
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfoxide hemithioindigo tweezers – visible light addressable capture and release†
Thomas
Bartelmann
a,
Frederik
Gnannt
b,
Max
Zitzmann
b,
Peter
Mayer
a and
Henry
Dube
 *ab
*ab
aLudwig-Maximilians-Universität München, Department of Chemistry, Center for Integrated Protein Science CIPSM, Butenandtstr. 5-13, 81377 München, Germany. E-mail: henry.dube@fau.de
bFriedrich–Alexander-Universität Erlangen–Nürnberg, Department of Chemistry and Pharmacy, Nikolaus-Fiebiger-Str. 10, 91058 Erlangen, Germany
First published on 14th January 2021
Abstract
Introducing responsive elements into supramolecular recognition systems offers great advantages for the control of intermolecular interactions and represents an important stepping stone towards multi-purpose and reprogrammable synthetic systems. Of particular interest is implementation of light-responsiveness because of the unique ease and precision of this signal. Here we present visible light responsive hemithioindigo-based molecular tweezers that bear a highly polar sulfoxide function as an additional recognition unit inside their binding site. Sulfur oxidation allows to simultaneously enhance all crucial properties of this receptor type i.e. photoswitching capability, thermal stability of individual switching states, binding affinity, and binding modulation upon switching. With a novel titration method the thermodynamic binding parameters were determined using reduced sample amounts. Employing these strongly enhanced molecular tweezers allowed to demonstrate photocontrol of intermolecular charge transfer in a reversible manner.
Introduction
The control of molecular recognition processes by external stimuli is a current focus of supramolecular chemistry as an important milestone on the way to programmable synthetic behavior and multi-purpose synthetic systems. In this context reversible control is highly desirable and many switchable molecular processes have been devised that allow regulation of supramolecular association by different signals.1–8 Of particular importance in this regard are capture and release systems with prospects in e.g. molecular machine building,5,9 switchable catalysis,10–15 drug delivery,16 or responsive molecular tools for chemical biology.17,18 A very promising signal is light irradiation because of its ease of application, high spatio-temporal resolution, waste-free nature, and straight-forward intensity regulation.6–8,10,11,15 These advantages have been recognized early and consequentially light controlled hydrophobic cyclodextrin binding or metal ion capture and release represent some of the first purely synthetic examples in the field.19–24 Despite a plethora of variants currently available (see ref. 25–49 for selected examples) challenges remain to this day even for fundamental light-controlled catch and release. Examples are the scarcity of visible light addressable systems, restrictions in the particular types of weak bonding interactions that can be invoked, unwanted thermal deterioration of switching states, or low ON/OFF modulation for the affinities upon switching.6,7,38,39,50 More specifically, for the majority of light-responsive host–guest systems stilbenes, azobenzenes, diarylethenes, or spiropyranes are used as photochromic unit requiring UV light for at least one photoswitching direction. Currently many efforts are therefore directed at establishing visible light responsive behavior in such host–guest systems.51–53 Typical Gibbs energies of activation for thermal cis-to-trans isomerization of azobenzenes and spiropyranes are in the 22–26 kcal mol−1 range. This leads to half-lives of the metastable switching states in the min to hours time regimes at ambient temperatures and thus unwanted spontaneous back-conversion. Additionally switching capacities are oftentimes <80% and residual binding of low affinity forms can still be sizable leading to diminished overall responsiveness. As to the types of weak interactions that can be used to drive association, pure aromatic interactions, halogen bonding, or anion–aromatic interactions are currently strongly underrepresented in light-responsive supramolecular associations. We have addressed some of these issues with the development of hemithioindigo (HTI) based supramolecular receptors that are responsive to visible light and allow ON/OFF switching of either weak polar aromatic interactions with electron deficient guest molecules (Fig. 1)27,28 or hydrogen bonding driven recognition.54 For example, we presented a bis-HTI receptor 1 that folds into a helix in its high affinity state in response to blue light irradiation and planarizes into a low affinity state after heating.27 Subsequently we devised two different molecular tweezers55–59 motives 2 and 3 that respond reversibly to the same wavelengths of light with opposite affinity changes.28 Tweezers 2 and 3 possess the same HTI core structure as switching unit making them responsive to visible light in both switching directions. The stilbene fragment (see Fig. 1 for assignment of the two fragments) is ring-fused to the photoisomerizable double bond to rigidify the structure and increase preorganization. The two electron rich biphenyl arms each bear two ortho-methyl substituents at their attachment points to the HTI core structure. The resulting sterical hindrance forces the biphenyls into a mutual parallel arrangement and thus preorganize them for the intended sandwich-type aromatic interactions. At the thioindigo fragment two different attachment points are used for tweezers 2 and 3 resulting in high affinity for the E configuration of 2 with both biaryl arms pointing to the same side of the molecule. In contrast the high affinity for tweezers 3 is obtained in the Z-configuration. In combination the tweezers 2 and 3 allowed to control supramolecular guest translocation by reduced signaling. Only one visible light signal orchestrates geometry changes, release from the first tweezers, and capture by the second. A different wavelength of visible light reverses the translocation of the guest molecule in this system. Although both molecular tweezers 2 and 3 responded to the same wavelengths of light and both were able to regulate capture and release in an ON/OFF fashion, tweezers 3 showed nonoptimal performance in their maximum affinity as well as in their switching ability. For both molecular tweezers 2 and 3 thermal stability of the switching states was also not very high with associated Gibbs energy of activation in the 25 kcal mol−1 range. In this work we present a synthetically straight-forward approach – one-step oxidation to the corresponding sulfoxide – to significantly improve all important characteristics of HTI tweezers (Fig. 1). With this approach it is possible to increase switching capabilities, affinity, as well as thermal stability of the switching states – all at the same time. In addition, we present a method enabling very convenient, precise, and repeatable assessment of association parameters for reversibly switchable supramolecular association phenomena – particularly if light is used as stimulus.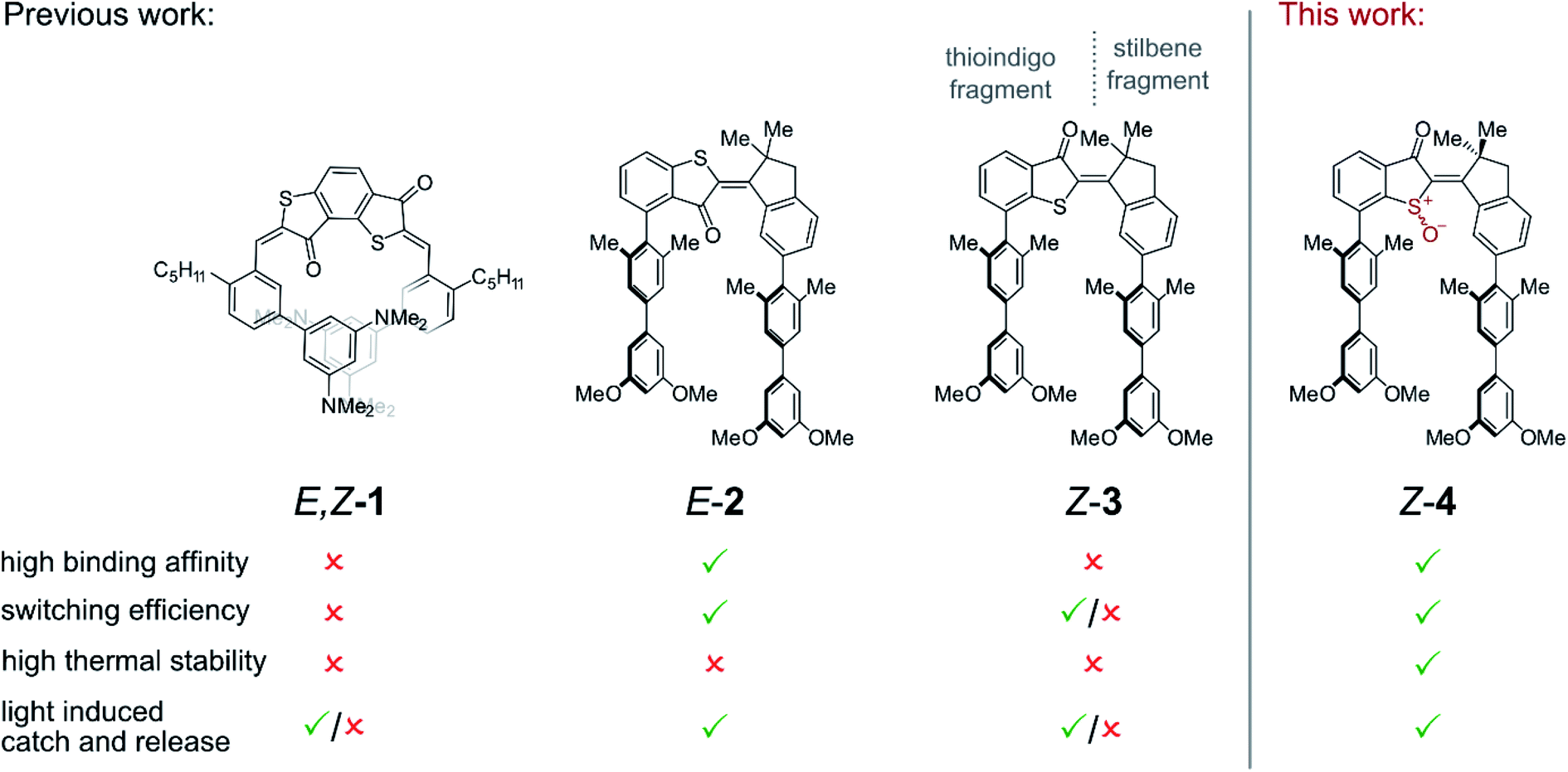 | ||
| Fig. 1 Progress in molecular receptors based on HTI for visible light photocontrol of host–guest polar aromatic interactions. | ||
Results and discussion
In this work we introduce another type of photoswitchable molecular tweezers 4, which are based on the earlier reported tweezers 3 but point a highly polar sulfoxide moiety into their binding site (Fig. 1). Tweezers 4 incorporate a central HTI60 moiety as photoresponsive element, which belongs to the class of indigoid chromophores61 currently emerging as promising alternative photoswitch motives.27,62–68 Two electron rich biphenyl side arms are attached on each, the stilbene and the thioindigo, molecular fragment. The high affinity “closed” state of 4 possesses Z configuration of the photoisomerizable double bond and arranges the two biphenyl side-arms in a parallel fashion on the same side of the molecule. The arms spatial orientation and distance are ideally suited for binding of an electron deficient planar aromatic guest molecule via polar aromatic interactions. Oxidation of the sulfur atom to the corresponding sulfoxide introduces a strongly negatively polarized group inside the binding cavity. In this way additional attractive interactions were projected to be available for binding of electron deficient guest molecules. Thus, increased binding affinities as compared to the related unoxidized tweezers 3 were expected. Upon Z to E photoisomerization the two side arms are spatially separated and project outwards at opposite sides of the central HTI core. As a result, the binding site opens up, available attractive interactions towards the guest molecule are reduced, and the guest molecule can effectively be released.A computational study on the DFT B3LYP/6-311G(d,p) level of theory was conducted to elucidate the molecular geometries of both isomers as well as electronic and polarity characteristics (Fig. 2a and c). In addition, the supramolecular complexation with an electron deficient aromatic guest was also studied theoretically to test especially interactions with the sulfoxide function. To be able to compare affinities with the previously reported HTI tweezers 3 the same aromatic guest molecule DTF (5) was used throughout this study. The optimized structures of Z-4 and E-4 are shown in Fig. 2a with the electrostatic potential surfaces (ESP) depicted as well. In the Z isomeric form the electron rich biphenyl arms reside at the same side of the sulfur atom atop of each other. The thioindigo-fragment and the stilbene-fragment are twisted with respect to each other by 40°, which leads to some displacement of the attached biphenyl arms. The distance between the two arms is 7.3 Å as measured center-to-center between the parallel methyl-substituted aromatics directly attached to the HTI core. This distance is ideally suited for intercalation of aromatic guest molecules. As can clearly be seen by the ESP the sulfoxide moiety establishes high electron density at the center of the binding site. It offers additional positive interactions to a bound guest molecule e.g. as hydrogen bond acceptor. Together with the electron rich biphenyl arms a highly electron rich pocket is thus created that should be ideally suited for binding of electron deficient aromatics via multiple noncovalent interactions. In the open E isomeric form the two biphenyl arms appear completely separated spatially and any synergistic chelating effects should thus be removed. When studying the binding of DTF guest 5 several minima structures were found for the associated complex by Monte Carlo conformer search on the MM3* level of theory. The main difference in these structures are the orientations of the DTF guest inside the tweezers cavity. The five lowest energy structures were further optimized on the DFT B3LYP-GD3BJ/6-311(d,p) level of theory. The energetically most stable structure was found to be 2.9 kcal mol−1 lower in energy than the next minimum structure and consequentially should be populated almost exclusively. For this reason, it is discussed in the following in more detail (see the ESI† for the other minimum structures). In the complex the DTF guest is intercalated between the two Z-4 tweezers arms in a sandwich-type fashion (Fig. 2d). The distances between guest and biphenyl arms are found to be below the van der Waals radii of the constituting carbon atoms thus directly reporting on favorable polar aromatic interactions. Additional close interactions between the positively polarized hydrogen atoms of DTF and the sulfoxide oxygen atom are observed in the complex. In fact, as judging by the ESP of DTF the most positively polarized edge of the guest molecule is found pointing towards the sulfoxide oxygen, which emphasizes the importance of additional hydrogen-bond type interactions. As a result high binding affinities are expected in the closed Z-4 form but significantly less affinities in the E-4 form. After this promising theoretical assessment of tweezers 4 binding capabilities, their synthesis and experimental characterization was conducted next.
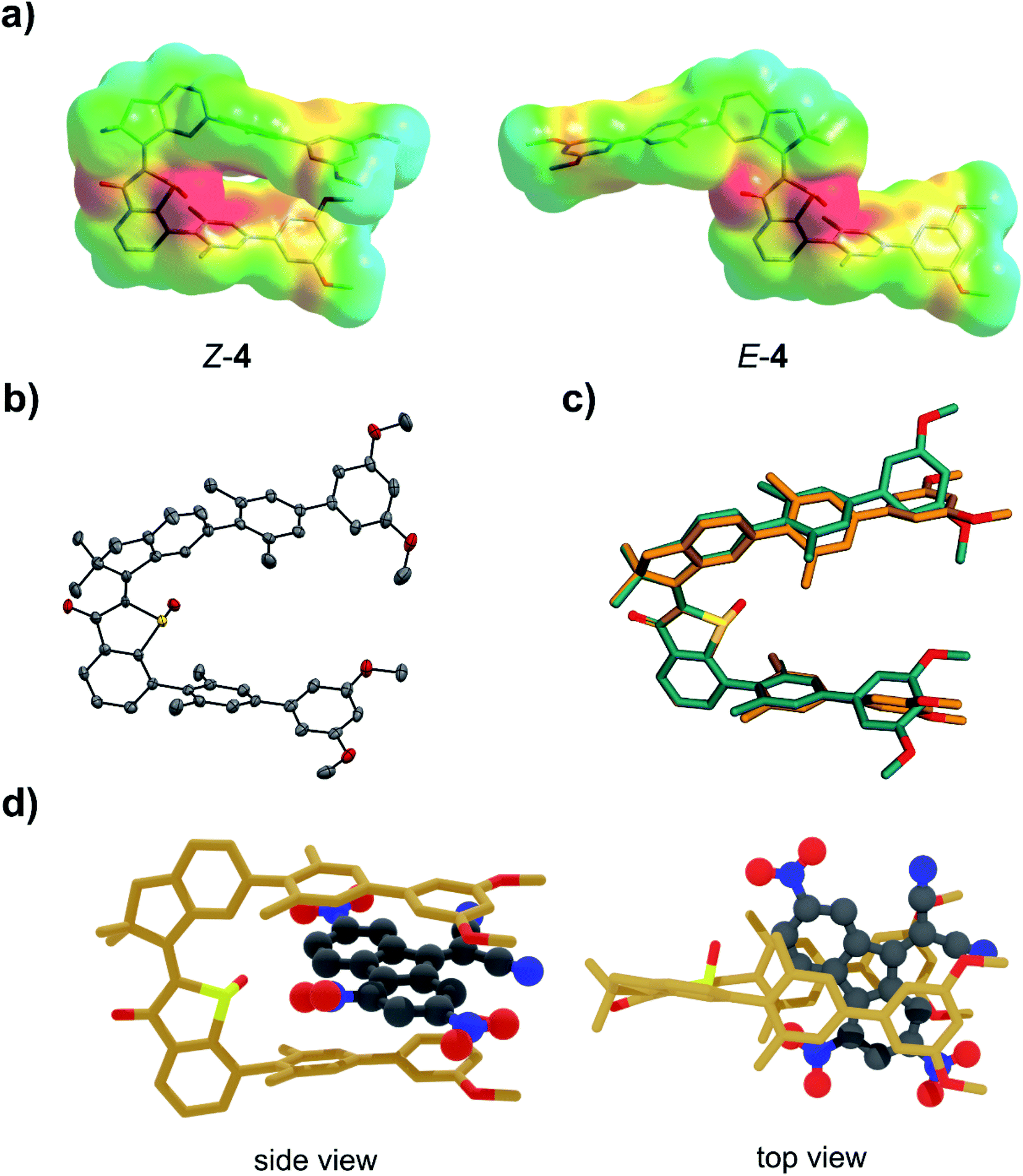 | ||
| Fig. 2 Molecular structures of tweezers 4 as assessed by theory (sub-figure (a) and (c)) and crystal structural analysis (sub-figure (b) and (c)) and calculated structure of the supramolecular complex of Z-4 with aromatic guest molecule 5 (sub-figure (d)). (a) Molecular structures of E-4 and Z-4 optimized on the DFT (B3LYP/6-311G(d,p)) level of theory. The corresponding ESPs quantify the overall polarity distribution. (b) Structure of Z-4 in the crystalline state. Thermal ellipsoids are drawn at a 50% probability level. (c) Comparison of the structures of Z-4 assessed by theory (orange) and crystal structure analysis (turquoise). (d) Structure of the lowest energy Z-4·5 complex calculated on the B3LYP-GD3BJ/6-311G(d,p) level of theory. | ||
Molecular tweezers 4 were synthesized by sulfur-oxidation of tweezers 3 to the corresponding sulfoxide as described in the ESI.† For the first time crystals suitable for X-ray structural analysis could be obtained for HTI tweezers, fortunately for the closed tweezers form Z-4 (Fig. 2b). The molecular structure of Z-4 as obtained in the crystal was found to be remarkably similar to the DFT-optimized structure. This can be seen especially when overlaying the two structures as shown in Fig. 2c. There is almost perfect superposition between theoretical and crystal structure in the HTI part of Z-4. Deviations are seen mostly on the outer methoxy-substituted aromatics of the biphenyl arms. As these moieties can rotate without significant barriers the overall agreement of the two structures is still surprisingly good. A subpopulation observed in the crystal possesses opposite stereo configuration at the sulfoxide but the rest of the structure remains exactly the same. This behavior emphasizes the effect of packing forces for the induced helicity about the isomerizable central double bond as well as for the biphenyl orientations. Because of the crystal structure the solution spectra of 4 could straightforwardly be assigned to the respective Z and E isomers facilitating study of their switching behavior and binding affinities.
Upon oxidation the (photo)physical and photochemical properties of 4 change significantly in comparison to 3. In agreement with a recent report from our group, oxidation of the sulfur atom to the corresponding sulfoxide leads to a hypsochromic shift of the absorption – and thus to a blue-shift of the switching wavelengths. Photoswitching of 4 was tested in CDCl3 solvent for a variety of wavelengths. Up to 88% of the E isomer could be accumulated in the photostationary state (pss) at 365 nm and up to 83% E isomer in the pss at 405 nm. Effective photoswitching can thus still be obtained with blue light, which renders these tweezers fully responsive to visible light. At 470 nm irradiation a pss containing 80% of the Z isomer was obtained, which could be increased to 83% at the same wavelength when using CD2Cl2 as solvent. Compared to the unoxidized tweezers 3 (maxima of 63% E isomer at 435 nm and 84% Z isomer at 530 nm were obtained) the photoswitching was therefore also improved significantly by oxidation of the sulfur atom albeit at the expense of hypsochromic absorption shifts (Fig. 3).
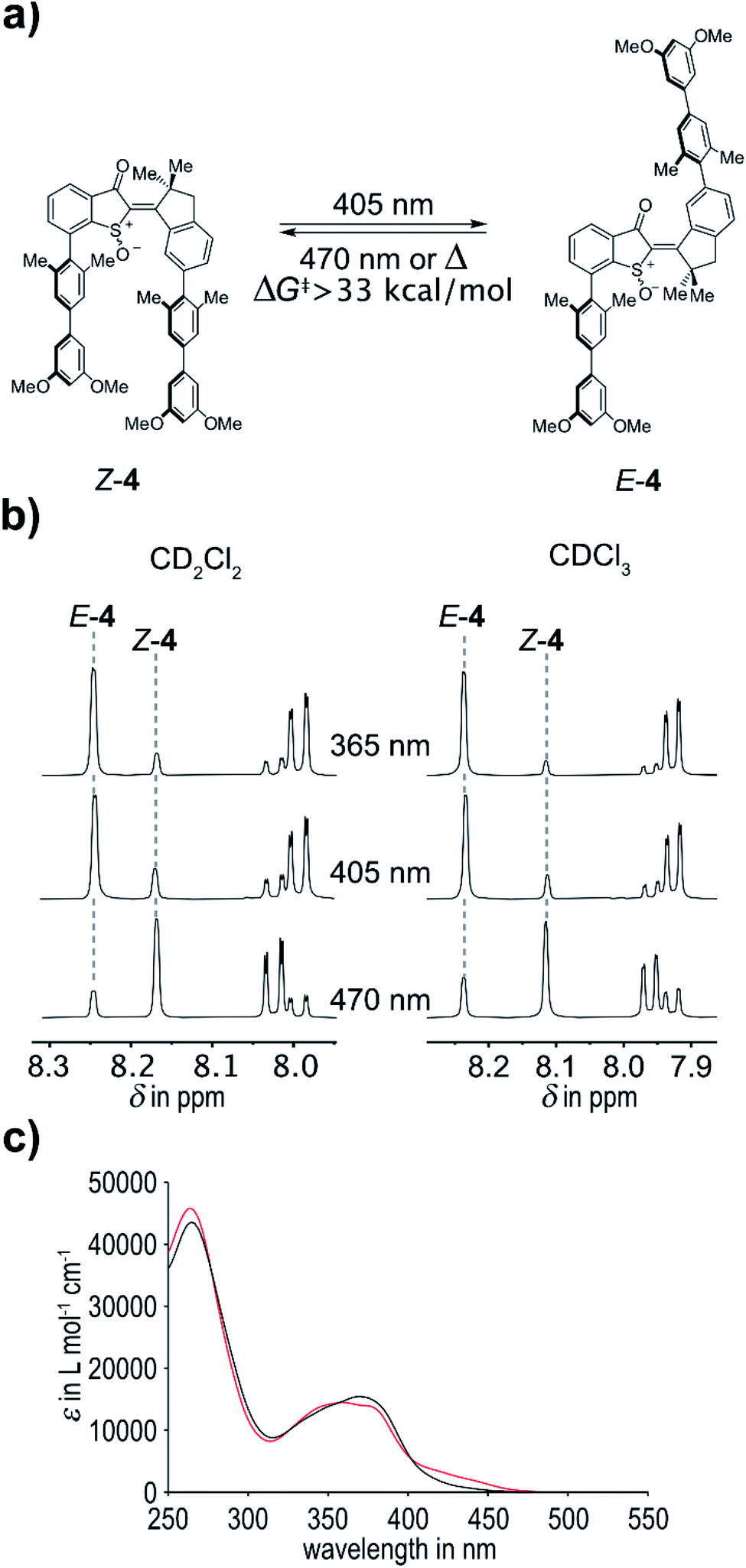 | ||
| Fig. 3 Switching behavior of tweezers 4. (a) Schematic representation of the switching process of 4. Z to E photoisomerization can be triggered by 405 nm light. E to Z photoisomerization can by induced by 470 nm light. The energy barrier associated with thermal E to Z isomerization is greater than 33 kcal mol−1 providing a highly bistable photoswitch. (b) Aromatic section of 1H-NMR spectra (400 MHz, 20 °C, left: CD2Cl2, right: CDCl3) of 4 recorded after irradiation with light of 365 nm, 405 nm, and 470 nm wavelengths until reaching the PSS. Indicative proton signals used to quantify the isomeric ratio in the respective PSS are highlighted. (c) Molar absorption coefficients of the pure isomers (black: Z isomer, red: E isomer) of 4 in CHCl3. | ||
For tweezers 4 the Z-isomeric state is the thermodynamically most stable state and spontaneous thermal relaxation of the metastable E isomer therefore needs to be investigated. After accumulation of E-4via blue light irradiation the thermal E to Z isomerization was attempted to be followed by 1H NMR spectroscopy in the dark. Toluene-d8 was used as solvent to facilitate measuring the associated kinetics on manageable time scales at higher temperatures. However, heating the sample to 100 °C for 27 h in the dark did not lead to a change in the isomer composition. It was thus possible to establish a lower limit for the associated Gibbs energy of activation ΔG‡ by assuming conservatively that 5% isomer conversion was obtained during the heating but was not detectable by the 1H NMR experiment. With this assumed conversion and applying first order kinetics for a fully converting system, a lower limit of ΔG‡ = 33.0 kcal mol−1 was obtained (see ESI† for details). Accordingly in chlorinated solvents such as CDCl3 or CD2Cl2 no thermal conversions were observed at the limited higher temperatures (up to 50 °C) available. With such high energy barrier truly bistable HTI tweezers are now available for the first time.
We then investigated the affinity of tweezers 4 for electron deficient aromatic guest molecule 5 with the aid of 1H NMR spectroscopy (see Fig. 4). In the open E isomeric form no binding affinity is observed as proton signals of the guest and tweezers E-4 show no visible shifts in the spectra. Likewise no signal shift of the DTF guest is observed in the presence of pure E-4. This behavior is similar to tweezers 3 in their low affinity open E isomeric form. The situation is quite different for the closed Z conformation of 4 where a binding constant K = 1090 L mol−1 for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry was measured at 20 °C by a newly developed and convenient in situ irradiation–titration method. For the latter an excess of tweezers 4 was added to solutions of the DTF guest molecule 5. Excess of guest could not be used because of its low solubility in CDCl3. By irradiation of the obtained solutions for varying duration times different ratios of binding Z-4 to guest 5 (with constant concentration) were obtained representing different titration points. The exact tweezers to guest ratios were determined by integration of suitable 1H NMR signals. Because of the full reversibility of the photoisomerization of 4 it is possible to collect as many titration points as desired using the same single sample. Analysis of the chemical shift differences occurring during titration then allowed us to determine the binding affinity of Z-4 with high precision. Another advantage of this titration method becomes evident when measuring the temperature dependence of binding constants. Again the same sample can be used for measuring at different temperatures, each time collecting as many titration points as desired by simple stepwise irradiations. In this way we have determined the enthalpy and entropy contributions to the binding affinity of Z-4 with respect to DTF 5. Based on this assessment the binding between Z-4 and DTF is mainly enthalpy driven with an enthalpy contribution of ΔH = −4.77 kcal mol−1 and an entropy of ΔS = −2.4 cal mol−1. When comparing the binding affinities of tweezers Z-4 (K = 4100 L mol−1 at −20 °C) with Z-3 (K = 2300 L mol−1 at −20 °C) for the same DTF guest a significant improvement of the binding for the former is observed.
:1 stoichiometry was measured at 20 °C by a newly developed and convenient in situ irradiation–titration method. For the latter an excess of tweezers 4 was added to solutions of the DTF guest molecule 5. Excess of guest could not be used because of its low solubility in CDCl3. By irradiation of the obtained solutions for varying duration times different ratios of binding Z-4 to guest 5 (with constant concentration) were obtained representing different titration points. The exact tweezers to guest ratios were determined by integration of suitable 1H NMR signals. Because of the full reversibility of the photoisomerization of 4 it is possible to collect as many titration points as desired using the same single sample. Analysis of the chemical shift differences occurring during titration then allowed us to determine the binding affinity of Z-4 with high precision. Another advantage of this titration method becomes evident when measuring the temperature dependence of binding constants. Again the same sample can be used for measuring at different temperatures, each time collecting as many titration points as desired by simple stepwise irradiations. In this way we have determined the enthalpy and entropy contributions to the binding affinity of Z-4 with respect to DTF 5. Based on this assessment the binding between Z-4 and DTF is mainly enthalpy driven with an enthalpy contribution of ΔH = −4.77 kcal mol−1 and an entropy of ΔS = −2.4 cal mol−1. When comparing the binding affinities of tweezers Z-4 (K = 4100 L mol−1 at −20 °C) with Z-3 (K = 2300 L mol−1 at −20 °C) for the same DTF guest a significant improvement of the binding for the former is observed.
In order to test if the observed improvements of binding performance upon oxidation is a general property of HTI-based molecular tweezers we have measured the photoswitchable host–guest performance for three additional guest molecules 6, 7, and 8 comparing both molecular tweezers 3 and 4 (see Table 1 as well as Fig. 4e for a comparison of the binding parameters for all guests and tweezers combinations). All titration experiments were done in independent duplicates to obtain error estimates for the obtained binding constants K. Because of solubility issues the binding constants for guests 6 and 8 were measured in CD2Cl2 solution instead of CDCl3. For all cases the same behavior was observed: high affinity for the closed Z-isomeric tweezers configuration and basically no affinity for the open E-isomeric form. Roughly a doubling of the binding constant for the oxidized tweezers 4 was found as compared to the unoxidized version 3 with the exception of guest 8, which was bound only 1.2 times better by 4. Additionally a remarkable selectivity for guest molecules with more extended π-surfaces was observed since the binding constants with guests 6 to 8 were significantly weaker compared to guest 5 by factors of 31–47 (values are compared at the same temperature of 293 K).
| Guest | Z-3 | Z-4 |
|---|---|---|
| a Average values obtained from two independent titration experiments with error margins given. | ||
| DTF (5) | 2380 (253 K, CDCl3) | 4140 (253 K, CDCl3) |
| 1830 (273 K, CDCl3) | ||
| 1090 ± 70 (293 K, CDCl3)a | ||
| TCNB (6) | 10 ± 1.5 (293 K, CD2Cl2)a | 26 ± 0.6 (293 K, CD2Cl2)a |
| PhA (7) | 16 ± 1.3 (293 K, CDCl3)a | 35 ± 4.7 (293 K), CDCl3a |
| 3NPN (8) | 18 ± 1.6 (293 K, CD2Cl2)a | 23 ± 0.04 (293 K, CD2Cl2)a |
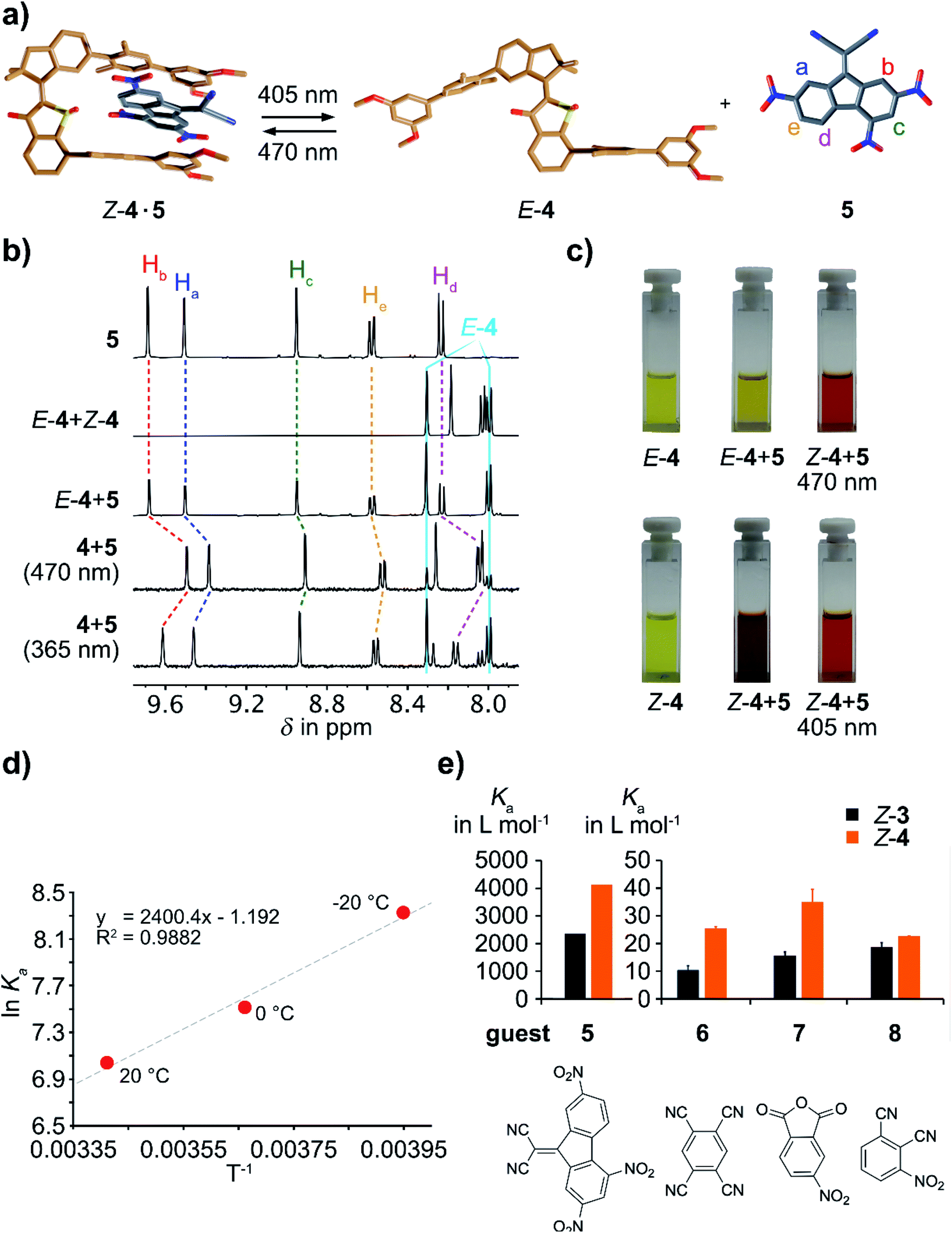 | ||
| Fig. 4 Photocontrolled capture and release of DTF (5) by molecular tweezers 4 and light control of charge transfer from the host to the guest. (a) Schematic representation of visible light induced capture and release of guest molecule 5 by tweezers 4. Light of 405 nm wavelength induces release of 5. 470 nm light induces capture of 5. (b) Aromatic section of 1H-NMR spectra (400 MHz, CDCl3, 20 °C) recorded to assess binding between 5 and 4. Spectra of pure guest 5 and a 1:1 Z:E isomeric mixture of pure tweezers 4 are shown for comparison. No proton signal shifts are observed when combining pure E-4 and 5. Photoswitching of E-4 with 470 nm light induces strong shifts of the proton signals of 5. Photoswitching of 4 with 365 nm light reverses the shift of the proton signals of 5 and restores them to their original position. (c) Photographs illustrating the color changes associated CT modulations upon catch and release of 5 by tweezers 4 in CHCl3 solution. Addition of 5 to a solution of pure Z-4 induces a strong color shift to red, originating from the CT band of the formed Z-4·5 complex. Addition of 5 to E-4 does not induce a color change. Photoisomerization of the respective solutions cause color changes to lighter and deeper orange. (d) Van't Hoff plot depicting the temperature dependence of the binding between Z-4 and 5 enabling determination of ΔH and ΔS contributions. (e) Comparison of the measured binding constants Ka for association of different guest molecules 5–8 with tweezers Z-3 (grey bars) and Z-4 (orange bars). Ka values for guests 5 and 7 were obtained in CDCl3 and for guests 6 and 8 in CD2Cl2 solutions. Ka values for guest 5 are compared at 253 K and for all other guests at 293 K. | ||
Therefore, it can be concluded that the additional introduction of a strongly negatively polarized residue at the binding site is indeed very effective in increasing the binding affinity in HTI-based molecular tweezers for electron deficient aromatic guests.
When binding of DTF (5) was studied, strongly visible changes in the color of the solutions were noticed. As shown in Fig. 4c addition of excess 5 to a CHCl3 solution of pure Z-4 results in a change from the initial yellow to a deep brown-red color. Such color change is indicative of a pronounced intermolecular charge transfer (CT) complex.69–72 No significant color change is observed for the mixture of 5 and pure E-4 except for a slight deepening of the yellow hue. Therefore, the strong ON/OFF modulation of binding between the two isomeric states of 4 is evident by the naked eye. Upon photoirradiation of the respective solutions reversible photoisomerization of 4 takes place and an orange coloration is observed. The less obvious changes visible to the naked eye upon reversible switching between the PSS at 405 nm and 470 nm irradiation can be explained by the less than 100% switching conversion in both directions and the excess of 5 used in the experiments. This leads to still significant color impression to the human eye even in the presence of only small amounts of remaining Z isomer. Nevertheless a deeper coloration is seen in Z-4 enriched solution as compared to the E-4 enriched one. In this way visible light photocontrol of intermolecular CT events was achieved by employing the herein reported molecular tweezers 4.
In summary we present a novel variant of HTI molecular tweezers bearing a highly polar sulfoxide within their binding site. As a result of the sulfur oxidation all properties of the tweezers are improved significantly. Photoswitching capabilities are enhanced and fully reversible >80% enrichment of either isomer is now possible with visible light irradiation. Thermal stability of the metastable E isomer is heightened and the associated energy barrier reaches beyond 33 kcal mol−1, which renders the oxidized tweezers truly bistable. Binding affinity of the Z isomer towards electron deficient aromatic guests is almost doubled as compared to the unoxidized tweezers, while for the E isomer no discernible binding affinity is present. Because of this favorable behavior a combined irradiation-titration method could be developed that allows repeated and precise measuring of the association constant and elucidation of its temperature dependence using the same sample. Overall very high ON/OFF modulation of supramolecular aromatic binding is observed with this receptor allowing to bring intermolecular CT under the control of visible light. In the future, further applications will be explored with special emphasis on achieving water solubility and recognition of biological substrates.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. Dube thanks the “Fonds der Chemischen Industrie” for a Liebig fellowship (Li 188/05) and the Deutsche Forschungsgemeinschaft (DFG) for an Emmy Noether fellowship (DU 1414/1-2). We further thank the Deutsche Forschungsgemeinschaft (SFB 749, A12) and the Cluster of Excellence ‘Center for Integrated Protein Science Munich’ (CIPSM) for financial support.References
- M. J. Langton and P. D. Beer, Rotaxane and catenane host structures for sensing charged guest species, Acc. Chem. Res., 2014, 47(7), 1935–1949 CrossRef CAS.
- C. J. Bruns and J. F. Stoddart, Rotaxane-based molecular muscles, Acc. Chem. Res., 2014, 47(7), 2186–2199 CrossRef CAS.
- X. Yan, F. Wang, B. Zheng and F. Huang, Stimuli-responsive supramolecular polymeric materials, Chem. Soc. Rev., 2012, 41(18), 6042–6065 RSC.
- A. J. McConnell, C. S. Wood, P. P. Neelakandan and J. R. Nitschke, Stimuli-Responsive Metal-Ligand Assemblies, Chem. Rev., 2015, 115(15), 7729–7793 CrossRef CAS.
- A. Goswami, S. Saha, P. K. Biswas and M. Schmittel, (Nano)mechanical Motion Triggered by Metal Coordination: from Functional Devices to Networked Multicomponent Catalytic Machinery, Chem. Rev., 2020, 120(1), 125–199 CrossRef CAS.
- H. Dube, F. Durola, D. Ajami and J. Rebek Jr, Molecular Switching in Nanospaces, J. Chin. Chem. Soc., 2010, 57, 595–603 CrossRef CAS.
- O. B. Berryman, H. Dube and J. Rebek Jr, Photophysics Applied to Cavitands and Capsules, Isr. J. Chem., 2011, 51(7), 700–709 CrossRef CAS.
- V. Ramamurthy and J. Sivaguru, Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition, Chem. Rev., 2016, 116(17), 9914–9993 CrossRef CAS.
- S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Artificial Molecular Machines, Chem. Rev., 2015, 115(18), 10081–10206 CrossRef CAS.
- S. F. Pizzolato, P. Štacko, J. C. M. Kistemaker, T. van Leeuwen and B. L. Feringa, Phosphoramidite-based photoresponsive ligands displaying multifold transfer of chirality in dynamic enantioselective metal catalysis, Nat. Catal., 2020, 3(6), 488–496 CrossRef CAS.
- R. Göstl, A. Senf and S. Hecht, Remote-controlling chemical reactions by light: towards chemistry with high spatio-temporal resolution, Chem. Soc. Rev., 2014, 43(6), 1982–1996 RSC.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, A photoswitchable catalyst system for remote-controlled (co)polymerization in situ, Nat. Catal., 2018, 1(7), 516–522 CrossRef CAS.
- V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44(15), 5341–5370 RSC.
- J. Choudhury, Recent developments on artificial switchable catalysis, Tetrahedron Lett., 2018, 59(6), 487–495 CrossRef CAS.
- R. Dorel and B. L. Feringa, Photoswitchable catalysis based on the isomerisation of double bonds, Chem. Commun., 2019, 55(46), 6477–6486 RSC.
- M. J. Webber and R. Langer, Drug delivery by supramolecular design, Chem. Soc. Rev., 2017, 46(21), 6600–6620 RSC.
- K. Petkau-Milroy and L. Brunsveld, Supramolecular chemical biology; bioactive synthetic self-assemblies, Org. Biomol. Chem., 2013, 11(2), 219–232 RSC.
- J. A. Finbloom and M. B. Francis, Supramolecular strategies for protein immobilization and modification, Curr. Opin. Chem. Biol., 2018, 46, 91–98 CrossRef CAS.
- S. Shinkai, T. Ogawa, T. Nakaji, Y. Kusano and O. Nanabe, Photocontrolled Extraction Ability of Azobenzene-Bridged Azacrown Ether, Tetrahedron Lett., 1979, 47, 4569–4572 CrossRef.
- S. Shinkai, T. Nakaji, Y. Nishida, T. Ogawa and O. Manabe, Photoresponsive Crown Ethers. 1. Cis-Trans Isomerism of Azobenzene as a Tool to Enforce Conformational Changes of Crown Ethers and Polymers, J. Am. Chem. Soc., 1980, 102, 5860–5865 CrossRef CAS.
- S. Shinkai, T. Nakaji, T. Ogawa, K. Shigematsu and O. Manabe, Photoresponsive Crown Ethers. 2. Photocontrol of Ion Extraction and Ion Transport by a Bis(crown ether) with a Butterfly-like Motion, J. Am. Chem. Soc., 1981, 103, 111–115 CrossRef CAS.
- U. Akihiko, S. Rumiko and O. Tetsuo, One Host-Two Guests Complexation Of Photosensitive Capped Cyclodextrin With Amino Acids, Chem. Lett., 1979, 8(7), 841–844 CrossRef.
- A. Ueno, H. Yoshimura, R. Saka and T. Osa, Photocontrol of binding ability of capped cyclodextrin, J. Am. Chem. Soc., 1979, 101(10), 2779–2780 CrossRef CAS.
- A. Ueno, K. Takahashi and T. Osa, Photoregulation of catalytic activity of β-cyclodextrin by an azo inhibitor, J. Chem. Soc., Chem. Commun., 1980, 17, 837–838 RSC.
- H. Dube, D. Ajami and J. Rebek Jr, Photochemical control of reversible encapsulation, Angew. Chem., Int. Ed., 2010, 49(18), 3192–3195 CrossRef CAS.
- H. Dube and J. Rebek Jr, Selective guest exchange in encapsulation complexes using light of different wavelengths, Angew. Chem., Int. Ed., 2012, 51(13), 3207–3210 CrossRef CAS.
- M. Guentner, E. Uhl, P. Mayer and H. Dube, Photocontrol of Polar Aromatic Interactions by a Bis-Hemithioindigo Based Helical Receptor, Chem.–Eur. J., 2016, 22, 16433–16436 CrossRef CAS.
- S. Wiedbrauk, T. Bartelmann, S. Thumser, P. Mayer and H. Dube, Simultaneous complementary photoswitching of hemithioindigo tweezers for dynamic guest relocalization, Nat. Commun., 2018, 9(1), 1456 CrossRef.
- G. H. Clever, S. Tashiro and M. Shionoya, Light-Triggered Crystallization of a Molecular Host-Guest Complex, J. Am. Chem. Soc., 2010, 132, 9973–9975 CrossRef CAS.
- M. Han, Y. Luo, B. Damaschke, L. Gomez, X. Ribas, A. Jose, P. Peretzki, M. Seibt and G. H. Clever, Light-Controlled Interconversion between a Self-Assembled Triangle and a Rhombicuboctahedral Sphere, Angew. Chem., Int. Ed., 2016, 55(1), 445–449 CrossRef CAS.
- R. J. Li, J. J. Holstein, W. Hiller, J. Andreasson and G. H. Clever, Mechanistic Interplay between Light-Switching and Guest-Binding in Photochromic [Pd2Dithienylethene4] Coordination Cages, J. Am. Chem. Soc., 2019, 141(5), 2097–2103 CrossRef CAS.
- N. Kishi, M. Akita, M. Kamiya, S. Hayashi, H. F. Hsu and M. Yoshizawa, Facile catch and release of fullerenes using a photoresponsive molecular tube, J. Am. Chem. Soc., 2013, 135(35), 12976–12979 CrossRef CAS.
- S. J. Wezenberg, M. Vlatkovic, J. C. Kistemaker and B. L. Feringa, Multi-state regulation of the dihydrogen phosphate binding affinity to a light- and heat-responsive bis-urea receptor, J. Am. Chem. Soc., 2014, 136(48), 16784–16787 CrossRef CAS.
- M. Vlatkovic, B. L. Feringa and S. J. Wezenberg, Dynamic Inversion of Stereoselective Phosphate Binding to a Bisurea Receptor Controlled by Light and Heat, Angew. Chem., Int. Ed., 2016, 55(3), 1001–1004 CrossRef CAS.
- T. van Leeuwen, G. H. Heideman, D. Zhao, S. J. Wezenberg and B. L. Feringa, In situ control of polymer helicity with a non-covalently bound photoresponsive molecular motor dopant, Chem. Commun., 2017, 53(48), 6393–6396 RSC.
- O. B. Berryman, A. C. Sather, A. Lledo and J. Rebek Jr, Switchable catalysis with a light-responsive cavitand, Angew. Chem., Int. Ed., 2011, 50(40), 9400–9403 CrossRef CAS.
- M. Samanta, A. Rananaware, D. N. Nadimetla, S. A. Rahaman, M. Saha, R. W. Jadhav, S. V. Bhosale and S. Bandyopadhyay, Light triggered encapsulation and release of C60 with a photoswitchable TPE-based supramolecular tweezers, Sci. Rep., 2019, 9(1), 9670 CrossRef.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Recent Progress in Photoswitchable Supramolecular Self-Assembling Systems, Adv. Opt. Mater., 2016, 4(9), 1322–1349 CrossRef CAS.
- D. H. Qu, Q. C. Wang, Q. W. Zhang, X. Ma and H. Tian, Photoresponsive Host-Guest Functional Systems, Chem. Rev., 2015, 115(15), 7543–7588 CrossRef CAS.
- G. Moncelsi, L. Escobar, H. Dube and P. Ballester, 2-(4'-Pyridyl-N-oxide)-Substituted Hemithioindigos as Photoresponsive Guests for a Super Aryl-Extended Calix[4]pyrrole Receptor, Chem.–Asian J., 2018, 13(12), 1632–1639 CrossRef CAS.
- X. Liao, G. Chen, X. Liu, W. Chen, F. Chen and M. Jiang, Photoresponsive pseudopolyrotaxane hydrogels based on competition of host-guest interactions, Angew. Chem., Int. Ed., 2010, 49(26), 4409–4413 CrossRef CAS.
- Y. Norikane, K. Kitamoto and N. Tamaoki, Novel Crystal Structure, Cis–Trans Isomerization, and Host Property of Meta-Substituted Macrocyclic Azobenzenes with the Shortest Linkers, J. Org. Chem., 2003, 68(22), 8291–8304 CrossRef CAS.
- T. S. C. MacDonald, B. L. Feringa, W. S. Price, S. J. Wezenberg and J. E. Beves, Controlled Diffusion of Photoswitchable Receptors by Binding Anti-electrostatic Hydrogen-Bonded Phosphate Oligomers, J. Am. Chem. Soc., 2020, 142(47), 20014–20020 CrossRef CAS.
- S. Lee and A. H. Flood, Photoresponsive receptors for binding and releasing anions, J. Phys. Org. Chem., 2013, 26(2), 79–86 CrossRef CAS.
- F. C. Parks, Y. Liu, S. Debnath, S. R. Stutsman, K. Raghavachari and A. H. Flood, Allosteric Control of Photofoldamers for Selecting between Anion Regulation and Double-to-Single Helix Switching, J. Am. Chem. Soc., 2018, 140(50), 17711–17723 CrossRef CAS.
- H. Zhao, S. Sen, T. Udayabhaskararao, M. Sawczyk, K. Kucanda, D. Manna, P. K. Kundu, J. W. Lee, P. Kral and R. Klajn, Reversible trapping and reaction acceleration within dynamically self-assembling nanoflasks, Nat. Nanotechnol., 2016, 11(1), 82–88 CrossRef CAS.
- T. Muraoka, K. Kinbara and T. Aida, Mechanical twisting of a guest by a photoresponsive host, Nature, 2006, 440(7083), 512–515 CrossRef CAS.
- H. Wang, F. Liu, R. C. Helgeson and K. N. Houk, Reversible photochemically gated transformation of a hemicarcerand to a carcerand, Angew. Chem., Int. Ed., 2013, 52(2), 655–659 CrossRef CAS.
- M. Baroncini, S. Silvi, M. Venturi and A. Credi, Reversible photoswitching of rotaxane character and interplay of thermodynamic stability and kinetic lability in a self-assembling ring-axle molecular system, Chem.–Eur. J., 2010, 16(38), 11580–11587 CrossRef CAS.
- Z. Yang, Z. Liu and L. Yuan, Recent Advances of Photoresponsive Supramolecular Switches, Asian J. Org. Chem., 2021, 10(1), 74–90 CrossRef CAS.
- H. Yan, Y. Qiu, J. Wang, Q. Jiang, H. Wang, Y. Liao and X. Xie, Wholly Visible-Light-Responsive Host-Guest Supramolecular Gels Based on Methoxy Azobenzene and beta-Cyclodextrin Dimers, Langmuir, 2020, 36(26), 7408–7417 CrossRef CAS.
- D. Wang, M. Wagner, H. J. Butt and S. Wu, Supramolecular hydrogels constructed by red-light-responsive host-guest interactions for photo-controlled protein release in deep tissue, Soft Matter, 2015, 11(38), 7656–7662 RSC.
- J. Wei, T.-T. Jin, Y.-F. Yin, X.-M. Jiang, S.-T. Zheng, T.-G. Zhan, J. Cui, L.-J. Liu, L.-C. Kong and K.-D. Zhang, Red-light-responsive molecular encapsulation in water: an ideal combination of photochemistry and host–guest interaction, Org. Chem. Front., 2019, 6(4), 498–505 RSC.
- K. Grill and H. Dube, Supramolecular Relay-Control of Organocatalysis with a Hemithioindigo-Based Molecular Motor, J. Am. Chem. Soc., 2020, 142(45), 19300–19307 CrossRef CAS.
- F.-G. Klärner and T. Schrader, Aromatic Interactions by Molecular Tweezers and Clips in Chemical and Biological Systems, Acc. Chem. Res., 2013, 46, 967–978 CrossRef.
- F.-G. Klärner and B. Kahlert, Molecular Tweezers and Clips as Synthetic Receptors. Molecular Recognition and Dynamics in Receptor-Substrate Complexes, Acc. Chem. Res., 2003, 36, 919–932 CrossRef.
- C.-W. Chen and H. W. Whitlock Jr, Molecular Tweezers: A Simple Model of Bifunctional Intercalation, J. Am. Chem. Soc., 1978, 100, 4921–4922 CrossRef CAS.
- S. C. Zimmerman and C. M. VanZyl, Rigid molecular tweezers: synthesis, characterization, and complexation chemistry of a diacridine, J. Am. Chem. Soc., 1987, 109(25), 7894–7896 CrossRef CAS.
- S. C. Zimmerman and W. Wu, A rigid molecular tweezers with an active site carboxylic acid: exceptionally efficient receptor for adenine in an organic solvent, J. Am. Chem. Soc., 1989, 111(20), 8054–8055 CrossRef CAS.
- S. Wiedbrauk and H. Dube, Hemithioindigo—an emerging photoswitch, Tetrahedron Lett., 2015, 56(29), 4266–4274 CrossRef CAS.
- C. Petermayer and H. Dube, Indigoid Photoswitches: Visible Light Responsive Molecular Tools, Acc. Chem. Res., 2018, 51(5), 1153–1163 CrossRef CAS.
- M. Guentner, M. Schildhauer, S. Thumser, P. Mayer, D. Stephenson, P. J. Mayer and H. Dube, Sunlight-powered kHz rotation of a hemithioindigo-based molecular motor, Nat. Commun., 2015, 6, 8406 CrossRef CAS.
- F. Kink, M. P. Collado, S. Wiedbrauk, P. Mayer and H. Dube, Bistable Photoswitching of Hemithioindigo with Green and Red Light: Entry Point to Advanced Molecular Digital Information Processing, Chem.–Eur. J., 2017, 23, 6237–6243 CrossRef CAS.
- A. Gerwien, P. Mayer and H. Dube, Photon-Only Molecular Motor with Reverse Temperature-Dependent Efficiency, J. Am. Chem. Soc., 2018, 140, 16442–16445 CrossRef CAS.
- A. Gerwien, T. Reinhardt, P. Mayer and H. Dube, Synthesis of Double-Bond Substituted Hemithioindigo Photoswitches, Org. Lett., 2018, 1, 232–235 CrossRef.
- A. Gerwien, M. Schildhauer, S. Thumser, P. Mayer and H. Dube, Direct evidence for hula twist and single-bond rotation photoproducts, Nat. Commun., 2018, 9(1), 2510 CrossRef.
- A. Gerwien, P. Mayer and H. Dube, Green light powered molecular state motor enabling eight-shaped unidirectional rotation, Nat. Commun., 2019, 10(1), 4449 CrossRef.
- E. Uhl, P. Mayer and H. Dube, Active and Unidirectional Acceleration of Biaryl Rotation by a Molecular Motor, Angew. Chem. Int. Ed., 2020, 59(14), 5730–5737 CrossRef CAS.
- A. Das and S. Ghosh, Supramolecular assemblies by charge-transfer interactions between donor and acceptor chromophores, Angew. Chem., Int. Ed., 2014, 53(8), 2038–2054 CrossRef CAS.
- M. H. Duker, H. Schafer, M. Zeller and V. A. Azov, Rationally designed calix[4]arene-pyrrolotetrathiafulvalene receptors for electron-deficient neutral guests, J. Org. Chem., 2013, 78(10), 4905–4912 CrossRef.
- N. L. Bill, O. Trukhina, J. L. Sessler and T. Torres, Supramolecular electron transfer-based switching involving pyrrolic macrocycles. A new approach to sensor development?, Chem. Commun., 2015, 51(37), 7781–7794 RSC.
- T. Ono, A. Taema, A. Goto and Y. Hisaeda, Switching of Monomer Fluorescence, Charge-Transfer Fluorescence, and Room-Temperature Phosphorescence Induced by Aromatic Guest Inclusion in a Supramolecular Host, Chem.–Eur. J., 2018, 24(66), 17487–17496 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2031007. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc04981f |
| This journal is © The Royal Society of Chemistry 2021 |