DOI:
10.1039/D0SC06186G
(Edge Article)
Chem. Sci., 2021,
12, 3599-3607
Rhodium-catalysed selective C–C bond activation and borylation of cyclopropanes†
Received
10th November 2020
, Accepted 14th January 2021
First published on 15th January 2021
Abstract
Transition metal (TM)-catalysed directed hydroboration of aliphatic internal olefins which facilitates the construction of complex alkylboronates is an essential synthetic methodology. Here, an efficient method for the borylation of cyclopropanes involving TM-catalysed directed C–C activation has been developed. Upon exposure to neutral Rh(I)-catalyst systems, N-Piv-substituted cyclopropylamines (CPAs) undergo proximal-selective hydroboration with HBpin to provide valuable γ-amino boronates in one step which are otherwise difficult to synthesize by known methods. The enantioenriched substrates can deliver chiral products without erosion of the enantioselectivities. Versatile synthetic utility of the obtained γ-amino boronates is also demonstrated. Experimental and computational mechanistic studies showed the preferred pathway and the origin of this selectivity. This study will enable the further use of CPAs as valuable building blocks for the tunable generation of C–heteroatom or C–C bonds through selective C–C bond activation.
Introduction
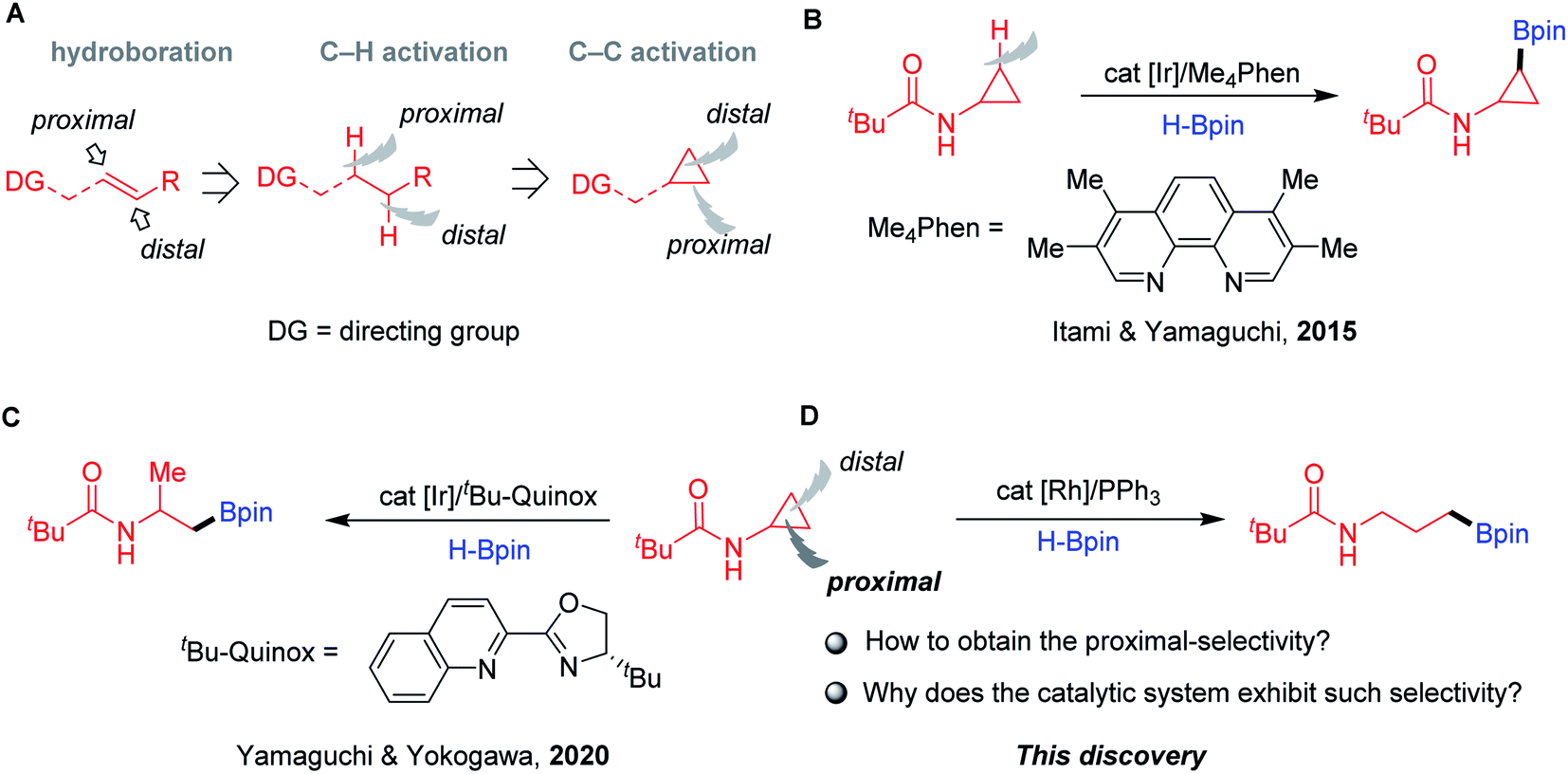
Alkylboronic esters1 are highly important in all facets of chemical science, ranging from synthetic chemistry2 to materials science3 to drug discovery.4 Traditionally, these compounds can be prepared by the well-known Brown hydroboration, in which the syn-addition of hydroboranes to alkenes follows an anti-Markovnikov manner.5 During the past decades, the TM-catalysed hydroboration of aliphatic internal olefins with tethered directing groups has shown interesting chemo-, regio-, and diastereoselectivity, which greatly complements the classical noncatalytic process (Fig. 1A).6 The coordination of directing groups with the TM catalysts can deliver hydroboration products with controllable distal7 and/or proximal8 selectivity. Recently, directed borylation of aliphatic C–H bonds with proximal9 and distal10 selectivity has also gained momentum. The functionalization of the C–C bond is one of the most attractive strategies in organic chemistry. Despite the significant advances, transformations that allow the selective borylation of single C–C bonds remain effectively unknown.
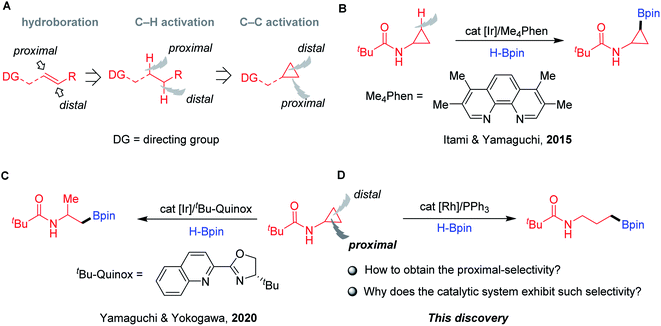 |
| | Fig. 1 TM-catalysed borylation of olefins and CPAs. (A) The development of TM-catalysed directed borylation. (B) Ir-catalysed directed C–H borylation of CPAs. (C) Ir-catalysed distal-selective hydroboration of CPAs. (D) Rh-catalysed proximal-selective hydroboration of CPAs. | |
CPAs are among the most prevalent molecules found in feedstock chemicals, drug molecules, and natural products.11 In this context, they have been exploited as robust building blocks in organic synthesis, including C–H and C–C bond functionalizations.12 Seminal work by Itami and Yamaguchi described the N-Piv-directed C–H borylation of CPAs with cis selectivity using phenanthroline-based ligands and a catalytic amount of an iridium salt (Fig. 1B).13 Inspired by the success of directed borylation of alkenes and alkanes, we envisioned that the borylation of CPAs by chelation-assisted C–C bond activation14 could provide a unique opportunity to access amino-containing boronates as potent protease inhibitors.15 During the preparation of this paper, Yamaguchi and Yokogawa reported a related transformation achieved by C–C hydroboration of CPAs using an iridium catalyst and tBu-Quinox as the ligand (Fig. 1C).16 Notably, this elegant chemistry exhibited excellent distal selectivity, resulting in β-methyl alkylboronates. In contrast, herein, we describe a simple and practical method to achieve hydroboration of CPAs via proximal C–C bond cleavage affording γ-amino boronates in the presence of a rhodium complex and a simple phosphine ligand (Fig. 1D). A series of mechanistic experiments and DFT calculations revealed the preferred pathway of this transformation, which provides high proximal-selectivity by the slightly catalytic system.
Results and discussion
Reaction design
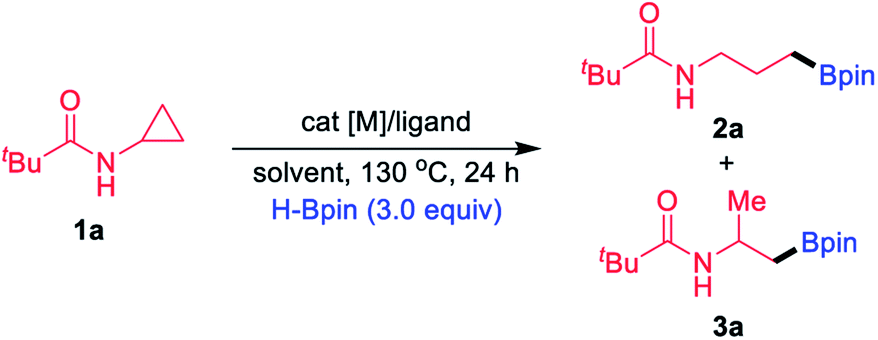
Examination and optimisation of the reaction parameters were explored using CPA 1a as the substrate and pinacolborane (HBpin) as the borylating source in conjunction with various TM catalysts and ligands (Table 1). As reported,16 when the reactions were performed with 5 mol% [Ir(cod)OMe]2 as the catalyst and 2.5 mol% tBu-Quinox as the ligand at 130 °C for 24 h in toluene, branched boronate 3a was formed in 52% yield together with a small amount of the linear byproduct 2a (2a/3a = 5/95, entry 1). In this study, we focused our attention on product 2a. Completely inverse selectivity (2a/3a > 99/1) was observed with 1 mol% Ir catalyst and 4 mol% PPh3, in which the desired product 2a was obtained in 42% yield (entry 2). Moreover, we found that Rh(I) complexes were more effective than the Ir catalyst. Upon switching the catalyst to [Rh(CO)2Cl]2, the yield of 2a further increased to 47% (entry 3). Other Rh(I) complexes, such as [Rh(coe)2Cl]2, provided the corresponding product 2a in 52% yield (entry 4), and [Rh(cod)Cl]2 was the most efficient, giving a 99% yield of 2a (entry 5). When Wilkinson's catalyst (Rh(PPh3)3Cl) was used instead of [Rh(cod)Cl]2 and PPh3, the reaction provided 2a in a much lower yield (entry 6). Under these reaction conditions, upon switching to a Rh(II) catalyst, the reaction became very sluggish (entry 7). The replacement of toluene by other solvents such as chexane and THF provided acceptable yields of 2a (entries 8 and 9). The effect of temperature was also examined, and 120 °C was found to be less optimal (entry 10). Furthermore, the use of HBpin is crucial to this borylation process; other classes of borylating sources such as B2pin2 did not afford any borylation product (entry 11). Furthermore, decreasing the Rh catalyst loading to 0.5 mol% (entry 12) or ligand loading to 2 mol% still resulted in good yields (entry 13). Finally, control experiments showed that PPh3 was required for high reactivity and the inhibition of side reactions (entry 14), and no product was observed in the absence of a Rh catalyst (entry 15).
Table 1 Optimisation of reaction conditions for the proximal-selective hydroboration of CPA 1aa
|
|
| Entry |
[M] (mol%) |
Ligand (mol%) |
Solvent |
2a/3ab |
Yield of 2ab (%) |
|
Reaction conditions: cat [M] (0.5–5 mol%), L (2–4 mol%), 1a (0.20 mmol), HBpin (0.60 mmol) in solvent (1.0 mL), 24 h, 130 °C, under Ar, in sealed Schlenk tubes.
Determined by GC-MS analysis.
Yield of purified compounds after chromatography.
At 120 °C.
Using B2pin2 instead of HBpin.
|
| 1 |
[Ir(cod)OMe]2 (5) |
t
Bu-Quinox (2.5) |
Toluene |
5/95 |
Trace |
| 2 |
[Ir(cod)OMe]2 (1) |
PPh3 (4) |
Toluene |
>99/1 |
42 |
| 3 |
[Rh(CO)2Cl]2 (1) |
PPh3 (4) |
Toluene |
>99/1 |
47 |
| 4 |
[Rh(coe)2Cl]2 (1) |
PPh3 (4) |
Toluene |
>99/1 |
52 |
|
5
|
[Rh(cod)Cl]
2
(1)
|
PPh
3
(4)
|
Toluene
|
>99/1
|
99 (91)
|
| 6 |
Rh(PPh3)3Cl (1) |
— |
Toluene |
>99/1 |
65 |
| 7 |
[Rh(OAc)2]2 (1) |
PPh3 (4) |
Toluene |
>99/1 |
4 |
| 8 |
[Rh(cod)Cl]2 (1) |
PPh3 (4) |
c
Hexane |
>99/1 |
76 |
| 9 |
[Rh(cod)Cl]2 (1) |
PPh3 (4) |
THF |
>99/1 |
70 |
| 10d |
[Rh(cod)Cl]2 (1) |
PPh3 (4) |
Toluene |
>99/1 |
73 |
| 11e |
[Rh(cod)Cl]2 (1) |
PPh3 (4) |
Toluene |
— |
0 |
| 12 |
[Rh(cod)Cl]2 (0.5) |
PPh3 (2) |
Toluene |
>99/1 |
85 |
| 13 |
[Rh(cod)Cl]2 (1) |
PPh3 (2) |
Toluene |
>99/1 |
79 |
| 14 |
[Rh(cod)Cl]2 (1) |
— |
Toluene |
— |
8 |
| 15 |
— |
PPh3 (4) |
Toluene |
— |
0 |
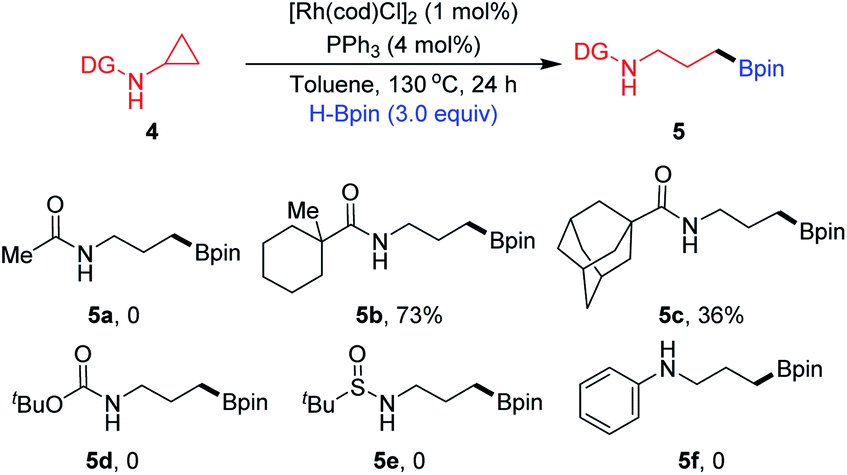
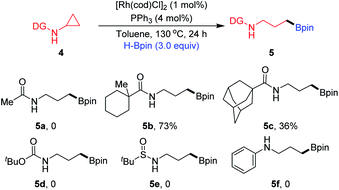
We next evaluated the influence of the steric hindrance of substituents on directing groups (Scheme 1). Changing the directing group to N-Ac (4a) displayed no reactivity for ring-opening. The directing group bearing a methylcyclohexane motif (4b) afforded the corresponding product 5b in 73% yield. When a sample of 4c containing a sterically hindered N-Ad moiety was subjected to the reaction conditions, the desired product 5c was formed in a much lower yield. CPAs bearing other directing groups such as N-Boc (4d) and N-SOtBu (4e), and substrate 4f without a directing group completely failed. These results indicate that the appropriate selection of the N-Piv directing group ensures high reactivity and selectivity for this transformation.
 |
| | Scheme 1 Evaluating different N-substituents. | |
Scope of the methodology
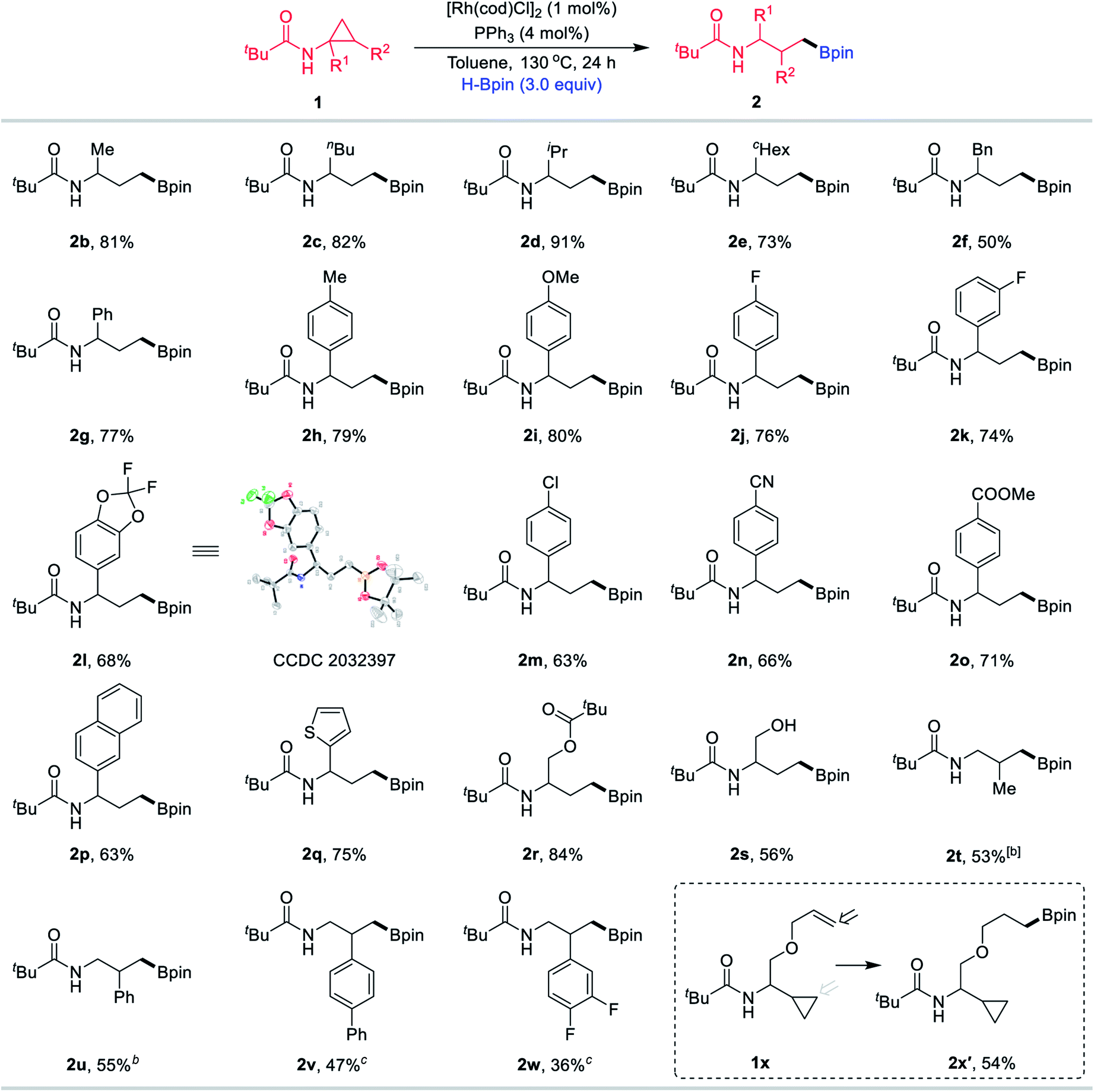
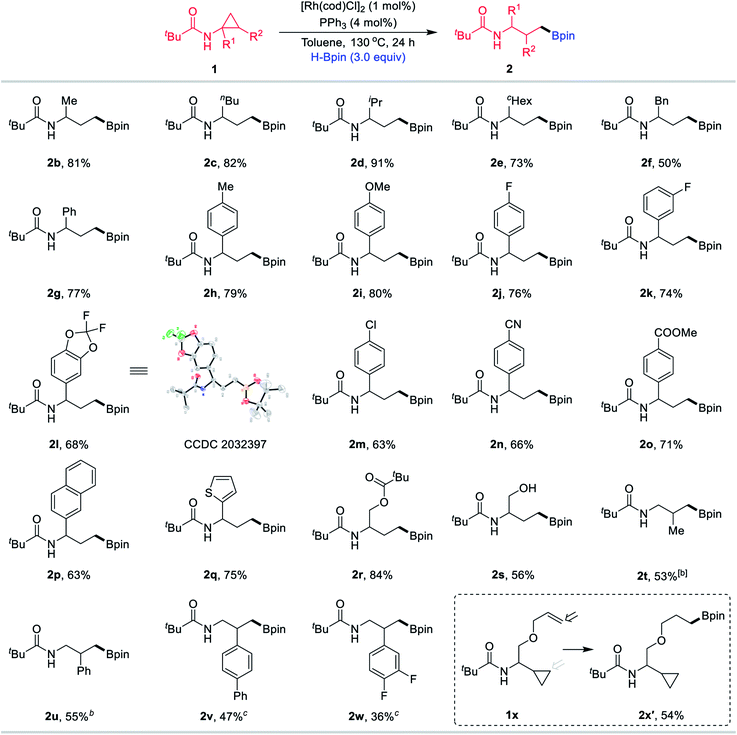
With the optimised conditions in hand, the scope of this methodology was subsequently explored (Table 2). The alkyl chains of the amide substituent R1, including methyl (1b), n-butyl (1c), isopropyl (1d), cyclohexyl (1e) and benzyl (1f), were converted into the corresponding boronic esters 2b–2f in good to high yields. Phenyl (1g) and other aryl substituents bearing methyl (1h), methoxy (1i), fluoro (1j–1l), chloro (1m), cyano (1n) and ester (1o) were compatible at the R1 position. Among them, product 2l was subjected to X-ray crystallographic analysis. Beyond the use of aryl-containing CPAs, we examined naphthyl and heterocyclic substrates. Naphthyl compound 1p was readily transformed, as was thiophene substrate 1q. The use of the CPA 1r with an O-Piv motif was also tested and the desired boronate 2r was obtained in 84% yield. Furthermore, substrate 1s bearing a hydroxyl group still resulted in the desired product 2s in 56% yield. In addition, this process was not limited to substrates bearing diverse R1 substituents, and CPAs with methyl (1t) and different aryl (1u–1w) groups at the R2 position were also suitable starting materials to effectively afford the desired products 2t–2w efficiently. Notably, when the substrate 1x containing a terminal olefin motif was employed for the reaction, olefin hydroboration product 2x′ was obtained in 54% yield and the ring-opening product 2x was not observed.
|
Reaction conditions: [Rh(cod)Cl]2 (1 mol%), PPh3 (4 mol%), 1 (0.20 mmol), HBpin (0.6 mmol), in toluene (1.0 mL), 24 h, 130 °C, under argon, in sealed Schlenk tubes, yield of purified compounds after chromatography.
Using [Rh(cod)Cl]2 (2.5 mol%), PPh3 (10 mol%), 36 h, 130 °C.
Using [Rh(cod)Cl]2 (2.5 mol%), PPh3 (10 mol%), 36 h, 150 °C.
|
|
Synthetic applications
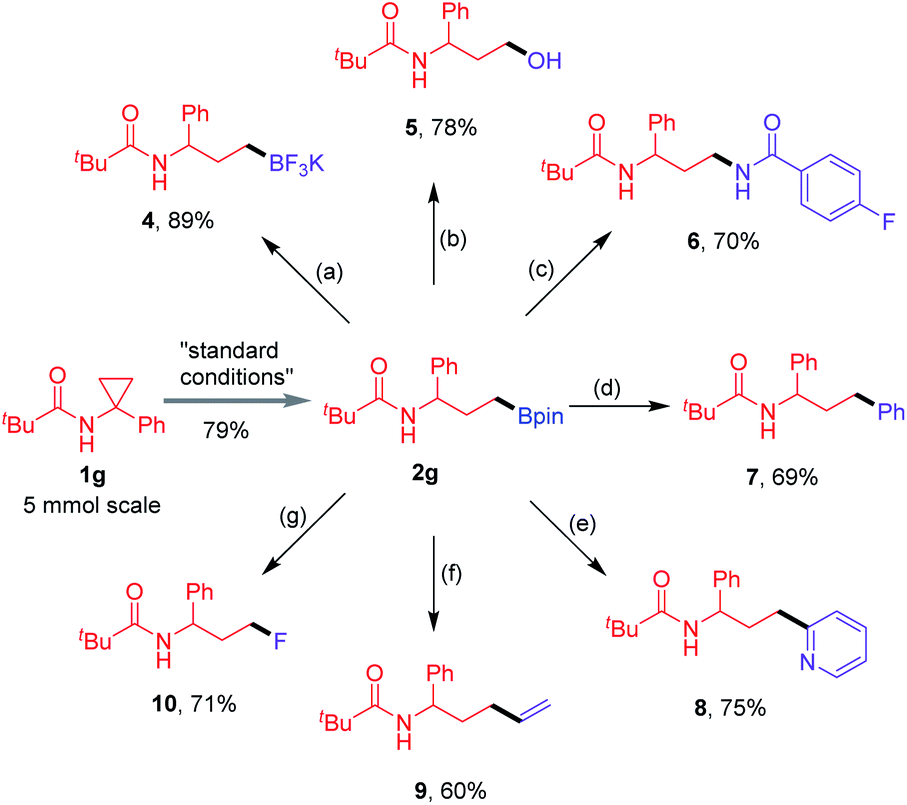
To showcase the synthetic utility of this proximal-selective hydroboration protocol, a 5.0 mmol reaction of CPA 1g was first conducted, enabling the gram-scale synthesis of the linear product 2g in 79% isolated yield (Scheme 2). The formed boronate group is an extremely versatile intermediate in organic synthesis, which can be converted into other functional substituents. Compound 2g could be transformed into potassium trifluoroborate salt 4 using KHF2 in an excellent yield. Subsequently, mild oxidation of 2g provided the corresponding alcohol 5 in 78% yield. The Cu-catalysed Chan–Lam coupling17 of 2g with 4-fluorobenzamide allowed the formation of 1,3-diamide derivative 6 in 70% yield. Furthermore, Pd-catalysed Suzuki–Miyaura coupling between substrate 2g and iodobenzene or 2-bromopyridine gave the desired products 7 and 8 efficiently.18 The Zweifel olefination of 2g with vinylmagnesium bromide furnished the terminal alkene product 9.19 In addition, the Ag-catalysed fluorination of 2g with Selectfluor® could give the monofluoroalkane 10 in a good yield based on a protocol developed by Li and co-workers.20
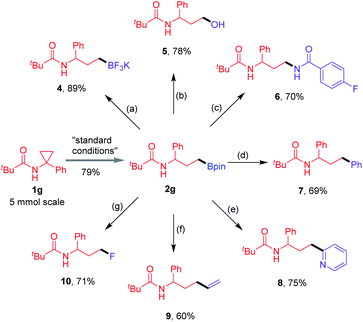 |
| | Scheme 2 Gram-scale synthesis and the follow-up transformation of compound 2g. Reagents and conditions: (a) aq. KHF2, MeOH, 3 h, rt; (b) NaBO3·4H2O (4.0 equiv.), H2O/THF, 6 h, rt; (c) 4-fluorobenzamide (1.0 equiv.), Cu(OAc)2 (10 mol%), 1,10-phen (10 mol%), NaOSiMe3 (1.1 equiv.), DTBP (3.0 equiv.), 4 Å MS, tBuOH, 24 h, 75 °C; (d) PhI (1.5 equiv.), PdCl2(dppf)·CH2Cl2 (8 mol%), Ag2O (1.5 equiv.), K2CO3 (3.0 equiv.), H2O/THF, 33 h, 80 °C to rt; (e) 2-bromopyridine (1.0 equiv.), Pd(OAc)2 (4 mol%), Ad2PnBu (12 mol%), LiOtBu (6.0 equiv.), H2O/THF, 24 h, 100 °C; (f) C2H3MgBr (6.0 equiv.), 0 °C to rt, 1 h; then I2 (6.0 equiv.) in MeOH, −78 °C, 30 min; then NaOMe (6.0 equiv.) in MeOH, −78 °C to rt, 1.5 h; (g) AgNO3 (20 mol%), Selectfluor® (3.0 equiv.), TFA (4.0 equiv.), H2O/DCM, 5 h, 50 °C. | |
Mechanistic studies
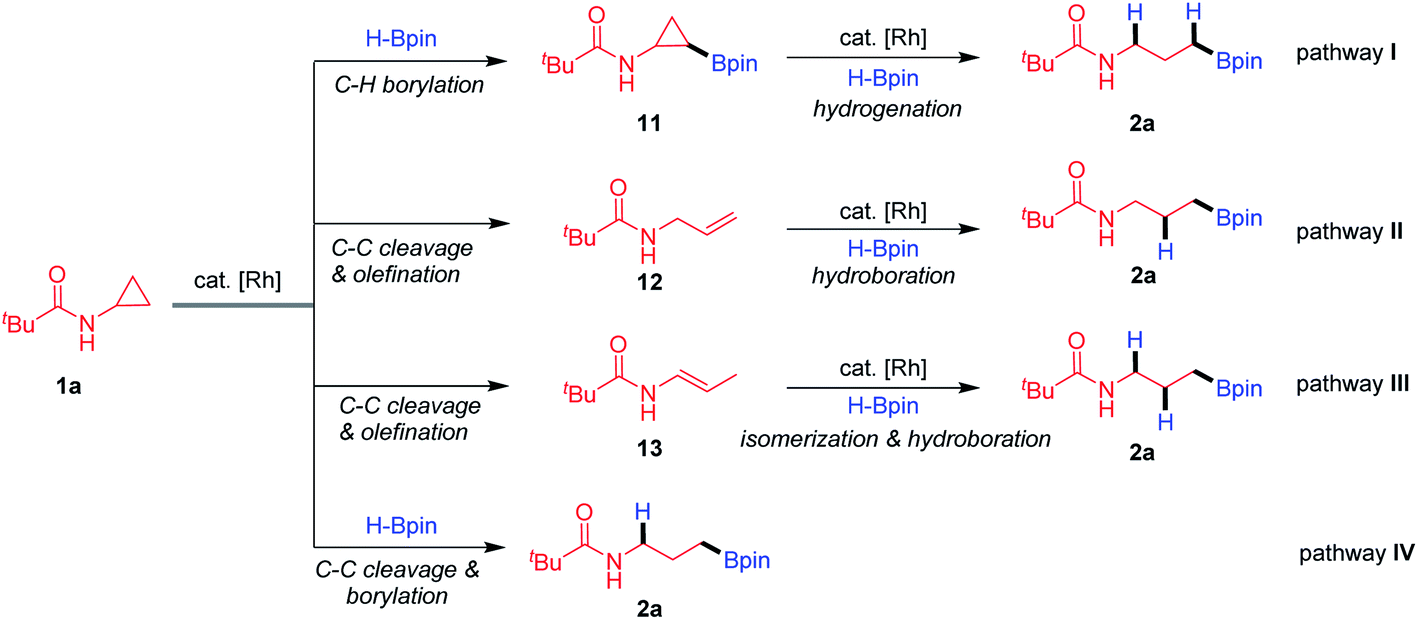
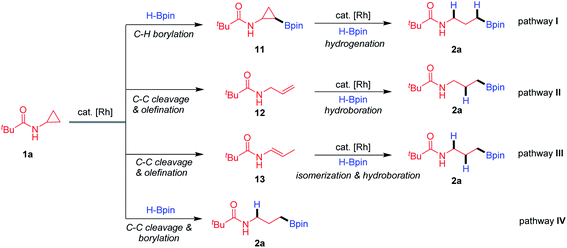
Based upon the above results and the reported studies, we proposed that the transformation proceeds through four possible pathways (Fig. 2). The Rh-catalysed C–H borylation proceeds first,16 followed by C–C activation and hydrogenation (pathway I). The Rh-catalysed C–C activation and β-H elimination generate an allylic amide, which further undergoes hydroboration (pathway II).21 Oxidative addition of Rh species to CPAs and β-H elimination generate an enamide, and subsequently olefin migration and hydroboration (pathway III) take place.22 The Rh-catalysed C–C activation and hydroboration deliver the final products directly (pathway IV).
 |
| | Fig. 2 Possible pathways for hydroboration of CPAs. | |
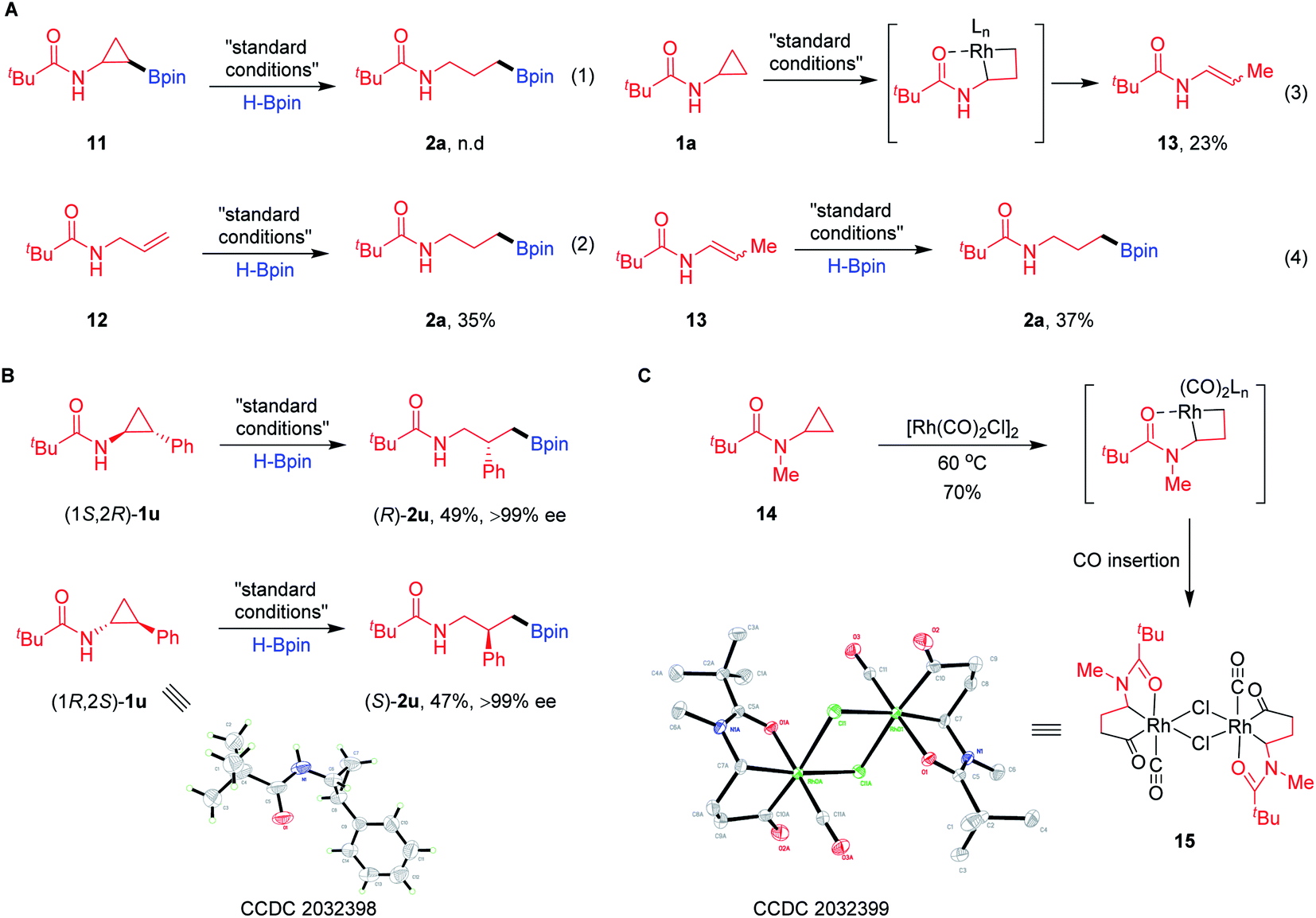
Several plausible intermediates were then investigated to provide insight about the potential mechanism of this transformation (Scheme 3). The C–H borylation product 11 failed to afford the desired product 2a, excluding pathway I (Scheme 3A-1). The reaction between pre-generated N-allylpivalamide (12) and HBpin could produce hydroboration product 2a in a modest yield (Scheme 3A-2). Impressively, treatment of substrate 1a in the system without the addition of HBpin was found to be applicable to the formation of enamide 13 through Rh oxidative addition to the hindered proximal C–C bond (Scheme 3A-3). Moreover, this compound 13 can convert into the corresponding product 2a by olefin isomerization and hydroboration (Scheme 3A-4). To further understand how these proximal-selective products formed, we investigated the reaction using the enantioenriched CPAs (Scheme 3B). The enantiomers (1S,2R)-1u (>99% ee) and (1R,2S)-1u (>99% ee) were, respectively, treated with HBpin under the standard conditions, giving the desired products (R)-2u and (S)-2u without erosion of the enantioselectivities. These results clearly excluded the insertion of Rh–H species onto the olefin intermediates through pathways II and III, suggesting that pathway IV involving direct C–C cleavage and hydroboration is possible. In order to support this proposal, we tried to isolate and characterize the key metallacyclic intermediate via Rh-mediated C–C bond activation of CPAs (Scheme 3C). Treatment of several CPAs with [Rh(cod)Cl]2 failed to provide any stable intermediate, but fortunately, heating CPA 14 with stoichiometric [Rh(CO)2Cl]2 under neat conditions could successfully generate a dimeric rhodacyclopentanone 15.23 When characterized by X-ray diffraction, the Rh(I) centre in the structure of complex 15 shows axial chelation assisted by the N-Piv directing group, leading to proximal selective C–C bond cleavage and CO insertion. This establishes that Rh(I) salts are effective for insertion into the proximal C–C bond of CPAs without the assistance of HBpin.
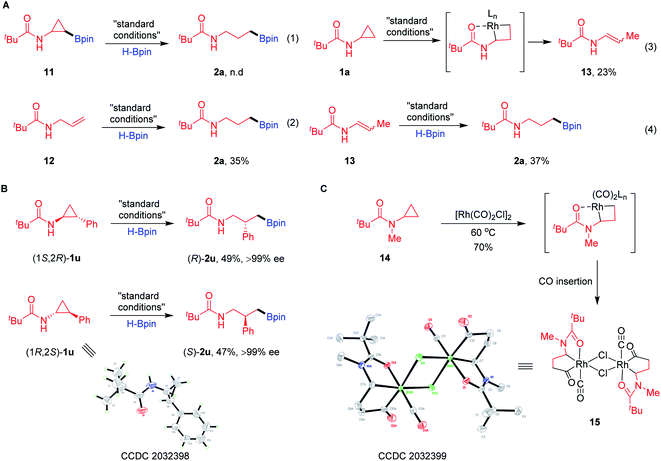 |
| | Scheme 3 Mechanistic experiments. (A) Investigation of the plausible intermediates. (B) Hydroboration of enantioenriched CPAs. (C) Synthesis of rhodacyclo complex 15. | |
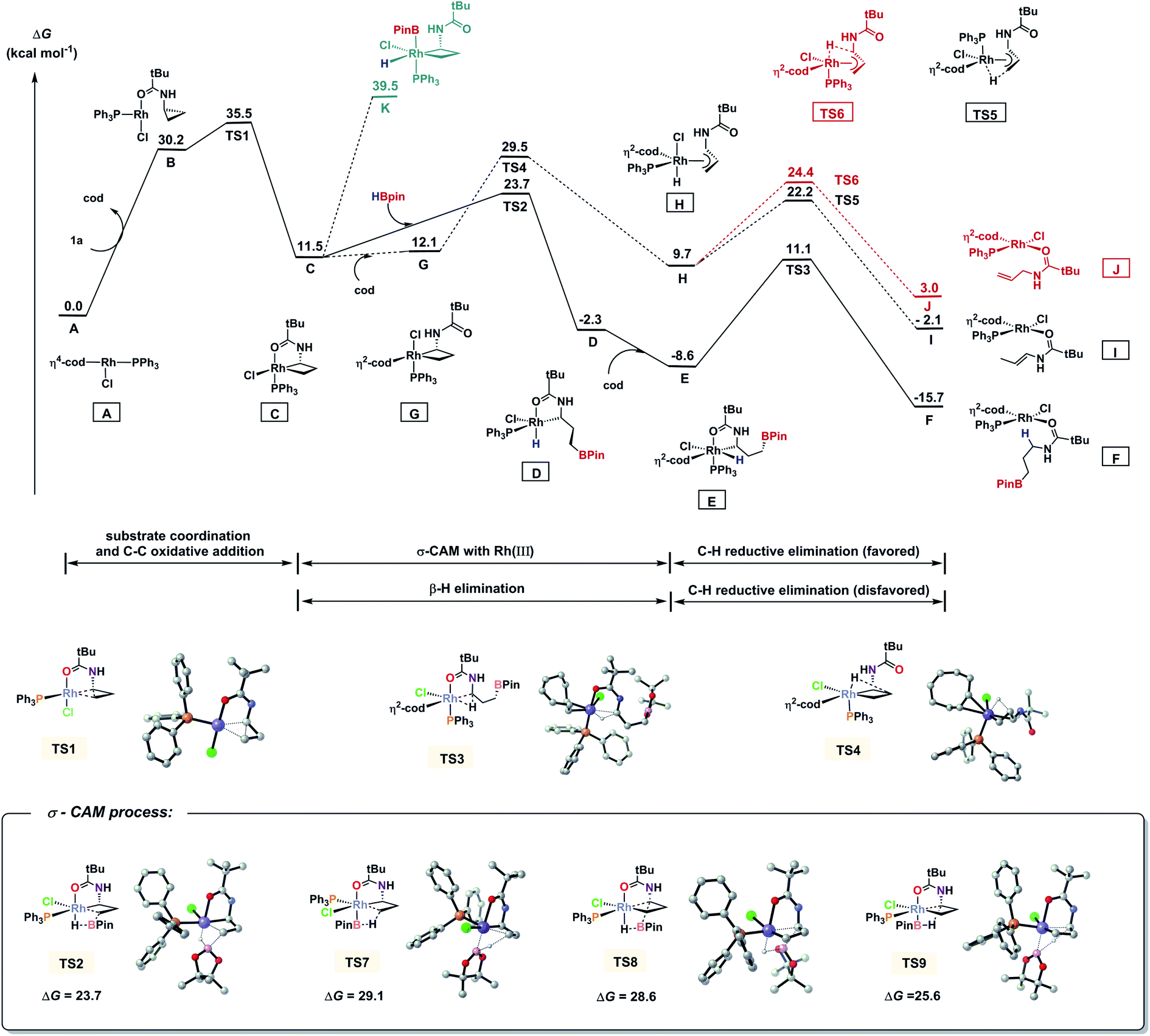
We also performed a series of density functional theory (DFT) calculations to obtain further mechanistic information of this transformation (for computational details, see the ESI†). Fig. 3 shows the Gibbs free energy profile of this reaction. The catalytic cycle begins with a ligand exchange that replaces a cod ligand in the stable intermediate A with substrate 1a. Directed by the amide oxygen, a subsequent proximal C–C bond cleavage of 1a takes place which affords C. This is the rate-limiting step in the catalytic cycle (ΔG≠ = 35.5 kcal mol−1). In this C–C bond cleavage step, we found that the non-directed transition states for both proximal and distal cleavage are unfavourable (for details, see the ESI†). Then we computed two possible pathways for hydroboration of C, oxidative addition and σ-complex assisted metathesis (σ-CAM).24 It was found that H–B oxidative addition to form Rh(V) species K is endergonic by 39.5 kcal mol−1. Alternatively, the σ-CAM pathway is much more favourable. The free energy of the four-membered cyclic σ-CAM transition state TS2 for the formation of intermediate D from intermediate C and Hbpin is 23.7 kcal mol−1. We also calculated other possible σ-CAM transition states (TS7–TS9), which would afford other hydroboration products, and found them to be higher in energy. From D, the re-coordination of the cod ligand leads to E, which reductively eliminates the C–H bond and forms the product-coordinated F. We also located a side pathway that corresponds to the formation of alkene products. From C, the directing group may dissociate from the metal centre to form intermediate G. Then, a β-H elimination (TS4) takes place which forms π-allyl–Rh(III) complex H. The C–H bond subsequently undergoes reductive elimination viaTS5 or TS6 to produce I or J, respectively. The enamide I is the kinetically favoured product, and its negative free energy (−2.1 kcal mol−1) supports the observation of 13 in Scheme 3A-3, where HBpin is not added. This also indicated that, under standard conditions, alkene 12 and 13 can be reversely converted into C (for 12, from JviaTS6 and TS4 with a barrier of 26.5 kcal mol−1; for 13, from IviaTS5 and TS4 with a barrier of 31.6 kcal mol−1), and then they enter the hydroboration catalytic cycle to afford 2a, as observed in Scheme 3A-3 and A-4.
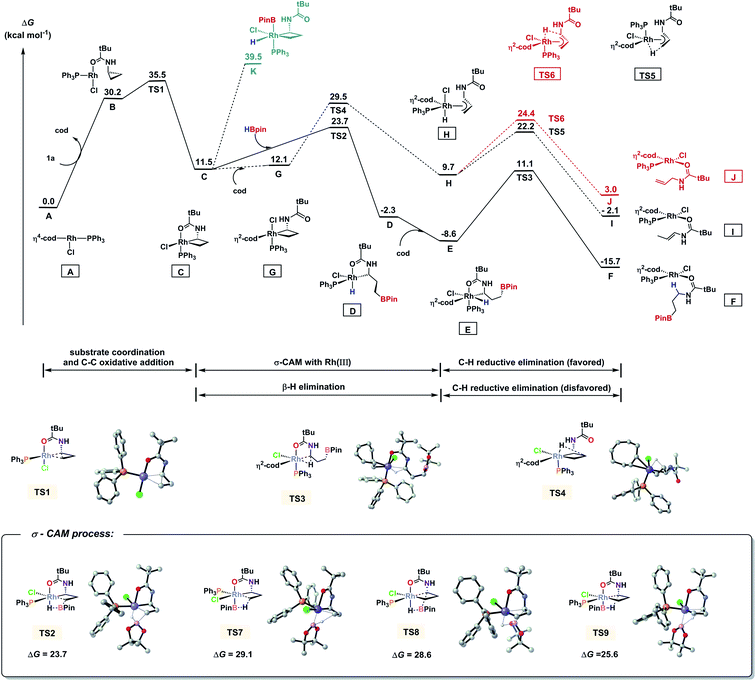 |
| | Fig. 3 Free energy diagram of hydroboration of CPAs using PPh3 as the ligand and DFT-computed four transition states for the σ-CAM pathways. Energies are in kcal mol−1. | |
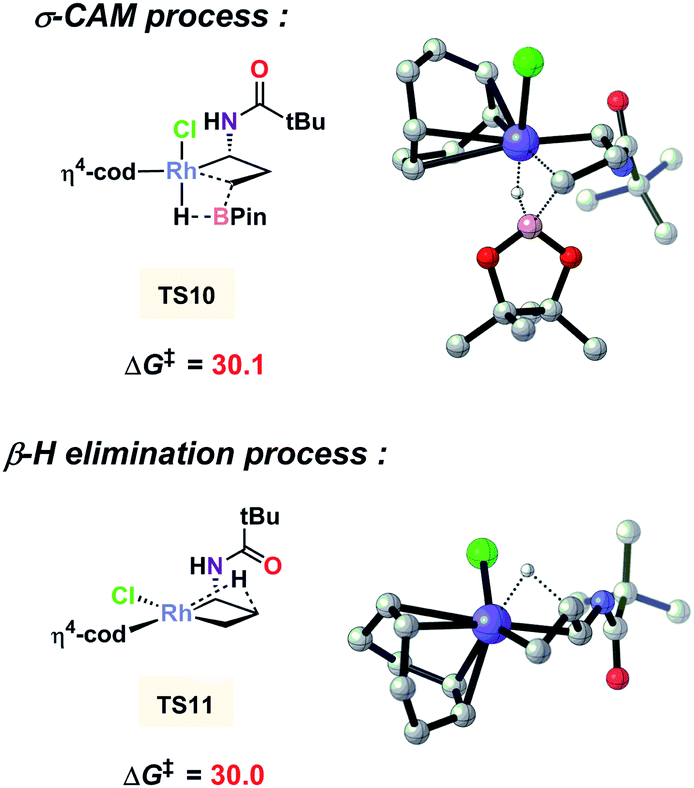
It is noted that ligand PPh3 plays an important role in regulating the competition of σ-CAM and β-H elimination from the C–C bond cleavaged intermediate. As shown in Fig. 3, the free energy of the β-H elimination pathway viaTS4 is 5.8 kcal mol−1 higher than that of the σ-CAM process through TS2 (29.5 versus 23.7 kcal mol−1). However, in the absence of PPh3, the β-H elimination transition state TS11 is even 0.1 kcal mol−1 lower in energy than the corresponding σ-CAM transition state TS10 (30.0 versus 30.1 kcal mol−1, Fig. 4). As a result, the efficiency of the desired hydroboration was very poor with many side reactions (Table 1, entry 14). These results imply that the electron-rich nature of the PPh3 ligand facilitates the σ-CAM process, inhibiting the formation of alkene byproducts.
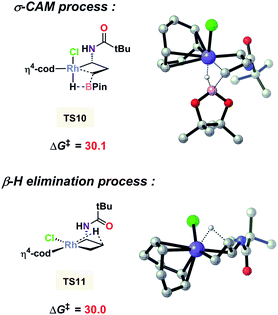 |
| | Fig. 4 DFT-computed free energies for the two competitive pathways without PPh3 as the ligand. The zero point is [Rh(cod)Cl]2. Energies are in kcal mol−1. | |
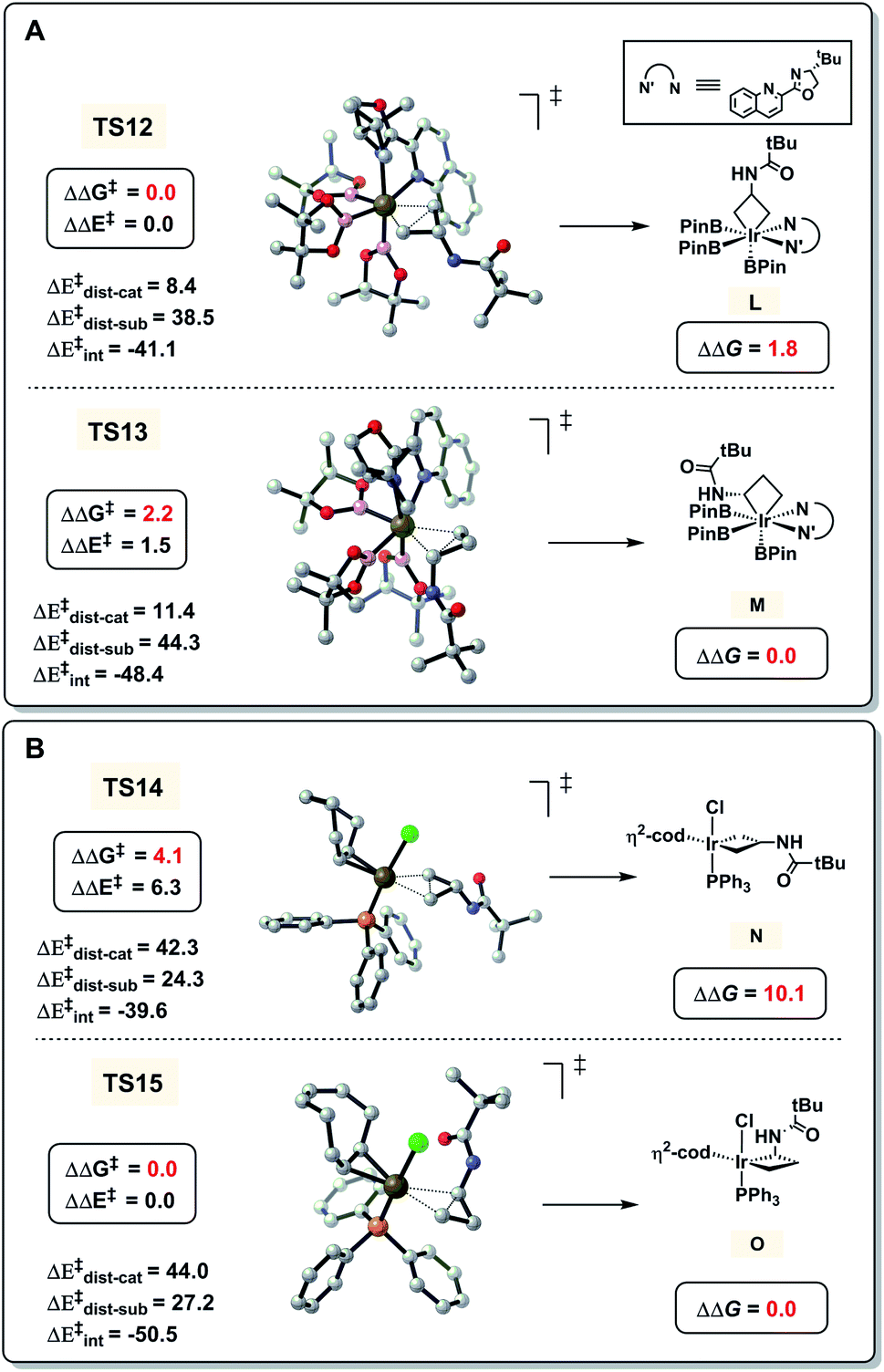
Finally, we investigated the opposite regioselectivity of hydroboration of CPAs in two Ir catalytic systems (Table 1, entries 1 and 2). Previous computations by Yamaguchi and Yokogawa showed that the real catalyst is Ir(III) species when [Ir(cod)OMe]2 and ligand tBu-Quinox are employed.16 As shown in Fig. 5A, our calculations indicate that the free energy of ring-opening transition state TS12 at the distal C–C bond is 2.2 kcal mol−1 lower than that of TS13 at the proximal C–C bond. However, the hydroboration of CPAs using [Ir(cod)Cl]2 and PPh3 involves Ir(I) species as the catalyst (Fig. 5B). The free energy of TS15 with proximal selectivity is 4.1 kcal mol−1 lower than that of TS14 with distal selectivity. These computational results are in good agreement with the observed selectivities in experiments. We also applied the distortion/interaction analysis on these transition states to understand the origins of the regioselectivity.25 In the Ir(III) system, the distortion, particularly the distortion of the catalyst, controls the regioselectivity. To activate the sterically more hindered proximal C–C bond viaTS13, the trigonal bipyramidal Ir(III) catalyst requires a distortion energy of 11.4 kcal mol−1, which is 3.0 kcal mol−1 higher than that in TS12 to break the less hindered distal C–C bond (11.4 versus 8.4 kcal mol−1, Fig. 5A), leading to the distal-selectivity. By contrast, the favorable interaction between 1a and the catalyst determines the regioselectivity in the Ir(I) system (Fig. 5B), which favors proximal-selective C–C bond activation significantly (−50.5 kcal mol−1 for TS15versus −39.6 kcal mol−1 for TS14). The tetrahedral structure of the Ir(I) catalyst makes the Ir center sterically less crowded compared with the trigonal bipyramidal Ir(III) catalyst. This favors the formation of the thermodynamically more stable intermediate O (versusN) with proximal-selectivity in the Ir(I) system, while less stable intermediate L (versusM) with distal-selectivity is preferred in the case of the Ir(III) catalyst.
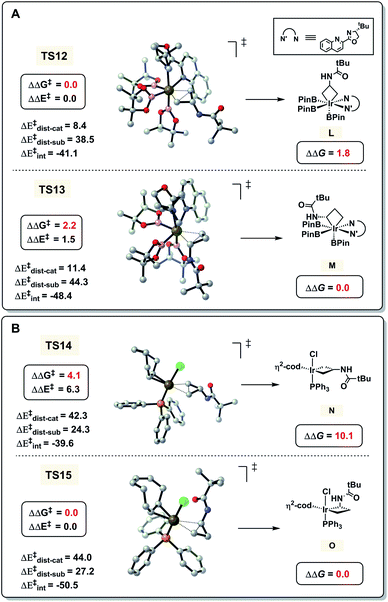 |
| | Fig. 5 DFT-computed energies and distortion/interaction analysis of C–C activation transition states. (A) [Ir(cod)OMe]2 and tBu-Quinox as ligands. (B) [Ir(cod)Cl]2 and PPh3 as ligands. Energies are in kcal mol−1. | |
Conclusions
In summary, we developed the first proximal-selective C–C bond activation followed by hydroboration of CPAs with HBpin leading to interesting γ-amino boronates enabled by the rhodium catalyst. The key to the high chemoselectivity and regioselectivity is the selection of PPh3 as the ligand. Significantly, experimental and computational mechanistic studies greatly provided insight into the reactivity and selectivity of this transformation. Further investigations of enantioselective variants and different types of C–C functionalizations of CPAs with tunable distal and proximal selectivity are currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank the National Natural Science Foundation of China (Grant 21672097, 21972064, and 21803030), the Fundamental Research Funds for the Central Universities, the National Natural Science Foundation of Jiangsu Province (Grant BK20170632 and BK20170631), the Excellent Youth Foundation of Jiangsu Scientific Committee (Grant BK20180007), and the “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province for their financial support.
Notes and references
-
(a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS
 ;
(b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC ;
(c) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS ;
(d) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC ;
(e) W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375 CrossRef CAS ;
(f) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS ;
(g) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415 RSC ;
(h) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS ;
(i) B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700 CrossRef CAS ;
(j) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348 CrossRef CAS .
;
(b) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992 RSC ;
(c) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS ;
(d) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229 RSC ;
(e) W. L. A. Brooks and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375 CrossRef CAS ;
(f) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091 CrossRef CAS ;
(g) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415 RSC ;
(h) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31 CrossRef CAS ;
(i) B. S. L. Collins, C. M. Wilson, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2017, 56, 11700 CrossRef CAS ;
(j) M. Wang and Z. Shi, Chem. Rev., 2020, 120, 7348 CrossRef CAS .
-
(a) K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228 CrossRef CAS ;
(b) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70 CrossRef CAS ;
(c) J. Hu, Y. Zhao, J. Liu, Y. Zhang and Z. Shi, Angew. Chem., Int. Ed., 2016, 55, 8718 CrossRef CAS ;
(d) Y. Huang, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2017, 56, 13314 CrossRef CAS ;
(e) M. Kischkewitz, K. Okamoto, C. Mück-Lichtenfeld and A. Studer, Science, 2017, 355, 936 CrossRef CAS ;
(f) L. Wang, T. Zhang, W. Sun, Z. He, C. Xia, Y. Lan and C. Liu, J. Am. Chem. Soc., 2017, 139, 5257 CrossRef CAS ;
(g) K. M. Logan, S. R. Sardini, S. D. White and M. K. Brown, J. Am. Chem. Soc., 2018, 140, 159 CrossRef CAS ;
(h) Z. Liu, H. Ni, T. Zeng and K. M. Engle, J. Am. Chem. Soc., 2018, 140, 3223 CrossRef CAS ;
(i) Y. Cheng, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2018, 140, 6221 CrossRef CAS ;
(j) R. Kojima, S. Akiyama and H. Ito, Angew. Chem., Int. Ed., 2018, 57, 7196 CrossRef CAS ;
(k) P. Gao, C. Yuan, Y. Zhao and Z. Shi, Chem, 2018, 4, 2201 CrossRef CAS ;
(l) J. Hu, Y. Zhao and Z. Shi, Nat. Catal., 2018, 1, 860 CrossRef CAS ;
(m) J. A. Myhill, C. A. Wilhelmsen, L. Zhang and J. P. Morken, J. Am. Chem. Soc., 2018, 140, 15181 CrossRef CAS ;
(n) J. Li, H. Wang, Z. Qiu, C.-Y. Huang and C.-J. Li, J. Am. Chem. Soc., 2020, 142, 13011 CrossRef CAS ;
(o) M. R. Jones, C. D. Fast and N. D. Schley, J. Am. Chem. Soc., 2020, 142, 6488 CrossRef CAS ;
(p) R. Oeschger, B. Su, I. Yu, C. Ehinger, E. Romero, S. He and J. Hartwig, Science, 2020, 368, 736 CrossRef CAS ;
(q) L. Wang, T. Zhao, W. Sun, Z. He, C. Xia, Y. Lan and C. liu, J. Am. Chem. Soc., 2017, 139, 5257 CrossRef CAS ;
(r) Y. Hu, W. Sun, T. Zhang, N. Xu, Y. Lan and C. Liu, Angew. Chem., Int. Ed., 2019, 58, 15813 CrossRef CAS ;
(s) C. Law, E. Kativhu, J. Wang and J. P. Morken, Angew. Chem., Int. Ed., 2020, 59, 10311 CrossRef CAS ;
(t) S. Namirembe and J. P. Morken, Chem. Soc. Rev., 2019, 48, 3464 RSC ;
(u) D. Ni, B. P. Witherspoon, H. Zhang, C. Zhou, K. N. Houk and M. K. Brown, Angew. Chem., Int. Ed., 2020, 59, 11432 CrossRef CAS ;
(v) D. Wang, C. Mück-Lichtenfeld and A. Studer, J. Am. Chem. Soc., 2020, 142, 9119 CrossRef CAS ;
(w) C. You and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 17245 CrossRef CAS .
- L. A. William and B. S. Sumerlin, Chem. Rev., 2016, 116, 1375 CrossRef .
-
(a) P. C. Trippier and C. McGuigan, MedChemComm, 2010, 1, 183 RSC ;
(b) H. S. ban and H. Nakamura, Chem. Rec., 2015, 15, 616 CrossRef CAS ;
(c) L. K. H. Leung, T. Brown Jr, C. J. Schofield and T. W. Claridge, MedChemComm, 2011, 2, 390 RSC ;
(d) K. M. E. Tehrani and N. Martin, MedChemComm, 2018, 9, 1439 RSC ;
(e) J. P. M. Antonio, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. P. Gois, Chem. Soc. Rev., 2019, 48, 3513 RSC .
-
(a) H. Brown and B. C. Rao, J. Org. Chem., 1957, 22, 1137 CrossRef CAS ;
(b) D. Wang, X.-S. Xue, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2018, 57, 16861 CrossRef CAS .
-
(a) J. D. Hewes, C. W. Kreimendahl, T. B. Marder and M. F. Hawthorne, J. Am. Chem. Soc., 1984, 106, 5757 CrossRef CAS ;
(b) D. Männig and H. Nöth, Angew. Chem., Int. Ed., 1985, 24, 878 CrossRef ;
(c) K. Burgess, W. A. van der Donk, S. A. Westcott, T. B. Marder, R. T. Baker and J. C. Calabrese, J. Am. Chem. Soc., 1992, 114, 9350 CrossRef CAS ;
(d) A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRef CAS ;
(e) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179 CrossRef CAS ;
(f) C. M. Crudden and D. Edwards, Eur. J. Org. Chem., 2003, 24, 4695 CrossRef ;
(g) C. A. Vogels and S. A. Westcott, Curr. Org. Chem., 2005, 9, 687 CrossRef CAS .
-
(a) G. Wang, X. Liang, L. Chen, Q. Gao, J.-G. Wang, P. Zhang, Q. Peng and S. Xu, Angew. Chem., Int. Ed., 2019, 58, 8187 CrossRef CAS ;
(b) X.-Y. Bai, W. Zhao, X. Sun and B.-J. Li, J. Am. Chem. Soc., 2019, 141, 19870 CrossRef CAS .
-
(a) D. A. Evans and G. C. Fu, J. Am. Chem. Soc., 1991, 113, 4042 CrossRef CAS ;
(b) S. M. Smith and J. M. Takacs, J. Am. Chem. Soc., 2010, 132, 1740 CrossRef CAS ;
(c) S. M. Smith, N. C. Thacker and J. M. Takacs, J. Am. Chem. Soc., 2008, 130, 3734 CrossRef CAS ;
(d) V. M. Shoba, N. C. Thacker, A. J. Bochat and J. M. Takacs, Angew. Chem., Int. Ed., 2016, 55, 1465 CrossRef CAS ;
(e) S. Chakrabarty and J. M. Takacs, J. Am. Chem. Soc., 2017, 139, 6066 CrossRef CAS ;
(f) Y. Xi and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 6703 CrossRef CAS ;
(g) G. L. Hoang and J. M. Takacs, Chem. Sci., 2017, 8, 4511 RSC ;
(h) H. Zhao, Q. Gao, Y. Zhang, P. Zhang and S. Xu, Org. Lett., 2020, 22, 2861 CrossRef CAS .
-
(a) R. L. Reyes, T. Iwai, S. Maeda and M. Sawamura, J. Am. Chem. Soc., 2019, 138, 6817 CrossRef ;
(b) Y. Shi, Q. Gao and S. Xu, J. Am. Chem. Soc., 2019, 141, 10599 CrossRef CAS ;
(c) R. L. Reyes, M. Sato, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2020, 139, 589 CrossRef ;
(d) L. Chen, Y. Yang, L. Liu, Q. Gao and S. Xu, J. Am. Chem. Soc., 2020, 142, 12062 CrossRef CAS .
- R. L. Reyes, M. Sato, T. Iwai, K. Suzuki, S. Maeda and M. Sawamura, Science, 2020, 369, 970 CrossRef CAS .
-
(a) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS ;
(b) A. Reichelt and S. F. Martin, Acc. Chem. Res., 2006, 39, 433 CrossRef CAS ;
(c) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC ;
(d) C. Ebner and E. M. Carreira, Chem. Rev., 2017, 117, 11651 CrossRef CAS .
-
(a) O. O. Sokolova and J. F. Bower, Chem. Rev., 2021, 121, 80 CrossRef CAS ;
(b) V. Pirenne, B. Muriel and J. Waser, Chem. Rev., 2021, 121, 227 CrossRef CAS .
- S. Miyamura, M. Araki, T. Suzuki, J. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 846 CrossRef CAS .
-
(a) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613 CrossRef CAS ;
(b) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410 CrossRef CAS ;
(c) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS ;
(d) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404 CrossRef CAS ;
(e) F. Song, T. Gou, B.-Q. Wang and Z.-J. Shi, Chem. Soc. Rev., 2018, 47, 7078 RSC ;
(f) H. Lu, T.-Y. Yu, P.-F. Xu and H. Wei, Chem. Rev., 2021, 121, 365 CrossRef CAS ;
(g) M. Murakami and N. Ishida, Chem. Rev., 2021, 121, 264 CrossRef ;
(h) Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600 CrossRef CAS .
-
(a) C. Li, J. Wang, L. M. Barton, S. Yu, M. Tian, D. S. Peters, M. Kumar, A. W. Yu, K. A. Johnson, A. K. Chatterjee, M. Yan and P. S. Baran, Science, 2017, 356, eaam7355 CrossRef ;
(b) A. Fawcett, J. Pradeilles, Y. Wang, T. Mutsuga, E. L. Myers and V. K. Aggarwal, Science, 2017, 357, 283 CrossRef CAS ;
(c) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155 CrossRef CAS .
- H. Kondo, S. Miyamura, K. Matsushita, H. Kato, C. Kobayashi, Arifin, K. Itami, D. Yokogawa and J. Yamaguchi, J. Am. Chem. Soc., 2020, 142, 11306 CrossRef CAS .
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311 CrossRef CAS .
- C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528 CrossRef CAS .
- R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 49, 3323 CrossRef CAS .
- Z. Li, Z. Wang, L. Zhu, X. Tan and C. Li, J. Am. Chem. Soc., 2014, 136, 16439 CrossRef CAS .
-
(a) N. G. McCreanor, S. Stanton and J. F. Bower, J. Am. Chem. Soc., 2016, 138, 11465 CrossRef CAS ;
(b) H. Kondo, K. Itami and J. Yamaguchi, Chem. Sci., 2017, 8, 3799 RSC .
-
(a) C. Romano, D. Fiorito and C. Mazet, J. Am. Chem. Soc., 2019, 141, 16983 CrossRef CAS ;
(b) X. Chen, Z. Cheng, J. Guo and Z. Lu, Nat. Commun., 2018, 9, 3939 CrossRef .
-
(a) M. H. Shaw, E. Y. Melikhova, D. P. Kloer, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2013, 135, 4992 CrossRef CAS ;
(b) M. H. Shaw, N. G. McCreanor, W. G. Whittingham and J. F. Bower, J. Am. Chem. Soc., 2015, 137, 463 CrossRef CAS ;
(c) G.-W. Wang and J. F. Bower, J. Am. Chem. Soc., 2018, 140, 2743 CrossRef CAS .
- R. N. Perutz and S. Sabo-Etienne, Angew. Chem., Int. Ed., 2007, 46, 2578 CrossRef CAS .
- F. M. Bickelhaupt and K. N. Houk, Angew. Chem., Int. Ed., 2017, 56, 10070 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2032397–2032399. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06186g |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Di
Wang
,
Yue
Zhao
*,
Di
Wang
,
Yue
Zhao