
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall molecules targeting the NEDD8·NAE protein–protein interaction†
Chen-Ming
Lin
 ,
Zhengyang
Jiang
,
Zhe
Gao
,
Maritess
Arancillo
and
Kevin
Burgess
*
,
Zhengyang
Jiang
,
Zhe
Gao
,
Maritess
Arancillo
and
Kevin
Burgess
*
Department of Chemistry, Texas A & M University, Box 30012, College Station, TX 77842, USA. E-mail: burgess@tamu.edu
First published on 14th December 2020
Abstract
Ubiquitination is a major controller of protein homeostasis in cells. Some ubiquitination pathways are modulated by a NEDDylation cascade, that also features E1 – 3 enzymes. The E1 enzyme in the NEDDylation cascade involves a protein–protein interaction (PPI) between NEDD8 (similar to ubiquitin) and NAE (NEDD8 Activating Enzyme). A small molecule inhibitor of the ATP binding site in NAE is in clinical trials. We hypothesized a similar effect could be induced by disrupting the NEDD8·NAE PPI, though, to the best of our knowledge, no small molecules have been reported to disrupt this to date. In the research described here, Exploring Key Orientations (EKO) was used to evaluate several chemotype designs for their potential to disrupt NEDD8·NAE; specifically, for their biases towards orientation of side-chains in similar ways to protein segments at the interface. One chemotype design was selected, and a targeted library of 24 compounds was made around this theme via solid phase synthesis. An entry level hit for disrupting NEDD8·NAE was identified from this library on the basis of its ability to bind NAE (Ki of 6.4 ± 0.3 μM from fluorescence polarization), inhibit NEDDylation, suppress formation of the corresponding E1 – 3 complexes as monitored by cell-based immunoblotting, and cytotoxicity to K562 leukemia cells via early stage apoptosis. The cell-based immunoblot assay also showed the compound caused NEDD8 to accumulate in cells, presumably due to inhibition of the downstream pathways involving the E1 enzyme. The affinity and cellular activities of the hit compound are modest, but is interesting as first in class for this mode of inhibition of NEDDylation, and as another illustration of the way EKO can be used to evaluate user-defined chemotypes as potential inhibitors of PPIs.
Introduction
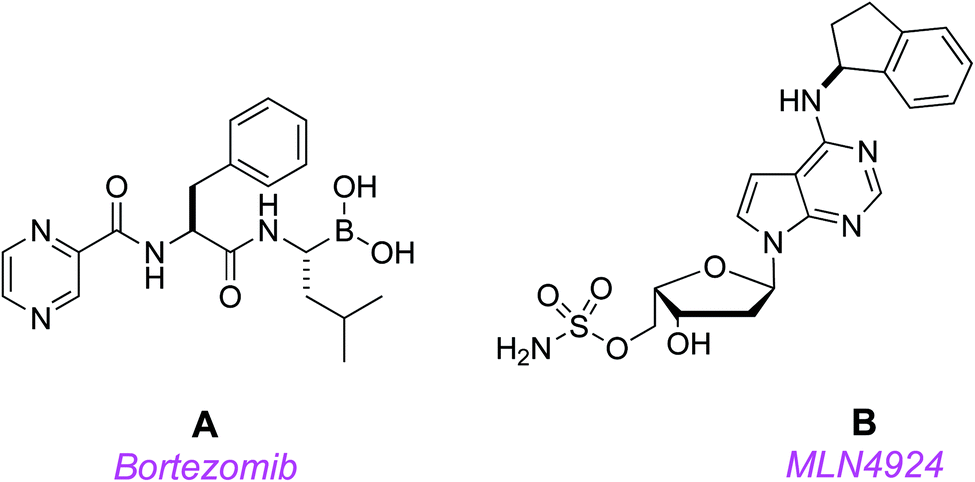
Protein homeostasis is essential for vital cellular processes, including cycle progression, signal transduction, and death.1 Aberrant protein degradation in cancer contributes to rapid progression, metastasis, and drug resistance,2 so counteracting this is an appealing therapeutic strategy. In protein homeostasis, the ubiquitin-proteasome system controls turnover of most short-lived intracellular proteins, while autophagy achieves bulk degradation of organelles and proteins with long half-lives.3 Consequently, small molecules that target the ubiquitin-proteasome system are potentially useful.Several potential cancer therapeutic agents that inhibit both ubiquitin-proteasome mediated protein turnover and autophagic degradation are in the clinic, or in clinical trials. The first proteasome inhibitor, bortezomib (Velcade®) A, received FDA approval for the treatment of multiple myeloma and mantle cell lymphoma.4 Bortezomib's success prompted efforts to develop agents that more specifically target key regulators of protein turnover to eliminate toxicities, particularly peripheral neuropathy, that result from global proteasomal inhibition.
Disrupting NEDD8-mediated protein turnover to perturb protein homeostasis is a promising new approach for chemotherapy.5–9 Overall, the NEDD8 conjugation pathway, “NEDDylation”, is the process of translocation of NEDD8 protein from its E1 enzyme (NEDD8 Activating Enzyme, NAE) through several steps to Cullin-RING Ligases (CRLs).10 The first step in this process involves activation of NEDD8 while bound to NAE; this process is mediated by Mg2+/ATP and affords an acyl adenylate intermediate, NEDD8-AMP. Subsequently, NEDD8 is transferred to a Cys residue in the active site to form NAE-NEDD8, and another molecule of NEDD8-AMP complexed to NAE is formed concomitantly as that transfer proceeds.11 That ternary complex passes off one molecule of NEDD8 to a E2 enzyme in the sequence (either UBC12 or UBE2F) via transthiolation. In the final step of the sequence, the thioester of the NEDD8·E2 complex acylates a Lys residue of the winged-helix B (WHB) subdomain of a CRLs to form an iso-peptide bond. That process stimulates ubiquitination, and the covalently attached NEDD8 suppresses binding of the protein CAND1 which would otherwise prevent NEDDylation.11,12
NEDDylation of CRLs regulates the ubiquitination system and ultimately controls degradation of proteins that have important roles in cell cycle progression (e.g. p27, cyclin E, c-Myc), tumor suppression (p53), DNA damage (CDT1), stress responses (NRF-2, HIF-1α), and signal transduction (phosphorylated inhibitor of nuclear factor κBα, pIκBα).13 Thus, overall, NEDD8 activating enzyme (NAE), the E1 enzyme in the NEDDylation cascade, is a key proximal regulator and a fine target for pharmacologic inhibition.14–16
Millennium's MLN4924 B17 is a first-in-class, potent, and selective small molecule NAE inhibitor;18,19 it is currently in phase 3 clinical trials (in combination with another anti-cancer drug, azacytidine; https://clinicaltrials.gov/ct2/show/NCT03268954).8,17 Targeting NAE with MLN4924 disrupts NEDD8-mediated protein turnover, induces apoptosis, and leads to stable disease regression in acute myeloid leukemia (AML) models.19,20 These promising findings have been recapitulated in the clinic; for instance, several patients with AML that were refractory to conventional therapy have achieved complete responses following treatment with MLN4924 in a phase I study.19,20
MLN4924 acts by competing for the ATP binding site of NAE; when it docks, the NEDD8 covalently binds the drug,21,22 inactivating the NEDDylation cascade. However, there are indications that problems could emerge with this approach because a treatment-emergent A171T mutation in the ATP-binding region of NAE conferred resistance to MLN4924-induced apoptosis in HCT116 colorectal carcinoma, and K562 and U937 leukemia cells.23–25
Post-MLN4924, several NAE inhibitors have been reported, and all but one bind the ATP binding site (compete with ATP), i.e. they also bind the NAE active site.26–31 As far as we are aware, there are no previous reports of small molecules that interrupt the NEDD8-NAE PPI. There is only one report of a compound series that disrupts PPIs in the NEDDylation cascade; these are peptidomimetics that bind DCN1 (Defective in Cullin NEDDylation 1) hence perturb the DCN1·UBC12 interaction (UBC12 is an E2 ligase).32–34
Discovery of elementary hits for the NEDD8·NAE target may facilitate a cascade of events involving co-crystallization, modeling to increase affinity, and extensive medicinal chemistry to optimize bioavailability, ultimately leading to a probe or pharmaceutical lead. Unfortunately, it is generally difficult to find even ground-level small molecule hits for PPIs. High throughput screening, for instance, has been used often for discovery of small molecules, but tends to give disappointing hit-rates relative to the cost and time expenditures involved.35–38
The following argument can be made that resistance to MLN4924 is more likely than it would be for a small molecule that perturbs the NEDD8·NAE protein–protein interaction (PPI). Only non-extensive mutations are required to block pharmaceuticals binding nucleotide triphosphate binding sites. Moreover, even though MLN4924 has been screened against a panel of kinases and adenosine receptors, and no serious off-target effects were detected,17 it is a mimic of ATP, hence is vulnerable to off-target effects involving other proteins that recognize NTPs. Conversely, the NEDD8·NAE interface is comparatively unique hence small molecule mimics that bind there will tend to be less vulnerable to off-target effects.
We hypothesized potential small molecules that evaluate positively in an EKO (Exploring Key Orientations)39 analysis of the NEDD8·NAE interaction might enable a ground-level hit to be identified from a small library consisting of only 10–20 compounds. In an EKO analysis, chemotype scaffolds are evaluated for their ability to place amino acid side-chains in the same orientations as small interface regions for either protein binding partner. To be effective, the chemotypes considered for EKO must be more rigid than corresponding peptides, present at least three amino acid side chains, but need not be peptidic or contain any amide bonds. Briefly, the EKO approach proceeds by comparing favored conformations of small molecules that present three amino acid side-chains to PPI interfaces, based on degree of fit of side-chain Cα and Cβ coordinates. Validation for EKO has been reported in just a few cases: HIV-1 protease dimer,39 antithrombin dimer,40 PCSK9·LDLR,41 and uPA·uPAR.42 Thus, at the onset of this work it was not clear that it would also work for NEDD8·NAE.
Results and discussion
Characteristics of the NEDD8·NAE
Fig. 1a shows the structure of NEDD8·NAE (1R4N).12 NAE is comprised of two fragments. One chain (red in Fig. 1a) is Amyloid Precursor Protein-Binding Protein 1, APPBP1, and the other is ubiquitin like modifier activating enzyme, UBA3 (yellow). NAE surrounds the NEDD8 substrate like a hand clenching a lollipop by the stick. Formation of this complex involves significant conformational changes of both protein components, the main one being reorientation of the NEDD8 C-terminal chain. In the complex, the C-terminus of NEDD8 penetrates into a thin groove in the fist towards the deeply embedded ATP binding site. At the end of the stick near the sweet part of the lollipop, NEDD8 presents a hydrophobic face consisting of a combination of Leu, Ile, and Val residues that interacts with UBA3.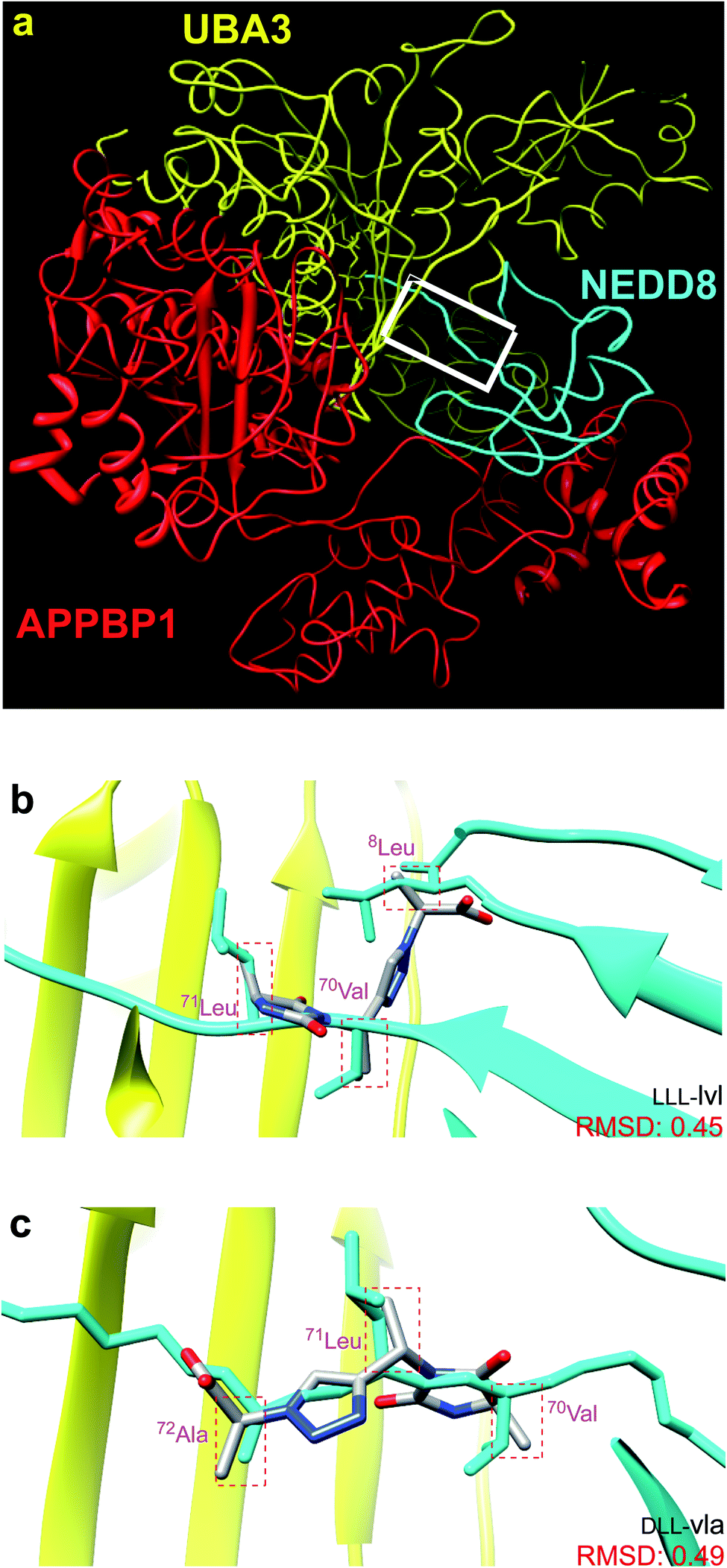 | ||
| Fig. 1 (a) NEDD8 is clenched at its C-terminus by a fist formed from NAE, which consists of UBA3 (yellow) and APPBP1 (red). (b and c) Overlay of the featured chemotypes on 8Leu, 70Val, 71Leu, 72Ala of NEDD8 (see Fig. S1† for all overlays). | ||
Chemotype design via EKO
Around 50 candidate chemotypes were evaluated using EKO for their potential to overlay side-chains in conformations that correspond to interface regions for NEDD8·NAE. Many of those chemotypes could not achieve appropriate conformations to do this. Others could populate suitable conformers, but their syntheses with the side-chains implicated by the overlay were likely to be time-consuming. However, one chemotype (corresponding to the colored parts of structures 1–3, see below) overlaid on that hydrophobic face alluded to above. That finding was appealing because the hydrophobic interface forms a gateway to the long-extended stick region of NEDD8 that extends into the NAE clenched fist; we hypothesized a small molecule that interacts there might prevent the NEDD8 C-terminus entering that cavity. Moreover, since the colored fragment overlays on hydrophobic residues of NEDD8, making the appropriate chemotype should be easier because the side-chains are not functionalized. These overlay regions are shown in Fig. 1b and c.Synthesis of chemotypes with functionalized side-chains tends to be orders of magnitude more difficult than ones with hydrocarbon appendages. Despite this, we usually prefer side-chain sets with some functionality in EKO analysis to avoid sticky, hydrophobic, non-selective interactions and poor aqueous solubility. However, the chemotype backbone in structures 1–3 has many polar functionalities and no benzenoid rings, so the overall combination was judged to be acceptable enough for further studies.
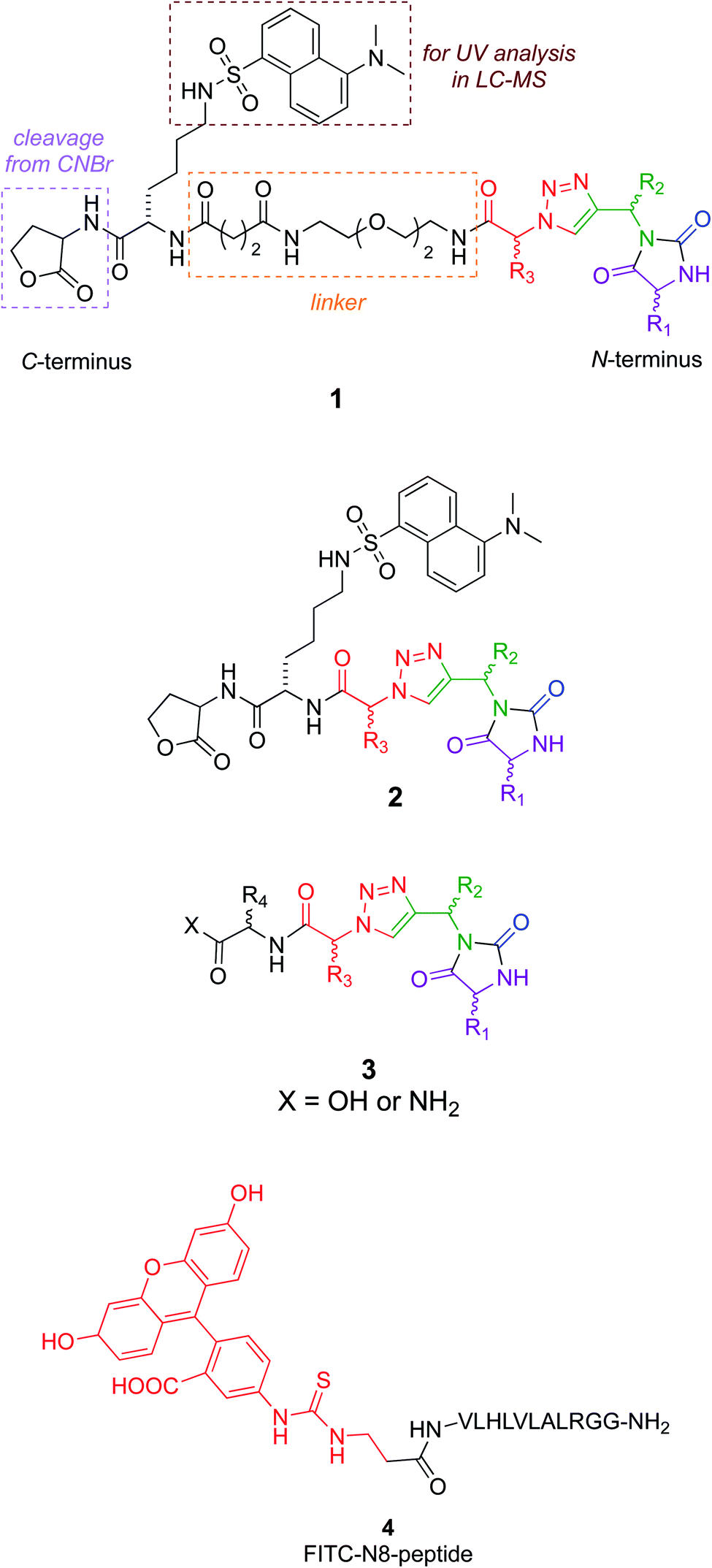
Several design considerations led to the non-colored peripheral fragments of structures 1–3. Structures 1 and 2 have a C-terminal lactone; this is because the tested compounds were eventually made via solid phase syntheses involving a cyanogen bromide cleavage, and this necessarily leaves a lactone appendix. It was anticipated that the lactone would be circumvented in iterations of the design as in structure 3, without negatively impacting binding. Secondly, a Lys(dansyl) residue was included next to the lactone, for the following reason. Solid phase synthesis methodology development of non-peptidic small molecules requires careful optimization of each step. During optimization, it is desirable to differentiate materials in the synthesis that are cleaved from the resin, from impurities that tend to be liberated from the support but do not involve the growing chain of interest. Inclusion of a Lys(dansyl) group facilitated observation of only the chain of interest by UV detection at 340 nm, because only the dansyl group absorbs there. Moreover, compounds containing the dansyl group are distinct from low molecular mass impurities in LC-MS analyses. Chemotypes 1 included a hydrophilic linker,43 to separate the lactone-Lys(dansyl) from the chemotype core. It was anticipated that linker might be critical if the chemotypes were to bind at the entrance to the NAE clenched fist region. Finally, the series 3 chemotypes each have an additional C-terminal amino acid residue. These residues were selected by taking the coordinates of the conformations that overlaid NEDD8 well in the EKO analyses, adding the side-chains corresponding to the NEDD8 at that particular section of the interface (EKO analyses uses only Ala side-chains for reasons already explained and justified), Glide minimization in the Schrödinger package, then selection of fragments to increase affinity using the CombiGlide routine in that same software (see ESI Glide section†).
A peptide corresponding to the NEDD8 C-terminus is known to inhibit the NEDDylation.44 Consequently, a fluorescein labeled derivative of this peptide (4) was synthesized to use as a positive control in a fluorescence polarization assay.45
Chemotype syntheses
Structures 1–3 are color-coded to indicate “warhead” regions derived from an amino acid (pink) amide-coupled to a propargylic amine derived from amino acids (green), clicked46,47 to azido-acids,48 also from amino acids (red). Propargylic amines for this synthesis were made from Boc-protected amino acids, as described in the ESI.† In the click reaction, excess CuI tends to bind to beads, turning them green. It was important to wash the beads with EDTA solution until they were colorless otherwise residual CuI leads to impurities wherein the triazole is iodinated. LC-MS analyses showed the crude products cleaved from the resin contained predominantly the desired materials (Fig. S2†); these were then purified further by preparative HPLC. Ultimately 24 compounds for testing were synthesized; eight in each series 1, 2, and 3. Throughout this paper, the nomenclature to number the compounds follows this convention: 1-letter abbreviation of N- to C-amino acid side-chains along with their configurations (L or D) (Scheme 1).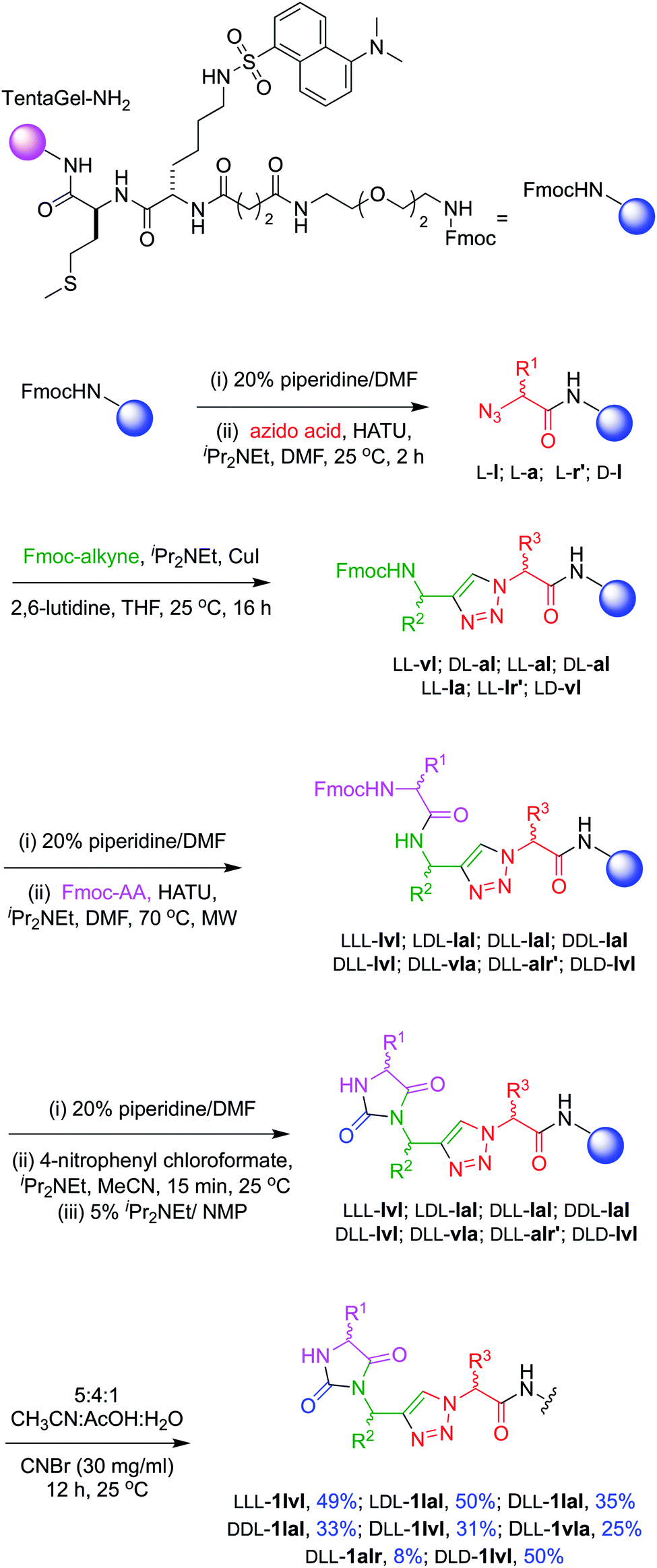 | ||
Scheme 1 Solid phase syntheses of chemotypes 1. In this route, LLD-1r′la was made slightly differently insofar as the final supported material was treated with 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 TFA:H2O:HSiEt3 for 2 h to remove the Mtr protecting group before CNBr cleavage. :2.5:2.5 TFA:H2O:HSiEt3 for 2 h to remove the Mtr protecting group before CNBr cleavage. | ||
Cell viability assays
Assays for NEDDylation tend to be complex relative to cytotoxicity screens. Consequently, we decided to begin with a “top-down” approach featuring a primary cytotoxicity screen; K562 leukemia line was used because these cells overexpress NEDD8.24 In retrospect, this approach was conservative (and we partially retreated from it later) since only three compounds of the 24 compounds showed cytotoxicity (Fig. 2a), and IC50 values could not be obtained for two of these, DLD-2lvl, and DLL-2vla, because they were insufficiently soluble at the concentrations required (Fig. S3†). However, reasonable data was obtained for the compound that emerged as the most promising hit: LLL-1lvl.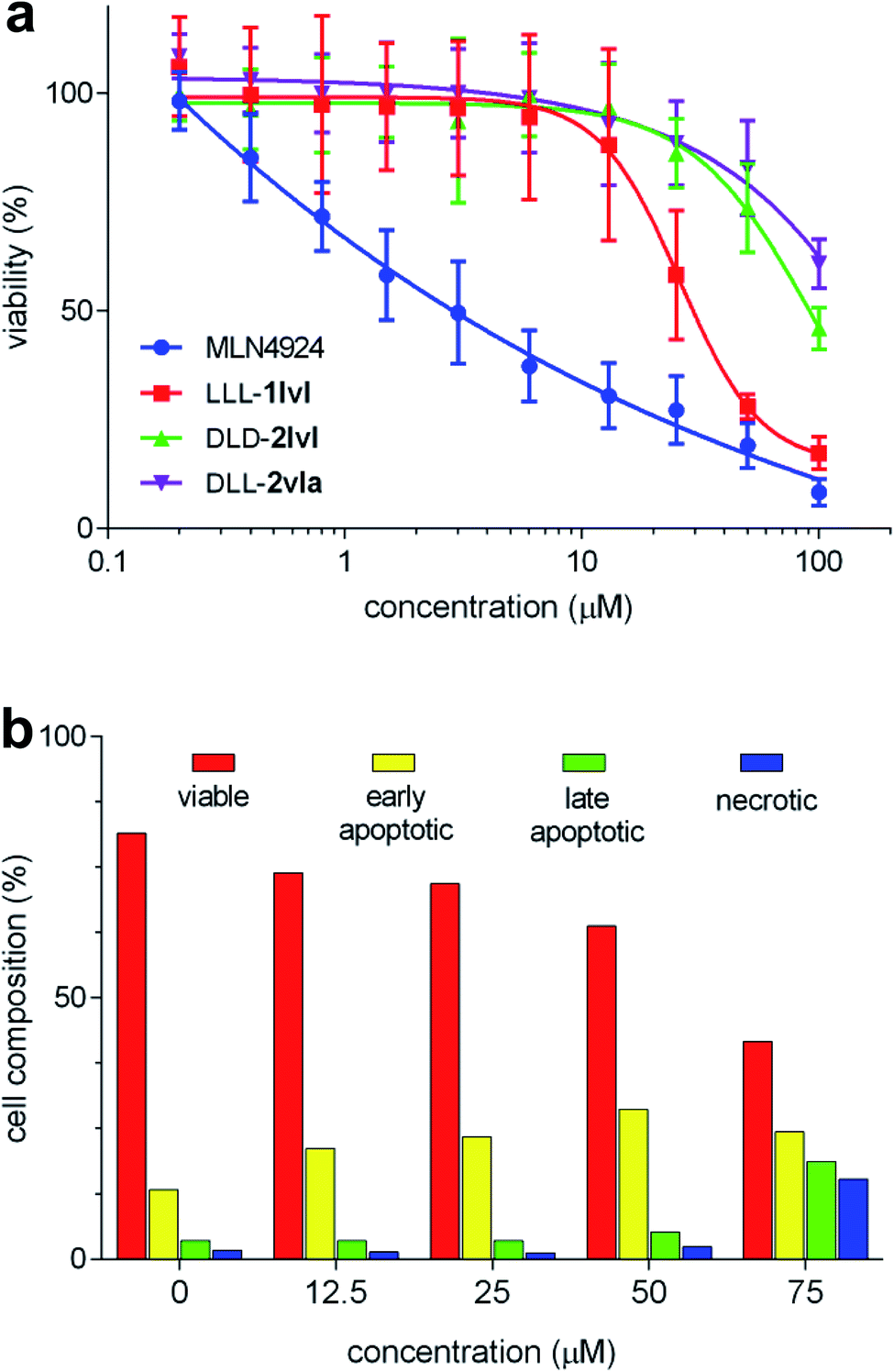 | ||
| Fig. 2 (a) Viability of K562 cells treated with LLL-1lvl, DLD-2lvl, DLL-2vla, and MLN4924 (IC50 of LLL-1lvl = 26.3 ± 4.6 μM; n = 3). (b) Onset of apoptosis and extent of necrosis as indicated by treatment of LLL-1lvl-treated cells with FITC annexin V and propidium iodide. Error bars represent standard deviations for n = 8. | ||
FACS assays featuring LLL-1lvl were performed to elucidate if its cytotoxicity to K562 leukemia cells was due to early or late stage apoptosis. Inhibition of NEDDylation most likely sends cells into apoptosis.49 To test this hypothesis here, the leukemia cells were treated with incubated LLL-1lvl then with dyes to test for cells in the apoptotic and necrotic states (FITC annexin V and propidium iodide, respectively). With the increased concentration of LLL-1lvl, the percentage of apoptotic cells was observed to increase (Fig. 2b). Confocal microscopy was used to observe K562 cells treated with 25 μM of LLL-1lvl at 24, 48 and 72 h. A progressively significant change in cell morphology was observed for some cells consistent with the onset of apoptosis (Fig. S8†).
In vitro NEDDylation protein assay
Compounds in series 3 share the same warhead region as chemotypes 1 and 2, consequently all eight chemotypes 3 were tested in an in vitro NEDDylation protein assay alongside the hits that showed some cytotoxicity: LLL-1lvl, DLD-2lvl, DLL-2vla (Fig. S5†). The objective of this experiment was to detect if NEDDylation could be inhibited by the compounds if cell permeability were not an issue. In this assay,49 NAE, Ubc12 (the E2 enzyme in the NEDDylation cascade), fluorescein-NEDD8, and the featured samples were incubated, then gel electrophoresis was used to quantify Ubc12-NEDD8 complex by fluorescence. Fig. 3 shows that four compounds, including LLL-1lvl, gave a credible dose–response for diminished formation of E2-NEDD8 (i.e. Ubc12-NEDD8) with increased concentration of the test samples. The four compounds indicated (and the N8-peptide positive control) impaired the NEDD8 cascade in a dose dependent manner. Overall, these data show that some compounds in series 3 do inhibit NEDDylation in vitro, and that they have about the same impact as the cytotoxic lead LLL-1lvl. We conclude from this that the extra amino acid that distinguishes chemotypes 3 from 1 and 2 is not significantly influencing the in vitro activity of the molecule with respect to inhibition of NEDDylation.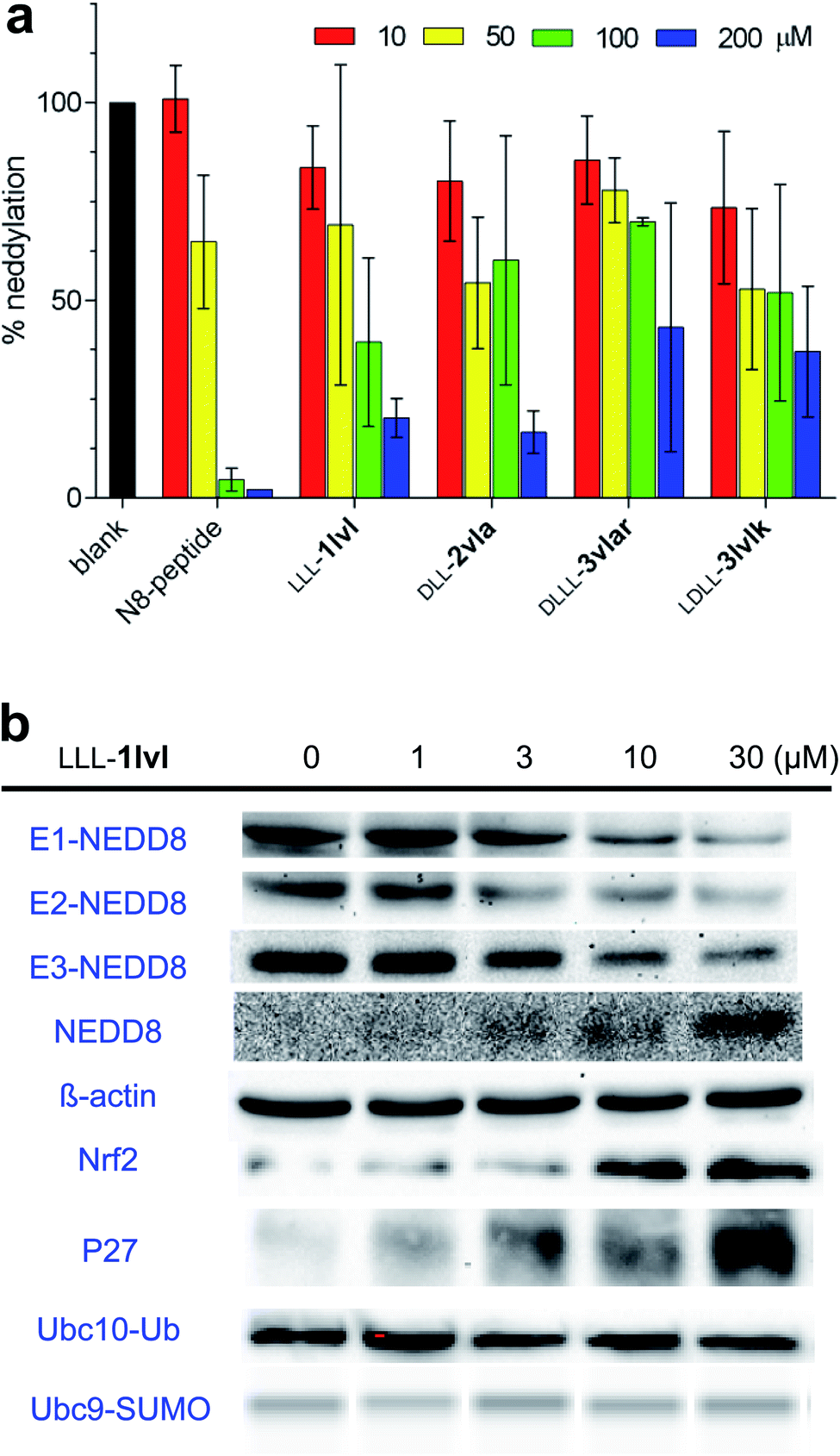 | ||
| Fig. 3 Influence of the featured compounds on the NEDDylation cascade. (a) Inhibition of covalent complex formation between the E2 enzyme (Ubc12) and NEDD8 by the four compounds shown as demonstrated via an in vitro protein assay. (b) Dose-dependence effects of LLL-1lvl incubated with live K562 cells on formation of E1-, E2-, and E3-NEDD8 complexes. Dose–response effects of LLL-1lvl on degradation of Nrf2 and P27 in live K562 cells. Lack of response to LLL-1lvl in formation of Ubc10-Ub or Ubc9-SUMO in the ubiquitination or SUMOlation cascade. Error bars shown represent standard deviations based on n = 3. | ||
It was possible that our compound bound the ATP-binding site. To test this possibility, an ATP competitive assay was carried out. In the event the lead compound LLL-1lvl did not compete with ATP binding for binding NAE (Fig. S5†).
Cell-based immunoblot
The cytotoxic hit compound, LLL-1lvl, was tested in immunoblot assays to determine if its effects on NEDDylation in vitro could be observed in live cells. Cell-based immunoblot assays for NEDD8 bound to E1 (UBA3), E2 (Ubc12), and E3 (cullins) enzymes in the NEDDylation cascade (Fig. 3b) demonstrated LLL-1lvl supressed formation of all three NEDD8 complexes (i.e. of the E1, E2, and E3 enzymes) in live K562 leukemia cells. Compound, LLL-1lvl also inhibited formation of NEDDylated Cullin3 in a dose–response manner (Fig. S6†). Curiously, levels of NEDD8 increased in response to the NEDDylation inhibitor LLL-1lvl (this experiment was repeated several times to check) indicating NEDD8 accumulates as the downstream pathways involving the E1 enzyme are inhibited.Experiments were performed to test the specificity of LLL-1lvl for inhibition of NEDDylation rather than ubiquitination. If LLL-1lvl inhibits NEDDylation it should prevent degration of substrate proteins downstream of CRLs in this cascade, e.g. Nrf2 and P27. Accumulation of Nrf2 and P27 could also be activated by other cell signaling transduction pathways including ones involving an autophagic Keap1 degradation50,51 and with G1 cell cycle arrrest.52–54 In the event, immunoblotting showed Nrf2 and P27 accumulated in a dose–response manner on treating the cells with LLL-1lvl, and consistent with inhibition of NEDDylation. Conversely, K562 cells treated with LLL-1lvl showed no reduction of Ubc10-Ub, Ubc9-SUMO or other E2-Ub expression indicating this compound did not suppress ubiquitination or SUMOlation (Fig. 3b and S6†).
NAE protein SiRNA knockdown
To further confirm LLL-1lvl disrupts the NEDD8·NAE interaction selectively, NAE expression in K562 cells were knocked down using siRNA prior to LLL-1lvl treatment (Fig. 4a). After knockdown, no NAE expression was detectable via blotting whereas it was before. We anticipated the dose-dependent response of E3-NEDD8 complex to LLL-1lvl in wild type K562 cells (Fig. 4b) should be abolished in cells where NAE was knocked down. Consistent with this expectation, we drastically increased the contrast to observe residual E3-NEDD8 levels and observed they were constant in NAE(−) cells (Fig. 4c) but decreased in wild type (compare Fig. 4b with c), indicating LLL-1lvl targets NAE selectively.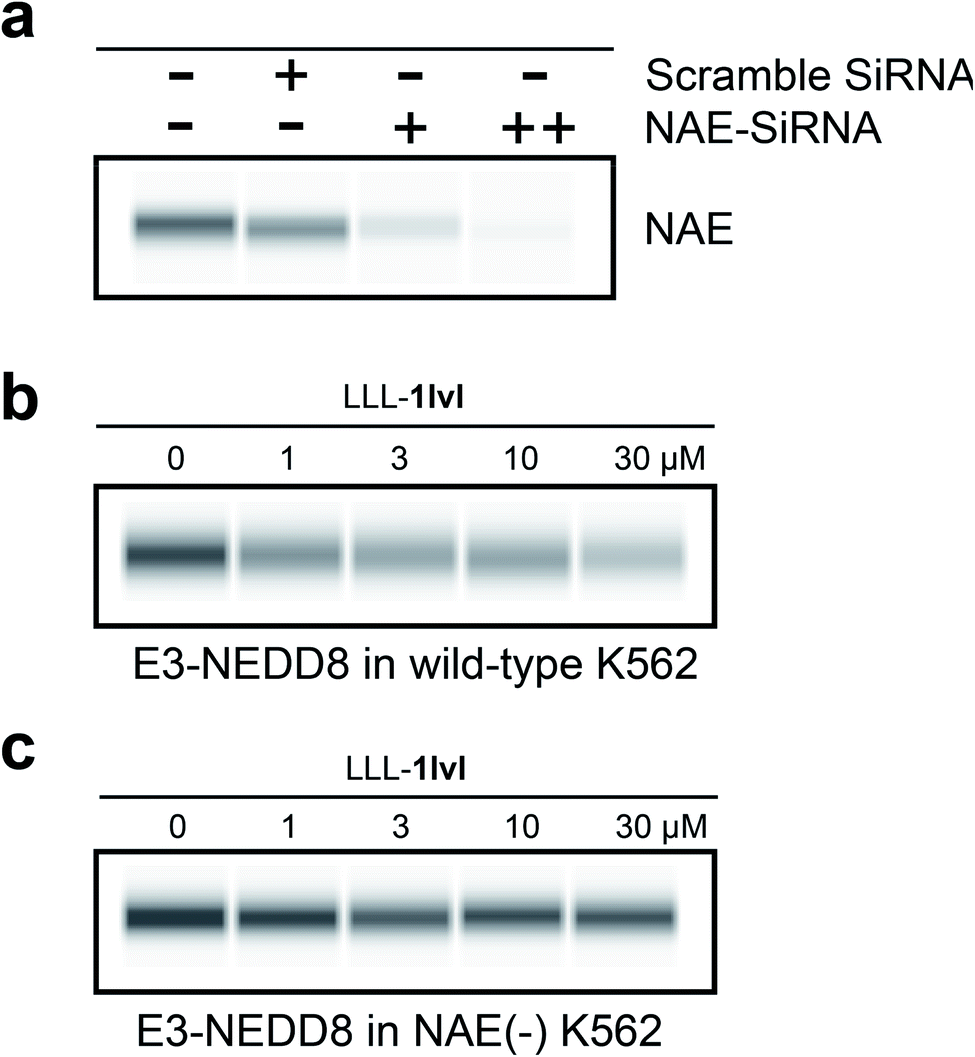 | ||
| Fig. 4 Cell-based immunoblots. (a) With an NAE antibody comparing untreated K562 cell lysate control (double −), scramble SiRNA (little effect on NAE expression) and increasing concentrations of NAE-SiRNA (+ for 30 pmol, ++ for 300 pmol; significantly reduced NAE expression in K562). (b) Dose-dependent response of E3-NEDD8 to LLL-1lvl in wild-type K562 cells. (c) E3-NEDD8 complex levels remains constant upon LLL-1lvl treatment in NAE(−) K562 cells. | ||
It is not surprising that the E3-NEDD8 conjugate can be still visualized via immunoblots after NAE knockdown because even a trace of residual NAE can catalytically trigger NEDDylation; additionally there could be compensation mechanisms featuring UAE-NEDD8 activation when levels of NEDD8 in cells are high, as observed by others.16
K i determination via fluorescence polarization
A series of experiments were performed to determine the dissociation constant for binding of LLL-1lvl to NAE via fluorescence polarization.45,55 However, direct labeling of small molecules like LLL-1lvl with relatively large fluors tends to impact their binding, so a different approach was used. Consequently, the Ki determination was performed in two steps: (i) measure Kd for fluorescein-N8 (labeled positive control peptide) with NAE via directing binding; then, (ii) determine the Ki of LLL-1lvl for NAE via competitive binding versus fluorescein-N8. A Z-factor55 was measured for the assay in step (i) to check its validity; the value was acceptable (0.52). Additionally, NAE binding affinities of two more compounds (DLLL-3klvl and LLL-fsi) were evaluated; DLLL-3klvl, which is similar to the featured lead LLL-1lvl, but only containing an extra lysine, and a negative control LLL-fsi designed for another target,42 wherein the side chains do not match the NEDD8 C-terminus.Addition of detergent in FP assays can be important to avoid some false positive outcomes.56 Here the FP assay was carried out with and without addition of 0.01% Tween-20 in buffer; the FP without 0.01% Tween-20 shown in Fig. 5 is essentially identical to that with addition of 0.01% Tween-20 (Fig. S8†).
 | ||
| Fig. 5 Fluorescence polarization binding assays for binding to NAE. (a) Direct binding of fluorescein-N8; and, (b) competitive binding of the three mimics indicated each with fluorescein-N8. Error bars represent standard deviations based on n = 6. | ||
In the direct binding assay, the Kd of fluorescein-N8 to NAE was determined to be 162 ± 16 nM (Fig. 5a). Subsequently, the competitive binding assay indicated LLL-1lvl binds to NAE with a Ki of 6.4 ± 0.3 μM (Fig. 5b), i.e. about an order of magnitude less than the labeled peptide C-terminus (fluorescein-N8). DLLL-3klvl and LLL-1lvl have the side-chains and orientations that EKO predicts will bind, but the partial control LLL-fsi has different side chains. In the event, both LLL-1lvl and the extended sequence DLLL-3klvl exhibited micromolar binding to NAE, but the partial negative control LLL-fsi did not bind, as expected. The cytotoxicity of LLL-fsi towards K562 cells was also checked, and none was observed, also as expected. These data are consistent with the assertion that EKO evaluations are effective for finding potential disruptors of NEDD8·NAE.
Further evidence for binding of LLL-1lvl to NAE was obtained via a native mass spectrometry assay (Fig. S10†). An analogous mass spectrometry experiment but using ubiquitin activating enzyme (UAE) in place of NAE did not show binding (Fig. S11†).
Intracellular imaging
Confocal microscopy was used to track the dansyl fluor in LLL-1lvl to determine if this compound internalized into cells and co-localized with Lysotracker Green and Nuclear Red. In eukaryotes, proteasomes are located in nuclei or in the cytoplasm. These experiments indicated the compound first internalized into the cytoplasm (no colocalization was seen in the nucleus after 1 h incubation, Fig. 6, or after 24 h {data not shown}).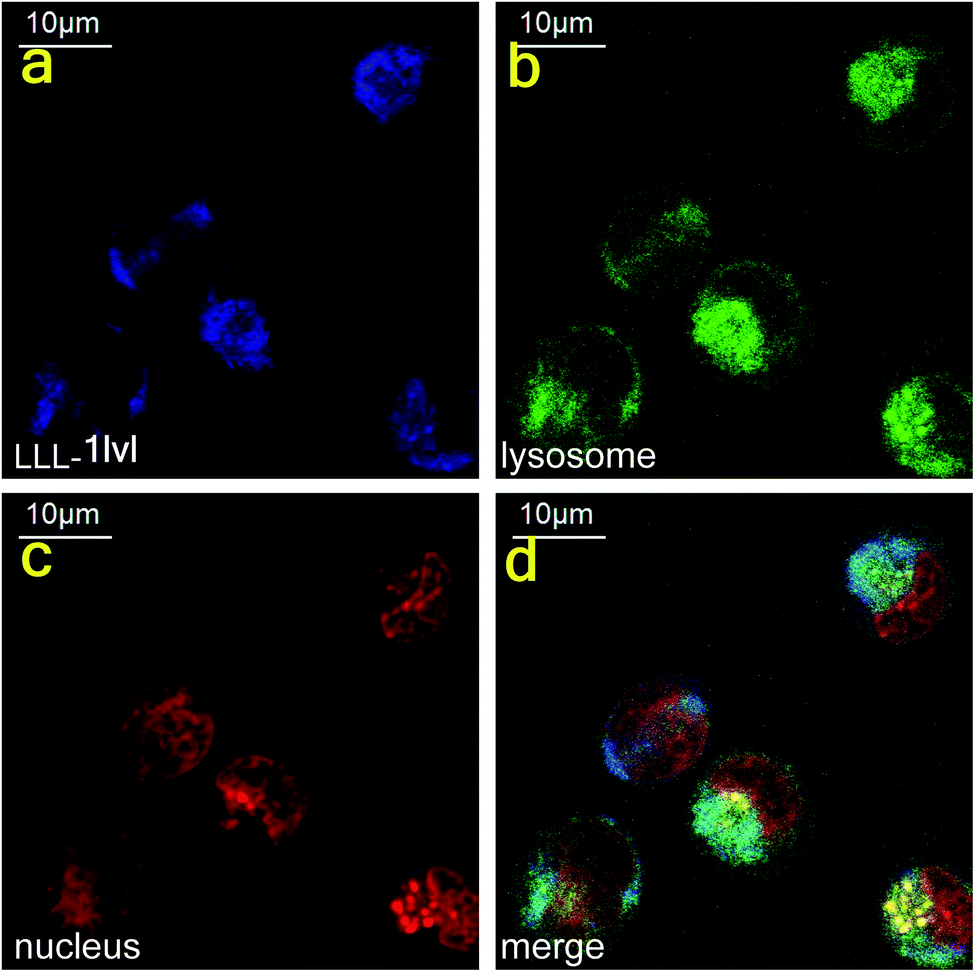 | ||
| Fig. 6 Dansyl fluorescence indicated the internalization of LLL-1lvl in cytoplasm of K562 cells within 1 h. | ||
Conclusions
We are unable to find prior reports of small molecules that disrupt NEDD8·NAE. The best hit found here, LLL-1lvl is cytotoxic to NEDD8-expressing cells (IC50 of LLL-1lvl = 26.3 ± 4.6 μM), and induced cell death predominantly via apoptosis, as anticipated. Compound LLL-1lvl perturbed the NEDD8·NAE interaction in vitro (enzyme assay) and in cellulo (cell-based immunoblot). Fluorescence polarization data indicated LLL-1lvl bound NAE with a Ki of 6.4 ± 0.3 μM. NAE knockdown experiments indicated this compound disrupts NEDD8·NAE by mimicking NEDD8, not NAE, in cells; this experiment also rules out other off-target effects.Compound LLL-1lvl is a ground level hit with modest efficacy and affinity, but it is important because it shows that the anticipated PPI disruption strategy is possible, and for providing insights for chemotype design against NEDD8·NAE. Our group is equally interested in these findings from the perspective of validation of the EKO approach. EKO analyses do no more than test if input chemotype scaffolds can project amino acid side chains in the same orientation as those in small PPI interface regions. Positive matches in EKO analyses do not guarantee binding or efficacy. Indeed, illustrative applications of the EKO strategy have only been reported in four papers (and we have another in preparation). Thus, it was not assured that any detectable binding or cellular efficacy would be observed in this study. In the event, only 24 compounds were prepared and tested and, in fact, these could be classified as three sets of only eight compounds, where members of each set are very similar. Thus, it is notable that some of these compounds appear to have the desired effect.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank Dr Arthur Laganowsky and Zahra Moghadamchargari of Texas A&M University for the mass spectrometry assay. We also thank Matthew D. Petroski and Dr Julia Toth of Sanford Burnham Prebys Medical Discovery Institute for helpful comments and for providing NAE protein. We thank DoD BCRP Breakthrough Award (BC141561), CPRIT (RP150559, RP170144, and RP180875), National Science Foundation (CHE-1608009), The Robert A. Welch Foundation (A-1121) and the NIH (R01EY029695) for support. The NMR instrumentation at Texas A&M University was supported by the Texas A&M University System and a grant from the National Science Foundation (DBI-9970232).References
- G. M. Burslem and C. M. Crews, Chem. Rev., 2017, 117, 11269–11301 CrossRef CAS
.
- R. Z. Orlowski and D. J. Kuhn, Clin. Cancer Res., 2008, 14, 1649–1657 CrossRef CAS
- A. L. Schwartz and A. Ciechanover, Annu. Rev. Pharmacol. Toxicol., 2009, 49, 73–96 CrossRef CAS
- A. M. Roccaro, T. Hideshima, P. G. Richardson, D. Russo, D. Ribatti, A. Vacca, F. Dammacco and K. C. Anderson, Curr. Pharm. Biotechnol., 2006, 7, 441–448 CAS
- I. R. Watson, M. S. Irwin and M. Ohh, Cancer Cell, 2011, 19, 168–176 CrossRef CAS
- M. Wang, B. C. Medeiros, H. P. Erba, D. J. DeAngelo, F. J. Giles and R. T. Swords, Expert Opin. Ther. Targets, 2011, 15, 253–264 CrossRef CAS
- T. A. Soucy, L. R. Dick, P. G. Smith, M. A. Milhollen and J. E. Brownell, Genes Cancer, 2010, 1, 708–716 CrossRef CAS
- T. A. Soucy, P. G. Smith and M. Rolfe, Clin. Cancer Res., 2009, 15, 3912–3916 CrossRef CAS
- J. W. Harper, Cell, 2004, 118, 2–4 CrossRef CAS
- A. C. O. Vertegaal, Chem. Rev., 2011, 111, 7923–7940 CrossRef CAS
- D. M. Duda, L. A. Borg, D. C. Scott, H. W. Hunt, M. Hammel and B. A. Schulman, Cell, 2008, 134, 995–1006 CrossRef CAS
- H. Walden, M. S. Podgorski, D. T. Huang, D. W. Miller, R. J. Howard, D. L. Minor Jr, J. M. Holton and B. A. Schulman, Mol. Cell, 2003, 12, 1427–1437 CrossRef CAS
- M. D. Petroski and R. J. Deshaies, Methods Enzymol., 2005, 398, 143–158 CAS
- V. N. Podust, J. E. Brownell, T. B. Gladysheva, R.-S. Luo, C. Wang, M. B. Coggins, J. W. Pierce, E. S. Lightcap and V. Chau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4579–4584 CrossRef CAS
- M. A. Read, J. E. Brownell, T. B. Gladysheva, M. Hottelet, L. A. Parent, M. B. Coggins, J. W. Pierce, V. N. Podust, R.-S. Luo, V. Chau and V. J. Palombella, Mol. Cell. Biol., 2000, 20, 2326–2333 CrossRef CAS
- R. I. Enchev, B. A. Schulman and M. Peter, Nat. Rev. Mol. Cell Biol., 2015, 16, 30–44 CrossRef CAS
- T. A. Soucy, P. G. Smith, M. A. Milhollen, A. J. Berger, J. M. Gavin, S. Adhikari, J. E. Brownell, K. E. Burke, D. P. Cardin, S. Critchley, C. A. Cullis, A. Doucette, J. J. Garnsey, J. L. Gaulin, R. E. Gershman, A. R. Lublinsky, A. McDonald, H. Mizutani, U. Narayanan, E. J. Olhava, S. Peluso, M. Rezaei, M. D. Sintchak, T. Talreja, M. P. Thomas, T. Traore, S. Vyskocil, G. S. Weatherhead, J. Yu, J. Zhang, L. R. Dick, C. F. Claiborne, M. Rolfe, J. B. Bolen and S. P. Langston, Nature, 2009, 458, 732–736 CrossRef CAS
- T. Nawrocki Steffan, P. Griffin, R. Kelly Kevin and S. Carew Jennifer, Expert Opin. Invest. Drugs, 2012, 21, 1563–1573 CrossRef CAS
- R. T. Swords, K. R. Kelly, P. G. Smith, J. J. Garnsey, D. Mahalingam, E. Medina, K. Oberheu, S. Padmanabhan, M. O'Dwyer, S. T. Nawrocki, F. J. Giles and J. S. Carew, Blood, 2010, 115, 3796–3800 CrossRef CAS
-
R. T. Swords, H. P. Erba, D. J. DeAngelo, P. G. Smith, M. D. Pickard, et al., ASH Annual Meeting, 2010, vol. 116, p. 658 Search PubMed
- J. E. Brownell, M. D. Sintchak, J. M. Gavin, H. Liao, F. J. Bruzzese, N. J. Bump, T. A. Soucy, M. A. Milhollen, X. Yang, A. L. Burkhardt, J. Ma, H.-K. Loke, T. Lingaraj, D. Wu, K. B. Hamman, J. J. Spelman, C. A. Cullis, S. P. Langston, S. Vyskocil, T. B. Sells, W. D. Mallender, I. Visiers, P. Li, C. F. Claiborne, M. Rolfe, J. B. Bolen and L. R. Dick, Mol. Cell, 2010, 37, 102–111 CrossRef CAS
- Y. Zhao, X. Xiong, L. Jia and Y. Sun, Cell Death Dis., 2012, 3, E386 CrossRef CAS
- J. I. Toth, L. Yang, R. Dahl and M. D. Petroski, Cell Rep., 2012, 1, 309–316 CrossRef CAS
- G. W. Xu, R. Hurren, N. Maclean, A. Sukhai Mahadeo, N. Bhattacharjee Rabindra, A. Goard Carolyn, D. Schimmer Aaron, I. Toth Julia, D. Petroski Matthew, R. da Silva Sara, S.-L. Paiva, T. Gunning Patrick, L. Lukkarila Julie and S. Dhe-Paganon, PLoS One, 2014, 9, e93530 CrossRef
- M. A. Milhollen, M. P. Thomas, U. Narayanan, T. Traore, J. Riceberg, B. S. Amidon, N. F. Bence, J. B. Bolen, J. Brownell, L. R. Dick, H.-K. Loke, A. A. McDonald, J. Ma, M. G. Manfredi, T. B. Sells, M. D. Sintchak, X. Yang, Q. Xu, E. M. Koenig, J. M. Gavin and P. G. Smith, Cancer Cell, 2012, 21, 388–401 CrossRef CAS
- K.-J. Wu, H.-J. Zhong, G. Li, C. Liu, H.-M. D. Wang, D.-L. Ma and C.-H. Leung, Eur. J. Med. Chem., 2018, 143, 1021–1027 CrossRef CAS
- P. Lu, Y. Guo, L. Zhu, Y. Xia, Y. Zhong and Y. Wang, Eur. J. Med. Chem., 2018, 154, 294–304 CrossRef CAS
- S. Ni, X. Chen, Q. Yu, Y. Xu, Z. Hu, J. Zhang, W. Zhang, B. Li, X. Yang, F. Mao, J. Huang, Y. Sun, J. Li and L. Jia, Eur. J. Med. Chem., 2020, 185, 111848 CrossRef CAS
- H.-J. Zhong, V. P.-Y. Ma, Z. Cheng, D. S.-H. Chan, H.-Z. He, K.-H. Leung, D.-L. Ma and C.-H. Leung, Biochimie, 2012, 94, 2457–2460 CrossRef CAS
- C.-H. Leung, D. S.-H. Chan, H. Yang, R. Abagyan, S. M.-Y. Lee, G.-Y. Zhu, W.-F. Fong and D.-L. Ma, Chem. Commun., 2011, 47, 2511–2513 RSC
- H.-J. Zhong, H. Yang, D. S.-H. Chan, C.-H. Leung, H.-M. Wang and D.-L. Ma, PLoS One, 2012, 7, e49574 CrossRef CAS
- H. Zhou, J. Lu, L. Liu, D. Bernard, C.-Y. Yang, E. Fernandez-Salas, K. Chinnaswamy, S. Layton, J. Stuckey, Q. Yu, W. Zhou, Z. Pan, Y. Sun and S. Wang, Nat. Commun., 2017, 8, 1–12 CrossRef
- H. Zhou, W. Zhou, B. Zhou, L. Liu, T.-R. Chern, K. Chinnaswamy, J. Lu, D. Bernard, C.-Y. Yang, S. Li, M. Wang, J. Stuckey, Y. Sun and S. Wang, J. Med. Chem., 2018, 61, 1934–1950 CrossRef CAS
- H. S. Kim, J. T. Hammill, D. C. Scott, Y. Chen, J. Min, J. Rector, B. Singh, B. A. Schulman and R. K. Guy, J. Med. Chem., 2019, 62, 8429–8442 CrossRef CAS
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nature, 2004, 3, 660–672 CAS
- D. A. Erlanson, A. C. Braisted, D. R. Raphael, M. Randal, R. M. Stroud, E. M. Gordon and J. A. Wells, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9367–9372 CrossRef CAS
- O. Sperandio, M. A. Miteva and B. O. Villoutreix, Methods Princ. Med. Chem., 2011, 48, 435–465 CAS
- L. M. C. Meireles and G. Mustata, Curr. Top. Med. Chem., 2011, 11, 248–257 CrossRef CAS
- E. Ko, A. Raghuraman, L. M. Perez, T. R. Ioerger and K. Burgess, J. Am. Chem. Soc., 2013, 135, 167–173 CrossRef CAS
- D. Xin, A. Holzenburg and K. Burgess, Chem. Sci., 2014, 5, 4914–4921 RSC
- J. Taechalertpaisarn, B. Zhao, X. Liang and K. Burgess, J. Am. Chem. Soc., 2018, 140, 3242–3249 CrossRef CAS
- C.-M. Lin, M. Arancillo, J. Whisenant and K. Burgess, Angew. Chem., Int. Ed., 2020, 59, 9398–9402 CrossRef CAS
- A. Song, X. Wang, J. Zhang, J. Marik, C. B. Lebrilla and K. S. Lam, Bioorg. Med. Chem. Lett., 2004, 14, 161–165 CrossRef CAS
- B. Zhao, K. Zhang, E. B. Villhauer, K. Bhuripanyo, H. Kiyokawa, H. Schindelin and J. Yin, ChemBioChem, 2013, 14, 1323–1330 CrossRef CAS
- A. M. Rossi and C. W. Taylor, Nat. Protoc., 2011, 6, 365–387 CrossRef CAS
- M. Meldal and C. W. Tornoe, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS
- P. B. Alper, S.-C. Hung and C.-H. Wong, Tetrahedron Lett., 1996, 37, 6029–6032 CrossRef CAS
- P. Lu, X. Liu, X. Yuan, M. He, Y. Wang, Q. Zhang and P.-k. Ouyang, ACS Chem. Biol., 2016, 11, 1901–1907 CrossRef CAS
- K. Taguchi, N. Fujikawa, M. Komatsu, T. Ishii, M. Unno, T. Akaike, H. Motohashi and M. Yamamoto, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 13561–13566 CrossRef CAS
- M. Furukawa and Y. Xiong, Mol. Cell. Biol., 2005, 25, 162–171 CrossRef CAS
- J. Liang, J. Zubovitz, T. Petrocelli, R. Kotchetkov, M. K. Connor, K. Han, J.-H. Lee, S. Ciarallo, C. Catzavelos, R. Beniston, E. Franssen and J. M. Slingerland, Nat. Med., 2002, 8, 1153–1160 CrossRef CAS
- C. Han, L. Jin, Y. Mei and M. Wu, Cell. Signal., 2013, 25, 144–149 CrossRef CAS
- S.-T. Wang, H. J. Ho, J.-T. Lin, J.-J. Shieh and C.-Y. Wu, Cell Death Dis., 2017, 8, e2626 CrossRef CAS
- J. Qi, M. Oppenheimer and P. Sobrado, Enzyme Res., 2011, 513905–513909 Search PubMed
- B. Y. Feng and B. K. Shoichet, Nat. Protoc., 2006, 1, 550–553 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Details of azido acid, Fmoc-alkyne, solid phase syntheses and characterizations, protocols for the biological assays (cell viability, flow cytometry, in vitro NEDDylation assay, cell-based immunoblot, NAE protein siRNA knockdown, fluorescence polarization), and confocal imaging. See DOI: 10.1039/d0sc00958j |
| This journal is © The Royal Society of Chemistry 2021 |