
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh(I)-catalyzed stereoselective desymmetrization of prochiral cyclohexadienones via highly exo-selective Huisgen-type [3 + 2] cycloaddition†‡
Krishna Kumar
Gollapelli
ab,
Vaibhav B.
Patil
ab,
Allam
Vinaykumar
ab and
Rambabu
Chegondi
 *ab
*ab
aDepartment of Organic Synthesis and Process Chemistry, CSIR-Indian Institute of Chemical Technology (CSIR-IICT), Hyderabad 500007, India
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201 002, India. E-mail: rchegondi@iict.res.in; Web: https://cramhcu.wixsite.com/rambabu-chegondi
First published on 27th November 2020
Abstract
A Rh(I)-catalyzed highly stereoselective desymmetrization of 2-alkynylbenzaldehyde-tethered cyclohexadienones triggered by intramolecular Huisgen-type [3 + 2] cycloaddition has been developed. This method enables convergent construction of complex epoxy-bridged polycyclic ring systems with five contiguous stereocenters with excellent exo-selectivity and broad substrate scope. The highly atom-economical process involves 6-endo-dig cyclization of carbonyl oxygen onto an activated alkyne resulting in a highly reactive metal–benzopyrylium intermediate, which readily undergoes intramolecular [3 + 2] annulation/hydration. Asymmetric induction is also achieved for the first time in Rh(I)-catalyzed 1,3-dipolar cycloaddition using an easily accessible chiral diene as the ligand.
Introduction
Highly diastereoselective and enantioselective synthesis of polycyclic scaffolds fused with six- or seven-membered carbocycles is of significant interest as a large number of biologically active molecules have this core structure.1,2 Transition-metal-catalyzed electrophilic activation of 2-enynylbenzaldehydes is a powerful and effective strategy for the formation of such complex frameworks via highly reactive metal–benzopyrylium intermediates (Scheme 1a).3,4 This transient species undergoes various [4 + 2] and [3 + 2] cycloadditions with alkenes to form the corresponding products with high skeletal diversity. A very limited number of [3 + 2] cycloaddition reactions are reported and commonly involve electron-rich olefins and platinum-bonded pyrylium ylides.4b–f Interestingly, there is only one report on the Rh(I)-catalyzed intramolecular [3 + 2] cycloaddition of 2-enynebezaldehydes with electron-rich olefins.4g However, an asymmetric version of Huisgen-type [3 + 2] cyclization has not been studied to date. Previously, Padwa et al. reported the formation of highly reactive six-membered carbonyl ylides from the decomposition of relatively stable o-alkyl-2-enoxycarbonyl-α-diazoacetophenones with rhodium(II) catalysts.5 These transient species subsequently undergo facile intramolecular dipolar cycloadditions to afford a similar type of polyoxacyclic skeleton. However, diazo compounds are often prepared from energetic reagents in multiple steps. Another possible mode of cationic Rh(I)-catalysis with 2-alkynylbenzaldehydes is the formation of a five-membered acylmetallacycle which subsequently undergoes various cycloaddition reactions to give six-membered carbocycles (Scheme 1b).6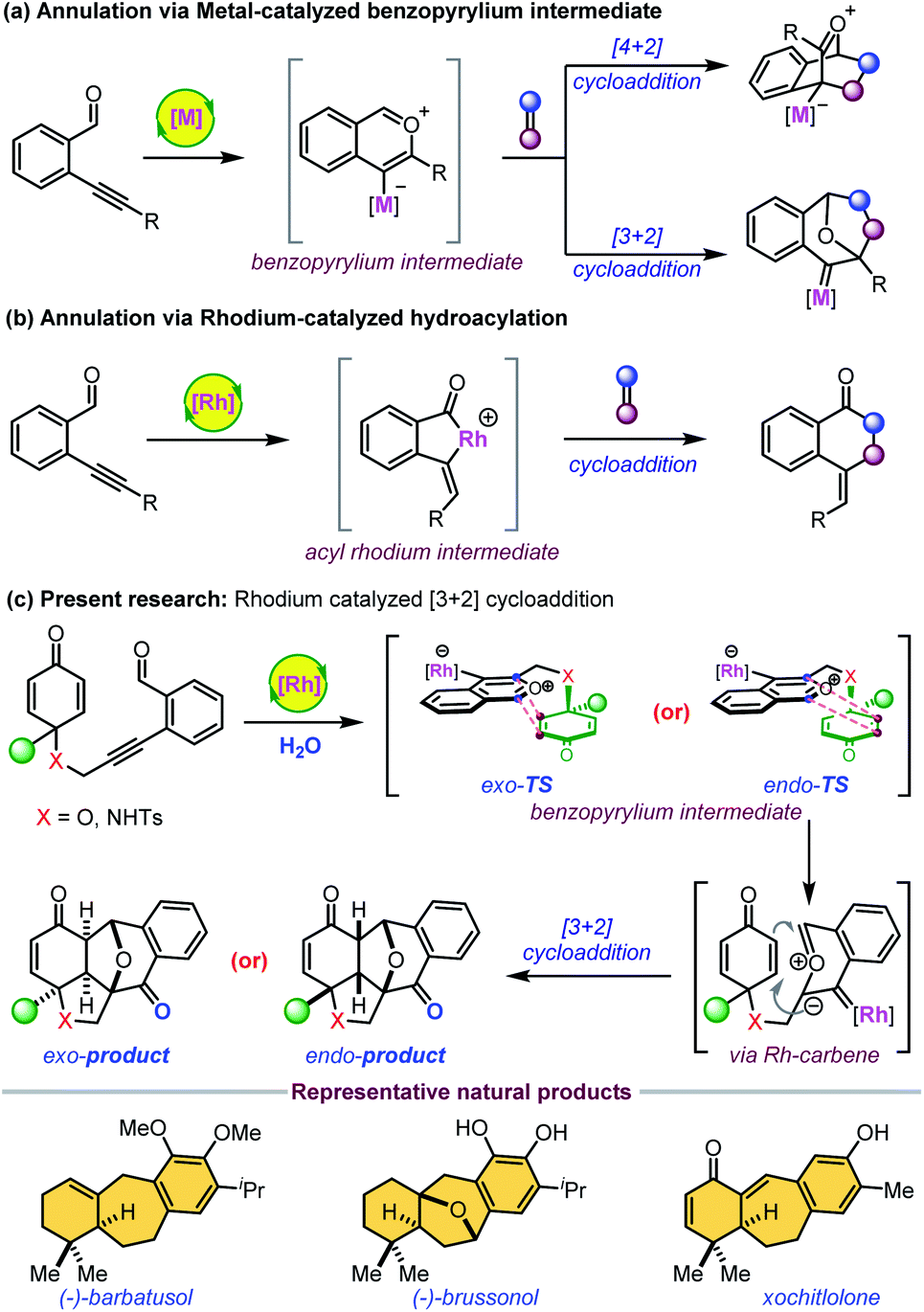 | ||
| Scheme 1 Previous and present reports on Rh(I)-catalysed annulations of ortho-alkynylbenzaldehydes. | ||
In continuation of our efforts in exploring new desymmetrization methods7,8 and the unique reactivity of ortho-alkynylbenzaldehydes,9 we envisaged that the rhodium-catalyzed Huisgen-type [3 + 2] cyclization10 of 2-alkynylbenzaldehyde-tethered cyclohexadienones would provide highly strained complex epoxy bridged polycycles with multiple stereocenters (Scheme 1c). This highly atom-economical reaction would generate seven-membered carbocycles fused with a phenyl ring, which is present in several natural products including barbatusol, pisiferin, brussonoll, and xochitlolone (Scheme 1).11 The starting substrates were easily accessible from dearomatization/Sonogashira coupling of the corresponding phenols in two steps (see the ESI‡). The electrophilic activation of an alkyne with Rh(I) gives a kinetically unstable metal–benzopyrilium intermediate that could undergo intramolecular [3 + 2] cycloaddition with an enone to form the desired product by adopting exo- or endo-configuration via a rhodium–carbene intermediate. The major challenges for this cascade reaction are: (1) the exo- or endo-selectivity of [3 + 2] cycloaddition; (2) competitive intramolecular [4 + 2] cycloaddition through the Rh-benzopyrilium intermediate or acylrhodium intermediate; (3) imparting enantioselectivity to the [3 + 2] cycloaddition.
Results and discussion
Our investigation towards intramolecular cycloaddition began using 2-alkynylbenzaldehyde 1a as the model substrate (Table 1). After extensive optimization of reaction conditions (see the ESI‡), we were delighted to afford the [3 + 2] annulation product exo-2a in 75% yield with complete diastereoselectivity in the presence of 5 mol% [Rh(COD)Cl]2 as the catalyst in H2O solvent at 100 °C for 1 h (Table 1, entry1). The exo-selectivity of [3 + 2]-cycloaddition and the molecular structure of product 2a were confirmed by X-ray crystallographic analysis (Fig. 1).12 The product formation through exo-TS would significantly reduce the steric repulsion between the metal–benzopyrilium intermediate and cyclohexadienone ring.13 Fortunately, we did not observe any [4 + 2] cycloaddition products from either rhodium–benzopyrilium intermediates or acylrhodium intermediates. Overall, this rigid and short tethered intramolecular annulation reaction ruled out the potential competitive endo-selective [3 + 2] and [4 + 2] cycloaddition probably due to such processes producing high ring-strain in the transition states. Using 5% aqueous xylene as a solvent at 120 °C, only 56% yield of 2a was obtained (Table 1, entry 2). The reaction performed in other solvents such as CH3CN, THF, dioxane and t-BuOH returned inferior yields, indicating that H2O is the best solvent for this reaction (entries 3–6). Moreover, reducing the reaction temperature to 80 °C led to a diminished yield of the polycyclic product even with a prolonged reaction time (entry 7). Additional optimization revealed that the reaction with other transition-metal (Rh, Ir and Ru) catalysts also gave desired product 2a in lower yields (entries 8–11).|
|
||
|---|---|---|
| Entry | Deviation from the standard conditions | Yieldc,d [%] |
| a Reaction conditions: 1a (0.3 mmol) and catalyst (5 mol%) in 3 mL of solvent under an inert atmosphere. b All commercial solvents used in the reaction. c Isolated yields of exo-2a. d Observed exclusive exo-selectivity. e 46% yield observed in the presence of commercial xylene. | ||
| 1 | None | 75 |
| 2 | 5% aq. xylene/120 °C instead of H2O/100 °C | 56e |
| 3 | CH3CN/80 °C instead of H2O/100 °C | 21 |
| 4 | THF/70 °C instead of H2O/100 °C | 32 |
| 5 | 1,4-Dioxane/100 °C instead of H2O/100 °C | 25 |
| 6 | t-BuOH/85 °C instead of H2O/100 °C | 31 |
| 7 | 80 °C instead of 100 °C | 48 |
| 8 | Rh(COD)2OTf/xylene instead of [Rh(COD)CI]2 catalyst/H2O | 16 |
| 9 | Rh(COD)2SbF6/xylene instead of [Rh(COD)CI]2 catalyst/H2O | 25 |
| 10 | [Ir(COD)CI]2/xylene instead of [Rh(COD)CI]2 catalyst/H2O | 21 |
| 11 | [Ru(p-cymene)CI2]2/xylene instead of [Rh(COD)CI]2 catalyst/H2O | 12 |
 | ||
| Fig. 1 ORTEP diagram of compound exo-(±)-2a. | ||
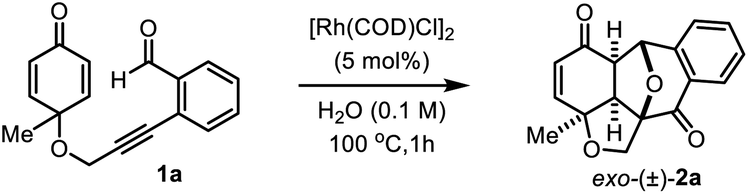
Initially, we explored the scope and limitations of O-tethered-cyclohexadienones under the optimal reaction conditions (Table 2). With substituents such as alkyl, benzyl, phenyl and methoxy groups at the substrate's quaternary carbon center, the reaction proceeded well with moderate to high yields and complete diastereoselectivity (entries 2a–2j). All meta-substituted and para-substituted 2-enynylbenzaldehydes regardless of the electronic effect of substituents at the phenyl ring afforded polycyclic products in very good yields (entries 2k–2s). The reaction of the ortho-fluoro substituted substrate gave the corresponding product 2t in lower yield. The annulation of a substrate containing a sec-butyl group at the cyclohexadienone prochiral center proceeded smoothly to give cycloadduct 2u/2u′ in 64% yield with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of diastereomers with respect to the stereocenter on the sec-butyl group. It is worth mentioning that the reaction with the methyl substituent at the α-position of the dienone also afforded annulation product 2v in 34% yield, but with a sterically more hindered tert-butyl group on the substrate's dienone ring it was unable to participate in the reaction (entry 2w). In addition, substrates 1x with the NHBoc group and o-alkynyl benzoate-tethered cyclohexadienone 1y were found to be inert in the [3 + 2] annulation reaction. Also, the C-tethered substrate 1z under the standard reaction conditions failed to give the desired product probably due to the absence of the Thorpe–Ingold effect.
:1 ratio of diastereomers with respect to the stereocenter on the sec-butyl group. It is worth mentioning that the reaction with the methyl substituent at the α-position of the dienone also afforded annulation product 2v in 34% yield, but with a sterically more hindered tert-butyl group on the substrate's dienone ring it was unable to participate in the reaction (entry 2w). In addition, substrates 1x with the NHBoc group and o-alkynyl benzoate-tethered cyclohexadienone 1y were found to be inert in the [3 + 2] annulation reaction. Also, the C-tethered substrate 1z under the standard reaction conditions failed to give the desired product probably due to the absence of the Thorpe–Ingold effect.

With these promising results in hand, next we sought to examine the reactivity of N-tethered cyclohexadienones (Table 3). The reactions still proceeded equally well with a wide range of NTs-linked 2-alkynylbenzaldehydes substituted with various groups on the phenyl ring (entries 4a–4g). However, substrates with electron-withdrawing substituents on the phenyl ring gave slightly lower yields compared to those with electron-donating substituents. A Boc-protected N-tethered substrate was unable to provide the required product, probably due to intervention from the carbonyl functionality of the Boc group in 6-endo-dig cyclization (entry 4a′). Notably, the phenyl group at the substrate's prochiral centre was also well tolerated in this electrophilic cyclization reaction to give the corresponding product 4h in 67% yield.
The process is not only limited to aryl aldehydes; 2-alkynylphenyl ketones 5 are also well tolerated in the annulation reaction (Table 4). Both alkyl and aryl substituted phenyl ketones smoothly underwent the [3 + 2] cycloaddition reaction irrespective of the electronic nature of substituents on the aryl ring to afford the corresponding products in higher yield with excellent diastereoselectivity (entries 6a–6e). It is important to mention that ketones 5 gave slightly higher yields than aldehyde substrates 1 in the annulation reaction. Significantly, N-tethered alkynylphenyl ketone was also converted to polycyclic enone 6f in 71% yield.

Late-stage functionalization (LSF) is a prevalent concept for the rapid generation of analogues from lead molecules and bioactive natural products. Herein, we successfully demonstrated the synthetic utility of the Rh(I)-catalyzed cascade annulation reaction for the late-stage functionalization of estrone (Table 5). Initially, estrone derivatives 7 (converted from estrone in two steps using a known procedure)14 were subjected to the standard reaction conditions. However, we observed poor reaction yields due to substrate insolubility in the water solvent, related to the highly hydrophobic nature of steroidal compounds. To our delight, the [3 + 2] cyclization of 7 in 5% aqueous xylenes as the solvent underwent smoothly to produce the corresponding polycyclic products 8a–d with five new stereogenic centers in 52–68% yield and excellent diastereoselectivity.
|
a Reaction conditions: 7 (0.2 mmol) in 5% aqueous xylene (2 mL, 0.1 M) under an inert atmosphere at 100 °C for 8 h.
b The diastereomeric ratio of 7a (12:1), 7b (15:1), 7c (9:1) and 7d (15:1) assigned by 1H NMR after column purification.
c Isolated yields of 8.
d The diastereomeric ratio of 8 (dr = >30:1) assigned by 1H NMR analysis of the crude aliquot.
|
|---|

|
Surprisingly, transition-metal catalyzed enantioselective transformations of benzopyryliums are limited and most of them are based on the use of bulky chiral Brønsted acids as a counter anion, presumably due to the planar structure and lack of appropriate coordination sites.15 To the best of our knowledge, so far there is no report on Rh(I)-catalyzed asymmetric [3 + 2] cycloaddition of 2-enynylbenzaldehydes. Encouraged by the results from the diastereoselective annulation reaction, we sought to investigate enantioselective Rh(I)-catalyzed Huisgen-type [3 + 2] cyclization using various chiral ligands (Table 6). Initially, chiral bisphosphine ligands (L1 and L2) were examined for the annulation of model substrate 1a in 5% aqueous xylene at 60 °C (entries 1 and 2). Unfortunately, these bidentate ligands were not effective to promote the asymmetric induction. Next, several varieties of chiral diene ligands (L4–L9)16 were evaluated and promising enantioselectivity was obtained with diene L4 (entry 4, 54% yield, 81:19 er). Additional optimization (entries 10–14) revealed that the reaction in THF as the solvent improved the reaction yield and enantiomeric ratio (entry 11). When the reaction was conducted in THF at 45 °C, the best result was obtained forming the product in 74% yield with a 91:9 er (entry 15).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand (L*) | Solvent | Yieldc [%] | erd |
| a Reaction conditions: 1a (0.3 mmol), [RhCl(C2H4)2]2 (2.9 mg, 2.5 mol%) and ligand L* (5 mol%) in solvent (3 mL, 0.1 M) under an inert atmosphere. b All commercial solvents used in the reaction. c Isolated yields of exo-2a. d er determined by HPLC analysis using a chiral stationary phase. | ||||
| 1 | (R)-BINAP, L1 | 5% aq. xylene | 34 | 56:44 |
| 2 | (S)-SEGPHOS, L2 | 5% aq. xylene | 21 | 55:45 |
| 3 | Diene, L3 | 5% aq. xylene | 59 | 55:45 |
| 4 | Diene, L4 | 5% aq. xylene | 54 | 81:19 |
| 5 | Diene, L5 | 5% aq. xylene | 51 | 61:39 |
| 6 | Diene, L6 | 5% aq. xylene | 53 | 65:35 |
| 7 | Diene, L7 | 5% aq. xylene | 47 | 61:39 |
| 8 | Diene, L8 | 5% aq. xylene | 71 | 57:43 |
| 9 | Diene, L9 | 5% aq. xylene | 58 | 59:41 |
| 10 | Diene, L4 | t-BuOH | 70 | 88:12 |
| 11 | Diene, L4 | THF | 75 | 89:11 |
| 12 | Diene, L4 | CH3CN | 55 | 81:19 |
| 13 | Diene, L4 | DMF | 32 | 78:22 |
| 14 | Diene, L4 | DCE | 47 | 76:24 |
| 15 | Diene, L4 | THF at 45 °C | 74 | 91:09 |

With the optimized reaction conditions for the enantioselective reaction in hand, the scope of representative alkyne-tethered cyclohexadienones was explored with various substituents on 1 or 5 (Table 7). With R1 substituents such as methyl, isopropyl, tert-butyl and phenyl groups, the reactions proceeded smoothly with good yields and good enantiomeric ratios (entries 2a, 2e, 2f and 2h). Both electron donating and withdrawing groups on aryl aldehyde were compatible with the enantioselective reaction and afforded the corresponding products 2m and 2r with high enantioselectivity. Notably, a methyl substituent at the α-position of the dienone was also well tolerated to give bridged polycycle 2v in good 62% yield with an 82:18 er. In addition, alkyl and aryl ketones were also amenable to furnish the annulation products (6a and 6b) in good yield, albeit with a moderate enantiomeric ratio.

To get further insight into the reaction mechanism, we subsequently conducted a 18O labelling study on the Rh(I)-catalyzed cyclization of 1a under the standard reaction conditions in anhydrous THF/H218O solvent which furnished 2a–18O confirmed by ESI-HRMS analysis (see the ESI‡). This reaction clearly suggests that the oxygen atom is from H2O molecules in the solvent system (Scheme 2).
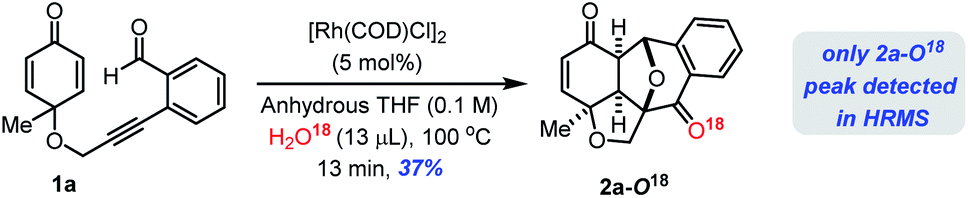 | ||
| Scheme 2 O18-Labelling study. | ||
On the basis of the above experimental results, a possible reaction mechanism was proposed as shown in Scheme 3. First, the alkyne moiety of 1a is activated through π-complex formation (I) to initiate 6-endo-dig cyclization. The intramolecular nucleophilic attack of the carbonyl oxygen on the activated triple bond results in intermediates IIA and IIB.4 The intramolecular [3 + 2] cycloaddition of transient carbonyl ylide II with the electron deficient dienone ring generates highly reactive rhodium–carbene complex III, which readily undergoes hydration to give the desired product 2avia intermediate IV with the regeneration of Rh(I) and liberation of molecular hydrogen.4g Here, the benzopyrylium intermediate has a planar 10π-electron aromatic structure and the chiral catalyst would be located on the favourable face and subsequently undergo cycloaddition from the opposite side.
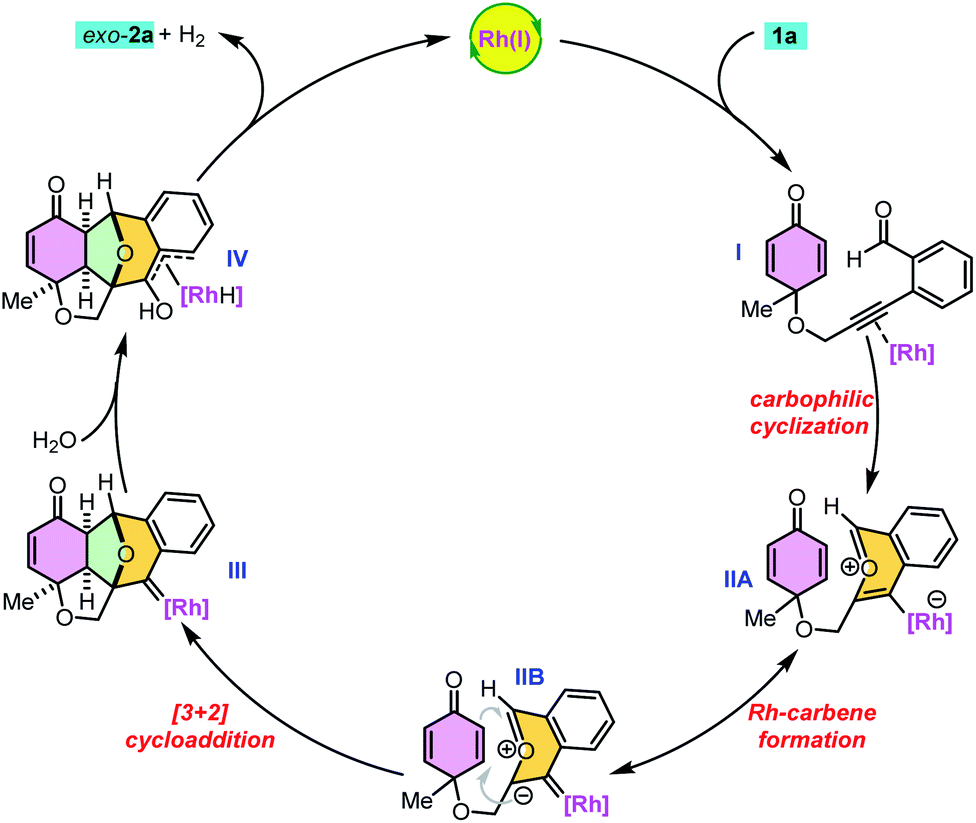 | ||
| Scheme 3 Plausible mechanism for the cascade cyclization reaction. | ||
To demonstrate the synthetic utility of Huisgen-type [3 + 2] cycloaddition, a gram-scale reaction of 1a was carried out with 2.5 mol% catalyst loading under standard reaction conditions. The reaction still proceeded well to afford (±)-2a with 72% yield (Scheme 4a). The complex polycyclic products have enone functionality, which can further undergo various transformations (Scheme 4b). Catalytic hydrogenation of 2a afforded 7 in 96% yield without affecting the highly strained epoxy-bridged polycyclic rings. The intermolecular 1,4-addition of 2a with indole and intramolecular 1,4-addition of 2j by desilylation gave corresponding products 8 and 9, respectively with exclusive diastereoselectivity due to the concave nature of starting substrates.
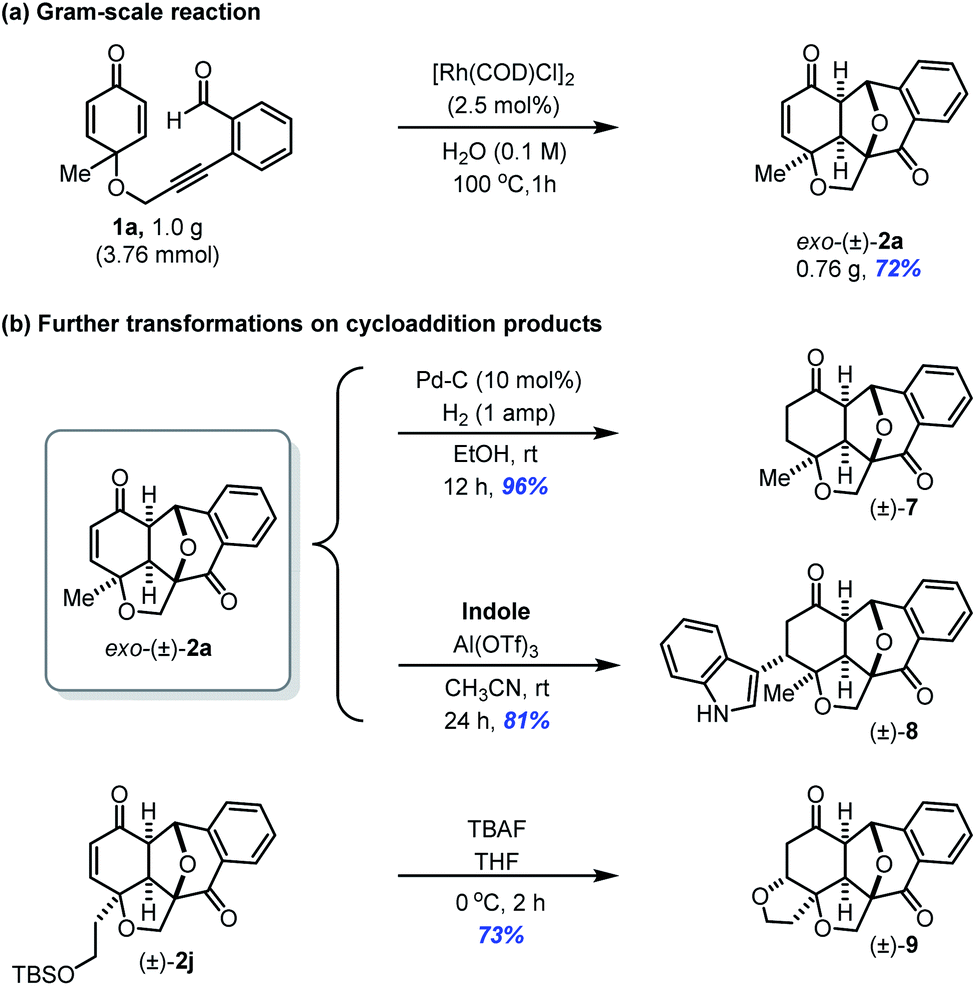 | ||
| Scheme 4 Gram scale reaction and subsequent transformations on cycloaddition products. | ||
Conclusions
In summary, we have developed a highly diastereoselective desymmetrization of prochiral ortho-alkynylbenzaldehyde-tethered cyclohexadienones by Rh(I)-catalyzed intramolecular Huisgen-type [3 + 2] cycloaddition. This atom economical reaction involves a highly reactive Rh–benzopyrylium intermediate that emanates from 6-endo-dig cyclization of 2-enynylbenzaldehyde, which readily undergoes exo-selective 1,3-dipolar cycloaddition. The reaction has broad substrate scope including 2-alkynylbenzaldehyes, 2-alkynylphenylketones and complex estrone derivatives giving highly strained epoxy-bridged polycyclic products bearing five contiguous stereocenters in high yields. The first enantioselective Rh(I)-catalyzed Huisgen-type [3 + 2] cycloaddition was achieved by using easily accessible chiral diene ligands. Further investigations on the design of new chiral ligands and applications are underway in our laboratory and will be reported in due course.Experimental
General procedure
A dried screw-cap vial was charged with 2-alkynylbenaldehyes 1 or 3 or 2-alkynylphenyl ketones 5 (0.3 mmol, 1.0 equiv.), and [Rh(COD)Cl]2 (7.4 mg, 5.0 mol%) in distilled H2O (1.5 mL, 0.1 M) under an inert atmosphere, and the reaction mixture was stirred at 100 °C for 1 hour (monitored by TLC). Then, it was cooled to room temperature and diluted with EtOAc (8 mL). The mixture was extracted with EtOAc (3 × 8 mL) and combined organic solvent was concentrated under reduced pressure. The residue was purified by silica gel column chromatography on silica gel with a gradient eluent of petroleum ether and EtOAc to afford the desired [3 + 2] cyclization product 2 or 4 or 6, respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the SERB, DST New Delhi, India (EMR/2017/001266) and the CSIR-IICT for financial support. KKG, VBP and AV thank the Council of Scientific and Industrial Research (CSIR), New Delhi for research fellowships. IICT Communication Number: IICT/Pubs./2020/151.Notes and references
- For selected reviews, see: (a) L. Yet, Chem. Rev., 2000, 100, 2963–3007 CrossRef CAS; (b) M. Lautens, W. Klute and W. Tam, Chem. Rev., 1996, 96, 49–92 CrossRef CAS ; for selected publications, see: ; (c) M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932–6933 CrossRef CAS; (d) I. D. G. Watson, S. Ritter and F. D. Toste, J. Am. Chem. Soc., 2009, 131, 2056–2057 CrossRef CAS.
- (a) M. B. Fraga, Nat. Prod. Rep., 2005, 22, 465–486 RSC; (b) D. G. I Kingston, H. Yuan, P. G. Jagtap and L. Samala, in The Chemistry of Organic Natural Products, ed. W. Herz, G. W. Kirby, R. E. Moore, W. Steglich and C. Tamm, Spinger-Verlag, New York, 2002, vol. 84 Search PubMed.
- For selected examples for [4+2] cycloaddition, see: (a) A. S. K. Raj and R.-S. Liu, Angew. Chem., Int. Ed., 2019, 58, 10980–10984 CrossRef CAS; (b) H. Luo, L. Liang, R. Chen, H. Jiang and S. Zhu, Org. Chem. Front., 2018, 5, 1160–1164 RSC; (c) T. Arto, P. Fernández, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2016, 52, 13405–13408 RSC; (d) K. Sakthivel and K. Srinivasan, Org. Biomol. Chem., 2014, 12, 269–277 RSC; (e) D. Malhotra, L.-P. Liu, M. S. Mashuta and G. B. Hammond, Chem.–Eur. J., 2013, 19, 4043–4050 CrossRef CAS; (f) S.-Y. Yu, H. Zhang, Y. Gao, L. Mo, S. Wang and Z.-J. Yao, J. Am. Chem. Soc., 2013, 135, 11402–11407 CrossRef CAS; (g) R. Umeda, K. Kaiba, S. Morishita and Y. Nishiyama, ChemCatChem, 2011, 3, 1743–1746 CrossRef CAS; (h) R.-Y. Tang and J.-H. Li, Chem.–Eur. J., 2010, 16, 4733–4738 CrossRef CAS; (i) Y.-C. Hsu, C.-M. Ting and R.-S. Liu, J. Am. Chem. Soc., 2009, 131, 2090–2091 CrossRef CAS; (j) N. Asao, H. Aikawa and Y. Yamamoto, J. Am. Chem. Soc., 2004, 126, 7458–7459 CrossRef CAS; (k) N. Asao, T. Kasahara and Y. Yamamoto, Angew. Chem., Int. Ed., 2003, 42, 3504–3506 CrossRef CAS; (l) G. Dyker, D. Hildebrandt, J. Liu and K. Merz, Angew. Chem., Int. Ed., 2003, 42, 4399–4402 CrossRef CAS.
- For selected examples for [3 + 2] cycloaddition, see: (a) R. Liang, T. Ma and S. Zhu, Org. Lett., 2014, 16, 4412–4415 CrossRef CAS; (b) C. H. Oh, M. S. Lee and C. S. Hong, Org. Lett., 2010, 12, 1308–1311 CrossRef CAS; (c) C. H. Oh, H. J. Yi, J. H. Lee and D. H. Lim, Chem. Commun., 2010, 46, 3007–3009 RSC; (d) C. H. Oh, J. H. Lee, S. J. Lee, J. I. Kim and C. S. Hong, Angew. Chem., Int. Ed., 2008, 47, 7505–7507 CrossRef CAS; (e) H. Kusama, H. Funami and N. Iwasawa, Synthesis, 2007, 2014–2024 CAS; (f) D. Hildebrandt, W. Hüggenberg, M. Kanthak, T. Plöger, I. M. Müller and G. A. Dyker, Chem. Commun., 2006, 2260–2261 RSC; (g) S. Shin, A. K. Gupta, C. Y. Rhim and C. H. Oh, Chem. Commun., 2005, 4429–4431 RSC; (h) H. Kusama, H. Funami, M. Shido, Y. Hara, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2005, 127, 2709–2716 CrossRef CAS.
- (a) A. Padwa, S. P. Carter and H. Nimmesgern, J. Org. Chem., 1986, 51, 1157–1158 CrossRef CAS; (b) A. Padwa, G. E. Fryxell and L. Zhi, J. Am. Chem. Soc., 1990, 112, 3100–3109 CrossRef CAS; (c) A. Padwa, L. Precedo and M. A. Semones, J. Org. Chem., 1999, 64, 4079–4088 CrossRef CAS.
- For reviews, see: (a) K. Tanaka, J. Synth. Org. Chem., Jpn., 2012, 70, 1134–1144 CrossRef CAS; (b) M. C. Willis, Chem. Rev., 2010, 110, 725–748 CrossRef CAS ; for selected references, see: ; (c) D. Hojo, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2009, 48, 8129–8132 CrossRef CAS; (d) K. Tanaka and G. C. A. Fu, Angew. Chem., Int. Ed., 2002, 41, 1607–1609 CrossRef CAS.
- For selected references from our group, see: (a) A. Munakala, K. K. Gollapelli, J. B. Nanubolu and R. Chegondi, Org. Lett., 2020, 22, 7019–7024 CrossRef CAS; (b) S. B. Thopate, S. B. Jadhav, J. B. Nanubolu and R. Chegondi, ACS Catal., 2019, 9, 10012–10019 CrossRef CAS; (c) S. B. Jadhav and R. Chegondi, Org. Lett., 2019, 21, 10115–10119 CrossRef CAS; (d) K. K. Gollapelli, S. Donikela, N. Manjula and R. Chegondi, ACS Catal., 2018, 8, 1440–1447 CrossRef CAS; (e) K. K. Gollapelli, S. Kallepu, N. Govindappa, J. B. Nanubolu and R. Chegondi, Chem. Sci., 2016, 7, 4748–4753 RSC.
- For selected references on transition-metal catalyzed desymmetrization of cyclohexadienones from other groups, see: (a) R. TelloAburto, K. A. Kalstabakken and A. M. Harned, Org. Biomol. Chem., 2013, 11, 5596–5604 RSC; (b) J. Keilitz, S. G. Newman and M. Lautens, Org. Lett., 2013, 15, 1148–1151 CrossRef CAS; (c) K. A. Kalstabakken and A. M. Harned, Tetrahedron, 2014, 70, 9571–9585 CrossRef CAS; (d) C. Clarke, C. A. Incerti-Pradillos and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 8068–8071 CrossRef CAS; (e) R. Kumar, Y. Hoshimoto, E. Tamai, M. Ohashi and S. Ogoshi, Nat. Commun., 2017, 8, 32 CrossRef; (f) J. Chen, X. Han and X. Lu, Angew. Chem., Int. Ed., 2017, 56, 14698–14701 CrossRef CAS; (g) X. Zhou, Y. Pan and X. Li, Angew. Chem., Int. Ed., 2017, 56, 8163–8167 CrossRef CAS; (h) B. Liu, J. Li, P. Hu, X. Zhou, D. Bai and X. Li, ACS Catal., 2018, 8, 9463–9470 CrossRef CAS; (i) Y.-X. Tan, F. Zhang, P.-P. Xie, S.-Q. Zhang, Y.-F. Wang, Q.-H. Li, P. Tian, X. X. Hong and G.-Q. Lin, J. Am. Chem. Soc., 2019, 141, 12770–12779 CrossRef CAS; (j) Q. Teng, N. Thirupathi, C.-H. Tung and Z. Xu, Chem. Sci., 2019, 10, 6863–6867 RSC; (k) C.-L. Duan, Y.-X. Tan, J.-L. Zhang, S. Yang, H.-Q. Dong, P. Tian and G.-Q. Lin, Org. Lett., 2019, 21, 1690–1693 CrossRef CAS; (l) Q. Teng, W. Mao, D. Chen, Z. Wang, C. H. Tung and Z. Xu, Angew. Chem., Int. Ed., 2020, 59, 2220–2224 CrossRef CAS.
- L. Li, D. Huang, C. Shi and G. Yan, Adv. Synth. Catal., 2019, 361, 1958–1984 CrossRef CAS.
- (a) R. Huisgen, G. Szeimies and L. Möbius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- (a) G. Majetich, R. Hicks, Y. Zhang, X. Tian, T. L. Feltman, J. Fang and S. Duncan, J. Org. Chem., 1996, 61, 8169–8185 CrossRef CAS; (b) G. Majetich and G. Zou, Org. Lett., 2008, 10, 81–83 CrossRef CAS.
- CCDC-2008483 (compound 2a) contains the supplementary crystallographic data for this paper..
- (a) H. Suga, H. Ishida and T. Ibata, Tetrahedron Lett., 1998, 39, 3165–3166l CrossRef CAS; (b) N. Shimada, T. Oohara, J. Krishnamurthi, H. Nambu and S. Hashimoto, Org. Lett., 2011, 13, 6284–6287 CrossRef CAS.
- Y.-X. Tan, X.-Q. Tang, P. Liu, D.-S. Kong, Y.-L. Chen, P. Tian and G.-Q. Lin, Org. Lett., 2018, 20, 248–251 CrossRef CAS.
- Catalytic enantioselective transformations of isobenzopyrylium ions: (a) H. Qian, W. Zhao, Z. Wang and J. Sun, J. Am. Chem. Soc., 2015, 137, 560–563 CrossRef CAS; (b) M. Terada, F. Li and Y. Toda, Angew. Chem., Int. Ed., 2014, 53, 235–239 CrossRef CAS; (c) S.-Y. Yu, H. Zhang, Y. Gao, L. Mo, S. Wang and Z.-J. Yao, J. Am. Chem. Soc., 2013, 135, 11402–11407 CrossRef CAS; (d) K. Saito, Y. Kajiwara and T. Akiyama, Angew. Chem., Int. Ed., 2013, 52, 13284–13288 CrossRef CAS; (e) M. Terada and Y. Toda, Angew. Chem., Int. Ed., 2012, 51, 2093–2097 CrossRef CAS; (f) D. Hojo, K. Noguchi and K. Tanaka, Angew. Chem., Int. Ed., 2009, 48, 8129–8132 CrossRef CAS; (g) S. Handa and L. M. Slaughter, Angew. Chem., Int. Ed., 2012, 51, 2912–2915 CrossRef CAS; (h) H. Suga, K. Inoue, S. Inoue and A. Kakehi, J. Am. Chem. Soc., 2002, 124, 14836–14837 CrossRef CAS.
- (a) F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 3364–3366 CrossRef CAS; (b) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS; (c) K. Okamoto, T. Hayashi and V. H. Rawal, Org. Lett., 2008, 10, 4387–4389 CrossRef CAS.
Footnotes |
| † Dedicated to Padma Bhushan Dr A. V. Rama Rao on the occasion of his 85th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2008483. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05543c |
| This journal is © The Royal Society of Chemistry 2021 |