
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(I)- and Rh(II)-catalyzed C–H alkylation of benzylamines with alkenes and its application in flow chemistry†
Amrita
Das
and
Naoto
Chatani
 *
*
Department of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: chatani@chem.eng.osaka-u.ac.jp
First published on 4th January 2021
Abstract
The Rh-catalyzed C–H alkylation of benzylamines with alkenes using a picolinamide derivative as a directing group is reported. Both Rh(I) and Rh(II) complexes can be used as active catalysts for this transformation. In addition, a flow set up was designed to successfully mimic this process under flow conditions. Several examples are presented under flow conditions and it was confirmed that a flow process is advantageous over a batch process. Deuterium labelling experiments were performed to elucidate the mechanism of the reaction, and the results indicated a possible carbene mechanism for this C–H alkylation process.
Introduction
Benzylamine derivatives are important core structures in various biologically active compounds and natural products.1 Hence, the functionalization of benzylamine derivatives via C–H bond activation strategies is one of the more important areas in organic chemistry.2 Direct functionalization and a directing group strategy have both been used for this type of derivatization. The ortho-functionalization of benzylamine derivatives, such as carbonylation,2a alkenylation,2b alkylation by using alkyl halides,2c trifluoromethylation,2d halogenation,2e arylation,2g sulfonylation,2k and hydroxylation2l using Pd complexes as catalysts have been reported (Scheme 1a). Apart from the use of palladium, only a few examples of the use of other metals as catalysts, such as Co2h–j or Rh2f for annulation or alkynylation reactions with alkynes have been reported. Given our continuous interest in C–H alkylation reactions,3 we were strongly motivated to investigate the ortho-alkylation of benzylamines with alkenes as alkyl sources, which, as of this writing, does not appear to have been reported (Scheme 1b). | ||
| Scheme 1 Functionalization of benzylamine. | ||
Flow chemistry is a growing field of research in organic chemistry. In industrial research, it is well known that flow chemistry has great advantages and is now a well-established process that has a variety of applications.4 In recent years flow transformation reactions have also found their way into academic research laboratories.5 Considering the advantages of flow chemistry over traditional batch reactions, such as reaction efficiency, safety, and environmental compatibility;5a–c we evaluated the potential of this process for use in the reaction described in this report. Because flow chemistry has not been extensively explored in the case of C–H bond activation (Scheme 1c),6 there is no apparent reason for why its flow conditions could not be used in this area of research (Scheme 1d). To our delight, we successfully optimized the conditions for our reaction even under flow. Moreover, the advantages of this process over batch reactions are also presented (Scheme 1e).
Results and discussion
We selected 2-methylbenzylamine and ethyl acrylate as model substrates to screen various directing groups7 using [Rh(OAc)(cod)]2 as a catalyst and pivalic acid as an additive in toluene as a solvent at 160 °C for 16 h (Table 1). An imine directing group failed to give the product 2aa. We then hypothesized that a bidentate directing group might promote the reaction. However, the imine prepared from 2-pyridinecarboxaldehyde also failed to provide the desired product 2ab. We next focused on the use of an amide directing group for this transformation. Our hypothesis was correct regarding the use of an amide directing group and the use of a picolinamide derivative promoted the reaction to a significant extent, with 2a being produced in a high yield, whereas in the absence of a nitrogen atom in the directing group, 2ac was not produced. Furthermore, to check the importance of a free NH moiety, we prepared a N–Me protected directing group. However, no 2ad was formed when the N–Me protected directing group was used, which supports the fact that the presence of a free NH group on the bidentate coordination group is crucial for the reaction to proceed.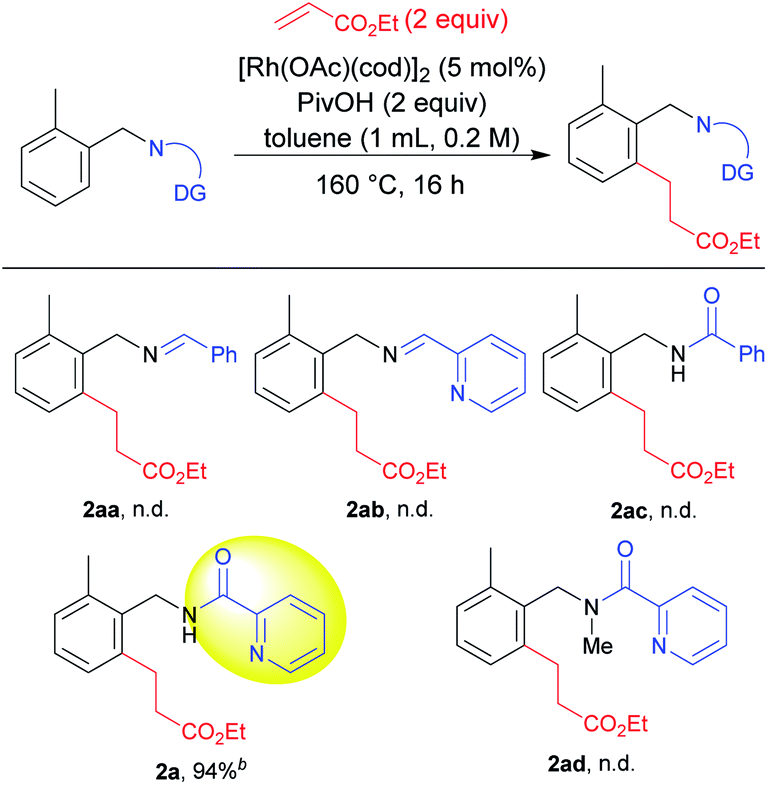
With a suitable directing group being identified, we proceeded to optimize the reaction conditions (Table 2). After screening a series of reaction temperatures, 160 °C was found to give a high yield of 2a (Table 2, entries 1–3). The amount of acid additive used in the reaction was also screened. Comparing entries 3–5, it was found that the use of 2 equiv. of pivalic acid was needed to achieve optimal conditions for this reaction (entry 3). The amount of ethyl acrylate was also found to be important for achieving a high yield. When it was decreased to 1.2 equiv. from the standard amount of 2 equiv., a lower product yield was obtained (entry 6). We further examined some other Rh catalysts such as Rh(I) (entries 7 and 8), Rh(II) (entry 9), and Rh(III) (entry 10) as catalysts for this transformation. The Rh(II) acetate dimer also provided the product in an excellent yield of 97% (entry 9). On the other hand, the Rh(III) catalyst was completely unreactive (entry 10). Finally, by slightly increasing the reaction temperature to 170 °C and using the Rh(II) acetate dimer, a quantitative yield of the desired product 2a was observed (entry 11). From the catalyst screening data, we obtained an interesting finding in that both Rh(I) (entry 3) and Rh(II) catalysts (entries 9 and 11) showed similar reactivities, giving high yields of the desired product. Finally, these conditions were determined to be the standard reaction conditions.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Temp. | Yield of 2ab (%) |
| a Reaction conditions: 1a (0.2 mmol), ethyl acrylate (0.4 mmol), catalyst (0.01 mmol), PivOH (0.4 mmol), toluene (1.0 mL) at specified temperature for 16 h. b Determined by 1H NMR analysis of the crude mixture. c Isolated yield. d No additive was used. e PivOH (1 equiv.) was used. f Ethyl acrylate (1.2 equiv.) was used. n.d. = not detected. | |||
| 1 | [Rh(OAc)(cod)]2 | 120 °C | 2 |
| 2 | [Rh(OAc)(cod)]2 | 150 °C | 26 |
| 3 | [Rh(OAc)(cod)]2 | 160 °C | 94(83)c |
| 4d | [Rh(OAc)(cod)]2 | 160 °C | 10 |
| 5e | [Rh(OAc)(cod)]2 | 160 °C | 62 |
| 6f | [Rh(OAc)(cod)]2 | 160 °C | 27 |
| 7 | [RhCl(cod)]2 | 160 °C | 11 |
| 8 | [RhCl(PPh3)3]2 | 160 °C | 58 |
| 9 | Rh2(OAc)4 | 160 °C | 97 |
| 10 | [Cp*RhCl2]2, AgSbF6 (20 mol%) | 160 °C | n.d. |
| 11 | Rh2(OAc)4 | 170 °C | 99 (85)c |
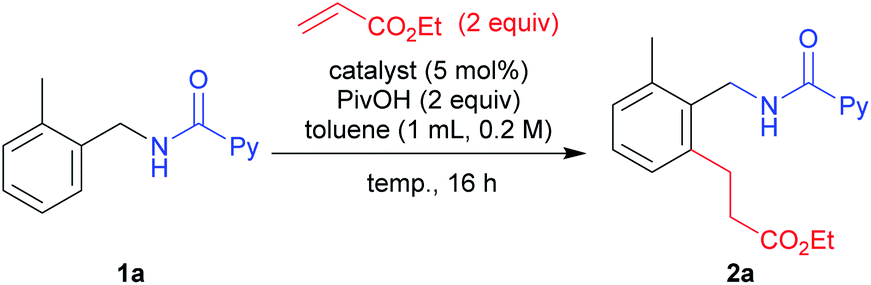
With the optimized conditions in hand, we next examined the substrate scope for the reaction (Table 3). The reaction of 1a with ethyl acrylate gave 2a in 83% isolated yield under the conditions shown in entry 3, Table 2. However, a lower isolated yield was obtained in the reaction with methyl acrylate (2b, 61%). By changing the reaction conditions to that used in entry 11, Table 2, the yield of 2b was sharply increased to 89% isolated yield. From this result, we decided to use mainly condition B for further investigations into the substrate scope. Not only ethyl (2a) and methyl (2b) but also n-butyl (2c) and more sterically congested acrylates such as benzyl (2d), phenyl (2e) and cyclohexyl (2f) acrylates all gave the corresponding products in good to high yields. A simple methyl vinyl ketone also gave the desired product 2g in moderate yield. With the successful identification of various acrylate coupling partners, we then examined C–H alkylation reactions with acrylates derived from biologically active compounds. When an acrylate derived from the essential animal sterol cholesterol was subjected to the alkylation reaction, the desired product 2h was produced in 79% isolated yield. Another candidate for biologically useful compounds was synthesized from oleyl alcohol which is an unnatural fatty alcohol and the reaction showed a high tolerance (2i). When diethyl vinylphosphonate was used, the desired product 2j was obtained in 90% yield. The maleimide derivative was also well tolerated under the reaction conditions to deliver the product 2k in high yield. The scope of styrene derivatives was also examined, where an increased amount of the styrene substrate and 7.5 mol% of Rh2(OAc)4 were needed to produce high yields of the desired products (2l–2q). We then moved our focus to testing the effect of substituent groups on the benzylamine moiety. Benzylamine derivatives bearing both electron-donating (2r) and electron-withdrawing (2s–2u) groups provided the expected products in high yields. To our delight, meta-substituted benzylamines resulted in a regioselective C–H alkylation to give the desired mono-alkylation products in moderate (2v) to high yields (2w). However, a starting material derived from 3-chloro-4-methoxybenzylamine resulted in the formation of the di-alkylation product in 28% yield, along with desired product 2x, probably due to the smaller sized substitution at the meta position. A di-substituted benzylamine moiety was also suitable, with the corresponding product 2y being produced in 71% yield. In the case of naphthylmethyl amine, the ortho-alkylation product 2z was formed as the sole product in excellent yield. The thiophen-2-ylmethylamine derivative reacted smoothly to give 2za in 71% yield, along with a minor amount of the alkenylated product. We also carried out the reaction on a gram scale to show practical utility of our catalytic system on a bulk scale and 2a was produced in high yield.
| a Reaction conditions: 1 (0.2 mmol), α,β-unsaturated compound (0.4 mmol), catalyst (0.01 mmol), PivOH (0.4 mmol), toluene (1.0 mL) at the specified temperature for 16 h. Isolated yields are reported in all cases. b Condition A was used. c Condition B was used. d 1.32 g of 2a was obtained from 5 mmol scale synthesis. e 7.5 mol% Rh2(OAc)4 and 5 equiv. styrene derivative were used. f A trace amount of the alkenylation product was observed. g 9% alkenylation product was observed. h A di-alkylation product was obtained in 28% yield. i 7.5 mol% of Rh2(OAc)4 was used. j An alkenylation product was obtained in 18% yield. |
|---|
|
We next turned our attention to optimizing the reaction conditions under a flow set up. A stock solution of the starting materials was prepared by dissolving 1a, ethyl acrylate (2 equiv.), [Rh(OAc)(cod)]2 (5 mol%), and PivOH (2 equiv.) in toluene (1 mL, 0.2 M). The solution was pumped using a dual pump at various flow rates and the solution was allowed to flow through a certain length of a stainless-steel loop that was immersed in an oil bath at 170 °C and further connected through a back-pressure regulator with a pressure of 1 MPa. After allowing 1 mL of the stock solution into the flow setup, an additional 15 mL of toluene was then allowed to flow after all of the stock solution had passed through the set-up, in order to collect the sample completely. We conducted a few optimizations for this set-up to obtain the optimal residence time for the flow reaction. It is noteworthy that we used [Rh(OAc)(cod)]2 as the catalyst because it is soluble in the reaction solvent which is crucial, since the reaction mixture needed to be completely homogeneous before being pumped through the flow pump. After screening several conditions, we obtained an optimal residence time of 196 minutes by using a 10 meter loop at a flow rate of 0.04 mL min−1 for this reaction towards full completion (Table 4, entry 3).
|
|
||||
|---|---|---|---|---|
| Entry | Loop length (xx m) | Flow rate (xx mL min−1) | Residence time (min) | Yield of 2ab (%) |
| a Reaction conditions: a stock solution containing 1a (0.2 mmol), ethyl acrylate (0.4 mmol), [Rh(OAc)(cod)]2 (0.01 mmol) and PivOH (0.4 mmol) in toluene (1.0 mL) was prepared and the resulting solution was allowed to flow through the stainless loop at a specified flow rate. b Determined by 1H NMR analysis of the crude mixture collected over three fractions to obtain the average value. | ||||
| 1 | 5 | 0.05 | 79 | 30 |
| 2 | 10 | 0.05 | 157 | 80 |
| 3 | 10 | 0.04 | 196 | 88 |
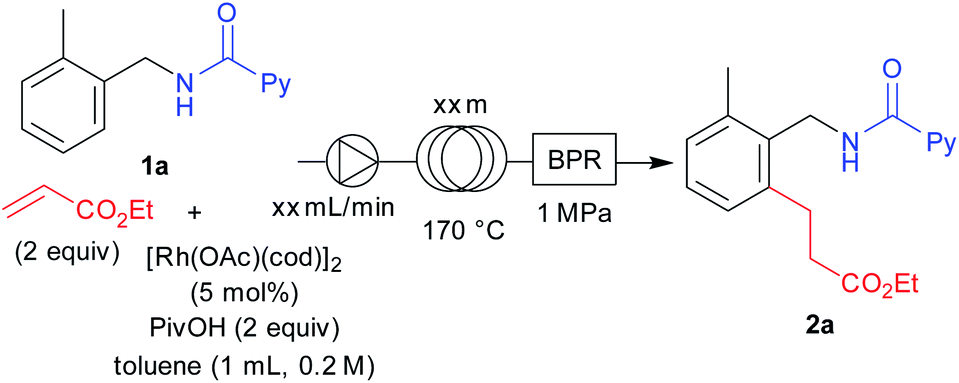
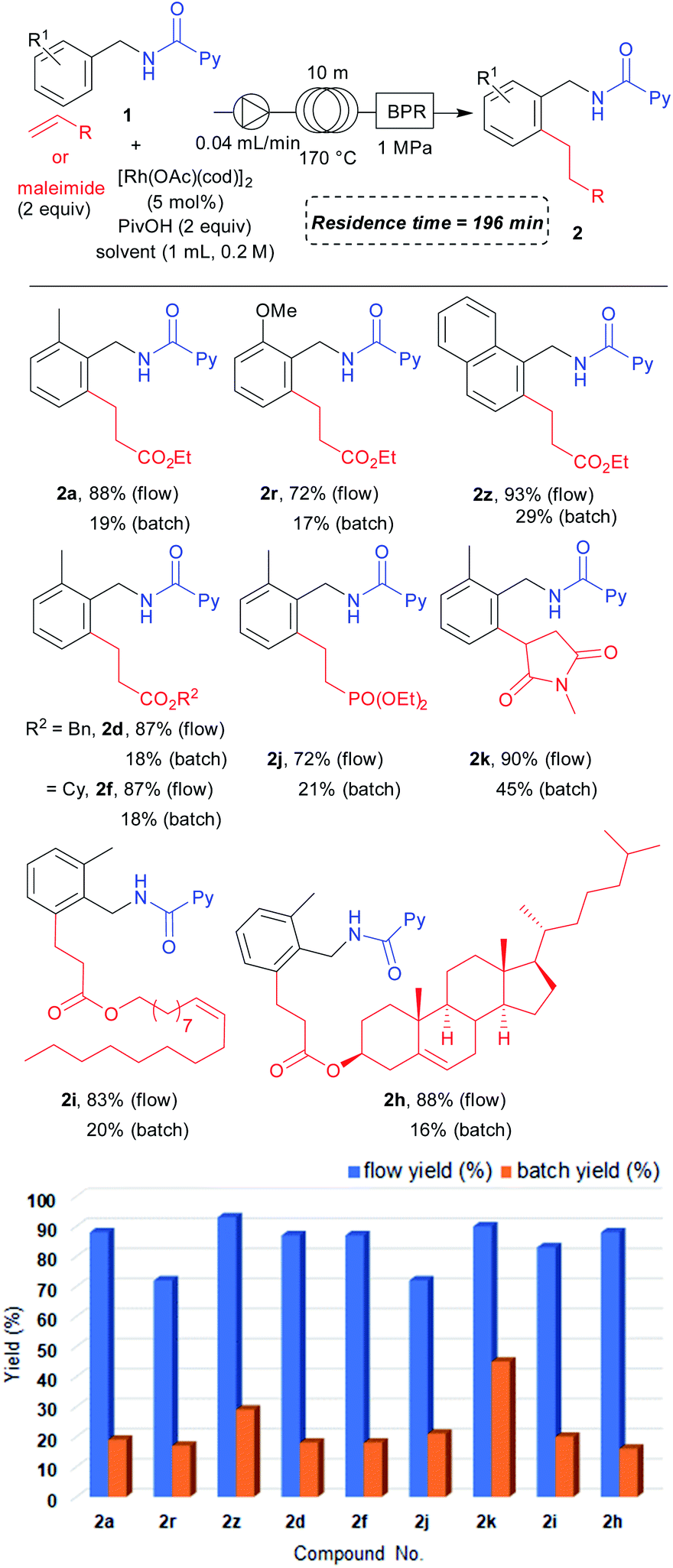
Having the optimized reaction under flow conditions in hand, we next examined the substrate scope under flow conditions (Table 5). Both ortho-methyl- (2a) and methoxy-substituted (2r) benzylamine derivatives provided the desired products in excellent yields. The naphthylmethyl amine derived substrate also provided 2z in an excellent yield. For the scope of alkenes, benzyl (2d), cyclohexyl (2f) acrylates and diethyl vinylphosphonate (2j) all furnished the desired products in high yields under flow conditions. We also obtained 2k in excellent yield in the reaction with N-methylmaleimide. Biologically active alcohol derived acrylates could also be used under the flow conditions to give high yields of 2i and 2h. We further conducted comparison reactions between flow and batch under similar reaction conditions (reaction time 196 minutes). To our delight, flow conditions provided the desired products in higher yields compared to batch reactions performed under a shorter reaction time. These data clearly show the advantages of flow conditions over batch reaction conditions in that the yield could be increased in only 196 minutes of reaction time. The reaction efficiency can be significantly improved by increasing the surface area to volume ratio in a flow process compared to that in a batch process which helps to distribute the heat transfer in the overall system more smoothly. This could be one of the explanations for the high efficiency of the reaction under flow conditions even when a shorter reaction time is used. However, this may not be the only reason for the significant improvement in reaction efficiency under flow conditions. Importantly, the reaction can be performed safely even when the reaction temperature is higher than the solvent's boiling point.
| a Reaction conditions for flow: 1 (0.2 mmol), α,β-unsaturated compound (0.4 mmol), [Rh(OAc)(cod)]2 (0.01 mmol) and PivOH (0.4 mmol) were dissolved in toluene (1.0 mL) to prepare a stock solution which was then allowed to flow through the stainless loop at a rate of 0.04 mL min−1. Determined by 1H NMR analysis of the crude mixture collected over three fractions to obtain the average value. Reaction conditions for batch: 1 (0.2 mmol), α,β-unsaturated compound (0.4 mmol), [Rh(OAc)(cod)]2 (0.01 mmol), PivOH (0.4 mmol), and toluene (1.0 mL) at 170 °C for 196 minutes. Determined by 1H NMR analysis of the crude mixture. |
|---|
|
To investigate the mechanistic aspects of this C–H alkylation reaction, deuterium labelling experiments were carried out. When 1a was treated with styrene-d8 for a shorter reaction time in the presence of the Rh(II) catalyst, 0.76 H was incorporated at the α position and 1.05 H at the β position in the product 3, suggesting that the mechanism is not a simple one (Scheme 2a). In addition, the ortho C–H bond in the recovered starting material contained 0.32 D. This result clearly indicates that the cleavage of the ortho C–H and C–D bonds is reversible. To determine whether the mechanism is the same or different, depending on the oxidation state of the Rh complexes, we also performed the above reaction using [Rh(OAc)(cod)]2 as the catalyst (Scheme 2b). A similar H/D scrambling was observed, where 0.73 H and 1.05 H were incorporated at the α position and the β position in 3′, respectively. In addition, a deuterium atom was also incorporated (0.25 D) into the starting material. These results imply that both the Rh(I) and Rh(II) catalytic systems appear to follow the same reaction mechanism. When the isotopically labelled substrate 1a-D was reacted with styrene, a 0.26 D incorporation was observed at the α position of the product 5 (Scheme 2c). On the other hand, no deuterium atom incorporation was detected at the β position. This suggests that a deuterium atom at the α position came from the ortho C–D bond of the substrate 1a-D. The recovered starting material contained 0.72 H at the ortho-position, again indicating that the cleavage of the ortho C–H bond is reversible. A deuterium atom was also incorporated only at the α position in the reaction between 1a-D and ethyl acrylate (Scheme 2d). The recovered starting material was also found to contain 0.52 H after the reaction. This result is similar to that observed in the case of the reaction with styrene, as shown in Scheme 2c.
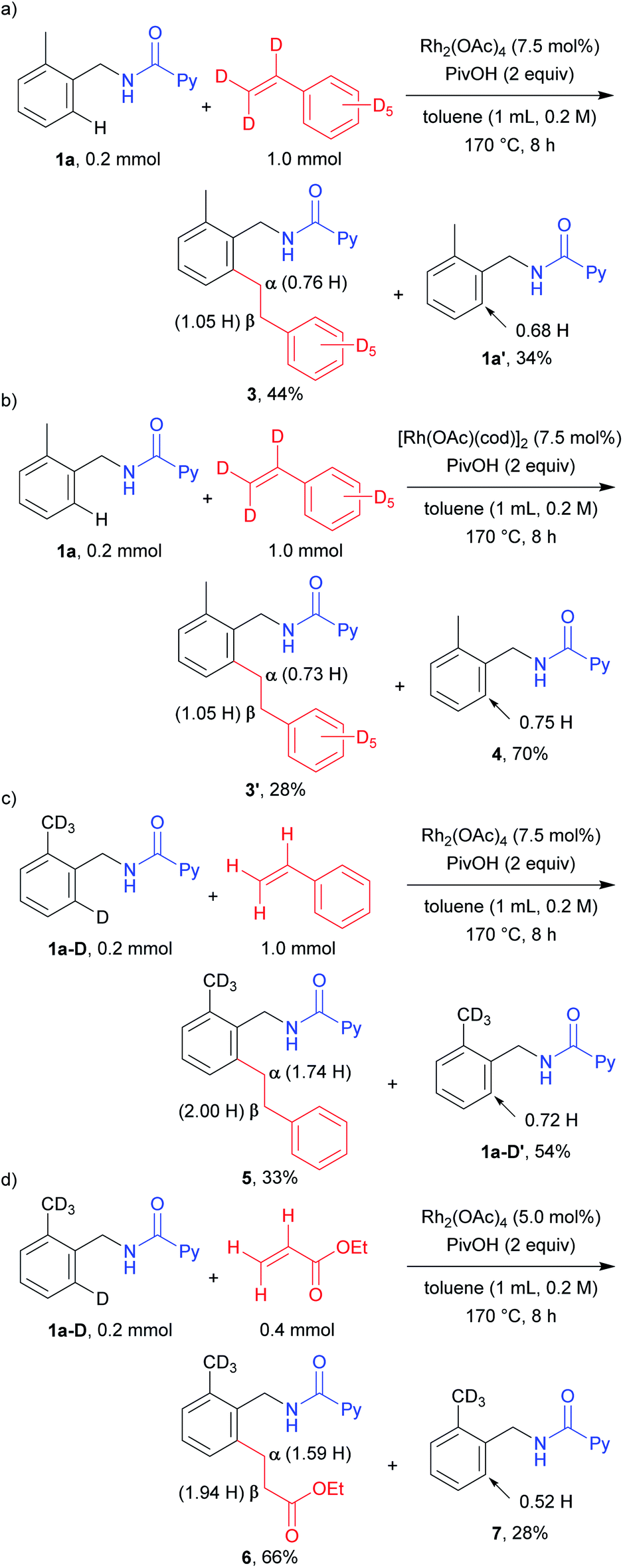 | ||
| Scheme 2 Deuterium labelling experiments (1). | ||
To collect additional mechanistic information regarding this alkylation, a deuterated substrate bearing a free ND bond (1a-ND) was reacted with ethyl acrylate in the presence of deuterated acid PivOD (Scheme 3a). In sharp contrast to the NH substrate, such as 1a and 1a-D, a deuterium atom was incorporated at the β position (1.60 H), suggesting that a deuterium atom is transferred from a nitrogen to the β position. However, the ratio of the deuterium incorporation was not so high because ND is easily converted into NH in the presence of the acid additive (the deuterated acid can be converted into the protic acid form through reversible ortho C–H activation of the substrate). When the deuterated substrate 1a-D was reacted in the absence of an alkene coupling partner for 8 h, H/D exchange was observed at the ortho-position (Scheme 3b). Thus, C–H activation is a reversible pathway.
 | ||
| Scheme 3 Deuterium labelling experiments (2). | ||
We also carried out a kinetic isotope effect (KIE) study by conducting two individual experiments containing 1a and the deuterated substrate 1a-D with styrene (Fig. 1). The kH/kD ratio from these experiments was found to be 1.02, which further supports the conclusion that the C–H activation step is not the rate determining one.8
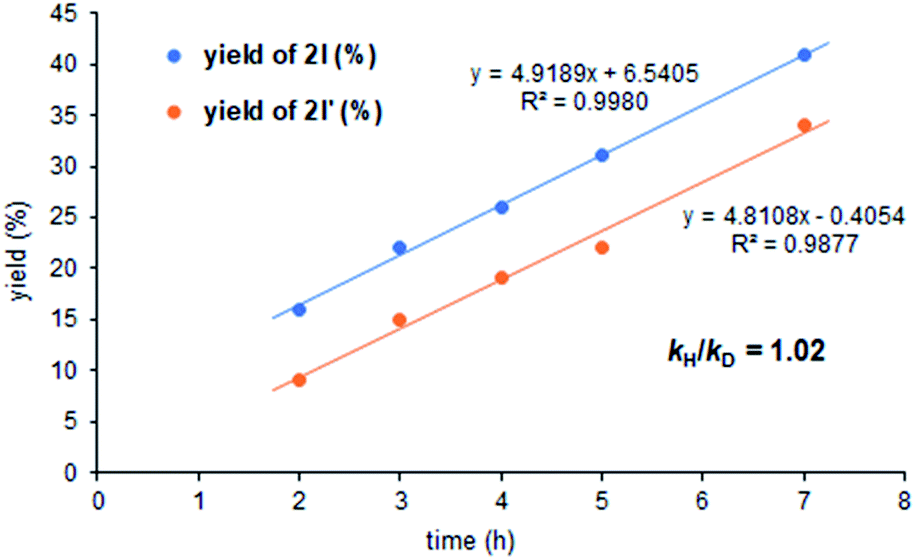 | ||
| Fig. 1 Determination of the kH/kD ratio. | ||
Based on the deuterium labelling experiments, the following plausible catalytic cycle is proposed (Scheme 4). Both catalysts [Rh(OAc)(cod)]2 and Rh2(OAc)4 were active for this transformation. The oxidative addition of an N–H bond to a Rh center provides Rh(III) or Rh(IV)9 species A (see ESI for details†) (the evidence of Rh(IV) formation was that the solution turned to a dark purple colour).10 In the next step, the insertion of an alkene into the Rh–H bond gives intermediate B, in which the NH hydrogen is transferred to the β position, as evidenced by deuterium labelling experiments (Scheme 3a). Two possible pathways are proposed from intermediate B. The activation of the ortho C–H bond generates intermediate Dvia a CMD mechanism (path a) or the elimination of the carboxylic acid (path b), which involves the α-elimination of the carboxylic acid to form a carbene intermediate C.3i,m,11 The final step for path b involves the migratory insertion of the ortho C–H bond into the carbene in C to afford D. Fast deuterium scrambling occurred at the ortho C–H bond, hence ruling out the possibility that C–H activation is the rate determining step. In fact, the kH/kD ratio was 1.02. Subsequently, in the presence of carboxylic acid, reductive elimination takes place from D which results in the formation of the product along with the regeneration of the [Rh] species which further continues the catalytic cycle. The most important result in the deuterium labelling experiments involved the finding that the ortho C–H bond is transferred only to the α-position of the product (Scheme 2c and d). If the reaction proceeded via path a, the ortho C–H bond would not be transferred to the product because it is eliminated as a carboxylic acid as a result of a CMD mechanism. Thus, path b is the most likely mechanism, however path a cannot be excluded completely.
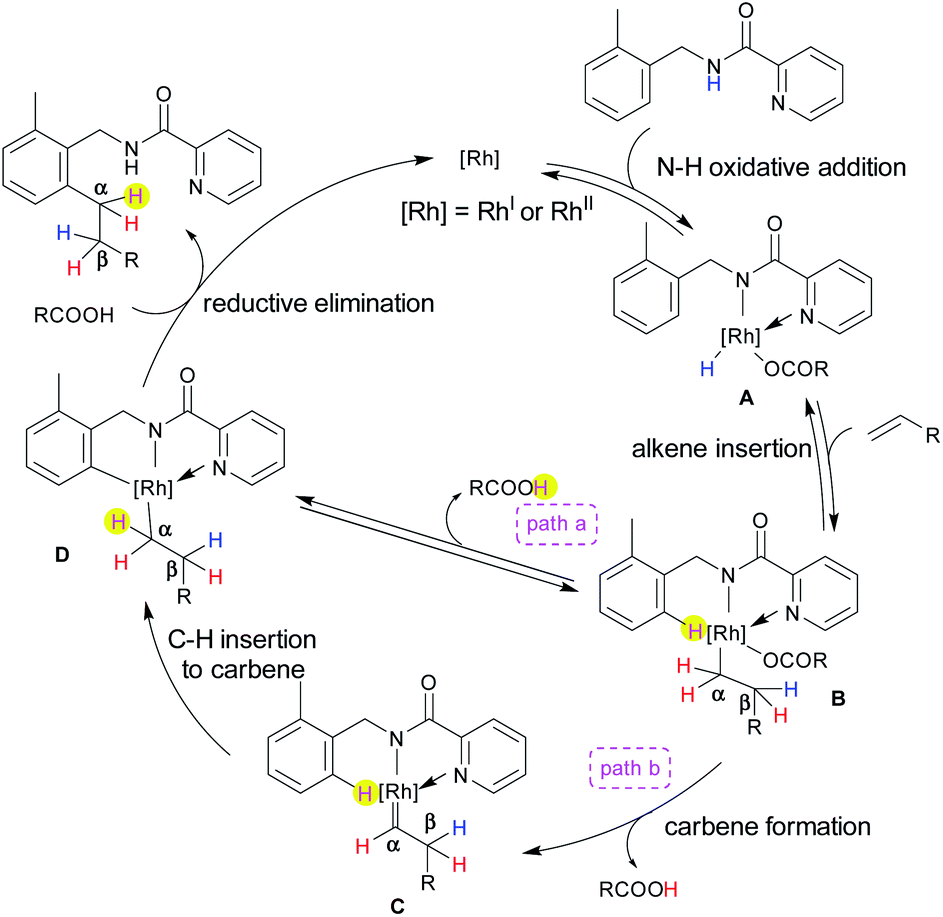 | ||
| Scheme 4 Proposed catalytic cycle. | ||
Conclusions
In summary, we report herein the C–H alkylation of benzylamine derivatives catalysed by both Rh(I) and Rh(II) catalysts. A picolinamide derivative as the directing group for this transformation was found to be crucial for the reaction to proceed. The reaction has a broad substrate scope for the alkenes being used including α,β-unsaturated compounds and styrene derivatives and various substituted benzylamines were well tolerated. To our delight, not only benzylamine but also other heterocycles such as a thiophene derived amine were also found to be suitable for the alkylation. The further novel applicability of this strategy involved the fact that the reaction is amenable to use under flow conditions. C–H activation chemistry under flow conditions is still a challenging and attractive field of research. Thus, our successful design of a flow set up for this alkylation process signifies its further scope and advantages. Mechanistic investigations suggest the possible involvement of a carbene mechanism for the reaction. Further expanding the scope of substrates for alkylation and other useful reactions under flow conditions is currently underway in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Grant in Aid for Specially Promoted Research by MEXT (No. 17H06091).Notes and references
- (a) Z. Zhang and D. B. Mccormick, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10407–10410 CrossRef CAS; (b) S. Maignan, J.-P. Guilloteau, S. Pouzieux, Y. M. Choi-sledeski, M. R. Becker, S. I. Klein, W. R. Ewing, H. W. Pauls, A. P. Spada and V. Mikol, J. Med. Chem., 2000, 43, 3226–3232 CrossRef CAS; (c) R. Kolhatkar, C. D. Cook, S. K. Ghorai, J. Deschamps, P. M. Beardsley, M. E. A. Reith and A. K. Dutta, J. Med. Chem., 2004, 47, 5101–5113 CrossRef CAS; (d) K.-J. Xiao, L. Chu, G. Chen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 7796–7800 CrossRef CAS; (e) Z. Jiang, W. D. Hong, X. Cui, H. Gao, P. Wu, Y. Chen, D. Shen, Y. Yang, B. Zhang, M. J. Taylor, S. A. Ward, P. M. O'Neill, S. Zhao and K. Zhang, RSC Adv., 2017, 7, 52227–52237 RSC; (f) N. Mibu, K. Yokomizo, M. Sano, Y. Kawaguchi, K. Morimoto, S. Shimomura, R. Sato, N. Hiraga, A. Matsunaga, J. Zhou, T. Ohata, H. Aki and K. Sumoto, Chem. Pharm. Bull., 2018, 66, 830–838 CrossRef CAS; (g) Y. Li, W. Yao, J. Lin, F. Li, Y. Wu and J. Xu, J. Heterocycl. Chem., 2019, 56, 2170–2178 CrossRef CAS.
- For selected examples of C–H functionalizations of benzylamine derivatives, see: (a) K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 CrossRef CAS; (b) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666–7673 CrossRef CAS; (c) Y. Zhao and G. Chen, Org. Lett., 2011, 13, 4850–4853 CrossRef CAS; (d) M. Miura, C.-G. Feng, S. Ma and J.-Q. Yu, Org. Lett., 2013, 15, 5258–5261 CrossRef CAS; (e) C. Lu, S.-Y. Zhang, G. He, W. A. Nack and G. Chen, Tetrahedron, 2014, 70, 4197–4202 CrossRef CAS; (f) Á. M. Martínez, J. Echavarren, I. Alonso, N. Rodríguez, R. Gómez-Arrayás and J. C. Carretero, Chem. Sci., 2015, 6, 5802–5814 RSC; (g) B. N. Laforteza, K. S. L. Chan and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 127, 11295–11298 CrossRef; (h) V. G. Landge, S. P. Midya, J. Rana, D. R. Shinde and E. Balaraman, Org. Lett., 2016, 18, 5252–5255 CrossRef CAS; (i) Á. M. Martínez, N. Rodríguez, R. Gómez-Arrayás and J. C. Carretero, Chem.–Eur. J., 2017, 23, 11669–11676 CrossRef; (j) C. Kuai, L. Wang, B. Li, Z. Yang and X. Cui, Org. Lett., 2017, 19, 2102–2105 CrossRef CAS; (k) U. Karmakar and R. Samanta, J. Org. Chem., 2019, 84, 2850–2861 CrossRef CAS; (l) C. Dai, Y. Han, L. Liu, Z.-B. Huang, D.-Q. Shi and Y. Zhao, Org. Chem. Front., 2020, 7, 1703–1708 RSC.
- For reviews on C–H alkylations with alkenes, see: (a) Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS; (b) N. Chatani, Bull. Chem. Soc. Jpn., 2018, 91, 211–222 CrossRef CAS . For selected examples from our group, see; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2013, 135, 5308–5311 CrossRef CAS; (d) G. Rouquet and N. Chatani, Chem. Sci., 2013, 4, 2201–2208 RSC; (e) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148–5151 CrossRef CAS; (f) K. Shibata, T. Yamaguchi and N. Chatani, Org. Lett., 2015, 17, 3584–3587 CrossRef CAS; (g) Y. Aihara, J. Wuelbern and N. Chatani, Bull. Chem. Soc. Jpn., 2015, 88, 438–446 CrossRef CAS; (h) Q. He, T. Yamaguchi and N. Chatani, Org. Lett., 2017, 19, 4544–4547 CrossRef CAS; (i) K. Shibata, S. Natsui, M. Tobisu, Y. Fukumoto and N. Chatani, Nat. Commun., 2017, 8, 1448 CrossRef; (j) S. Rej and N. Chatani, ACS Catal., 2018, 8, 6699–6706 CrossRef CAS; (k) Q. He and N. Chatani, J. Org. Chem., 2018, 83, 13587–13594 CrossRef CAS; (l) S. Rej and N. Chatani, Chem. Commun., 2019, 55, 10503–10506 RSC; (m) T. Yamaguchi, S. Natsui, K. Shibata, K. Yamazaki, S. Rej and Y. Ano, Chem.–Eur. J., 2019, 25, 6915–6919 CrossRef CAS; (n) C. Wang, S. Rej and N. Chatani, Chem. Lett., 2019, 48, 1185–1187 CrossRef CAS; (o) N. Ohara, S. Rej and N. Chatani, Chem. Lett., 2020, 49, 1053–1057 CrossRef CAS.
- (a) B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS; (b) S. A. May, J. Flow Chem., 2017, 7, 137–145 CrossRef CAS; (c) D. Dallinger and C. O. Kappe, Curr. Opin. Green Sustain. Chem., 2017, 7, 6–12 CrossRef; (d) A. R. Bogdan and A. W. Dombrowski, J. Med. Chem., 2019, 62, 6422–6468 CrossRef CAS; (e) M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24(10), 1802–1813 CrossRef CAS.
- For a book chapter, see: (a) T. F. Jamison and G. Koch, Science of Synthesis: Flow Chemistry in Organic Synthesis, 1st edn, Georg Thieme Verlag KG: Stuttgart, 2018. DOI:10.1055/b-006-161272. For recent reviews, see; (b) D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675–680 RSC; (c) R. Porta, M. Benaglia and A. Puglisi, Org. Process Res. Dev., 2016, 20, 2–25 CrossRef CAS; (d) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, Chem. Rev., 2017, 117, 11796–11893 CrossRef CAS; (e) S. Santoro, F. Ferlin, L. Ackermann and L. Vaccaro, Chem. Soc. Rev., 2019, 48, 2767–2782 RSC; (f) S. Govaerts, A. Nyuchev and T. Noël, J. Flow Chem., 2020, 10, 13–71 CrossRef CAS . For selected examples, see:; (g) M. Christakakou, M. Schön, M. Schnürch and M. D. Mihovilovic, Synlett, 2013, 24, 2411–2418 CrossRef CAS; (h) H. P. L. Gemoets, G. Laudadio, K. Verstraete, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2017, 56, 7161–7165 CrossRef CAS; (i) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS; (j) X. Wei, I. Abdiaj, C. Sambiagio, C. Li, E. Zysman-colman, J. Alcázar and T. Noël, Angew. Chem., Int. Ed., 2019, 58, 13030–13034 CrossRef CAS; (k) G. Laudadio, Y. Deng, K. Van Der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS; (l) Z. Wen, A. Maheshwari, C. Sambiagio, Y. Deng, G. Laudadio, K. V. Aken, Y. Sun, H. P. L. Gemoets and T. Noël, Org. Process Res. Dev., 2020, 24, 2356–2361 CrossRef CAS.
- (a) H. P. L. Gemoets, V. Hessel and T. Noël, Org. Lett., 2014, 16, 5800–5803 CrossRef CAS; (b) J. Zakrzewski, A. P. Smalley, M. A. Kabeshov, M. J. Gaunt and A. A. Lapkin, Angew. Chem., Int. Ed., 2016, 55, 8878–8883 CrossRef CAS; (c) H. Wang, F. Pesciaioli, J. C. A. Oliveira, S. Warratz and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 15063–15067 CrossRef CAS; (d) U. K. Sharma, H. P. L. Gemoets, F. Schröder, T. Noël and E. V. Van der Eycken, ACS Catal., 2017, 7, 3818–3823 CrossRef CAS; (e) F. Ferlin, S. Santoro, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 2510–2514 RSC; (f) F. Ferlin, L. Luciani, S. Santoro, A. Marrocchi, D. Lanari, A. Bechtoldt, L. Ackermann and L. Vaccaro, Green Chem., 2018, 20, 2888–2893 RSC; (g) C. Zhu, J. C. A. Oliveira, Z. Shen, H. Huang and L. Ackermann, ACS Catal., 2018, 8, 4402–4407 CrossRef CAS; (h) A. Bouchard, V. Kairouz, M. Manneveau, H. Xiong, T. Besset, X. Pannecoucke and H. Lebel, J. Flow Chem., 2019, 9, 9–12 CrossRef CAS; (i) W.-J. Kong, L. H. Finger, A. M. Messinis, R. Kuniyil, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2019, 141, 17198–17206 CrossRef CAS.
- (a) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS; (b) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843–895 RSC; (c) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107–1295 RSC; (d) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (e) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (f) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS; (g) J. I. Higham and J. A. Bull, Org. Biomol. Chem., 2020, 18, 7291–7315 RSC.
- (a) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef; (b) Y. Xia, Z. Liu, S. Feng, Y. Zhang and J. Wang, J. Org. Chem., 2015, 80, 223–236 CrossRef CAS.
- S. B. Sinha, D. Y. Shopov, L. S. Sharninghausen, D. J. Vinyard, B. Q. Mercado, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2015, 137, 15692–15695 CrossRef CAS.
- (a) S. A. Macgregor, Organometallics, 2001, 20, 1860–1874 CrossRef CAS; (b) J. Zhao, A. S. Goldman and J. F. Hartwig, Science, 2005, 307, 1080–1083 CrossRef CAS; (c) I. Mena, M. A. Casado, P. García-Orduña, V. Polo, F. J. Lahoz, A. Fazal and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 11735–11738 CrossRef CAS; (d) E. Vélez, M. P. Betoré, M. A. Casado and V. Polo, Organometallics, 2015, 34, 3959–3966 CrossRef.
- (a) N. D. Jones, G. Lin, R. A. Gossage, R. Mcdonald and R. G. Cavell, Organometallics, 2003, 22, 2832–2841 CrossRef CAS; (b) T. Cantat, M. Demange, N. Mezailles, L. Ricard, Y. Jean and P. Le Floch, Organometallics, 2005, 24, 4838–4841 CrossRef CAS; (c) H. Heuclin, X. F. Le Goff and N. Mezailles, Chem.–Eur. J., 2012, 18, 16136–16144 CrossRef CAS; (d) F. Hu, Y. Xia, C. Ma, Z. Yan and J. Wang, Chem. Commun., 2015, 51, 7986–7995 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05813k |
| This journal is © The Royal Society of Chemistry 2021 |