
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sterically constrained tricyclic phosphine: redox behaviour, reductive and oxidative cleavage of P–C bonds, generation of a dilithium phosphaindole as a promising synthon in phosphine chemistry†‡
Alexander
Brand§
a,
Stephen
Schulz§
 b,
Alexander
Hepp
a,
Jan J.
Weigand
*b and
Werner
Uhl
*a
b,
Alexander
Hepp
a,
Jan J.
Weigand
*b and
Werner
Uhl
*a
aInstitut für Anorganische und Analytische Chemie der Universität Münster, Corrensstraße 30, D-48149 Münster, Germany. E-mail: uhlw@uni-muenster.de
bAnorganische Molekülchemie, Fakultät für Chemie und Lebensmittelchemie, Technische-Universität Dresden, D-01069 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
First published on 18th January 2021
Abstract
The redox behaviour of sterically constrained tricyclic phosphine 3a was investigated by spectroelectrochemistry. The data suggested a highly negative reduction potential with the reversible formation of a dianionic species. Accordingly, 3a reacted with two equivalents of Li/naphthalene by reductive cleavage of a P–C bond of one of the PC4 heterocycles. The resulting dilithium compound 5 represents a phosphaindole derivative with annulated aromatic C6 and PC4 rings. It is an interesting starting material for the synthesis of new heterocyclic molecules, as was shown by treatment with Me2SiCl2 and PhPCl2. The structures of the products (6 and 7) formally reflect ring expansion by insertion of silylen or phosphinidene fragments into a P–C bond of 3a. Treatment of 3a with H2O2 did not result in the usually observed transfer of a single O atom to phosphorus, but oxidative cleavage of a strained PC4 ring afforded a bicyclic phosphinic acid, R2PO2H.
Introduction
The highly interesting chemical properties of sterically constrained bicyclic or tricyclic phosphines (1 to 3; Scheme 1) have attracted considerable attention in recent research.1–7 These compounds are accessible through facile routes and have in most cases heteroatoms such as N, O, S or P bound to their central P atoms. Only recently we found facile access to the first derivatives (3) which had the P atoms coordinated in a homoleptic fashion by three C atoms.1,7,8 The latter compounds were obtained by treatment of the dimeric or trimeric trilithium compounds 4 (ref. 7 and 9) with PCl3 and salt elimination. The steric strain of these molecules became evident from flattened molecular structures with unusually large Cvinyl–P–Cvinyl angles of about 128°, very short P–C bonds to the ipso-C atoms of the bridging aryl rings (∼175 pm) and a considerable deviation of the α-C atoms of the vinyl groups from the planes of the central aryl rings. All phosphines 1 to 3 show a fascinating chemical reactivity which comprises normal phosphine reactions such as oxidative addition to the P atoms or formation of transition metal complexes by retention of the molecular constitution.1 The first type of reactions2–5,7,8,10,11 afforded e.g. dihalogenophosphoranes, which allowed the synthesis of unusual dimethyl substituted phosphor(V) species.2a,8 More fascinating are reactions which are clearly favoured by the minimization of ring strain and result in ring cleavage or insertion of atoms into endocyclic P–X bonds by ring expansion. Such transformations comprise the activation of H–H,12 O–H2,4 or N–H bonds5 and the insertion of O,10a S7 and Se atoms7 or of CO molecules.13 Ring strain should also result in a unique and highly promising redox behaviour of such compounds. | ||
| Scheme 1 Sterically constrained bi- and tricyclic phosphines (1–3) and the trilithium starting compound (4). | ||
Not only electron precise species may be formed, but one-electron reduction or oxidation may result in the formation of radical species which may be stabilized by delocalisation of electron density into the molecular backbone. However, investigations into the redox chemistry of 1 to 2 are limited and have not been elucidated systematically. Treatment with a strong reductant (KC8) resulted in P–P bond formation,10a while oxidation with silver cations Ag+ afforded a relatively persistent radical cation.14 The stable and nonpolar P–C bonds of compounds 3 may help to stabilize unusual products of electron transfer reactions. The constitution of 3 may allow the facile delocalisation of spin density or charges into the π-systems of adjacent aromatic rings and alkenyl moieties and contribute to the stability and isolability of reactive secondary products. In this article we report on comprehensive studies into the redox properties of phosphine 3a by cyclovoltammetry combined with spectroelectrochemistry and on first successful attempts to transfer the results into a preparative scale.
Results and discussion
Cyclic voltammetry and square-wave voltammetry of phosphine 3a
Analytical cyclic voltammetry (CV) was used to elucidate the electrochemical behaviour of tricyclic phosphine 3a. Based on the sterically constrained structure of phosphine 3a oxidation reactions may lead to the formation of kinetically stabilized radical cations,15 while reductions are expected to result in P–C bond cleavage16 driven by release of ring strain. In situ UV-vis-spectroelectrochemical methods like UV-vis CV as well as UV-vis multi pulse chrono amperommetry (in situ UV-vis MPCA) in semi-preparative measurement cells were used to explore the stability of formed species and to probe for preparative accessible compounds. CV of phosphine 3a at a platinum disc electrode was optimized in fluorobenzene (see ESI Fig. SI.1.‡), acetonitrile (see ESI Fig. SI.2.–4.‡) and tetrahydrofuran (see ESI Fig. SI.5. and 6.‡) as solvents. A low diffusion coefficient and, therefore, low intense and broad peaks in the CV hampered an use of electrochemically reductive stable fluorobenzene as an electrolyte. Acetonitrile enabled the determination of the oxidation (E1/2(SWV) = 1.15 V)17 and reduction potential (E1/2(SWV) = −2.38 V)17 but also showed follow-up reactions of the oxidized (ESI Fig. SI.3.3‡) and reduced (ESI Fig. SI.3.4‡) species. Tetrahydrofuran represents a good alternative with a wider potential window for the reduction reaction, while allowing for an acceptable peak shape due to the diffusion coefficient of phosphine 3a. Oxidation of neutral phosphine 3a does not lead to stable (PhF, CH3CN) or accessible (THF) species.Cyclic voltammetry of phosphine 3a in THF with [nBu4N][OTf] as a supporting electrolyte shows a non-reversible reduction process at a very low peak-potential of EP(red I) = −2.63 V (vs. E1/2(Cp2Fe/Cp2Fe+)) in the first cycle (Fig. 1). The second cycle reveals a non-reversible re-oxidation process at EP(re-ox II) = −0.74 V in a potential region where the starting material 3a was electrochemically not active. Square-wave voltammetry (SWV) allowed to assign a half-wave potential of E1/2 = −2.58 V for the substrate-based reduction reaction I, while the re-oxidation process II is not being found in accordance with the necessity of the formation of the product from the reduction I. In situ UV-vis CV spectroelectrochemistry was used to gain mechanistic insights for the reduction I of 3a and the re-oxidation II of the product of the reduction I.
In situ UV-vis-NIR spectroelectrochemistry of phosphine 3a
CV measurement of phosphine 3a in THF with in situ UV-vis monitoring under optimized conditions in double-compartment cuvette-cell lead to a CV curve (Fig. 2, right) comparable to the conventional CV (Fig. 1). In situ UV-vis spectroscopy required a high concentration of phosphine 3a by compromising peak shapes (see ESI Fig. SI.7.‡ for a low concentration measurement, comparable to Fig. 1). The UV-vis spectrum (Fig. 2, red horizontal line) of the reduction product I shows a visible absorption maximum at λ = 486 nm. At the re-oxidation peak II the UV-vis spectrum indicates a new product with an absorption at λ = 380 nm decreasing (Fig. 2, black horizontal line) till the reduction I potential is reached in the second cycle again. The absorption-based CV curve intensity for the product of the reduction I (red line, bottom right) increases at the reduction I but also rapidly decreases at the re-oxidation II. This finding indicates a spectroscopically reversible reduction of phosphine 3a to a reduction product which is formed back at the re-oxidation II. An intermediate of the re-oxidation II (black line) is evolving at the re-oxidation II and decreases slowly under formation of the phosphine 3a. In situ UV-vis CV is executed solely for the reduction reaction I in order to investigate the stability of the reduction product. Fig. 3 shows the formation of the product of reaction I as indicated by the UV-vis spectrum (red horizontal line) and the intensive peak I in the first CV cycle. Further CV cycles around the reduction process I indicates no additional signs of decay (UV-vis, black line). Absorbance fluctuations after the first cycle (red line, vertical) are caused by diffusion from the thin-layer to the bulk area of the cuvette-cell. Based on the SEC results an electrode reaction mechanism (Scheme 2) is proposed. The electrochemically non-reversible reduction reaction I may lead to the formation of unstable intermediate [3a]2− by a two-electron reduction (vide infra) which than reacts under P–C bond cleavage to the dianion [3a′]2−. The electron transfer with its follow-up reaction is best described by an EEC mechanism scheme. Comparing the peak potentials of the re-oxidation process II (EP = −0.74 V) to the peak potential reported for the oxidation of vinyl lithium (EP = −0.06 V)18 and benzophospholide lithium salts (Ep ≈ 1.19 V)19 results in the assumption of a two-electron (vide supra) reduction thus leading to the intermediate 3a′ with a preserved phospholide moiety and a α-silyl vinyl cation by two-electron reduction. In a homogenous follow-up reaction intermediate 3a′ is undergoing a ring-closure reaction forming phosphine 3a back in accordance to the spectroscopic reversibility observed for the full process. To proof the two-electron transfer steps followed by a slow chemical follow-up reaction as an EEC mechanism a further spectroelectrochemical proof of the ring-closure reaction without an electron transfer involved has been realized. Fig. 4 shows an in situ UV-vis CV with a first cycle fully converting phosphine 3a by the reduction Ivia an EEC mechanism to the stable dianion [3a′]2− (red horizontal spectrum). Due to the slow scan rate the concentration of the dianion [3a′]2− and, therefore, its absorption becomes steady in time dependent (red vertical line) and potential dependent representation (black line, right diagram). Potential sweeps around the re-oxidation II range fully converts dianion [3a′]2− to the intermediate 3a′ based on the decreasing absorption of dianion [3a′]2−. Within the 2nd to 5th cycle no significant current is observed in the CV. The UV-vis spectrum shows the characteristic absorptions of intermediate 3a′ (black horizontal spectrum) decreasing over time (blue shaded area) by the chemical follow-up reaction forming phosphine 3a. A half-life time determination (ESI, Fig. S8.‡) for the ring-closure reaction to form phosphine 3a was performed giving t1/2 = 278 s regarding the intermediate 3a′. | ||
| Fig. 1 Cyclic voltammogram of 3a (2.32 mM) in THF/0.1 M [nBu4N][OTf] at a Pt disc electrode (1.6 mm; 0.1 V s−1). iR compensated by R = 4750 Ω. | ||
 | ||
| Fig. 2 Spectroelectrochemical CV measurement of the reduction I and reoxidation II (v = −10 mV s−1) of phosphine 3a (2.50 mM) in THF/0.1 M [nBu4N][OTf] at a platinum grid electrode in a double compartment cuvette cell. Left contour plot: relative UV-vis spectral changes during 3 cycles of CV; middle vertical plot: time dependent UV-vis absorption; top left: projected relative UV-vis spectra after reduction I (red line) and after reoxidation II (black line); top right: in situ CV, bottom right: absorption based CV. | ||
 | ||
| Fig. 3 Spectroelectrochemical UV-vis CV (v = −15 mV s−1) measurement of the reduction I of phosphine 3a (1.00 mM) in THF/0.1 M [nBu4N][OTf] at a platinum grid electrode in double compartment cuvette cell. Left contour plot: relative UV-vis spectral changes during 3 cycles of CV; middle vertical plot: time dependent relative UV-vis absorbance; top left: projected relative UV-vis spectra after reduction in the 1st CV cycle (red line) and after the 2nd CV cycle (black line); right: in situ CV. | ||
 | ||
| Scheme 2 Mechanism for the two-electron reduction I of tricyclic phosphine 3a to anion [3a′]2− by an EEC mechanism und re-oxidation II mechanism via an EEC sequence involving a meta-stable intermediate 3a′. | ||
 | ||
| Fig. 4 Spectroelectrochemical CV measurement of the reduction I and exhaustive re-oxidation II (v = −10 mV s−1) of phosphine 3a (2.50 mM) in THF/0.1 M [nBu4N][OTf] at a platinum grid electrode in a double compartment cuvette-cell. Left contoure plot: relative UV-vis spectral changes during 5 cycles of CV; middle vertical plot: time dependent relative UV-vis absorption; top left: projected relative UV-vis spectra after reduction I cycle and multiple reoxidation II cycles; top right: in situ CV; bottom right: absorption based CV. | ||
Finally, the number of electrons involved in the EEC processes concerning the reduction I of phosphine 3a to dianion [3a′]2− and its re-oxidation II is determined by the use of in situ UV-vis multi pulse amperommetry (Fig. 5). Applying repetitive reduction and re-oxidation pulses (1st diagram Fig. 5) in a calibrated volume double-compartment cuvette-cell allows for the determination of the number of electrons consumed from the chronocoulogram (3rd diagram Fig. 5). Multiple reversible switching between phosphine 3a and dianion [3a′]2− (red line, 4th diagram Fig. 5) over intermediate 3a′ (black line, 4th diagram Fig. 5) further proved the reversibility as well as the two-electron nature of the process after correction of diffusion effects (compare ESI Fig. S9.‡).
 | ||
| Fig. 5 In situ UV-vis multi pulse chrono amperommetry (in situ UV-vis MPCA) of phosphine 3a (2.50 mM) in THF/0.1 M [nBu4N][OTf] at a platinum grid electrode in a double compartment cuvette-cell (d = 0.5 mm). Left, bottom: 2D-plot of relative in situ UV-vis spectra during the MPCA measurement (reduction I and re-oxidation II pulses); left, top: relative UV-vis spectra of the reduction product [3a′]2− (red line) and the intermediate 3a′ (black line) of the re-oxidation; right top to bottom: potential profile, chonoamperogramm, chronocoulogramm, chronoabsorptiometry. | ||
The redox properties of tricyclic phosphine 3a does not allow to access a product of its oxidation, while the reduction reaction at highly negative potentials leads to the formation of stable dianion [3a′]2−. In situ UV-vis CV and MPCA reveals a two-electron process for the reduction via an EEC sequence without a detectable intermediate (CV at 10 V s−1, ESI Fig. S10.‡) pointing towards a fast, chemical follow-up reaction to form dianion [3a′]2−. Furthermore, it was demonstrated that the reduction sequence from this dianion is chemically reversible proceeding over a metastable intermediate [3a′]2− after the two-electron reduction (EE part) with a slow follow-up reaction (C part) back to phosphine 3avia a total EEC sequence. Therefore, suitable reduction reagents should allow for preparative synthesis of dianion [3a′]2− while the intermediate 3a′ is only accessible in situ.
Reductive ring cleavage with the tricyclic phosphine 3a
Electrochemical investigations revealed a very high negative potential necessary for the reduction of the tricyclic phosphine 3a and suggested a challenging search for suitable reductants, which would allow the transfer of these results into a preparative scale. Treatment of 3a with potassium graphite afforded dark red mixtures. NMR spectra were unspecific and showed only broad and low intensity resonances, which did not allow a reasonable assignment. All attempts to isolate a crystalline material by recrystallization of the crude product from various organic solvents failed (Electrosynthesis: see ESI Chapter 1.4‡). Metal naphthalenides are strong reducing agents and have potentials of about −3.0 V.20 We, therefore, treated 3a with in situ prepared Li naphthalenide in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 in THF at −78 °C (Scheme 3). Slow warming to room temperature, filtration, evaporation and recrystallization of the solid residue from toluene afforded yellow crystals of compound 5 in 70% yield. Crystal structure determination (see below) confirmed the reductive cleavage of a P–C bond of one of the five-membered PC4 heterocycles by transfer of electrons to the C atom of the C–SiMe2Ph group and to the P atom.
:2 in THF at −78 °C (Scheme 3). Slow warming to room temperature, filtration, evaporation and recrystallization of the solid residue from toluene afforded yellow crystals of compound 5 in 70% yield. Crystal structure determination (see below) confirmed the reductive cleavage of a P–C bond of one of the five-membered PC4 heterocycles by transfer of electrons to the C atom of the C–SiMe2Ph group and to the P atom.
 | ||
| Scheme 3 Synthesis of 5 [i: 2 Li, 2C10H8, THF; R = SiMe2Ph]; numbering scheme used for assignment of NMR data. | ||
Once crystallized, the solubility of 5 in toluene or benzene decreased dramatically. The NMR spectra were recorded in THF. The P atom of 5 shows a signal at δ = 45.1 ppm in the 31P NMR spectrum, which is shifted to a higher field compared to that of 3a (δ = 68.8 ppm). The typical range of lithium phosphides is at about −120 ppm.21 The 3JPH coupling constant to the H atom attached to the intact ring is comparatively small (11.3 Hz versus 18.3 Hz in 3a) and may indicate a reduced s-character in the P–C bonding orbitals in accordance with Bent's rules22 or a reduced bond order of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The usual chemical shift of δ = 7.47 ppm is observed for this H atom, while the signal of the vinylic H atom at the opened ring (H–C7) in β-position to Li is shifted to a lower field (δ = 9.02 ppm). The latter value corresponds to that of the vinylic H atoms in the trilithium compounds 4.7 The C atoms attached to P have 13C NMR signals at δ = 145.1 (C10) and 150.1 (C1) with 1JPC coupling constants of 54.0 and 34.6 Hz. Also the remaining two C atoms of the PC4 ring show resonances [δ = 144.8 ppm (C2) and 128.1 ppm (C9)] similar to those of the annulated, aromatic C6 ring. The resonance of the carbanionic C atom C8 is shifted to a lower field (δ = 198.5 ppm) and similar to the chemical shift observed for 4 (δ = 192.1 ppm). The coupling constant of C8 to the P atom is surprisingly large with 34.5 Hz. Only a broad resonance was detected in the 7Li NMR spectrum at δ = 1.0 ppm.
C bond. The usual chemical shift of δ = 7.47 ppm is observed for this H atom, while the signal of the vinylic H atom at the opened ring (H–C7) in β-position to Li is shifted to a lower field (δ = 9.02 ppm). The latter value corresponds to that of the vinylic H atoms in the trilithium compounds 4.7 The C atoms attached to P have 13C NMR signals at δ = 145.1 (C10) and 150.1 (C1) with 1JPC coupling constants of 54.0 and 34.6 Hz. Also the remaining two C atoms of the PC4 ring show resonances [δ = 144.8 ppm (C2) and 128.1 ppm (C9)] similar to those of the annulated, aromatic C6 ring. The resonance of the carbanionic C atom C8 is shifted to a lower field (δ = 198.5 ppm) and similar to the chemical shift observed for 4 (δ = 192.1 ppm). The coupling constant of C8 to the P atom is surprisingly large with 34.5 Hz. Only a broad resonance was detected in the 7Li NMR spectrum at δ = 1.0 ppm.
5 forms centrosymmetric dimers in the solid state (Fig. 6) with the Li atoms Li2 and Li2′ in the bridging positions of a double sandwich type molecule. The CC distance in the vinylic group of the opened ring (C7–C8) is with 135.6(2) pm in the typical range of CC double bonds. The C–C distances in the PC4 ring are longer [138.7(3) (C9–C10) to 143.4(2) pm (C1–C2)] and similar to the remaining ones of the annulated aromatic ring [137.1(3) and 142.5(3) pm]. The P–C bond lengths are similar [178.3(2) and 178.8(2) pm] and shorter than usually observed for P–C single bonds. The PC4 ring is almost planar with the maximum deviation of 0.3 pm for the atom C9 and is coplanar to the C6 ring (angle between the normals of the planes 2.6°). These structural parameters resemble those of phospholyl anions23–25 with delocalized π-electrons. The NICS(0) value23k of 5 was calculated for the optimized structure (HF/def2-TZVP//PBE0/def2-TZVP)26 to −11.44, it confirms the aromatic character of the PC4 ring and is quite similar to values of related phospholyl anions reported in the literature.23l,m Hence, reduction of the tricyclic phosphine resulted in an unexpected structural motif which may best be compared to that of phosphaindoles. Two comparable compounds have been reported, in which the five-membered heterocycles were coordinated to Li or transition metal atoms.25 Our interpretation is supported by the results of 31P and 13C NMR spectra, in particular the 31P NMR chemical shift is in the typical range.23–25 Similar to the few lithium phospholides reported in the literature, the Li atom Li2 has relatively short contacts to all four C atoms of the intact PC4 ring [Li2–C9′ 235.4(4) to Li2–C1′ 257.0(4) pm] and to P1′ [261.6(3) pm]. Li2 is further coordinated to three atoms of the opened ring in the second molecular half to form the dimeric formula unit. The relevant distances are 277.9(3) pm (Li2–P1), 217.4(4) pm (Li2–C8) and 249.5(4) (Li2–C7); the other Li–C distances are at least 25 pm longer. The atoms Li2, Li2′, P1 and P1′ form a Li2P2 ring. The second Li atom (Li1) has coordination number three [Li1–O1 187.1(4); Li1–C8 210.9(4); Li1–P1 246.8(3) pm] in an almost ideal planar surrounding (sum of the angles 350.9°). The Li–P distance compares well to values reported for lithium phosphides.27 The η1-coordination of the P atom of a phospholyl anion to Li has not been observed previously. Cleavage of P–C bonds by Li is known for phenyl- or allylphosphines,28 but 5 is the only isolated compound, which has the resulting anionic components combined in a single molecule.24
 | ||
| Fig. 6 Molecular structure and numbering scheme of 5. Displacement ellipsoids are drawn at the 40% level; T = 100 K. H atoms and the SiMe2Ph substituents (with exception of the Si atoms) are omitted. Dashed lines indicate short distances between Li2 and atoms of the phosphine backbones. Important bond lengths (pm) and angles (°): Li1–P1 246.8(3), Li1–O1 187.1(4), Li1–C8 210.9(4), Li2–P1 277.9(3), Li2–P1′ 261.6(3), Li2–C1 277.1(4), Li2–C1′ 257.0(4), Li2–C2′ 247.0(4), Li2–C9′ 235.4(4), Li2–C10′ 236.7(4), Li2–C7 249.5(4), Li2–C8 217.4(4), P1–C1 178.3(2), P1–C10 178.8(2), C1–P1–C10 91.45(8); symmetry equivalent atoms generated by −x + 1, −y + 1, −z + 2. | ||
Reactions of the dilithium compound with element dihalides
The dilithium phosphaindol 5 is a highly interesting starting material for the synthesis of various secondary products by salt elimination. As a proof of principle we treated it with Me2SiCl2 and PhPCl2, in order to obtain new tricyclic compounds with a heteroatom embedded in one of the rings. The proposed transformations correspond formally to a ring expansion of the starting phosphine 3a. THF solutions of isolated or in situ generated samples of 5 were treated with the neat element dihalides at room temperature or at −78 °C (Scheme 4). All volatiles were removed in vacuum at room temperature, the residues were extracted with n-pentane or n-hexane, and the resulting suspensions were filtered. Removal of the solvent from the filtrate afforded the Si compound 6 as a colorless, viscous liquid, which could not be obtained as a solid material from various solvents, while the diphosphorus compound 7 crystallized after concentration of the filtrate at room temperature and was isolated as yellow crystals in 85% yield.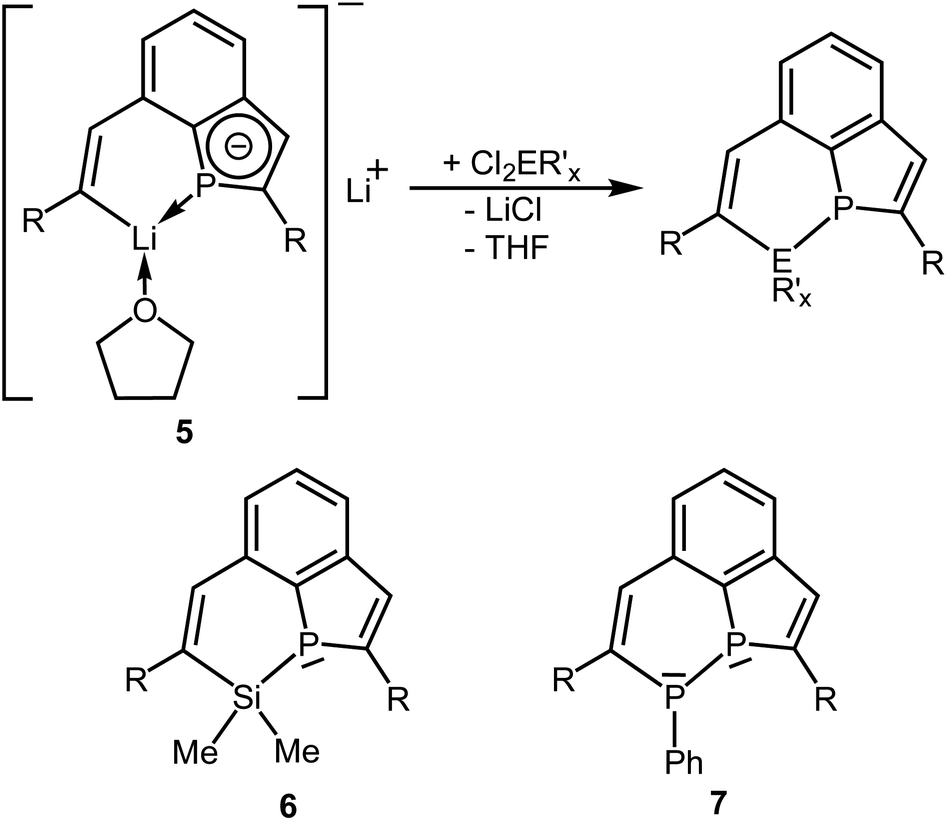 | ||
Scheme 4 Reactions of 5 with  (E = Si, P; R′ = Me, Ph; R = SiMe2Ph; x = 2 or 1) and schematic drawings of 6 and 7. (E = Si, P; R′ = Me, Ph; R = SiMe2Ph; x = 2 or 1) and schematic drawings of 6 and 7. | ||
Crystal structure determination of the diphosphorus compound 7 confirmed its molecular constitution. The tricyclic molecule consists of a six-membered, aromatic C6 homocycle and two annulated heterocycles, a six-membered P2C4 and a five-membered PC4 ring (Fig. 7). The heterocycles contain localized CC double bonds with standard bond lengths of 134.3(7) (C7–C8) and 136.0(3) pm (C9–C10). The P–P distance corresponds with 219.59(9) pm to values reported in the literature.29,30 The P atoms have trigonal pyramidal surroundings and are stereogenic centers.
 | ||
| Fig. 7 Molecular structure and numbering scheme of 7. Displacement ellipsoids are drawn at the 40% level; T = 100 K. H atoms are omitted. Important bond lengths (pm) and angles (°) (main component; see text): P1–P2 219.59(9), P1–C1 179.8(3), P1–C10 184.1(2), P2–C8 185.3(8), P2–C31 184.0(3), C7–C8 134.3(7), C9–C10 136.0(3), C1–P1–C10 90.0(1), C1–P1–P2 93.0(1), C8–P2–P1 95.2(3). | ||
The P atoms are disordered over two positions, and all atoms of the molecular centers were refined on split positions (0.72:0.28; only the structural parameters of the main component are discussed). The two superimposed molecules represent the enantiomeric diastereomers with the configurations (R,S) and (S,R) at the P atoms. The phenyl group attached to P2 is in an exo-position (see for comparison the structure of the W complex discussed below). The endocyclic angles C1–P1–C10 and P–P–C are small [90.0(1) and 94.1(av)°], the exocyclic ones are larger (>95°). The C–P–P–C torsion angles of 161.5 (C1–P1–P2–C31) and −147.0° (C10–P1–P2–C8) indicate an approach of the lone pairs at P to an antiperiplanar orientation, which has previously been observed for diphosphines.29b
Compound 6 shows the expected simple NMR spectra. The 31P{1H} NMR signal (δ = −40.1 ppm) is considerable shifted to a higher field compared to that of the starting phosphine 3a (δ = 68.8 ppm), which may be caused by relaxation of ring strain and the bonding to the relatively electropositive Si atom. Three doublets were observed in the 29Si NMR spectrum. A relatively large coupling constant (1JPSi = 33.8 Hz) was characteristic of the Si atom of the PSiC4 heterocycle. Smaller values resulted expectedly for the Si atoms of the SiMe2Ph groups (2JPSi = 26.9 Hz; 3JPSi = 5.5 Hz). The vinylic H atoms showed 1H NMR signals at δ = 7.73 ppm (intact PC4 ring) and 7.86 ppm (PSiC4 ring) with 3JPH coupling constants of 16.0 (similar to 3b) and 2.7 Hz. The spectra of 7 were much more complicated. The 31P{1H} NMR spectrum revealed two doublets of doublets for the diastereomeric (R,S/S,R) and (S,S/R,R) forms, which showed remarkable differences in their chemical shifts. The (R,S) form resonated at δ = −5.2 ppm (P–Ph) and −16.3 ppm (PC4 ring) with a 1JPP coupling constant of 156.1 Hz, while the (S,S) form gave two doublets at δ = −20.6 ppm (PC4 ring) and −77.6 ppm (P–Ph) with a large 1JPP coupling constant of 354.3 Hz.29,31 The molar ratio between both isomers [(R,S)/(S,R) to (R,R)/(S,S)] in CDCl3 depends on temperature and was calculated from the integration ratios of VT 1H NMR spectra. It changes from about 1:0.41 at room temperature to 1:0.26 at 230 K. The 1H and 31P NMR resonances became broad upon warming to 330 K indicating a fast equilibration at elevated temperature. DFT calculations for both diastereomeric molecules [PBE0 def2-TZVP D3BJ CPCM(CHCl3]26 reproduced the differences in chemical shifts and coupling constants [(S,R): δ = −27.1 ppm (P1) and −12.9 ppm (P2), 1JPP = 113.0 Hz; (S,S): δ = −30.5 and −99.8, 323.7 Hz]. The optimized geometries revealed that in both diastereomers the lone pair of electrons at the P atom of the PC4 heterocycle is oriented almost perpendicularly to the plane of the sp2-hybridized C atoms, which may result in deshielding by anisotropy of the π-orbitals and the ring current of the aromatic ring and in similar chemical shifts for both isomers. The lone pair at the P atom attached to the Ph group of the (S,R) isomer is almost in an antiperiplanar arrangement with respect to the lone pair of the P atom of the PC4 ring (∼180°). This arrangement results in similar 31P NMR shifts. For the (S,S) isomer the lone pairs at the P atoms approach a gauche conformation (torsion angle ∼30°) with the lone pair at the phenyl substituted P atom almost in the nodal plane of the vinylic π-orbital. The approach to the gauche orientation of the lone pairs may not only influence the chemical shifts, but may also result in an additional through-space coupling and may help to understand the increased 1JPP coupling constant. For the gas phase a relatively small energetic difference was calculated between both diastereomers with a slight preference of the (RR,SS) over the (RS,SR) form by 9.3 kJ mol−1. The calculated activation barriers for inversion of configuration via planar transition states differ with 112.5 kJ mol−1 at P2 (P–Ph) and 70.4 kJ mol−1 at the P atom of the PC4 heterocycle. The values are related on the (SR,RS) form and may reflect the better delocalisation of the lone pair at P into the PC4 ring. Systematic conformational studies on the influence of the relative orientation of the lone pairs at the P atoms of diphosphines on the 1JPP coupling constants afforded similar results.29b A comparable effect has been reported for the coupling between fluorine atoms.32
Coordination compounds of 6 and 7
Compound 6 was only isolated as a highly viscous liquid which could not be purified by crystallization. Coordination to a transition metal atom should help to generate a crystalline material for purification and further characterization. We treated 6 with an in situ generated solution of [Cr(CO)5(thf)] in THF at room temperature (Scheme 5). Removal of the solvent, extraction of the residue with n-hexane, filtration and concentration of the filtrate afforded yellow crystals of complex 8 in 82% yield. The results of elemental analysis and mass spectrometry were in accordance with the constitution given in Scheme 5. | ||
| Scheme 5 Syntheses of the complexes 8 and 9 (R = SiMe2Ph). | ||
The resonance of the P atom (δ = −12.2 ppm) of 8 is shifted to a higher field compared to that of the starting phosphine 6, and the 3JPH coupling constant to the vinylic H atom at the PC4 ring increased to 29.3 Hz, which is in accordance with the higher coordination number of four at P. The 4JPH coupling constant to the H atom attached to the PSiC4 heterocycle is, in contrast, with 2.1 Hz almost unchanged compared to that one of 6. The chiral surrounding of the P atom results in a diastereotopic splitting of the resonances of all three pairs of Me groups. Three doublets were observed in the 29Si NMR spectrum, which are shifted to a lower field compared to the resonances in uncoordinated 6 (δ = 5.2 versus 2.1 ppm for the SiMe2 groups in the heterocycle). The NMR parameters of both compounds are very similar and confirm the identity of their constitution. 8 crystallized in very thin platelets. The obtained X-ray data were of insufficient quality and did not allow the refinement to reasonable R values and displacement parameters. But they allowed the unambiguous identification of the molecular constitution as depicted in Scheme 5. The analogous W complex was not obtained in a pure form.
In a similar reaction the tricyclic diphosphine 7 was treated with an in situ generated solution on [W(CO)5(thf)] in THF at room temperature. All volatiles were removed in vacuum, and the crude product was purified by column chromatography (SiO2; n-hexane/diethyl ether 8:1). Recrystallisation from Et2O/n-hexane afforded colorless crystals of the W complex 9 (Scheme 5) in a moderate yield of 55%. Only two doublets were observed in the 31P NMR spectrum. Resonances of a second diastereomer were not found. The relatively large 1JPP coupling constant (313.8 Hz) corresponds to the value observed for the uncoordinated (R,R) diastereomer of 7.
Also the chemical shifts of the P atoms of 9 are closer to those of the (R,R) form of 7. They have the characteristically large difference with δ = −11.9 ppm for the bridgehead P atom P1 and δ = −65.7 ppm for the atom P2 attached to the phenyl group. The coordination to the W(CO)5 fragment is confirmed by a large 1JPW coupling constant to the atom P1 of 211.3 Hz. P2 does not show a coupling to the W atom. In accordance with the increased coordination number at P, the 3JPH coupling constant to the vinylic H atom at the PC4 ring is relatively large with 35.0 Hz.
The molecular structure of 9 shows the intact diphosphine coordinated to W(CO)5via a W–P bond to the bridgehead P atom P1, as predicted by NMR spectroscopy (Fig. 8). The P1–W1 distance [252.9(1) pm] is in the typical range.33 The P1–C1 distance [177.9(2) pm] is shorter than that one in 7 [179.8(3) pm], the other P–C distances approach the standard value and are between 182.3(2) and 185.2(2) pm. In contrast to the molecular structure of 7, the phenyl group attached to P2 adopts an endo-position, and the SiMe2Ph groups change their orientation with all three phenyl groups in neighboring positions (compare Fig. 2 and 3). Short intramolecular distances between atoms of the aryl groups indicate weak interactions (C36–C27 355.1, C36–H27 316, C36–C28 351.8, C36–H28 309, C36 to C13–C18 376.1 to 395.1 pm). The observed diastereomer has the configuration (S,R), with its enantiomer generated by the crystallographic center of symmetry of space group P21/c. This configuration verifies, that complex 9 contains surprisingly the (R,R) diastereomer of 7 which was detected in solution by NMR spectroscopy as the minor component. This interpretation is in accordance with the NMR spectroscopic findings. Obviously, the coordination to the metal atom favors the shift of the dynamic equilibrium in solution and the rearrangement of the diastereomers of the ligand from (R,S/S,R) to (R,R/S,S). Rearrangement may be favored by the minimization of steric repulsion between the substituents of the phosphine ligand and the W(CO)5 group. This assumption is supported by the determination of the “percent buried volumes” % Vbur,34 which were calculated based on the optimized geometries of the diasteromers of 7. P1 was experimentally observed as the binding partner of the metal atom, and we obtained values of 40.4 (S,R) and 32.8% (S,S) which confirmed the smaller steric demand of the (S,S/R,R) couple and the resulting smaller steric stress in complex 9.
 | ||
| Fig. 8 Molecular structure and numbering scheme of 9. Displacement ellipsoids are drawn at the 40% level. H atoms are omitted; T = 100 K. Important bond lengths (pm) and angles (°): P1–P2 219.93(9), W1–P1 252.94(7), P1–C1 177.9(2), P1–C10 182.3(2), P2–C8 185.2(2), P2–C31 182.5(3), C7–C8 134.8(3), C9–C10 135.0(3), C1–P1–C10 92.1(1), C1–P1–P2 100.64(8), C8–P2–P1 95.61(8). | ||
Reaction of 3a with H2O2 – oxidative ring cleavage
Ag+ and a strained phosphine of type 2 (Scheme 1) afforded by oxidation a remarkable radical cation.14 As shown by preliminary investigations, 3a did, in contrast, not react with Ag+ by oxidation, but by formation of a simple adduct with the Ag cation coordinated to the P atom. This behaviour resembles that one of Au+.1,7 The oxidant H2O2 reacts with tertiary phosphines by oxidation of the P atoms, formation of phosphine oxides and release of water.35,36 Recently the highly selective synthesis of Ph3PO in a 100% yield was achieved on such a route.36 The highly strained ring system of compounds 3 may result in another reaction course, and we, therefore, treated a cooled (−78 °C) solution of 3a in n-pentane with a concentrated H2O2 solution in water (Scheme 6). The mixture was slowly warmed to room temperature, all volatiles were removed in vacuum, and the residue was recrystallized from a mixture of CH2Cl2 and Et2O. Compound 10 was isolated as colorless crystals in 79% yield. | ||
| Scheme 6 Reaction of 3b with H2O2. | ||
Crystal structure determination (Fig. 9) confirmed cleavage of a P–C bond of one of the PC4 rings of 3a, the oxidation of the P atom and the formation of a phosphinic acid, R2PO2H, by formal insertion of the P atom into the O–O bond of the peroxide. Ring cleavage and H transfer resulted in an alkenyl side-chain with the trans-arrangement of two H atoms at the CC bond. Hence, the reaction of 3a with H2O2 is completely different from those usually observed with tertiary phosphines. 10 is dimeric in the solid state with a central, non-planar P2O4H2 heterocycle. The O–H distances (∼125 pm) are in a narrow range and indicate together with the short O–O distances of 250.0 pm on average two symmetric O–H–O hydrogen bonds.37 The P–O bond lengths [151.6(2) to 153.7(2) pm] are in the typical range of phosphinic acids, for which hydrogen bonding results in various structural motifs with dimeric, oligomeric and polymeric species in the solid state.38 The dimer of 10 is not centrosymmetric, both molecular halfes are instead related by a pseudo-twofold rotational axis perpendicular to the P2O4H4 ring. The P atom of 10 resonated at δ = 62.8 ppm in the 31P{1H) NMR spectrum in C6D6. The vinylic H atom at the PC4 ring showed a doublet at δ(1H) = 6.93 ppm with a large 3JPH coupling constant of 51.6 Hz indicating the increased coordination number and oxidation state of P. The protons of the vinylic side-chain were detected at δ = 8.15 ppm (α to the central aryl ring) and 6.75 (H–C–Si), the 3JHH coupling constant of 18.9 Hz confirmed their trans-arrangement. The vinylic C atoms showed the usual 13C NMR shifts between δ = 133.6 and 153.0 ppm. The proton of the P–OH group was detected as a broad singlet. Its chemical shift strongly depended on concentration and varied between 10.00 and 12.60 ppm, which may correlate with an increasing dissociation of the dimeric formula units into monomeric fragments in diluted solution. Mass spectrometry confirmed the constitution of 10 by the characteristic molecular ion peak at m/z = 459 for the monomeric formula unit.
 | ||
| Fig. 9 Molecular structure and numbering scheme of 10. Displacement ellipsoids are drawn at the 40% level; T = 100 K. H atoms with exception of H1 and H2 and the SiMe2Ph groups except the central Si atoms are omitted. Important bond lengths (pm) and angles (°): P1–O11 153.7(2), P1–O12 151.6(2), P2–O21 153.4(2), P2–O22 151.8(2), P–C 179.6(av), O11–P1–O12 113.00(9), O21–P2–O22 112.85(9). | ||
Conclusions
The sterically constrained tricyclic phosphine 3a with annulated five- and six-membered rings and the P atom homoleptically bound to C atoms shows fascinating chemical and redox reactivity in various transformations, which resulted in coordination to metal atoms, oxidative addition to the P atoms and the generation of dimethyl phosphoranes, insertion into one of the rings by ring expansion etc. We reported here on reductive or oxidative ring cleavage. Applying cyclic voltammetry, square-wave voltammetry combined with mechanistic studies by the use of in situ UV-vis CV and multi-pulse chronoamperometry allowed detailed investigation of the reduction reaction. Reduction of phosphine 3a leads to dianion [3a′]2−via an EEC reaction mechanism consisting of a two-electron transfer followed by a fast ring-opening reaction to dianion [3a′]2−. The electrochemical reduction is a reversible reaction proceeding via a metastable intermediate 3a′ under ring closure back to phosphine 3a. Electrochemical investigations predicted the formation of a stable dianion [3a′]2− by a two-electron reduction at E1/2 = −2.58 V (vs. E1/2(Cp2Fe/Cp2Fe+)). Inspired by these results we treated phosphine 3a with Li and isolated the unique dilithium compound 5 by cleavage of one of the PC4 heterocycles. 5 represents a phosphaindole derivative with annulated aromatic C6 and PC4 rings and has a promising perspective for an application in secondary processes. Preliminary reactions with SiMe2Cl2 or PhPCl2 afforded by salt elimination new ring systems, in which Si or P atoms were incorporated into six-membered heterocycles and which formally represent products of a ring expansion of 3a. The resulting diphosphine (7) afforded a mixture of diastereomers, which showed a dynamic equilibrium in solution. The (R,S) form was detected by crystal structure determination. Configuration change was observed upon coordination to W(CO)5. Minimization of steric repulsion seem to favour rearrangement to yield exclusively the (R,R) form of 7 coordinated to the metal atom. Hydrogen peroxide reacts usually with triorganylphosphines by transfer of one O atom and formation of triorganylphosphine oxides. In contrast, the strained ring system of 3a led to cleavage of a P–C bond and formation of a phosphinic acid derivative by formal insertion of the P atom into the O–O bond of the peroxo group. Once again these reactions underscore impressively the fascinating capability of these tricyclic phosphines to be applied in various chemical transformations and in the generation of unprecedented structural motifs.Experimental section
General considerations
All manipulations were carried out under purified argon, using standard Schlenk techniques. Solvents were distilled from drying agents and degassed (THF, diethyl ether and toluene over sodium/benzophenone; n-pentane and n-hexane over LiAlH4). NMR spectra were recorded in C6D6, d8-THF and CDCl3 using the following Bruker instruments: Avance I (1H, 400.13; 13C, 100.62; 31P, 161.98), Avance III (1H, 400.03; 13C, 100.60; 31P, 161.93 MHz) and referenced internally to residual solvent resonances (chemical shift data in δ). 13C and 31P NMR spectra were usually proton-decoupled. The assignment of NMR spectra is based on HSQC, HMBC, DEPT135, DEPT19.5 and H,H-ROESY data. Elemental analyses were determined by the microanalytic laboratory of the Westfälische Wilhelms Universität Münster. IR spectra were recorded as KBr pellets on a Shimadzu Prestige 21 spectrometer, electron impact mass spectra on a Finnigan MAT 95 mass spectrometer, HRMS (ESI-TOF) on Orbitrap LTQXL. Commercially available Me2SiCl2, PhPCl2, [Cr(CO)6] and [W(CO)6] were used as purchased. The starting tricyclic phosphine 3a,7 [Cr(CO)5(thf)] and [W(CO)5(thf)]39 were synthesized according to literature procedures.Cyclic voltammetry (CV)
All electrochemical experiments were performed in a nitrogen filled glovebox with specially dried (see SI) solvents and [nBu4N][OTf] as a supporting electrolyte by a PGSAT302 (Metrohm Autolab, Netherlands). CV measurements were performed in custom made 10 mL cells with platinum disk working electrodes (d = 1.6 mm diameter, ALS Co. Ltd., Japan) and a platinum wire coil (200 × 0.5 mm) as a counter electrode. Working electrodes were wet polished (1 μm polishing diamond, 0.05 μm polishing alumina) on different polishing pads before use. Reference electrodes of first-order, consisting of a silver wire in 0.01 M AgNO3 solution in acetonitrile with 0.1 M [nBu4N][OTf] as supporting electrolyte are used. Working and counter electrodes were employed without any separation, while a Vycor glass cylinder (D = 3 mm, L = 4 mm, ALS Co. Ltd., Japan) was used to separate the reference electrolyte from the substrate solution. Electrochemical measurements with disk electrodes in analytical cells are referenced by adding an equimolar amount of Cp2Fe, calibrating the E1/2(Cp2Fe/Cp2Fe+) by cyclic voltammetry as a zero point according to the IUPAC procedure.40 Peak potentials and half wave potentials of the substrates where measured internally in case no reaction with ferrocene takes place. Spectroelectrochemical measurements in cuvette-cell were externally referenced by calibration of the reference electrode in an analytical cell or by calibrating the reversible half-wave potentials to measurements at disk electrodes.In situ UV-vis spectroelectrochemistry (SEC) in transmission geometry
In situ UV-vis SEC was performed with a Maya 2000PRO (Ocean Optics, Ostfildern, Germany) diode array spectrometer equipped with a 25 μm slit, grating JC-1 (300 lines per mm), resulting wavelength resolution of 1.1 nm and a DH-2000-BAL (Ocean Optics Ostfildern, Germany) balanced and adjustable deuterium-halogen light source. Transmission measurements were performed in a 0.5 mm pathway fused silica cuvette (ALS, Japan) equipped with a platinum gauze (80 mesh, 6 × 7 mm) electrode as working electrode and counter electrode. Working and counter electrode meshes were spot-welded to a Pt wire (0.08 mm) and embedded into perforated office lamination foil by heat treatment in special developed procedures. Mesh electrodes were conditioned by 30 min galvanostatic cycling between ±500 mA for 30 s in 1.0 M nitric acid. A standard cell holder installed in the glovebox with adjustable collimation lenses attached to 600 μm solarization resistant optical fibers were used to focus the light beam onto the working electrode. All in situ spectroelectrochemical cells were equipped with the same reference electrode used for disk electrode measurements. UV-vis SEC spectra in in situ measurements were recorded as relative absorbance data with the start material as a reference. All spectral data were recorded after optimization of the halogen light intensity relative to the deuterium lamp. Minimized integration times (t = 34–50 ms) are used with an averaging of 15 scans for each spectrum.Dilithium phosphide 5
Phosphine 3a (0.30 g, 0.70 mmol), lithium (0.010 g, 1.44 mol) and naphthalene (0.18 g, 1.40 mmol) were cooled to −78 °C and suspended in 15 mL of THF. The mixture was slowly warmed to room temperature overnight. Filtration and removal of all volatiles in vacuo afforded a solid material, which was carefully evacuated to about 5 × 10−2 mbar and dissolved in a small quantity of toluene at 70 °C. The solution was slowly cooled to 3 °C to afford 5 as yellow blocks (0.25 g, 70%). Mp: 175 °C (dec.). The assignment of NMR resonances is based on the numbering scheme given in Scheme 3. 1H NMR (d8-THF, 300 K, 400 MHz): δ 9.02 (s, 1H, 7-H), 7.57 (overlap, m, 4H, o-H 8- and 10-Si-Ph), 7.50 (d, 3JHH = 7.7 Hz, 1H, 3-H), 7.47 (d, 3JPH = 11.3 Hz, 1H, 9-H), 7.23 (m, 2H, m-H 8-Si-Ph), 7.17 (m, 1H, p-H 8-Si-Ph), 7.16 (overlap, m, 3H, m- and p-H 10-Si-Ph), 6.85 (pseudo-t br., 3JHH = 7.3 Hz, 1H, 4-H), 6.75 (d, 3JHH = 6.7 Hz, 1H, 5-H), 0.54 (s, 6H, 10-Si-CH3), 0.36 (s, 6H, 8-Si-CH3). 13C{1H} NMR (d8-THF, 300 K, 101 MHz): δ 198.5 (d, 2JPC = 34.5 Hz, 8-C), 164.4 (d, 3JPC = 5.5 Hz, 7-C), 150.1 (d, 1JPC = 34.6 Hz, 1-C), 145.9 (s, i-C 8-Si-Ph), 145.1 (overlap, d, 1JPC = 54.0 Hz, 10-C), 144.8 (overlap, s, 2-C), 144.3 (s, i-C 10-Si-Ph), 142.7 (d, 2JPC = 3.2 Hz, 6-C), 135.0 (s, o-C 10-Si-Ph), 134.9 (s, o-C 8-Si-Ph), 128.4 (s, p-C 8- and 10-Si-Ph), 128.1 (s br., 9-C), 127.7 (s, m-C 8-Si-Ph), 127.6 (s, m-C 10-Si-Ph), 124.3 (d, 3JPC = 8.5 Hz, 5-C), 122.1 (s, 3-C), 118.4 (s, 4-C), 0.5 (d, 3JPC = 4.2 Hz, 10-Si-CH3), −0.2 (s, 8-Si-CH3). 29Si{1H} NMR (d8-THF, 300 K, 79 MHz): δ −12.0 (d, 2JPSi = 34.7 Hz, 10-Si), −14.1 (d, 3JPSi = 9.2 Hz, 8-Si). 31P{1H} NMR (d8-THF, 300 K, 162 MHz): δ = 45.1. 7Li{/} NMR (d8-THF, 300 K, 156 MHz): δ = 1.00 (br.). IR (KBr, cm−1): 3063 w, 3044 m, 3019 w, 2999 w, 2951 s, 2889 m, 2837 vw ν(CH); 1954 vw, 1892 vw, 1817 vw, 1651 vw, 1591 w, 1562 w, 1541 vw, 1528 vw, 1489 w, 1454 w, 1423 s, 1406 w ν(CC), phenyl; 1356 vw, 1304 w, 1285 w, 1246 vs δ(CH3); 1175 w, br., 1113 s, 1065 vw, 1038 m, 986 m, 968 m, 955 m, 916 vw, 887 vw, 833 vs, 812 vs, 773 s, 729 vs ν(CC), ν(CO), ρ(SiCH3); 698 vs, 652 w, 613 w ν(SiC), phenyl; 590 vw, 509 m, 469 m ν(PC), δ(CC). Anal. calcd for C30H35Li2OPSi2 (512.6 for the monomer): C, 70.3, H, 6.9. Found: C, 70.5; H, 6.9.
Synthesis of 6 (SiMe2)
5 (0.15 g, 0.29 mmol of the monomer) was dissolved in 5 mL of THF, and Cl2SiMe2 (39 μL, 0.041 g, 0.32 mmol; density: 1.06 g mL−1) was added at room temperature. The yellow color of 5 disappeared immediately, and the colorless solution was stirred for 1 h. All volatiles were removed in vacuo, and the residue was treated with 10 mL of n-pentane. The suspension was filtered, and the solvent of the filtrate removed in vacuo. 6 was obtained as a colourless oil, which did not crystallize from various solvents (0.12 g, 85%). The assignment of NMR resonances is based on the numbering scheme given in Scheme 3. 1H NMR (C6D6, 300 K, 400 MHz): δ 7.86 (d, 4JPH = 2.7 Hz, 1H, 7-H), 7.73 (d, 3JPH = 16.0 Hz, 1H, 9-H), 7.54 (m, 2H, o-H 10-Si-Ph), 7.50 (m, 2H, o-H 8-Si-Ph), 7.48 (d, 3JHH = 7.4 Hz, 1H, 3-H), 7.19 (t, 3JHH = 7.6 Hz, 1H, 4-H), 7.16 (overlap, m, 6H, o- and m-H 8- and 10-Si-Ph), 6.91 (dd, 3JHH = 7.3 Hz, 4JPH = 3.6 Hz, 1H, 5-H), 0.53 (s, 6H, 10-SiCH3Ph), 0.40 (s, 6H, 8-Si-CH3Ph), −0.21 (d, 3JPH = 5.0 Hz, 6H, ring SiMe2). 13C{1H} NMR (d8-THF, 300 K, 101 MHz): δ 156.6 (d, 3JPC = 1.4 Hz, 7-C), 147.1 (d, 1JPC = 3.6 Hz, 1-C), 147.0 (d, 2JPC = 12.0 Hz, 2-C), 145.8 (d, 1JPC = 48.4 Hz, 10-C), 145.5 (s, 9-C), 139.0 (s, i-C 8-Si-Ph), 138.8 (d, 3JPC = 1.6 Hz, i-C 10-Si-Ph), 138.0 (d, 2JPC = 6.9 Hz, 6-C), 136.2 (d, 2JPC = 12.7 Hz, 8-C), 134.5 (s, o-C 8-Si-Ph), 134.4 (s, o-C 10-Si-Ph), 129.5 (s, p-C 10-Si-Ph), 129.4 (s, p-C 8-Si-Ph), 128.2 (overlap, s, m-C 8- and 10-Si-Ph), 126.1 (s, 4-C), 124.1 (s, 3-C), 123.5 (d, 3JPC = 4.2 Hz, 5-C), −1.1 (d, 3JPC = 3.4 Hz, 10-Si-CH3), −1.3 (s, 8-Si-CH3), −2.5 (d, 2JPC = 11.1 Hz, ring SiMe2). 29Si{1H} NMR (d8-THF, 300 K, 79 MHz): δ 2.1 (d, 1JPSi = 33.8 Hz, ring SiMe2), −5.4 (d, 3JPSi = 5.5 Hz, 8-Si), −10.9 (d, 2JPSi = 26.9 Hz, 10-Si). 31P{1H} NMR (d8-THF, 300 K, 162 MHz): δ −40.1 (s). IR (KBr, cm−1): 3067 m, 3049 m, 3011 w, 2955 s ν(CH); 1954 vw, 1819 vw, 1775 vw, 1722 vw, 1688 vw, 1653 vw, 1524 s ν(CC), phenyl; 1425 m, 1406 w, 1306 w, 1248 vs δ(CH); 1190 vw, 1159 vw, 1111 s, 1040 m, 959 s, 907 s, 833 vs, 812 vs, 790 vs, 777 vs, 731 vs ν(CC), ρ(SiCH3); 700 vs, 654 m, 598 vw ν(SiC), phenyl; 484 w, 471 m, 453 w, 422 w ν(PC), δ(CC). MS (EI, 20 eV, 303 K): m/z (%) = 484 (100) [M]+, 135 (10) [SiMe2Ph]+. HRMS (ESI-ORBITRAP) m/z: [M + H]+ calcd for C28H34Si3P 485.1700; found 485.1709; [M + OH]+ C28H34OSi3P 501.1650; found 501.1651.
Synthesis of 7 (PPh)
A solution of compound 5 in 10 mL of THF was prepared from 3a (0.15 g, 0.35 mmol), Li (4.9 mg, 0.71 mmol) and naphthalene (0.090 g, 0.70 mmol) as described above. It was cooled to −78 °C and directly treated with PhPCl2 (57 μL, 0.075 g, 0.42 mmol; density: 1.32 g mL−1). The cooling bath was removed and the mixture stirred at room temperature for 3 h. All volatiles were removed in vacuo, and the residue was extracted with 10 mL of n-hexane. After filtration, concentration of the filtrate and addition of a seed crystal 7 crystallised at room temperature as yellow blocks (0.16 g, 85%). Alternatively, the solvent of the filtrate was completely removed in vacuo and the residue treated with refluxing methanol (7 mL), until a clear solution was formed. Cooling the solution to room temperature led to the separation of pure 7 as a yellow oil, which was isolated and recrystallized from n-hexane. Mp: 89 °C. The assignment of NMR resonances is based on the numbering scheme given in Scheme 3. NMR data of the R,S/S,R diastereomer: signals of the para-C- and -H atoms of the silicon bound phenyl rings overlapped and were not assigned. 1H NMR (CDCl3, 300 K, 400 MHz): δ 7.66 (overlap, m, 2H, o-H P-Ph), 7.63 (overlap, m, 1H, 9-H), 7.44 (overlap, m, 1H, 3-H), 7.34 (overlap, m, 1H, p-H P-Ph), 7.31 (overlap, m, 2H, o-H 10-Si-Ph), 7.29 (overlap, m, 1H, 4-H), 7.28 (overlap, m, 2H, o-H 8-Si-Ph), 7.26 (overlap, m, 4H, m-H 8- and 10-Si-Ph), 7.24 (d, 3JPH = 14.0 Hz, 7-H), 7.17 (t, 3JHH = 7.5 Hz, 2H, m-H P-Ph), 6.83 (dd, 3JHH = 7.3 Hz, 4JPH = 3.1 Hz, 5-H), 0.19 (overlap, s, 6H, 8- and −10-Si-CH31), 0.14 (s, 3H, 10-Si-CH32), 0.13 (s, 3H, 8-Si-CH32). 13C{1H} NMR (CDCl3, 300 K, 101 MHz): δ 151.0 (dd br., 1JPC = 55.0 Hz, 2JPC = 8.9 Hz, 10-C), 149.5 (s br., 9-C), 147.0 (dd br., 3JPC = 9.0 Hz, 2JPC = 2.0 Hz, 7-C), 146.2 (dd br., 2JPC = 12.3 Hz, 3JPC = 2.0 Hz, 2-C), 144.5 (d br., 1JPC = 9.1 Hz, 1-C), 138.8 (dd, 1JPC = 50.8 Hz, 2JPC = 5.2 Hz, 8-C), 137.3 (dd, 2JPC = 9.1 Hz, 3JPC = 5.3 Hz, 6-C), 137.0 (dd, nJPC = 22.2 Hz, nJPC = 10.7 Hz, o-C P-Ph), 137.0 (s, i-C 8- and 10-Si-Ph), 134.0 (s, o-C 8-Si-Ph), 133.9 (s, o-C 10-Si-Ph), 133.8 (overlap, m, i-C P-Ph), 129.9 (s, p-C P-Ph), 128.8 (d, 3JPC = 8.3 Hz, m-C P-Ph), 128.4 (s, 4-C), 127.7 (overlap, m-C 8- and 10-Si-Ph), 123.7 (s, 3-C), 121.7 (d, 3JPC = 4.8 Hz, 5-C), −1.1 (s, 8-Si-CH32), −1.2 (s, 8-Si-CH31), −1.7 (d, 3JPC = 2.0 Hz, 10-Si-CH32), −2.1 (d, 3JPC = 2.8 Hz, 10-Si-CH31). 29Si{1H} NMR (CDCl3, 300 K, 79 MHz): δ −4.5 (dd, 3JPSi = 8.5 Hz, 2JPSi = 4.1 Hz, 8-Si), −10.7 (d, 2JPSi = 25.3 Hz, 10-Si). 31P{1H} NMR (CDCl3, 300 K, 162 MHz): δ −5.2 (d, 1JPP = 156.1 Hz, P-Ph), −16.3 (d, 1JPP = 156.1 Hz, PC4 heterocycle). NMR data of the R,R/S,S diastereomer: signals of the ortho-, meta- and para-C and -H atoms of the silicon bound phenyl rings overlapped and were not assigned. 1H NMR (CDCl3, 300 K, 400 MHz): δ 7.83 (d, 3JPH = 16.0 Hz, 1H, 7-H), 7.39 (overlap, m, 1H, 9-H), 7.31 (overlap, m, 1H, 3-H), 7.27 (overlap, m, 1H, 4-H), 7.06 (dd, 3JHH = 7.4 Hz, 4JHP = 3.0 Hz, 1H, 5-H), 6.91 (t, 3JHH = 6.9 Hz, p-H P-Ph), 6.76 (pseudo-t, 3JHH = 7.2 Hz, 2H, m-H P-Ph), 6.40 (pseudo-t, 3JHH = 7.1 Hz, 3JPH = 8.0 Hz, 2H, o-H P-Ph), 0.67 (s, 3H, 10-Si-CH31), 0.64 (s, 3H, 10-Si-CH32), 0.56 (s, 3H, 8-Si-CH31), 0.51 (s, 3H, 8-Si-CH32). 13C{1H} NMR (CDCl3, 300 K, 101 MHz): δ 150.3 (dd br., 3JPC = 11.0 Hz, 2JPC = 6.0 Hz, 7-C), 149.4 (s, 9-C), 145.2 (d br., 1JPC = 62.0 Hz, 10-C), 143.5 (d, 3JPC = 18.2 Hz, 2JPC = 2.0 Hz, 2-C), 143.2 (m br., 1-C), 138.2 (s br., i-C 10-Si-Ph), 137.2 (overlap, m, i-C 8-Si-Ph), 135.5 (dd, nJPC = 8.5 Hz, nJPC = 3.7 Hz, 6-C), 131.1 (d, nJPC = 19.8 Hz, o-C P-Ph), 130.6 (d br., 1JPC = 58.0 Hz, 8-C), 128.0 (s, p-C P-Ph), 127.0 (s, 4-C), 127.0 (m, m-C P-Ph), 124.2 (s, 3-C), 122.5 (d, 3JPC = 4.6 Hz, 5-C), −1.0 (s, 10-Si-CH31), −1.1 (s, 10-Si-CH32), −2.6 (d br., 3JPC = 6.0 Hz, 8-Si-CH31), −3.0 (d br., 3JPC = 6.0 Hz, 8-Si-CH32). 29Si{1H} NMR (CDCl3, 300 K, 79 MHz): δ −4.5 (overlap, m, 8-Si), −10.7 (overlap, m, 10-Si). 31P{1H} NMR (CDCl3, 300 K, 162 MHz): δ −20.6 (dd, 1JPP = 354.3 Hz, PC4 heterocycle), −77.6 (d, 1JPP = 354.3 Hz, P-Ph). IR (KBr, cm−1): 3063 w, 3044 w, 3021 w, 3011 w, 2955 m, 2899 vw ν(CH); 1958 vw, 1931 vw, 1896 vw, 1877 vw, 1813 vw, 1769 vw, 1653 vw, 1584 vw, 1564 vw, 1522 w ν(CC), phenyl; 1489 w, 1429 m, 1404 w, 1331 vw, 1296 w, 1246 s δ(CH); 1186 vw, 1159 vw, 1111 s, 1042 m, 995 w, 953 s, 883 vs, 833 vs, 806 vs, 779 vs, 735 vs ν(CC), ρ(SiCH3); 698 vs, 656 m, 623 w ν(SiC), phenyl; 590 vw, 527 m, 500 w, 484 vw, 461 m, 434 vw ν(PC), δ(CC). MS (EI, 20 eV, 353 K): m/z (%) = 534 (100) [M]+, 135 (32) [SiMe2Ph]+. Anal. calcd for C32H32P2Si2 (534.7): C, 71.9, H, 6.0. Found: C, 71.4; H, 6.2.
Synthesis of the Cr complex 8
[Cr(CO)6] (0.19 g, 0.86 mmol) was dissolved in 10 mL of THF and irradiated by a mercury vapor lamp for 6 h. The orange solution was added to neat 6 (0.34 g, 0.70 mmol) at room temperature, and the mixture was stirred overnight. All volatiles were removed in vacuo at 40 °C. The residue was treated with 10 mL of n-hexane. The suspension was filtered. The product crystallised from the concentrated filtrate at room temperature (0.39 g, 82%). Mp: 110 °C. The assignment of NMR resonances is based on the numbering scheme given in Scheme 3. 1H NMR (CDCl3, 300 K, 400 MHz): δ 7.69 (d, 4JPH = 2.1 Hz, 1H, 7-H), 7.48 (m, 2H, o-H 10-Si-Ph), 7.40 (d, 3JPH = 29.3 Hz, 1H, 9-H), 7.37 (m, 2H, o-H 8-Si-Ph), 7.15 (overlap, m, 4H, m-H 8- and 10-Si-Ph), 7.14 (overlap, m, 2H, p-H 8- and 10-Si-Ph), 7.04 (d, 3JHH = 7.5 Hz, 1H, 3-H), 6.98 (t, 3JHH = 7.5 Hz, 1H, 4-H), 6.68 (dd, 3JHH = 7.4 Hz, 4JPH = 4.4 Hz, 1H, 5-H), 0.61 (s, 6H, 10-Si-CH3), 0.50 (s, 3H, 8-Si-CH31), 0.45 (s, 3H, 8-Si-CH32), 0.40 (d, 3JPH = 8.6 Hz, P–Si–CH31), −0.71 (d, 3JPH = 4.6 Hz, P–Si–CH32). 13C{1H} NMR (CDCl3, 300 K, 101 MHz): δ 222.0 (d, 2JPC = 6.6 Hz, Cr–CO axial), 217.3 (d, 3JPC = 10.0 Hz, Cr–CO equatorial), 155.8 (d, 3JPC = 2.0 Hz, 7-C), 149.8 (d, 1JPC = 16.8 Hz, 10-C), 149.5 (d, 2JPC = 2.9 Hz, 9-C), 147.4 (d, 1JPC = 24.2 Hz, 1-C), 145.5 (d, 3JPC = 15.8 Hz, 2-C), 138.3 (d, 2JPC = 5.0 Hz, 6-C), 138.0 (s, i-C 8-Si-Ph), 137.5 (d, 3JPC = 2.7 Hz, i-C 10-Si-Ph), 137.0 (d, 2JCP = 7.9 Hz, 8-C), 134.6 (s, o-C 8-Si-Ph), 134.4 (s, o-C 10-Si-Ph), 129.8 (s, p-C 10-Si-Ph), 129.7 (s, p-C 8-Si-Ph), 129.5 (s, 4-C), 128.4 (s, m-C 10-Si-Ph), 128.3 (s, m-C 8-Si-Ph), 125.2 (d, 3JPC = 5.8 Hz, 5-C), 124.9 (d, 3JPC = 4.6 Hz, 3-C), −0.9 (s, 10-Si-CH31), −1.2 (s, 8-Si-CH32), −1.5 (d, 3JPC = 2.6 Hz, 10-Si-CH32), −1.7 (s, 8-Si-CH31), −2.5 (d, 2JPC = 15.3 Hz, P–Si–CH31), −4.3 (d, 2JPC = 1.1 Hz, P–Si–CH32). 29Si{1H} NMR (CDCl3, 300 K, 79 MHz): δ 5.2 (d, 1JPSi = 13.5 Hz, P–Si), −4.4 (d, 3JPSi = 7.3 Hz, 8-Si), −10.7 (d, 2JPSi = 22.3 Hz, 10-Si). 31P{1H} NMR (CDCl3, 300 K, 162 MHz): δ −12.2 (s). IR (KBr, cm−1): 3048 vw, 2961 w ν(CH); 2058 s, 1981 m, 1931 vs, 1906 vs ν(CO); 1522 w ν(CC), phenyl; 1445 vw, 1423 w, 1406 w, 1306 w, 1248 m δ(CH); 1161 vw, 1109 w, 1036 w, 951 w, 901 m, 833 m, 810 m, 777 m, 731 w ν(CC), ρ(SiCH3); 698 m, 656 s ν(SiC), phenyl; 527 vw, 513 vw, 455 w ν(PC), δ(CC). MS (EI, 20 eV, 353 K): m/z (%) = 676 (1) [M]+, 620 (2) [M − 2CO]+, 592 (4) [M − 3CO]+, 536 (29) [M − 5CO]+, 484 (100) [6].+ Anal. calcd for C33H33O5PSi3Cr (676.9): C, 58.6, H, 4.9. Found: C, 58.4; H, 4.8.
Synthesis of the W complex of 9
[W(CO)6] (0.17 g, 0.45 mmol) was dissolved in 10 mL of THF and irradiated with a mercury vapor lamp for 7 h. The orange solution was added to neat 7 (0.17 g, 0.32 mmol) at room temperature. The mixture was stirred overnight. All volatiles were removed at 40 °C in vacuo, and the crude product was purified by column chromatography (SiO2; n-hexane/Et2O 8:1). The solvents were removed in vacuo, and the product was recrystallised from a saturated mixture in n-hexane and Et2O at room temperature (0.15 g, 55%). Mp: 160 °C. 1H NMR (CDCl3, 300 K, 400 MHz): δ 7.80 (dd, 3JPH = 18.2 Hz, 4JPH = 1.5 Hz, 1H, 7-H), 7.44 (overlap, m, 3H, 4-H and o-H 10-Si-Ph), 7.43 (overlap, m, 3-H), 7.30 (overlap, m, 2H, p-H 8- and 10-Si-Ph), 7.23 (overlap, m, 5H, 5-H and m-H 8- and 10-Si-Ph), 7.19 (overlap, d, 3JPH = 35.0 Hz, 1H, 9-H), 7.17 (overlap, m, 2H, o-H 8-Si-Ph), 7.08 (t, 3JHH = 7.4 Hz, 1H, p-H P-Ph), 6.81 (pseudo-t, 3JHH = 7.5 Hz, 2H, m-H P-Ph), 6.44 (dd, 3JPH = 9.0 Hz, 3JHH = 7.6 Hz, 2H o-H P-Ph), 0.69 (s, 3H, 10-Si-CH31), 0.63 (s, 3H, 8-Si-CH31), 0.51 (s, 3H, 10-Si-CH32), 0.43 (s, 3H, 8-Si-CH32). 13C{1H} NMR (CDCl3, 300 K, 101 MHz): δ 198.0 (dd, 2JPC = 19.7 Hz, 3JPC = 2.1 Hz, W–CO axial), 196.5 (dd, 2JPC = 6.3 Hz, 3JPC = 2.6 Hz, W–CO equatorial), 150.8 (s, 9-C), 147.6 (dd, 2JPC = 13.7 Hz, 3JPC = 6.0 Hz, 7-C), 147.0 (dd, 1JPC = 13.3 Hz, 2JPC = 4.5 Hz, 10-C), 143.5 (dd, 2JPC = 18.2 Hz, 3JPC = 2.0 Hz, 2-C), 138.7 (dd, 1JPC = 30.5 Hz, 2JPC = 3.6 Hz, 1-C), 137.2 (d, 3JPC = 3.7 Hz, i-C 10-Si-Ph), 135.7 (d, 3JPC = 4.1 Hz, i-C 8-Si-Ph), 135.5 (dd, 2JPC = 8.5 Hz, 3JPC = 3.7 Hz, 6-C), 134.4 (s, o-C 10-Si-Ph), 134.0 (s, o-C 8-Si-Ph), 132.6 (dd, 2JPC = 22.8 Hz, 3JPC = 4.3 Hz, o-C P-Ph), 132.2 (dd, 1JPC = 63.4 Hz, 2JPC = 4.1 Hz, 8-C), 130.6 (dd, 1JPC = 34.4 Hz, 2JPC = 5.8 Hz, i-C P-Ph), 130.3 (s, 4-C), 129.9 (s, p-C P-Ph), 129.5 (s, p-C 10-Si-Ph), 129.2 (s, p-C 8-Si-Ph), 128.1 (dd, 3JPC = 7.8 Hz, 4JPC = 1.5 Hz, m-C P-Ph), 127.8 (s, m-C 10-Si-Ph), 127.7 (s, m-C 8-Si-Ph), 125.5 (d, 3JPC = 5.4 Hz, 3-C), 124.8 (d, 3JPC = 6.6 Hz, 5-C), −0.3 (s, 10-Si-CH32), −1.1 (s, 10-Si-CH31), −2.0 (d, 3JPC = 5.2 Hz, 8-Si-CH31), −2.7 (d, 3JPC = 5.0 Hz, 8-Si-CH32). 29Si{1H} NMR (CDCl3, 300 K, 79 MHz): δ −2.6 (dd, 2JPSi = 44.7 Hz, 3JPSi = 12.3 Hz, 8-Si), −10.2 (d, 2JPSi = 21.3 Hz, 10-Si). 31P{1H} NMR (CDCl3, 300 K, 162 MHz): δ −11.9 (d, 1JPP = 313.8 Hz, 1JWP = 211.3 Hz, PC4 heterocycle, −65.7 (d, 1JPP = 313.8 Hz, P-Ph). IR (KBr, cm−1): 3065 vw, 2961 vw ν(CH); 2068 m, 1981 w, 1948 vs, 1923 sh, 1911 vs ν(CO); 1659 vw, 1562 vw ν(CC, phenyl; 1425 vw, 1300 vw, 1248 w δ(CH); 1157 vw, 1111 w, 1038 vw, 999 vw, 959 vw, 876 w, 833 w, 810 w, 779 w, 729 w ν(CC), ρ(SiCH3); 694 w, 652 vw ν(SiC), phenyl; 594 w, 577 w, 519 w, 463 vw ν(PC), δ(CC). MS (EI, 20 eV, 403 K): m/z (%) = 858 (1) [M]+, 830 (62) [M − CO]+, 802 (5) [M − 2CO]+, 774 (50) [M − 3CO]+, 746 (46) [M − 4CO]+, 718 (58) [M − 5CO]+, 534 (100) [7]+. Anal. calcd for C37H32O5P2Si2W (858.6): C, 51.8, H, 3.8. Found: C, 51.6; H, 3.8.
Synthesis of 10
3a (0.14 g, 0.33 mmol) was dissolved in 20 mL of n-pentane and cooled to −78 °C. A solution of H2O2 in H2O (35.5%, 28 μL, 0.32 mmol) was added. The mixture was slowly warmed to room temperature overnight. All volatiles were removed in vacuo, and the residue was dissolved in a mixture of dichloromethane and diethyl ether. The solution was concentrated and cooled to 7 °C to afford 10 as colourless crystals (0.12 g, 79%). Mp: 124 °C. 1H NMR (C6D6, 300 K, 400 MHz): δ 12.55 (s br., 1H, P–OH), 8.15 (d, 3JHH = 18.9 Hz, 1H, 7-H), 7.61 (m, 2H, o-H 10-Si-Ph), 7.54 (m, 2H, o-H 8-Si-Ph), 7.33 (dd, 3JHH = 8.0 Hz, 4JPH = 5.4 Hz, 1H, 5-H), 7.23 (overlap, m, 3H, m-H and p-H 10-Si-Ph), 7.18 (overlap, m, 3H, m-H and p-H 8-Si-Ph), 6.93 (d, 3JPH = 51.6 Hz, 1H, 9-H), 6.90 (td, 3JHH = 7.6 Hz, 5JPH = 1.3 Hz, 1H, 4-H), 6.75 (d, 3JHH = 18.9 Hz, 1H, 8-H), 6.41 (dd, 3JHH = 7.3 Hz, 4JPH = 2.9 Hz, 3-H), 0.72 (s, 6H, 10-Si-CH3), 0.46 (s, 6H, 8-Si-CH3). 13C{1H} NMR (C6D6, 300 K, 101 MHz): δ 153.0 (d, 2JPC = 13.1 Hz, 9-C), 141.6 (d, 2JPC = 45.3 Hz, 2-C), 141.6 (d, 3JPC = 5.4 Hz, 7-C), 140.6 (d, 2JPC = 8.4 Hz, 6-C), 139.1 (s, i-C 8-Si-Ph), 137.0 (d, 3JPC = 4.3 Hz, i-C 10-Si-Ph), 136.3 (d, 1JPC = 89.6 Hz, 10-C), 134.7 (s, o-C 10-Si-Ph), 134.3 (s, o-C 8-Si-Ph), 133.6 (s, 8-C), 133.0 (d, 4JPC = 1.9 Hz, 4-C), 129.7 (s, p-C 10-Si-Ph), 129.2 (s, p-C 8-Si-Ph), 128.7 (overlap, d, 1JPC = 125.5 Hz, 1-C), 128.3 (overlap, s, m-C 10-Si-Ph), 128.1 (s, m-C 8-Si-Ph), 126.3 (d, 3JPC = 8.8 Hz, 5-C), 124.1 (d, 3JPC = 13.2 Hz, 3-C). 29Si{1H} NMR (C6D6, 300 K, 79 MHz): δ −10.2 (d, 2JPSi = 14.8 Hz, 10-Si), −10.5 (s, 8-Si). 31P (1H) NMR (C6D6, 300 K, 162 MHz): δ 62.8 (s). IR (KBr, cm−1): 3431 vw ν(OH); 3162 vw, 3065 m, 3049 m, 3011 w, 2955 m, 2895 w ν(CH); 2583 vw, 2286 w, 2156 vw; 1954 vw, 1883 vw, 1819 vw, 1794 vw, 1769 vw, 1651 w, 1614 w, 1591 w, 1562 m, 1539 s ν(CC), phenyl; 1485 w, 1456 s, 1425 s, 1412 m, 1379 vw, 1333 vw, 1317 w, 1261 vs, 1250 vs δ(CH); 1219 w, 1204 m, 1188 w, 1152 s, 1113 vs, 1045 w, 1007 vs, 991 vs, 953 vs, 837 vs, 810 vs, 804 vs, 779 vs, 737 vs, 700 vs ν(CC), ρ(SiCH3), δ(OH); 677 m, 658 m, 635 m, 611 m ν(SiC), phenyl; 575 m, 540 w, 511 vs, 469 s, 430 w ν(PC), ν(PO), δ(CC). MS (EI, 20 EV, 393 K): m/z (%) = 459 (47) [M − H]+, 382 (100) [M − Ph − H]+, 367 (20) [M − CH3 − Ph − H]+. Anal. calcd for C26H29O2PSi2 (460.7): C, 67.8, H, 6.3. Found: C, 67.6; H, 6.2.
X-ray crystallography
Crystals suitable for X-ray crystallography were obtained by crystallisation from toluene (5, 3 °C), n-hexane (7, room temperature), a mixture of diethyl ether/n-hexane (9, room temperature) and CH2Cl2/Et2O (10, 7 °C). Intensity data was collected on a Bruker D8 Venture diffractometer with monochromated Mo Kα radiation. The collection method involved ω-scans. Data reduction was carried out using the program SAINT+.41 The crystal structures were solved by direct methods using SHELXTL.42 Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculation based on F2 using SHELXTL.42 H atoms were positioned geometrically and allowed to ride on their respective parent atoms. 5 crystallizes with two molecules of toluene per dimer. The P2C4 rings of 7 were disordered; the atoms of the molecular centre were refined on split positions (0.72:0.28). A further minor disorder led to the superposition of PC4 and P2C4 rings; the P atoms of the minor component were refined with site occupancy factors of 0.036 and 0.023. The crystals of 10 enclosed a molecule of CH2Cl2; its Cl atoms were refined on split positions (0.66:0.34).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (IRTG-2027) and the European Regional Development Fond, the Free State of Saxony (ERDF-InfraPro, GEPARD-100326379) is gratefully acknowledged.Notes and references
- Review: A. Brand and W. Uhl, Chem.–Eur. J., 2019, 25, 1391–1404 CrossRef CAS.
- (a) A. J. Arduengo III, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef; (b) S. A. Culley and A. J. Arduengo III, J. Am. Chem. Soc., 1984, 106, 1164–1165 CrossRef CAS.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., 2015, 127, 13962–13967 ( Angew. Chem Int. Ed. , 2015 , 54 , 13758–13763 ) CrossRef.
- W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS.
- (a) G. Baccolini, E. Mezzina, P. E. Todesco and E. Foresti, J. Chem. Soc., Chem. Commun., 1988, 304–305 RSC; (b) G. Baccolini, E. Mezzina and P. E. Todesco, J. Chem. Soc., Perkin Trans. I, 1988, 3281–3283 RSC; (c) G. Baccolini, S. A. Mosticchio, E. Mezzina, C. Rizzoli and P. Sgarabotto, Heteroatom Chem, 1993, 4, 319–322 CrossRef CAS.
- A. Hentschel, A. Brand, P. Wegener and W. Uhl, Angew. Chem., 2018, 130, 840–843 ( Angew. Chem., Int. Ed. , 2018 , 57 , 832–835 ) CrossRef.
- A. Brand, A. Hentschel, A. Hepp and W. Uhl, Eur. J. Inorg. Chem., 2020, 361–369 CrossRef CAS.
- W. Uhl, D. Kovert, M. Layh and A. Hepp, Chem.–Eur. J., 2011, 17, 13553–13561 CrossRef CAS.
- (a) T. R. Robinson, D. De Rosa, S. Aldridge and J. M. Goicoechea, Chem.–Eur. J., 2017, 23, 15455–15465 CrossRef CAS; (b) A. J. Arduengo III, J. Breker, F. Davidson and M. Kline, Heteroatom Chem, 1993, 4, 213–221 CrossRef.
- S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 135, 4640–4650 CrossRef.
- J. Cui, Y. Li, R. Ganguly and R. Kinjo, Chem.–Eur. J., 2016, 22, 9976–9985 CrossRef CAS.
- A. Brand, P. Wegener, A. Hepp and W. Uhl, Organometallics, 2020, 39, 1384–1392 CrossRef CAS.
- A. J. Pistner, H. W. Moon, A. Silakov, H. H. Yennawar and A. T. Radosevich, Inorg. Chem., 2017, 56, 8661–8668 CrossRef CAS.
- K. Schwedtmann, S. Schulz, F. Hennerdorf, T. Strassner, E. Dmitrieva and J. J. Weigand, Angew. Chem., 2015, 54, 11054–11058 CrossRef CAS.
- S. Y. S. Schulz, F. Hennersdorf, S. Kumar, R. Fischer and J. J. Weigand, Organometallics, 2018, 37, 748–754 CrossRef.
- Half-wave potential determinated from square-wave voltammetry because of the non-reversible CV.
- D. Gudat, M. Schrott, A. W. Holderbger, V. Bajorat and M. Nieger, Phosphorus, Sulfur Silicon Relat. Elem., 1999, 144, 457–460 CrossRef.
- B. Jaun, J. Schwarz and R. Breslow, J. Am. Chem. Soc., 1980, 102, 5741–5748 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- Few recent examples: (a) K. Izod, P. Evans and P. G. Waddell, Dalton Trans., 2017, 46, 13824–13834 RSC; (b) K. Izod, J. Stewart, E. R. Clark, W. Clegg and R. W. Harrington, Inorg. Chem., 2010, 49, 4698–4707 CrossRef CAS.
- H. A. Bent, Chem. Rev., 1961, 61, 275 CrossRef CAS.
- (a) S. Du, W.-X. Zhang and Z. Xi, Organometallics, 2018, 37, 2018–2022 CrossRef CAS; (b) L. Xu, Y. Chi, S. Du, W.-X. Zhang and Z. Xi, Angew. Chem., Int. Ed., 2016, 55, 9187–9190 CrossRef CAS; (c) T. Douglas and K. H. Theopold, Angew. Chem., Int. Ed., 1989, 28, 1367–1368 CrossRef; (d) F. Paul, D. Carmichael, L. Ricard and F. Mathey, Angew. Chem., Int. Ed., 1996, 35, 1125–1127 CrossRef CAS; (e) J. Chojnacki, E. Baum, I. Krossing, D. Carmichael, F. Mathey and H. Schnöckel, Z. Anorg. Allg. Chem., 2001, 627, 1209–1212 CrossRef CAS; (f) K. Wimmer, C. Birg, R. Kretschmer, T. M. A. Al-Shboul, H. Görls, S. Krieck and M. Westerhausen, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 1360–1368 CAS; (g) F. Nief and L. Ricard, J. Organomet. Chem., 2002, 642, 208–211 CrossRef CAS; (h) M. Westerhausen, M. W. Oßberger, A. Keilbach, C. Gückel, H. Piotrowski, M. Suter and H. Nöth, Z. Anorg. Allg. Chem., 2003, 629, 2398–2407 CrossRef CAS; (i) M. Westerhausen, C. Gückel, H. Piotrowski, P. Mayer, M. Warchold and H. Nöth, Z. Anorg. Allg. Chem., 2001, 627, 1741–1750 CrossRef CAS; dibenzophospholyl: (j) A. Decken, M. A. Neil and F. Bottomley, Can. J. Chem., 2001, 79, 1321–1329 CrossRef CAS; (k) P. von Rague Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikemma Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef; studies into the aromaticity of phospholyl anions: (l) A. Dransfeld, L. Nyulaszi and P. von Rague Schleyer, Inorg. Chem., 1998, 37, 4413–4420 CrossRef CAS; (m) L. Nyulaszy, Chem. Rev., 2001, 101, 1229–1246 CrossRef.
- A dilithium compound was postulated based on NMR experiments: D. Gudat, V. Bajorat, S. Häp, M. Nieger and G. Schröder, Eur. J. Inorg. Chem., 1999, 1169–1174 CrossRef CAS.
- (a) E. Niecke, M. Nieger and P. Wenderoth, Angew. Chem., Int. Ed., 1994, 33, 353–354 CrossRef; (b) M. Ogasawara, S. Arae, S. Watanabe, V. Subbarayan, H. Sato and T. Takahashi, Organometallics, 2013, 32, 4997–5000 CrossRef CAS.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed; (c) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS; (d) J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS; (e) C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS; (f) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC; (g) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104-1–154104-19 CrossRef; (h) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS; (i) G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13, 554–562 CrossRef CAS.
- Few examples: (a) K. Izod, P. Evans and P. G. Waddell, Dalton Trans., 2017, 46, 13824–13834 RSC; (b) C. Appelt, J. C. Slootweg, K. Lammertsma and W. Uhl, Angew. Chem., Int. Ed., 2012, 51, 5911–5914 CrossRef CAS; (c) W. Weng, L. Yang, B. M. Foxman and O. V. Ozerov, Organometallics, 2004, 23, 4700–4705 CrossRef CAS; (d) M. Winkler, M. Lutz and G. Müller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2279–2281 CrossRef; (e) J. Langer, K. Wimmer, H. Görls and M. Westerhausen, Dalton Trans., 2009, 2951–2957 RSC; (f) A. Pape, M. Lutz and G. Müller, Angew. Chem., Int. Ed. Engl., 1994, 33, 2281–2283 CrossRef; (g) P. B. Hitchcock, M. F. Lappert, P. P. Power and S. J. Smith, J. Chem. Soc., Chem. Commun., 1984, 1669–1670 RSC; (h) R. A. Jones, A. L. Stuart and T. C. Wright, J. Am. Chem. Soc., 1983, 105, 7459–7450 CrossRef CAS.
- (a) J. Dogan, J. B. Schulte, G. F. Swiegers and S. B. Wild, J. Org. Chem., 2000, 65, 951–957 CrossRef CAS; (b) V. L. Blair, M. A. Stevens and C. D. Thompson, Chem. Commun., 2016, 52, 8111–8114 RSC; (c) X. Yu, Y. Li, H. Fu, X. Zheng, H. Chen and R. Li, Appl. Organomet. Chem., 2018, 32, e4277 CrossRef; (d) G. W. Luther III, G. Beyerle, D. Cox and K. Cohn, Inorg. Synth., 1977, 17, 186–188 Search PubMed.
- (a) Z. Hu, Z. Li, K. Zhao, R. Tian, Z. Duan and F. Mathey, Org. Lett., 2015, 17, 3518–3520 CrossRef CAS; (b) N. Szynkiewicz, Ł. Ponikiewski and R. Grubba, Dalton Trans., 2018, 47, 16885–16894 RSC.
- (a) A. S. Ionkin, W. J. Marshall, B. M. Fish, M. F. Schiffhauer and C. N. McEwen, Chem. Commun., 2008, 5432–5434 RSC; (b) S. L. Hinchley, C. A. Morrison, D. W. H. Rankin, C. L. B. Macdonald, R. J. Wiacek, A. H. Cowley, M. F. Lappert, G. Gundersen, J. A. C. Clyburne and P. P. Power, Chem. Commun., 2000, 2045–2046 RSC; (c) R. W. Alder, C. Ganter, C. J. Harris and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1992, 1170–1172 RSC; (d) M. Baba and T. Mizuta, Polyhedron, 2015, 92, 30–36 CrossRef CAS; (e) C. Fave, M. Hissler, T. Kárpáti, J. Rault-Berthelot, V. Deborde, L. Toupet, L. Nyulászi and R. Réau, J. Am. Chem. Soc., 2004, 126, 6058–6063 CrossRef CAS; (f) J. Fischer, A. Mitschler, F. Mathey and F. Mercier, J. Chem. Soc., Dalton Trans., 1983, 841–845 RSC; (g) T. Mizuta, T. Nakazono and K. Miyoshi, Angew. Chem., Int. Ed., 2002, 41, 3897–3998 CrossRef CAS; (h) D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS; (i) S. Wang, K. Samedov, S. C. Serin and D. P. Gates, Eur. J. Inorg. Chem., 2016, 4144–4151 CrossRef CAS; (j) S. L. Hinchley, H. E. Robertson, K. B. Borisenko, A. R. Turner, B. F. Johnston, D. W. H. Rankin, M. Ahmadian, J. N. Jones and A. H. Cowley, Dalton Trans., 2004, 2469–2476 RSC; (k) A. Dashti-Mommertz and B. Neumüller, Z. Anorg. Allg. Chem., 1999, 625, 954–960 CrossRef CAS; (l) C. J. Harlan, R. A. Jones, S. U. Koschmieder and C. M. Nunn, Polyhedron, 1990, 9, 669–679 CrossRef CAS.
- (a) N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS; (b) K. M. Doxsee, N. P. Wood, E. M. Hanawalt and T. J. R. Weakley, Heteroatom Chem, 1996, 7, 383–389 CrossRef CAS; (c) H. Klöcker, J. C. Tendyck, L. Keweloh, A. Hepp and W. Uhl, Chem.–Eur. J., 2019, 25, 4793–4807 CrossRef; (d) A. Schnepf, G. Stößer, D. Carmichael, F. Mathey and H. Schnöckel, Angew. Chem., Int. Ed., 1999, 38, 1646–1649 CrossRef CAS; (e) A. Zagidullin, V. Miluykov, O. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Heteroatom Chem, 2014, 25, 28–34 CrossRef CAS.
- (a) J. Hilton and L. H. Sutcliffe, Prog. Nucl. Mag. Res. Spectr., 1975, 10, 27–39 CrossRef; (b) T. Tuttle, J. Gräfenstein and D. Cremer, Chem. Phys. Lett., 2004, 394, 5–13 CrossRef CAS.
- (a) B. Hoge, T. Herrmann, C. Thösen and I. Pantenberg, Inorg. Chem., 2003, 42, 3623–3632 CrossRef CAS; (b) J. Wong, Y. Li, Y. Hao, R. Tian and F. Mathey, Angew. Chem., Int. Ed., 2015, 54, 12891–12893 CrossRef CAS; (c) A. R. Kenaree, E. R. Sauvé, P. J. Ragogna and J. B. Gilroy, Dalton Trans., 2016, 45, 2859–2867 RSC; (d) J. H. Reibenspies, D. Darensbourg and E. Atnip, Z. Kristallogr. - Cryst. Mater., 1994, 209, 759–760 CAS; (e) B. Ndiaye, S. Bhat, A. Jouaiti, T. Berclaz, G. Bernadinelli and M. Geoffroy, J. Phys. Chem. A, 2006, 110, 9736–9742 CrossRef CAS; (f) X. Zhao, Z. Lu, Q. Wang, D. Wei, Y. Lu, Z. Duan and F. Mathey, Organometallics, 2016, 35, 3440–3443 CrossRef CAS; (g) P. G. Jones, C. Wismach, W.-W. du Mont and G. Dobreva, CSD communication 2015 Search PubMed.
- (a) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 CrossRef CAS; (c) https://www.molnac.unisa.it/OMtools/sambvca2.0/index.html .
- (a) B. Borup, J. C. Huffman and K. G. Caulton, J. Organomet. Chem., 1997, 536–537, 109–113 CrossRef; (b) A. Bowden, S. J. Coles, M. B. Pitak and A. W. G. Platt, Polyhedron, 2014, 68, 258–264 CrossRef CAS; (c) M. Gonschorowsky, K. Merz and M. Driess, Eur. J. Inorg. Chem., 2006, 455–463 CrossRef CAS; (d) M. Bendle, K. Huynh, M. F. Haddow and I. Manners, Inorg. Chem., 2011, 50, 10292–10302 CrossRef CAS; mechanism: (e) A. Chellmani and R. Suresh, React. Kinet. Catal. Lett., 1988, 37, 501–505 CrossRef.
- L. Dubován, A. Pöllnitz and C. Silvestru, Eur. J. Inorg. Chem., 2016, 1521–1527 CrossRef.
- (a) T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS; (b) G. Desiraju, Acc. Chem. Res., 1991, 24, 290–296 CrossRef CAS; (c) G. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS; (d) T. Steiner, Cryst. Rev., 1996, 6, 1–57 CrossRef CAS.
- Few recently published structures of phosphinic acids: (a) V. L. Capel, K. B. Dillon, A. E. Goeta, J. A. K. Howard, P. K. Monks, M. R. Probert, H. J. Shepherd and N. V. Zorina, Dalton Trans., 2011, 40, 1808–1816 RSC; (b) N. V. Dubrovina, H. Jiao, V. I. Tararov, A. Spannenberg, R. Kadyrov, A. Monsees, A. Christiansen and A. Börner, Eur. J. Org. Chem., 2006, 3412–3420 CrossRef CAS; (c) R. A. Burrow, D. H. Farrar, A. J. Lough, M. R. Siqueira and F. Squizani, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, e357–e358 CrossRef; (d) E. V. Goud, D. Das, A. Sivaramakrishna, K. Vijayakrishna, V. Sabareesh, G. Gopakumar, C. V. S. B. Rao, M. Y. Lone and P. C. Jha, Polyhedron, 2016, 117, 741–748 CrossRef CAS; (e) L. Feng, J. S. Maass and R. L. Luck, Inorg. Chim. Acta, 2011, 373, 85–92 CrossRef CAS; (f) R. P. Shekurov, V. A. Miluykov, D. R. Islamov, D. B. Krivolapov, O. N. Kataeva, T. P. Gerasimova, S. A. Katsyuba, G. R. Nasybullina, V. V. Yanilkin and O. G. Sinyashin, J. Organomet. Chem., 2014, 766, 40–48 CrossRef CAS; (g) M. Granitzka, P. Stollberg and D. Stalke, Z. Naturforsch., B: J. Chem. Sci., 2014, 69, 1429–1440 CAS; (h) D. A. Tatarinov, M. V. Kundina, A. B. Dobrynin and V. F. Mironov, Russ. J. Gen. Chem., 2018, 88, 90–95 CrossRef CAS; (i) P. V. Ioannou, Monatsh. Chem., 2012, 143, 1349–1356 CrossRef CAS; (j) S. Chatterjee, L. Hintermann, M. Mandal, A. Achari, S. Gupta and P. Jaisankar, Org. Lett., 2017, 19, 3426–3429 CrossRef CAS.
- (a) W. Strohmeier and K. Gerlach, Chem. Ber., 1961, 94, 398–406 CrossRef CAS; (b) F. E. Hahn and M. Tamm, J. Organomet. Chem., 1993, 456, C11–C14 CrossRef CAS.
- G. Gritzner and J. Kuta, Pure Appl. Chem., 1984, 56, 461–466 Search PubMed.
- (a) Saint+, Version 6.02 (includes Xprep and Sadabs), Bruker AXS INC., Madison, Wisconsin, USA, 1999 Search PubMed; (b) G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- (a) Shelxtl-Plus, REL. 4.1, Siemens Analytical X-RAY Instruments Inc., Madison, WI, 1990 Search PubMed; (b) G. M. Sheldrick, SHEXL-97 and SHELXL-2013, Program for the Refinement of Structures; Universität Göttingen, Germany, 1997 and 2013 Search PubMed; (c) G. M. Sheldrick, Acta Cryst, 2008, A64, 112–122 CrossRef; (d) G. M. Sheldrick, Acta Cryst, 2015, C71, 3–8 Search PubMed.
Footnotes |
| † In Memorial of François Mathey. |
| ‡ Electronic supplementary information (ESI) available: Additional electrochemical and spectroelectrochemical measurements of phosphine 3a NMR spectra of all compounds and results of quantum chemical calculations for 7. CCDC 2014280 (5), 2014281 (7), 2014283 (9) and 2014282 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06155g |
| § Both authors contributed equally and share first authorship. |
| This journal is © The Royal Society of Chemistry 2021 |