
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cofacial metal–organic framework based photocathode for carbon dioxide reduction†
Bowen
Ding
a,
Bun
Chan
 b,
Nicholas
Proschogo
a,
Marcello B.
Solomon
a,
Cameron J.
Kepert
a and
Deanna M.
D'Alessandro
*a
b,
Nicholas
Proschogo
a,
Marcello B.
Solomon
a,
Cameron J.
Kepert
a and
Deanna M.
D'Alessandro
*a
aSchool of Chemistry, The University of Sydney, Sydney, New South Wales 2006, Australia. E-mail: deanna.dalessandro@sydney.edu.au; Fax: +61 3 9351 3329; Tel: +61 2 9351 3777
bGraduate School of Engineering, Nagasaki University, Bunkyo 1-14, Nagasaki-shi, Nagasaki 852-8521, Japan
First published on 4th January 2021
Abstract
Innovative and robust photosensitisation materials play a cardinal role in advancing the combined effort towards efficient solar energy harvesting. Here, we demonstrate the photocathode functionality of a Metal–Organic Framework (MOF) featuring cofacial pairs of photo- and electro-active 1,4,5,8-naphthalenediimide (NDI) ligands, which was successfully applied to markedly reduce the overpotential required for CO2 reduction to CO by a well-known rhenium molecular electrocatalyst. Reduction of [Cd(DPNDI)(TDC)]n (DPNDI = N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide, H2TDC = thiophene-2,5-dicarboxylic acid) to its mixed-valence state induces through-space Intervalence Charge Transfer (IVCT) within cofacial DPNDI units. Irradiation of the mixed-valence MOF in the visible region generates a DPNDI photoexcited radical monoanion state, which is stabilised as a persistent species by the inherent IVCT interactions and has been rationalised using Density Functional Theory (DFT). This photoexcited radical monoanion state was able to undergo charge transfer (CT) reduction of the rhenium molecular electrocatalyst to effect CO generation at a lower overpotential than that required by the discrete electrocatalyst itself. The exploitation of cofacial MOFs opens new directions for the design philosophy behind light harvesting materials.
The development of photocathode materials for CO2 reduction and hydrogen evolution catalyses has traditionally focussed on photosensitising transition metal complexes or nanostructured solid state semiconductors.1,2 At the nascent frontier between robust solid state semiconductors and synthetically protean metal complexes are photo-/electro-active Metal–Organic Frameworks (MOFs) that consolidate the flexibility of homogeneous systems into the robust heterogeneous phase.3 Contrasting with reported MOF examples, natural photosynthesis remains one of the most efficient light harvesting systems.4 One common reaction centre adopted in photosynthesis features a redox-active cofacial dimer of chlorophyll pigment molecules.5 This cofacial moiety stabilises the photoexcited charge separated state through intra-dimer Intervalence Charge Transfer (IVCT) interactions, enabling the trapping and conversion of light to chemical energy. Recently, we characterised IVCT interactions upon reduction to the mixed-valence state in the MOF [Zn2(TDC)2(DPPTzTz)2]n (DPPTzTz = 2,5-bis(4-(4-pyridyl)phenyl)thiazolo[5,4-d]thiazole and H2TDC = thiophene-2,5-dicarboxylic acid) featuring cofacial dimers of the thiazolothiazole redox-active core, and probed its structure–activity dependence computationally and experimentally.6–9 Subsequently, we sought design a new MOF featuring cofacial pairs of the photo- and redox-active N,N′-di(4-pyridyl)-1,4,5,8-naphthalenediimide (DPNDI) ligand, as a conceptually neoteric photosensitiser for incorporation into systems relevant towards artificial photosynthesis.
The naphthalene diimide (NDI) core was selected for its photoactive radical monoanion state.10 For a number of discrete systems, Wasielewski and coworkers have computationally and experimentally demonstrated the ability to photoexcite the easily accessible NDI radical monoanion using visible light, facilitating its transient photoelectrochemical reduction of Re based catalytic CO2 reduction sites.2,11–14 Recently, Goswami et al. synthesised a Zr NDI-based MOF, applying this as a radical state heterogeneous photosensitiser to decompose dichloromethane.15
Here, we describe the synthesis of a new photo- and redox-active MOF [Cd(DPNDI)(TDC)]n, denoted csiMOF-6 (cofacial stacked IVCT), featuring cofacial dimers of the DPNDI ligand. Cofacial DPNDI MOFs have been reported previously by Takashima et al.16 and Sikdar et al.,17 where guest dependent charge transfer (CT) and neutral state photoexcitation behaviours were examined. Dinolfo et al. also incorporated DPNDI into a rhenium based cofacial complex, where its mixed-valence IVCT behaviour was probed using electrochemical and spectroelectrochemical (SEC) techniques.18 We envisaged that the cofacial NDI units in csiMOF-6 would stabilise its photoexcited radical monoanion state by IVCT interactions, akin to cofacial moieties in natural photosynthsesis processes. This strengthens the persistence of the NDI photoexcited radical monoanion state, thereby improving its efficacy at photoelectrochemical reduction of catalytically active sites. Effectiveness of the cofacial design principle behind csiMOF-6 photocathodes was verified using a combined experimental and computational approach. The successful photocathode performance of csiMOF-6 under broad band visible light irradiation encompassed its photoelectrochemical reduction of the [Re(bipy-tBu)(CO)3Cl] (bipy-tBu = 4,4′-di-tert-butyl-2,2′-bipyridine, developed by Smieja et al.19) CO2 reduction electrocatalyst, resulting in CO generation at reduced overpotential requirements.
Results and Discussion
The crystal structure of csiMOF-6 was solved and refined in the orthorhombic space group Pbam with unit cell parameters of a = 12.8322(3), b = 19.9878(3), c = 20.0440(5) Å. The solvent accessible void space was calculated using PLATON20 to comprise 55% of the total volume. csiMOF-6 is constructed of 2D sheets of TDC co-ligands and CdII nodes extending within the a,b-plane and pillared along the c-axis by dimeric cofacial units of DPNDI, which is similar to other cofacial MOFs reported in the literature (Fig. 1 in addition to Fig. S1–S3 and Table S1 in the ESI†).6–9,16,17,21 Within each 2D sheet, the coordination of the carboxylate groups from TDC co-ligands guide the assembly of CdII nodes into dinuclear clusters, templating the cofacial arrangement of DPNDI ligands. The NDI cores within each DPNDI cofacial dimer exhibit positional disorder, which was refined with a two component system (Fig. S1c†). This disorder results in a variable stacking interaction-with a minimal cofacial distance of 3.3 Å and a maximum distance of 3.5 Å−suggesting that there is localised flexibility at the NDI cores in csiMOF-6 to facilitate improved cofacial interactions.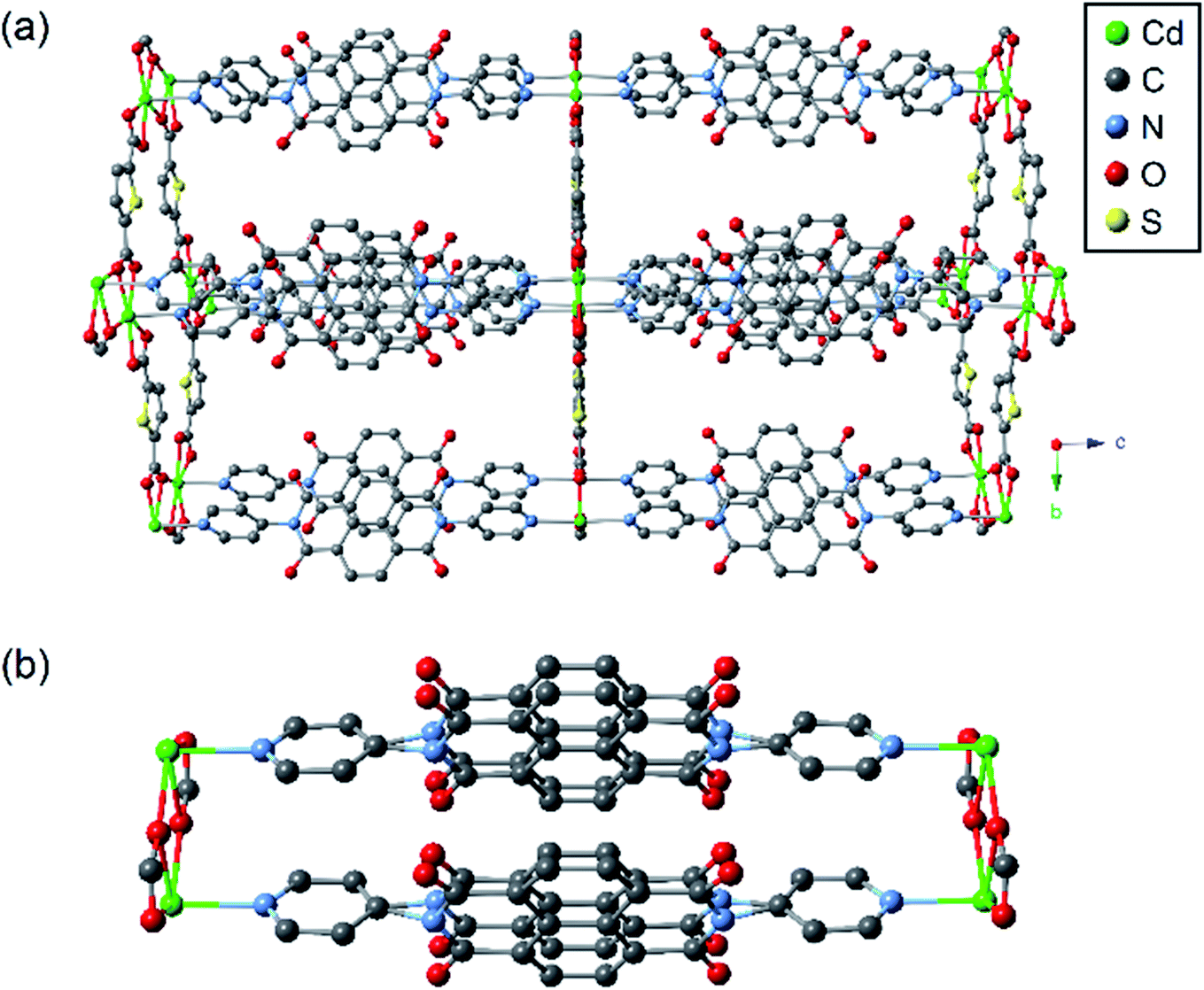 | ||
| Fig. 1 Representation of the as-synthesised crystal structure of csiMOF-6 showing (a) the overall structure, and (b) the cofacial dimers of DPNDI. Hydrogen atoms are omitted for clarity. | ||
In its neutral state, csiMOF-6 displays a long-lived response to visible light photoexcitation using a 100 W Hg arc lamp with UV cut off filter (all irradiation in experiments were conducted with this light source). The UV-Vis-NIR spectrum of light irradiated csiMOF-6 (Fig. S14†) includes a near-infrared (NIR) band at 6500 cm−1 which is assigned to IVCT within cofacial DPNDI pairs, while the bands in the visible region correspond to those for the NDI radical monoanion state. The presence of bands correlated to the NDI radical suggests the occurrence of CT interactions between TDC co-ligand donors and photoexcited NDI acceptors in csiMOF-6, forming radical species at the NDI core (Scheme 1).17 This photogenerated radical induces a mixed-valence state at the NDI cofacial unit, explaining the NIR IVCT band. An organic radical signal at g = 2.0030 was observed in the EPR spectrum upon in situ light irradiation of csiMOF-6, supporting the formation of NDI radical species (Fig. S15†). Post irradiation decay experiments were also conducted (Fig. S16†), whereupon the organic radical signal was observed to halve in intensity after 3 h. The stability of the photogenerated radical may be correlated with disorder at the NDI cores (Fig. 1b), which improves cofacial stacking interactions leading to enhanced IVCT interactions in the photogenerated NDI radical species. It is very interesting to note that in the NDI dimer reported by Wasielewski and Stoddart,22 which featured cofacially stacked NDI units (at a distance of 3.5 Å) separated by 1,2-cyclohexanediamine units, denoted 2NDI, fluorescence spectroscopy revealed a significantly enhanced lifetime for the photoexcited electron compared with a single NDI unit. The observation was attributed to the sharing of the photoexcited electron between the NDI units in the cofacial system. The authors electrochemically and chemically generated a mixed-valence species 2NDI which confirmed that the electron was delocalised between the two NDI units.
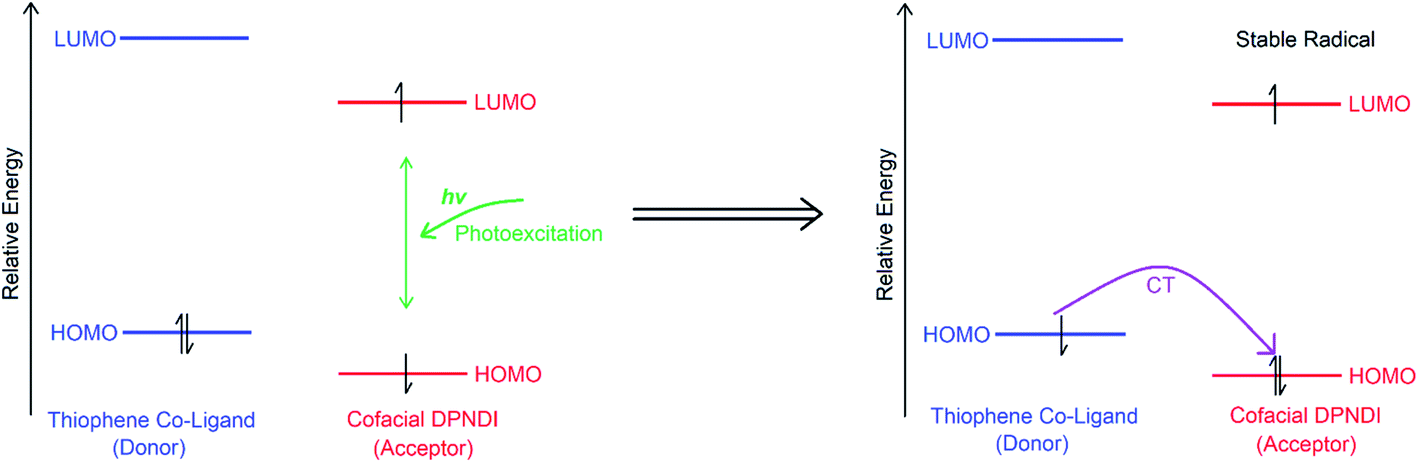 | ||
| Scheme 1 Photoexcitation of as-synthesised csiMOF-6 (left) results in stable NDI radical monoanion generation (right) through internal CT from the TDC donors (blue) to the cofacial DPNDI acceptors (red). Green arrow indicates photoexcitation and purple arrow indicates CT. | ||
The ability of csiMOF-6 to accommodate radical and excited state species was further probed by intercalation with the electron rich guest N,N′-dimethylaniline (DMA). Previous studies have demonstrated the formation of a partial CT interaction between DMA and the NDI core.16 Single crystals of csiMOF-6 were soaked in DMA for 24 h, resulting in a colour change to black. Successful incorporation of DMA into the pores of csiMOF-6 was confirmed by 1H NMR of TFA-d digested samples (Fig. S8†) that revealed ca. 50% incorporation of DMA per DPNDI ligand (i.e., one DMA molecule per cofacial DPNDI unit). Both the UV-Vis-NIR and EPR spectra of in situ DMA intercalated csiMOF-6 (Fig. S17 and S18†) demonstrated the formation of a partial CT species between the NDI core and DMA guest in accordance with previous studies.16 The generation of radical species at the NDI cores resulting from partial CT with DMA also invoked a mixed-valence IVCT interaction within DPNDI cofacial dimers, as characterised by a NIR band at 6500 cm−1 observed in the UV-Vis-NIR spectrum of csiMOF-6 with DMA (Fig. S17†). Interestingly, an IVCT band centred at 5714 cm−1 (1750 nm) was also detected in the singly reduced cofacial NDI dimer [2NDI]˙− reported by Wasielewski and Stoddart, and was attributed to moderately strong electronic delocalisation between the NDI units.22
The crystal structure of csiMOF-6 incorporating DMA was solved and refined in the orthorhombic space group Pbam, as for the as-synthesised sample. A change in unit cell parameters to a = 12.9936(1), b = 19.8725(1), c = 20.0366(3) Å was found, corresponding to an increased cell volume of 5173.76(9) Å3, compared with 5141.03(19) Å3 for the as-synthesised framework (Table S2†) at the same temperature. The structure of the DMA guest exchanged csiMOF-6 closely resembles that of the as-synthesised form, except for the structure at the NDI cores where more significant disorder was encountered (Fig. S1d†). The NDI disorder was best represented using two partially occupied NDI units (50/50 occupancy), which in contrast to the as-synthesised structure, were optimised to exhibit a more substantial rotational and translational separation (Fig. S1d and S1e†). The disorder at the NDI cores resulted in a minimal cofacial stacking distance of 3.3 Å and a maximal cofacial stacking distance of 3.5 Å (Fig. S1e†). The correlations between the NDI radical state, access to reduced cofacial stacking distances (intensified cofacial interactions) and occurrence of IVCT, collectively infers flexibility at the NDI core in csiMOF-6 to stabilise radical (and excited) states by enhanced mixed-valence IVCT interactions.
The non-irradiated electrochemical behaviour of csiMOF-6 in 0.1 M [n-Bu4N]PF6/MeCN was indicative of DPNDI ligand-based processes, which suggested good electron communication within the cofacial ligand pairs (Fig. 2a and S19–S22†). Upon scanning to a cathodic potential of −2.4 V vs. Fc/Fc+, two quasi-reversible processes were observed in the cyclic voltammogram (CV) at −0.8 and −1.4 V vs. Fc/Fc+. These two processes were attributed to accessing the radical monoanion state of one, followed by both NDI cores within each DPNDI cofacial pair. The irreversible process beyond −1.6 V vs. Fc/Fc+ was attributed to the generation of the dianion species of DPNDI. The assignments of the first two reduction processes were confirmed using solid state UV-Vis-NIR and EPR SEC (Fig. 2b and c) in 0.1 M [n-Bu4N]PF6/MeCN. A NIR band at 8000 cm−1 was observed at an applied potential of −1.3 V vs. Fc/Fc+ and was attributed to a mixed-valence IVCT interaction (Fig. S23†). By holding the applied potential at this point whilst irradiating the sample with broad band white light (with UV cut-off filter), new blue-shifted bands formed at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500, 18000 and 21500 cm−1, along with an intensification of the NIR IVCT band at 8000 cm−1 and a reduction in the NDI radical monoanion band at 17000 cm−1 (Fig. 2b). The appearance of blue-shifted visible region bands upon csiMOF-6 irradiation signify photoexcitation of the NDI radical monoanion to an electronic configuration at a higher energy level than the NDI LUMO. The stabilisation of this photoexcited radical state by increased IVCT interactions between cofacial DPNDI units explains the intensification of the NIR band upon irradiation. Application of the same potential in the (non-irradiated) EPR SEC experiment resulted in the appearance of an organic signal at g = 2.0025, attributed to the NDI radical monoanion (Fig. 2c). Remarkably, returning the applied potential to −0.1 V vs. Fc/Fc+ resulted in the slow decay of the radical (Fig. S24†), with the intensity of the signal halving only after 1 day, demonstrating the stability of the radical species generated in csiMOF-6.
500, 18000 and 21500 cm−1, along with an intensification of the NIR IVCT band at 8000 cm−1 and a reduction in the NDI radical monoanion band at 17000 cm−1 (Fig. 2b). The appearance of blue-shifted visible region bands upon csiMOF-6 irradiation signify photoexcitation of the NDI radical monoanion to an electronic configuration at a higher energy level than the NDI LUMO. The stabilisation of this photoexcited radical state by increased IVCT interactions between cofacial DPNDI units explains the intensification of the NIR band upon irradiation. Application of the same potential in the (non-irradiated) EPR SEC experiment resulted in the appearance of an organic signal at g = 2.0025, attributed to the NDI radical monoanion (Fig. 2c). Remarkably, returning the applied potential to −0.1 V vs. Fc/Fc+ resulted in the slow decay of the radical (Fig. S24†), with the intensity of the signal halving only after 1 day, demonstrating the stability of the radical species generated in csiMOF-6.
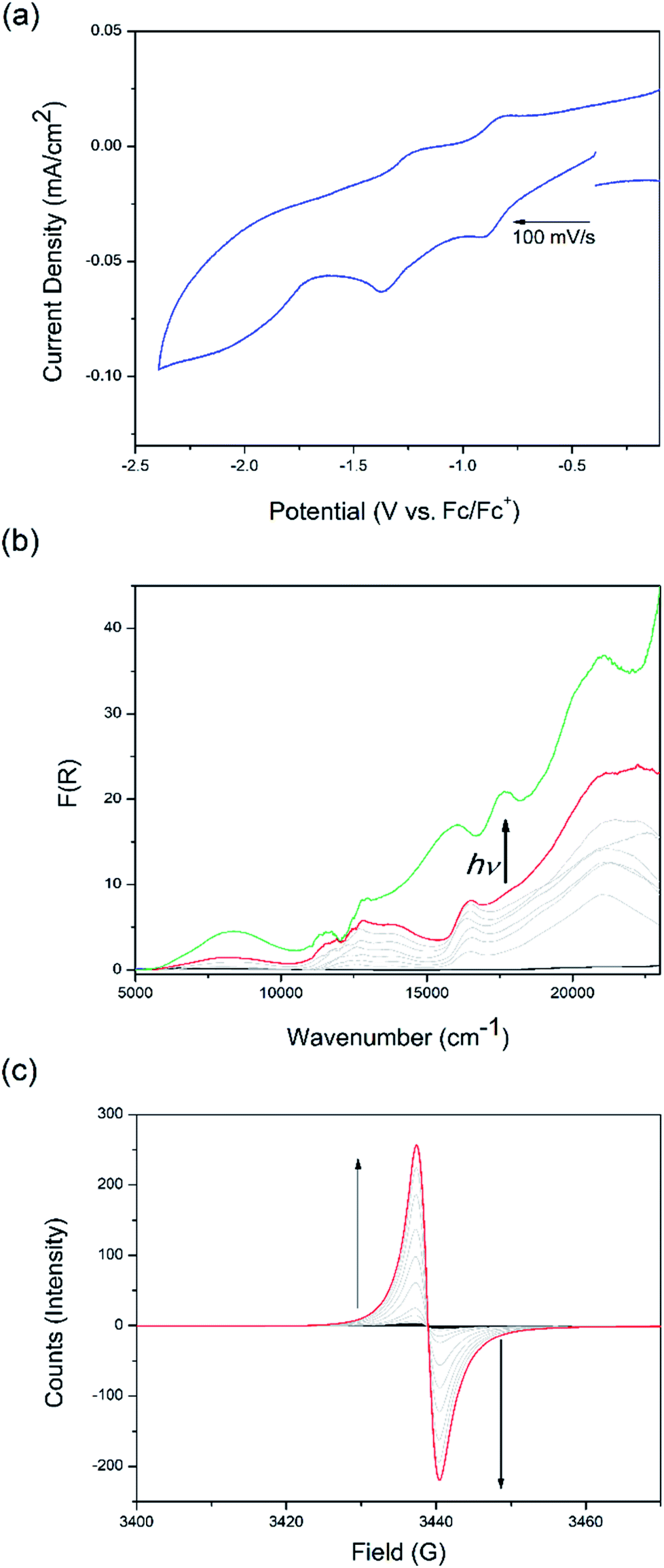 | ||
| Fig. 2 Electrochemical, spectroelectrochemical and photoelectrochemical properties of csiMOF-6 probed by (a) CV, (b) solid state UV-Vis-NIR SEC, and (c) solid state EPR SEC at −1.3 V vs. Fc/Fc+. Spectral colours: black = starting spectrum; grey = spectral progression showing electrochemical generation of the radical monoanion; red = maximum of the spectral reduction; and green = photoexcited radical generated upon in situ light irradiation in the UV-Vis-NIR SEC cell. Arrows indicate sweep direction or spectral changes. | ||
To further characterise the photoexcited radical state of the DPNDI cofacial unit, DFT calculations were performed using (F–Li–DPNDI–Li−F)2−1 (denoted dimer(−1) in Fig. S25 and S26†) as a model system. We note that our computational model bears resemblance to the cofacial NDI dimer system reported experimentally by Wasielewski and Stoddart.22 The calculations, which reproduce key experimental observations, supported the stabilising influence of the cofacial dimer DPNDI structure within the MOF on the lifetime of the photoexcited radical state, and alluded to the photoelectrochemical reduction capabilities of csiMOF-6 (vide infra). TD-DFT calculations indicate that some of the bands in the 15000–30000 cm−1 range are associated with originating orbitals located on NDI and target orbitals with considerable contributions from the pyridyl (Py) moieties of the DPNDI ligands. Irradiation in this range would thus lead to charge transfer from the NDI core to the terminal Py units. Indeed, for the band at 21943 cm−1 (Fig. S25,† experimentally at 21500 cm−1 in Fig. 2b) that corresponds to a predominantly NDI-to-Py transition, there is such a charge transfer of 0.45 electrons when we compare the excited state density to the ground-state density. This photoexcitation induced NDI-to-Py CT in csiMOF-6 precedents its applicability as a photocathode for the photoelectrochemical reduction of catalytic systems.
To understand the potency of the photoexcited csiMOF-6 radical state as a reductant, the energetic consequences of the NDI-to-Py CT transitions within the material were calculated using DFT calculations. Considering the pairs NDI−1 + Py–Li–F and NDI + (Py–Li–F)−1 as representations for, respectively, the reduced ground-state mixed-valence MOF and its photoexcited charge-transfer form, this simple model suggests that a full charge transfer from the reduced NDI core to a Py unit would increase its potency as a reductant by 200 kJ mol−1 (i.e., +180 to −20 kJ mol−1, corresponding to ∼2.1 V, and ∼1.7 V for the dimer pairs, respectively). We can see a stabilisation of the radical monoanion through charge delocalisation between two units, as indicated by the Electron Affinity (EA) of the dimers being more positive than the corresponding monomers (Table 1). Notably, dimerisation stabilises (Py–Li–F)−1 to a greater extent (ΔEA = +83 kJ mol−1) than it does NDI−1 (+46 kJ mol−1). This framework effect contributes to stabilising the NDI-to-Py CT state to the extent that it can be observed in stable state experiments. Fine tuning of these component quantities could enhance the reduction potency of the material or further improve the excited-state lifetime.
| Py–Li–F | NDI | F–Li–DPNI–Li–F | |
|---|---|---|---|
| Monomer | −20 | +180 | +310 |
| Dimer | +63 | +226 | +411 |
| ΔEA | +83 | +46 | +101 |
In order to verify the photocathode applicability of csiMOF-6, a photoelectrochemical CO2 reduction system was devised, employing csiMOF-6 as a radical state photosensitiser for the discrete CO2 reduction electrocatalyst [Re(bipy-tBu)(CO)3Cl] (Scheme 2). In this system, the stable csiMOF-6 photoexcited radical state was used to reduce [Re(bipy-tBu)(CO)3Cl] to its catalytically active state via outer-sphere electron transfer. [Re(bipy-tBu)(CO)3Cl] was selected due to its high efficiency for the selective reduction of CO2 to CO; however, this electrocatalyst exhibits high overpotential demands such that no CO2 electrocatalysis is observed at potentials more anodic than −1.7 V vs. Fc/Fc+ in 0.1 M [n-Bu4N]PF6/MeCN (Fig. S31† shows peak performance at −2.3 V vs. Fc/Fc+). We envisaged that using csiMOF-6 as a heterogeneous radical state photosensitiser (photocathode) may significantly reduce the overpotential requirements for CO2 photoelectrochemical reduction (to that corresponding to generation of the NDI radical monoanion).
 | ||
| Scheme 2 Schematic showing the photocathode role of csiMOF-6 in our CO2 photoelectrochemical reduction system employing a rhenium electrocatalyst. Green arrow indicates photoexcitation, purple arrow indicates CT and black arrow signifies CO2 reduction to CO. | ||
Electrochemical studies in 0.1 M [n-Bu4N]PF6/MeCN electrolyte under 100 W Hg arc lamp irradiation with a UV cut off filter were conducted on the photoelectrochemical reduction system using csiMOF-6 grown onto glassy carbon (GlCa) as the photocathode (Fig. 3a and S27–S30†). CV studies of csiMOF-6 up to a cathodic potential of −1.6 V vs. Fc/Fc+ at 20 mV s−1 in an Ar atmosphere resulted in current enhancement at −1.2 V vs. Fc/Fc+ upon addition of [Re(bipy-tBu)(CO)3Cl] into the electrolyte. This was attributed to photoelectrochemical reduction of [Re(bipy-tBu)(CO)3Cl] to give the active five coordinate species, [Re(bipy-tBu)˙−(CO)3].19 Upon CO2 saturation of the electrolyte, further current enhancement around −1.2 V vs. Fc/Fc+ was observed and was attributed to CO2 reduction by the photoelectrochemically reduced [Re(bipy-tBu)˙−(CO)3] species (Fig. S27 and S28†). The absence of light led to minimal current enhancements, attributed to diminished [Re(bipy-tBu)(CO)3Cl] and subsequent CO2 reduction by csiMOF-6.
CO2 reduction with our system was further investigated with EPR SEC. Previous solution state EPR SEC studies on the CO2 reduction electrocatalyst [Re(bipy)(CO)3Cl] by Scheiring et al. revealed hyperfine coupling features attributed to 185,187Re upon reduction to [Re(bipy)˙−(CO)3].23 For [Re(bipy-tBu)(CO)3Cl], we also demonstrated similar EPR SEC behaviour at potentials more cathodic than −1.7 V vs. Fc/Fc+ (Fig. S32†). Furthermore, all reduced six coordinate catalytically inactive species (i.e., pre chloride dissociation or degraded dimer species) were not observed to exhibit hyperfine coupling features. With this distinction in the EPR SEC behaviour established, solid state EPR SEC was conducted with the csiMOF-6 photocathode, using [Re(bipy-tBu)(CO)3Cl] doped 0.1 M [n-Bu4N]PF6/MeCN electrolyte and 100 W Hg arc lamp irradiation with a UV cut off filter (Fig. 3b). Under a saturated CO2 environment, the system displayed hyperfine coupling features upon the application of a cathodic potential of −1.3 V vs. Fc/Fc+ to access the NDI radical monoanion (g = 2.0039), signalling the occurrence of CO2 photoelectrochemical reduction. Purging the solution with Ar to remove CO2 at this potential resulted in a broadening of the hyperfine coupling (Fig. S33 and S34†). This observation was attributed to the gradual catalyst degradation as seen in the homogeneous state. Blank irradiation EPR SEC experiments either with [Re(bipy-tBu)(CO)3Cl] in the electrolyte but under Ar, or without [Re(bipy-tBu)(CO)3Cl] and under CO2, did not result in hyperfine coupling at −1.3 V vs. Fc/Fc+ (Fig. 3b inset, S35 and S36†). These results affirm the success of our csiMOF-6 photocathode system in enabling CO2 photoelectrochemical reduction at smaller overpotentials than those required for the electrocatalytic reduction of CO2 by [Re(bipy-tBu)(CO)3Cl].
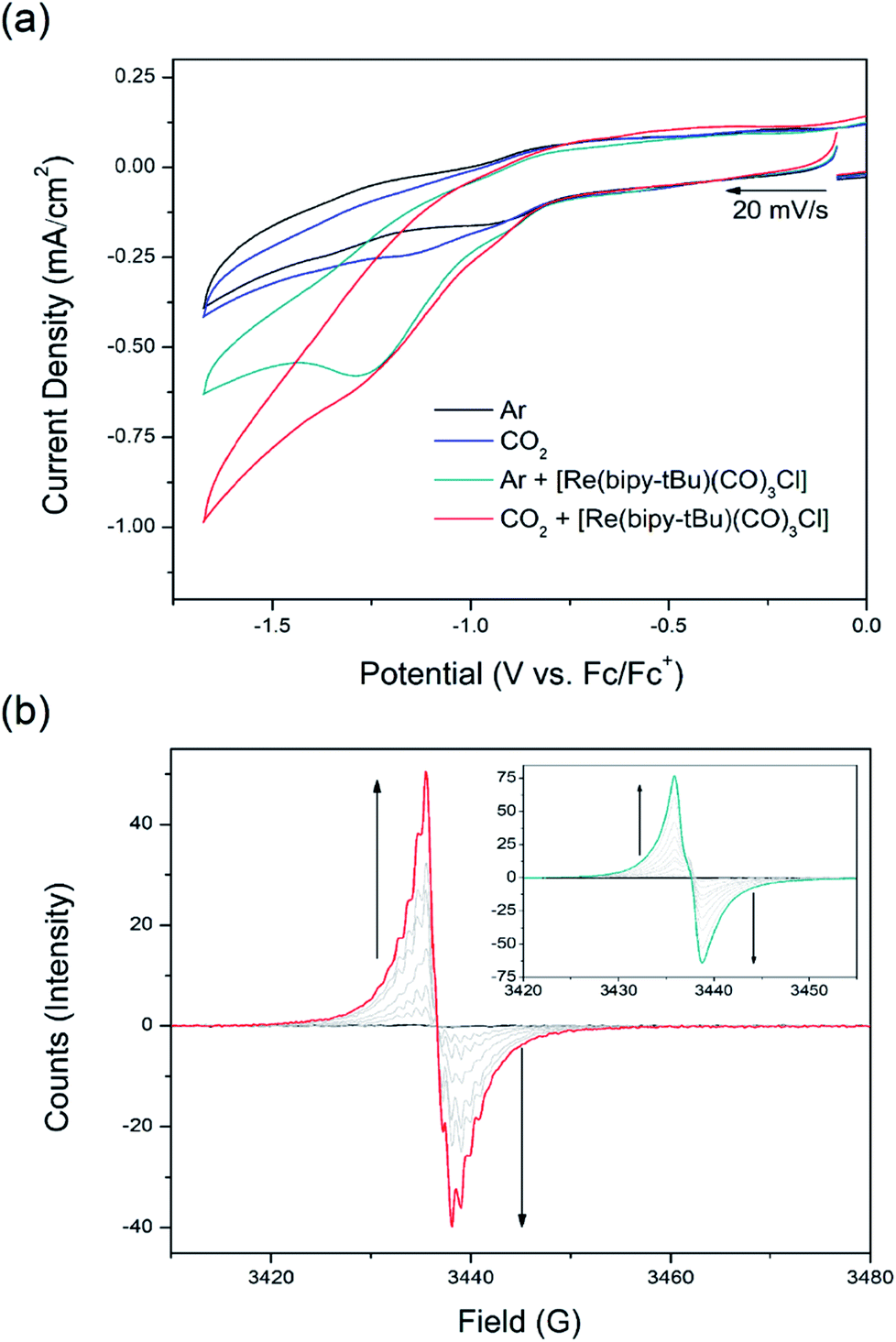 | ||
| Fig. 3 CO2 photoelectroreduction performed with csiMOF-6 in 0.1 M [n-Bu4N]PF6/MeCN with [Re(bipy-tBu)(CO)3Cl] electrocatalyst, demonstrated using (a) CV, and (b) EPR SEC with CO2 saturated electrolyte (inset: under Ar in the absence of CO2). Arrows indicate sweep direction or spectral changes. | ||
To quantify the amount of evolved CO from our csiMOF-6 system, 100 W Hg arc lamp (with UV cut off filter) irradiated controlled potential electrolysis (CPE) studies were conducted whilst holding the potential at −1.3 V vs. Fc/Fc+ with a gas tight three electrode electrochemical cell (csiMOF-6 grown on GlCa photocathode) using an electrolyte solution of 0.1 M [n-Bu4N]PF6/MeCN with 0.7 mM [Re(bipy-tBu)(CO)3Cl]. Product analysis using GC-MS revealed the generation of 0.03 mmol CO over 2 h, corresponding to 78% faradaic efficiency and a turnover number (TON) of 7 (Fig. S37†). Blank irradiated measurements with bare GlCa and [Re(bipy-tBu)(CO)3Cl] under CO2 (Fig. S38†) as well as csiMOF-6 on GlCa under CO2 without Re catalyst, did not result in CO generation (Fig. S39†). Upon completion of CPE and EPR SEC experiments, powder X-ray diffraction analysis of the used csiMOF-6 samples confirmed the retention of crystallinity (Fig. S11†). ICP-MS analysis of the electrolyte post CPE showed an insignificant amount of CdII ions (0.8 ppm), confirming that framework decomposition had not occurred.
Conclusion
In conclusion, we have presented the new framework csiMOF-6 featuring cofacially stacked dimers of the photo- and redox-active DPNDI ligand. This material displayed an excellent capability to stabilise both radical and photoexcited radical states through mixed-valence IVCT interactions within cofacial NDI units. The persistence of the photoexcited NDI-to-Py radical state in csiMOF-6 served as a key prerequisite for its application as a radical state photosensitiser (i.e. photocathode). The applicability of csiMOF-6 as a photocathode was verified upon its incorporation into a photoelectrochemical CO2 reduction system involving the [Re(bipy-tBu)(CO)3Cl] electrocatalyst. Photoexcitation of the csiMOF-6 photocathode in its radical monoanion state at −1.3 V vs. Fc/Fc+ by broad band visible light promoted its successful photoelectrochemical reduction of the Re electrocatalytic complex to the CO2 reduction active state. Correspondingly, CO2 reduction to CO was observed to occur with our system, with 78% faradaic efficiency and a TON of 7, at lower reduction potentials than that required by the molecular catalyst itself. This study paves the way for the future growth of the application of cofacial MOFs as photocathodes. Ongoing research in our group is working towards understanding the mechanistic details of catalysis within these systems, developing strategies to achieve the tuning of photoactive MOF energy levels and exploring the heterogenization of already established catalytic systems.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors gratefully acknowledge funding from the Australian Research Council (FT170100283), the Japan Society for the Promotion of Science (Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers), and computer time from RIKEN ACCC (Project Q18266) and the National Computational Infrastructure Australia. Helpful advice from Drs Martina Lessio (University of New South Wales) and Tiesheng Wang (Shanghai Jiao Tong University) is also very gratefully acknowledged.References
- P. Ding, T. Jiang, N. Han and Y. Li, Mater. Today Nano, 2020, 10, 100077 CrossRef.
- A. Sinopoli, N. T. La Porte, M. R. Wasielewski and M. Sohail, Photosensitisers for CO2 photoreduction: from metal complexes to rylenes, an overview, in Organometallic Chemistry, The Royal Society of Chemistry, 2019, vol. 42, p. 80 Search PubMed.
- R. Murase, B. Ding, Q. Gu and D. M. D'Alessandro, Philos. Trans. R. Soc., A, 2019, 377, 20180226 CrossRef CAS.
- D. R. Whang and D. H. Apaydin, ChemPhotoChem, 2017, 2, 148 CrossRef.
- L. L. Shipman, T. M. Cotton, J. R. Norris and J. J. Katz, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 1791 CrossRef CAS.
- C. Hua, P. W. Doheny, B. Ding, B. Chan, M. Yu, C. J. Kepert and D. M. D'Alessandro, J. Am. Chem. Soc., 2018, 140, 6622 CrossRef CAS.
- B. Ding, C. Hua, C. J. Kepert and D. M. D'Alessandro, Chem. Sci., 2019, 10, 1392 RSC.
- P. W. Doheny, J. K. Clegg, F. Tuna, D. Collison, C. J. Kepert and D. M. D'Alessandro, Chem. Sci., 2020, 11, 5213 RSC.
- D. A. Sherman, R. Murase, S. G. Duyker, Q. Gu, W. Lewis, T. Lu, Y. Liu and D. M. D'Alessandro, Nat. Commun., 2020, 11, 2808 CrossRef CAS.
- S. V. Bhosale, C. H. Jani and S. J. Langford, Chem. Soc. Rev., 2008, 37, 331 RSC.
- N. T. La Porte, J. F. Martinez, S. Hedstrom, B. Rudshteyn, B. T. Phelan, C. M. Mauck, R. M. Young, V. S. Batista and M. R. Wasielewski, Chem. Sci., 2017, 8, 3821 RSC.
- J. F. Martinez, N. T. La Porte and M. R. Wasielewski, J. Phys. Chem. C, 2018, 122(5), 2608 CrossRef CAS.
- J. F. Martinez, N. T. La Porte, S. Chaudhuri, S. Alessandro, Y. J. Bae, M. Sohail, V. S. Batista and M. R. Wasielewski, J. Phys. Chem. C, 2019, 123(16), 10178 CrossRef CAS.
- J. F. Martinez, N. T. La Porte and M. R. Wasielewski, J. Photochem. Photobiol., A, 2019, 372, 21 CrossRef CAS.
- S. Goswami, J. N. Nelson, T. Islamoglu, Y.-L. Wu, O. K. Farha and M. R. Wasielewski, Chem. Mater., 2018, 30, 2488 CrossRef CAS.
- Y. Takashima, S. Furukawa and S. Kitagawa, CrystEngComm, 2011, 13, 3360 RSC.
- N. Sikdar, K. Jayaramulu, V. Kiran, K. V. Rao, S. Sampath, S. J. George and T. K. Maji, Chem.–Eur. J., 2015, 21, 11701 CrossRef CAS.
- P. H. Dinolfo, M. E. Williams, C. L. Stern and J. T. Hupp, J. Am. Chem. Soc., 2004, 126, 12989 CrossRef CAS.
- J. M. Smieja and C. P. Kubiak, Inorg. Chem., 2010, 49, 9283 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9 CrossRef CAS.
- T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 6312 CrossRef CAS.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476 CrossRef CAS.
- T. Scheiring, A. Klein and W. Kaim, J. Chem. Soc., Perkin Trans. 2, 1997, 2569 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1868581, 1868582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04691d |
| This journal is © The Royal Society of Chemistry 2021 |