R. Karimov, A. Sharma and J. F. Hartwig, ACS Cent. Sci., 2016, 2, 715 CrossRef CAS
- C. Zhu, C. Wang, Q. Qin, S. Yruegas, C. D. Martin and H. Xu, ACS Catal., 2018, 8, 5032 CrossRef CAS
-
(a) Y. Zhang, X. Han, J. Zhao, J. Qian, T. Li, Y. Tang and H.-Y. Zhang, Adv. Synth. Catal., 2018, 360, 2659 CrossRef CAS
-
(a) G. Dagousset, A. Carboni, E. Magnier and G. Masson, Org. Lett., 2014, 16, 4340 CrossRef CAS
-
(a) E. Merino and C. Nevado, Chem. Soc. Rev., 2014, 43, 6598 RSC
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS
-
(a) J. K. Ruff, Inorg. Chem., 1965, 4, 567 CrossRef CAS
- N. Kamigata, O. Kawakita, A. Lzuoka and M. Kobayashi, J. Org. Chem., 1985, 50, 398 CrossRef CAS
-
(a) Y. Xia, L. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 12940 CrossRef CAS
- K. Jana, A. Bhunia and A. Studer, Chem, 2020, 6, 512 CAS
- For recent remarkable examples of bifunctional reagents:
(a) J. Yu, Z. Wu and C. Zhu, Angew. Chem., Int. Ed., 2018, 57, 17156 CrossRef CAS
-
(a) D. Zhong, D. Wu, Y. Zhang, Z. Lu, M. Usman, W. Liu, X. Lu and W.-B. Liu, Org. Lett., 2019, 21, 5808 CrossRef CAS
-
(a) G. Sheng, K. Huang, Z. Chi, H. Ding, Y. Xing, P. Lu and Y. Wang, Org. Lett., 2014, 16, 5096 CrossRef CAS
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317 CrossRef CAS
- A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8950 CrossRef CAS
-
(a) C. Walling, Tetrahedron, 1985, 41, 3887 CrossRef CAS
-
(a) X. Su, H. Huang, Y. Yuan and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 1338 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06473d |
| This journal is © The Royal Society of Chemistry 2021 |