
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAbsorption and emission of light in red emissive carbon nanodots†
Neeraj
Soni‡§
ab,
Shivendra
Singh‡¶
ab,
Shubham
Sharma
ab,
Gayatri
Batra||
ab,
Kush
Kaushik
 ab,
Chethana
Rao
ab,
Navneet C.
Verma§
ab,
Bhaskar
Mondal
*a,
Aditya
Yadav
*ab and
Chayan K.
Nandi
*abc
ab,
Chethana
Rao
ab,
Navneet C.
Verma§
ab,
Bhaskar
Mondal
*a,
Aditya
Yadav
*ab and
Chayan K.
Nandi
*abc
aSchool of Basic Sciences, Indian Institute of Technology Mandi, H.P. 175075, India. E-mail: bhaskarmondal@iitmandi.ac.in; adityayadavg@gmail.com; chayan@iitmandi.ac.in
bAdvanced Materials Research Centre, Indian Institute of Technology Mandi, H.P. 175075, India
cBioX Centre, Indian Institute of Technology Mandi, H.P. 175075, India
First published on 14th January 2021
Abstract
The structure–function relationship, especially the origin of absorption and emission of light in carbon nanodots (CNDs), has baffled scientists. The multilevel complexity arises due to the large number of by-products synthesized during the bottom-up approach. By performing systematic purification and characterization, we reveal the presence of a molecular fluorophore, quinoxalino[2,3-b]phenazine-2,3-diamine (QXPDA), in a large amount (∼80% of the total mass) in red emissive CNDs synthesized from o-phenylenediamine (OPDA), which is one of the well-known precursor molecules used for CND synthesis. The recorded NMR and mass spectra tentatively confirm the structure of QXPDA. The close resemblance of the experimental vibronic progression and the mirror symmetry of the absorption and emission spectra with the theoretically simulated spectra confirm an extended conjugated structure of QXPDA. Interestingly, QXPDA dictates the complete emission characteristics of the CNDs; in particular, it showed a striking similarity of its excitation independent emission spectra with that of the original synthesized red emissive CND solution. On the other hand, the CND like structure with a typical size of ∼4 nm was observed under a transmission electron microscope for a blue emissive species, which showed both excitation dependent and independent emission spectra. Interestingly, Raman spectroscopic data showed the similarity between QXPDA and the dot structure thus suggesting the formation of the QXPDA aggregated core structure in CNDs. We further demonstrated the parallelism in trends of absorption and emission of light from a few other red emissive CNDs, which were synthesized using different experimental conditions.
Introduction
Fluorescent carbon nanodots (CNDs), an intriguing carbon-based nanomaterial (typical size 2–5 nm) owing to their superior optical properties, low-toxicity and cell penetrability, have tremendous applications in bioimaging, photovoltaics, drug delivery, optoelectronics, and various other fields.1–4 One of the fascinating properties of CNDs is their optical origin, especially the excitation dependent photoluminescence, which has baffled scientists across the globe.5–8 In addition, recent reports on the bottom-up synthesis of CNDs have raised numerous questions and pointed out erroneous conclusions.9–11 Several reports showed that the observed fluorescence originates from molecular fluorophores and/or their aggregated structure, quasi CNDs (molecular fluorophores attached to the core of CNDs) or polymer dots, which are produced as by-products or even as the sole product during CND synthesis.12–19 Moreover, the typical excitation dependent photoluminescence was not observed in a large number of recent studies.For example, an organic molecular fluorophore, namely TPDCA {(5-oxo-3,5-dihydro-2H-thiazolo[3,2a]pyridine-7-dicarboxylic acid)}, was actually found to be responsible for all the optical properties of the CNDs synthesized using citric acid and L-cysteine as the precursor molecules by utilizing the hydrothermal method.20 Upon rigorous purification, ∼98 wt% TPDCA and only ∼2 wt% real CNDs were observed in the CND solution. Interestingly, while excitation independent photoluminescence was observed both in the CND solutions and in TPDCA, a small fraction of the real CNDs showed excitation dependent photoluminescence. Similarly, a substantial amount of another molecular fluorophore, IPCA {(imidazole [1,2-a]pyridine-7-carboxylic acid, 1,2,3,5-tetrahydro-5-oxo-)}, was obtained when CNDs were synthesized at a lower temperature by using citric acid and ethylenediamine as precursor molecules.21 On increasing the temperature, the amount of CNDs increased and a clear shift from excitation independent to dependent photoluminescence with a decrease in quantum yield (QY) was observed.22 Several pieces of evidence suggest that the aggregation of the molecular fluorophore, especially via hydrogen bonding, either in the ordered crystalline or amorphous form, can mimic the CND structure and its optical properties.23 In addition, at elevated concentration, the aggregated structure of citrazinic acid mimicked the dot-like structure with a size of 4.5 nm as observed under a transmission electron microscope (TEM). On the other hand, some groups also proposed that CNDs were basically the drying mediated crystalline aggregation of a sole product of a molecular fluorophore, methylenesuccinic acid.24 It should be emphasized here that, to date, CNDs were synthesized mainly from citric acid as the precursor molecule.19,20,25 Phenylenediamine (PDA), another class of precursor molecule, has recently been used to synthesize red to near infrared emissive CNDs.26–30 Efforts were further made for the solvent dependent emission color change in CNDs.27–29
Here, for the first time, we reveal that the red emission in CNDs synthesized from o-phenylenediamine (OPDA) utilizing the hydrothermal method actually originates from a molecular fluorophore, quinoxalino[2,3-b]phenazine-2,3-diamine (QXPDA). By performing systematic chromatographic separation and characterization using various techniques, we identified three major components, which emit in the blue, green and red regions, respectively. NMR and mass spectral data tentatively confirmed the structure of QXPDA. The extended conjugation of QXPDA has been verified by the close resemblance of the theoretically simulated spectra with the experimental vibronic progression and the mirror symmetry of the absorption and emission spectra. On the other hand, blue emissive species obtained after purification showed the CND like structure with a typical size of ∼3 nm under a TEM. Both excitation dependent and independent emission spectra were observed in this blue emissive species. The resemblance in Raman spectroscopic data for QXPDA and the dot structure suggested the formation of the QXPDA aggregated core structure in CNDs. The amount of the blue emissive CND like dot structure was found to be substantially low (∼7 wt%), while the amount of red emissive QXPDA was found to be very high (∼80 wt%). As a result, even with the enormously high quantum yield (QY ∼ 55%) of the blue emissive dot structure, the presence of a vast amount of red emissive QXPDA (with a low QY of ∼9%) dictates the actual red emission of the complete CND solution. We further showed that the formation of QXPDA, in terms of the optical signature, is a general phenomenon in the synthesis of red emissive CNDs, irrespective of any other secondary molecule used along with OPDA.
Results and discussion
Optical characterization of the synthesized unpurified CNDs
Red emissive CNDs were synthesized by following a reported method with a minor modification.26 In brief, the synthesis involved the complete dissolution of 0.108 g OPDA in 10 ml of water under vigorous stirring followed by the addition of 5 ml of 5% hydrochloric acid (HCl) solution (Fig. 1a). The mixture was then vigorously stirred for 5 minutes and transferred to an autoclave hydrothermal reactor and heated at 180 °C for 10 h. Once the reaction was completed, it was cooled down to room temperature and then centrifuged at 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 minutes. The obtained black precipitate was washed several times with water to remove any unreacted precursor material and finally dissolved in ethanol for further characterization. It should be pointed out that there are several reports on CND synthesis using OPDA as the sole precursor or mixed with other precursor molecules for the synthesis of green to red emissive CNDs as presented in Table S1.† However, for an easy understanding and to avoid much complexity, we synthesized the red emissive CNDs using only OPDA in water with a small amount of HCl.
000 rpm for 20 minutes. The obtained black precipitate was washed several times with water to remove any unreacted precursor material and finally dissolved in ethanol for further characterization. It should be pointed out that there are several reports on CND synthesis using OPDA as the sole precursor or mixed with other precursor molecules for the synthesis of green to red emissive CNDs as presented in Table S1.† However, for an easy understanding and to avoid much complexity, we synthesized the red emissive CNDs using only OPDA in water with a small amount of HCl.
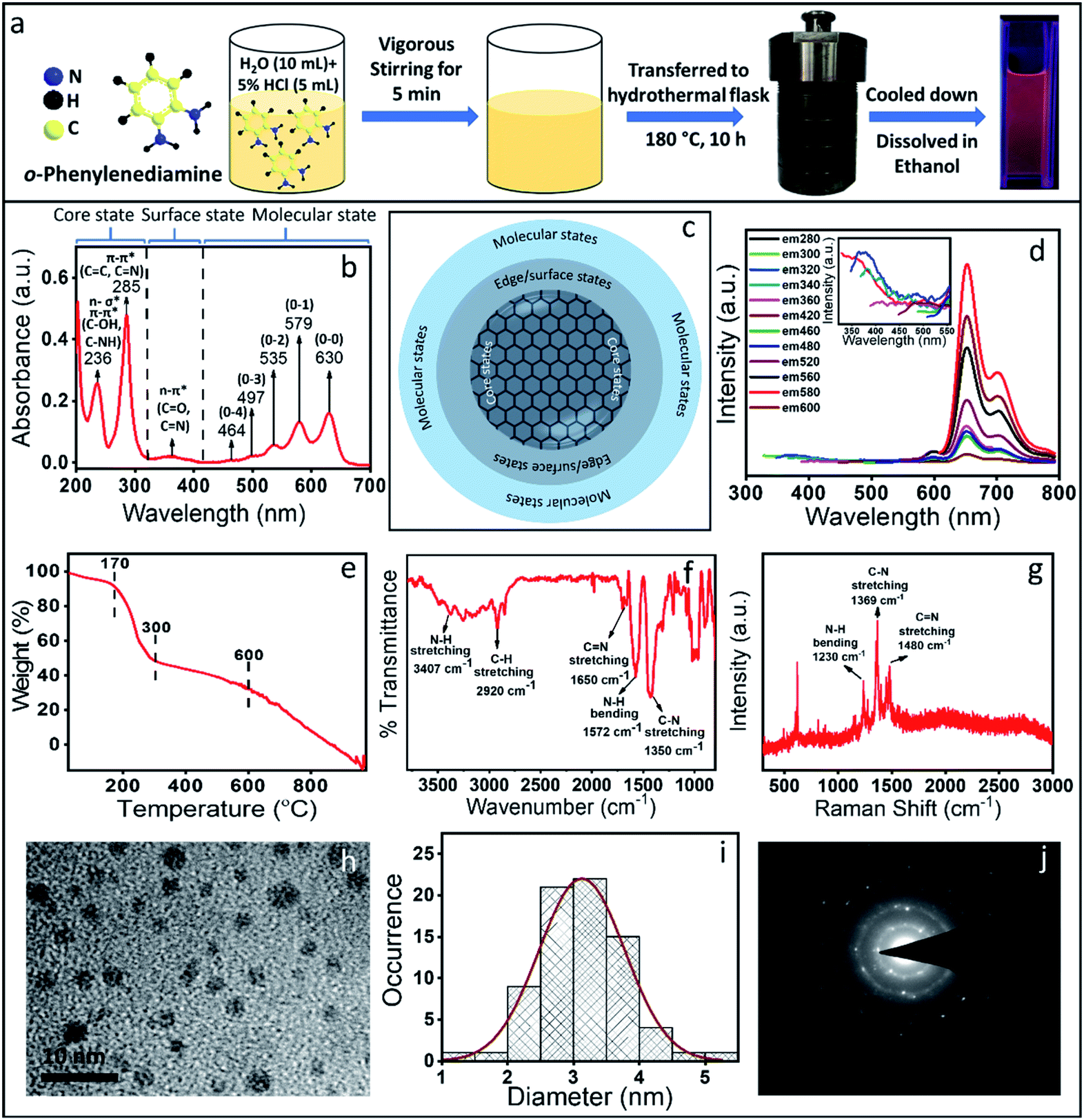 | ||
| Fig. 1 (a) Schematics of the synthesis of the red emissive CNDs using OPDA as a precursor molecule. (b) Absorption spectrum of the synthesized crude sample showing the core state, surface state and molecular state transition. (c) Schematic of a commonly accepted CND structure. (d) Fluorescence emission spectra of the red emissive CNDs show excitation independent emission; the inset shows the excitation dependent emission in the blue region. (e) TGA data show at least two species with different thermal stability. (f) FTIR spectrum with the designated vibrational modes. (g) Raman spectrum shows the signature of normal vibrational modes with no signature of D and G bands which are typically observed in graphitic material. (h & i) TEM image and the corresponding size distribution. The average size obtained was ∼3 nm. (j) SAED pattern confirming the polycrystalline nature of the CNDs with a d-spacing of 0.26 and 0.45 nm, which do not match with graphitic 001 or 002 planes. | ||
The measured absorption spectrum of the as-synthesized CNDs could be categorically separated into three regions (Fig. 1b). The peaks below 300 nm could be defined as the core state, the peaks within 300–400 nm could be assigned to the surface state and peaks above 400 nm correspond to the molecular state transition. Considering the earlier reports, the peak at 236 nm in the absorption spectrum could be assigned to either n–σ* (C–NH/C–OH) transition and the peak at 285 nm could be assigned to the π–π* transition of the carbon core (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN) network.31–33 The absorption peak at ∼360 nm could be assigned as n–π* transition of the surface states (C–O and CO). Interestingly, the peaks in the molecular state region showed a nice progression, which could be defined as vibronic overtone bands with the (0–0) transition at 630 nm and with a decrease in energy difference between two overtone bands. Similar overtone transitions have exclusively been observed in conjugated aromatic molecules such as anthracene, pentacene and rubrene.34–36 From the absorption spectrum and considering CND synthesis, the structure could be hypothesized to be a carbon-like core along with its surface state and molecule states, which is a well-accepted structure of the CNDs (Fig. 1c).37,38
C and CN) network.31–33 The absorption peak at ∼360 nm could be assigned as n–π* transition of the surface states (C–O and CO). Interestingly, the peaks in the molecular state region showed a nice progression, which could be defined as vibronic overtone bands with the (0–0) transition at 630 nm and with a decrease in energy difference between two overtone bands. Similar overtone transitions have exclusively been observed in conjugated aromatic molecules such as anthracene, pentacene and rubrene.34–36 From the absorption spectrum and considering CND synthesis, the structure could be hypothesized to be a carbon-like core along with its surface state and molecule states, which is a well-accepted structure of the CNDs (Fig. 1c).37,38
CNDs usually show excitation dependent emission i.e., the emission maximum shifts when the excitation wavelength is changed. Interestingly, in the present case, excitation independent red emission was observed with the maximum intensity at 650 nm (when excited at 579 nm) with the other three peaks at around 595 nm, 710 nm and 770 nm.26,39,40 Similar to the absorption spectrum, these bands also could be assigned as the excited state overtone transitions (Fig. 1d). Interestingly, very weak excitation dependent emission spectra with a peak maximum at around 350 nm were also observed (inset, Fig. 1d). The excitation independent emission and the overtone bands observed both in the absorption and emission spectra led us to the following questions: (1) are we really observing CNDs with excitation independent emission and (2) are there distinctly separated species present in the synthesis mixture? To answer these questions, we carried out thermogravimetric analysis (TGA), which provides information on the mass loss of the sample upon heating. TGA works on the principle of thermal stability of the materials. Fig. 1e shows a significant mass loss (more than 50%) within 300 °C (starting from 170 °C) and then the second mass loss was observed till 600 °C. Finally, complete mass loss was observed from 600 °C to 1000 °C. The extensive loss of mass at such a lower temperature (within 300 °C) indicates the presence of less thermally stable volatile organic compounds. On the other hand, the thermally more stable species could be attributed to the formation of a stable CND like structure or the aggregated core structure of organic compounds. It should be mentioned here that the TGA data can't suggest whether volatile organic compounds are exclusively free or attached to the core structure. Nonetheless, this data indicates the presence of at least two or more species with different thermal stability. Interestingly, the obtained TEM results (Fig. 1h and i) showed the CND like structure with a typical size of ∼4 nm. The Selected Area Electron Diffraction (SAED) pattern (Fig. 1j) showed few bright spots with a diffused ring, indicating the polycrystalline nature of the sample. The interplanar distance was calculated to be 0.45 nm and 0.26 nm, neither of which match the 002 plane or the 001 planes of the graphitic structure. In fact, the Raman spectroscopic data also don't show the D and G bands typically observed in graphitic CNDs (Fig. 1g). All these observations suggest that the dots, which were observed under a TEM may be an aggregated structure of the organic compound.
Purification and characterization of the components of the CND solution
The above observations motivated us to carry out a systematic column chromatographic purification of the synthesized solution. Unfortunately, a single solvent was not enough to elute out all the samples and we had to use a series of solvent mixtures of different polarities to separate out each of the components. Three major components with distinct red, green and blue color came out of the column when 10% ethyl alcohol in 90% ethyl acetate (most polar), 50% ethyl acetate and 50% hexane and finally 10% ethyl acetate in 90% hexane (least polar) were used, respectively (Fig. 2b). It is interesting to note here that the red component has the maximum product yield of ∼80%, the green component has ∼13% and the blue component has the least product yield of ∼7%.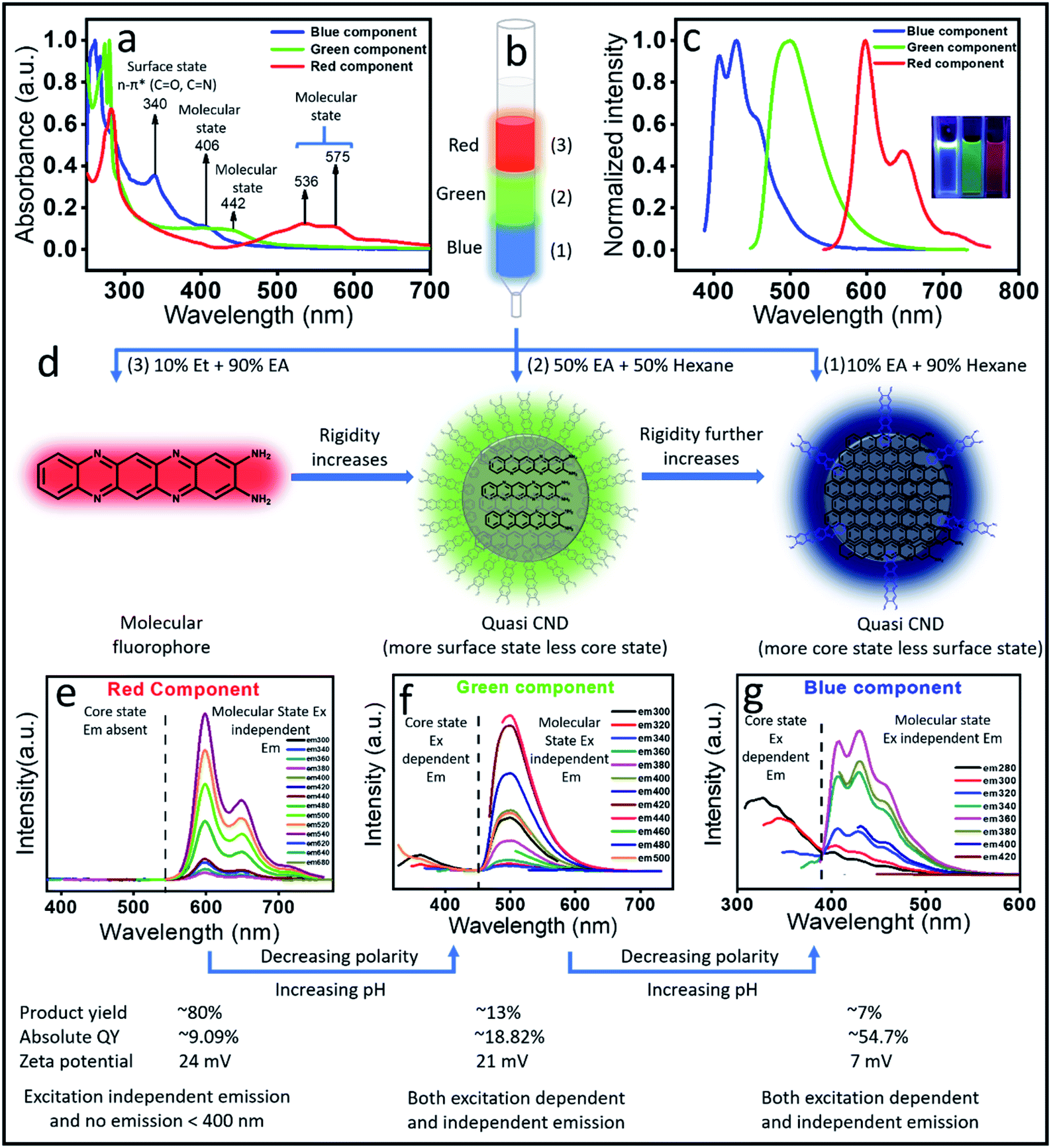 | ||
| Fig. 2 (a) Absorption spectra of the three separated components; (b) schematics of the separation using column chromatography of the red, green and blue components. (c) The emission spectra of the column separated components (inset shows the UV light emission of each component). (d) Possible structure of the red, green and blue emissive components. The red component has a molecular fluorophore structure, while the green and blue components show the quasi CND structure with more core formation in the blue component. (e–g) Emission spectra recorded by changing the excitation position for red, green and blue components, respectively. The red emissive component shows only excitation independent emission, while the green and blue components show both excitation dependent and independent emission. | ||
Next, we carried out both optical and chemical characterization to understand the nature of these components. Interestingly, all the three components depicted different absorption spectral signatures, which suggests their different ground state structures (Fig. 2a). To our surprise, the red component showed mainly molecular state absorption at 536 nm and 575 nm. It also showed a strong absorption peak at around 280 nm. The (0–0) transition for the red component is approximately 50 nm blue shifted compared to the molecular fluorophore transition in the unpurified sample (Fig. 1b). On the other hand, with a further blue shift, the green component also showed molecular state transition at around 442 nm and the blue component showed it at around 406 nm. Remarkably, an additional intense peak at around ∼340 nm is visible for the blue component. This was also present in the absorption spectrum of the as-synthesized sample (Fig. 1b). This peak most probably suggested the formation of a surface state with CO and CN bonds, which is a common observation in CNDs synthesized by the bottom-up approach. The observation of the surface state in the blue component suggests the formation of a CND like structure. On the other hand, the absence of any surface state peak in red emissive species affirmed the molecular signature.
The measured emission spectra from each of these components showed their respective fluorescence color (Fig. 2c and inset). Similar to the absorption spectrum, the emission spectra of the red component also showed vibronic progressions with a maximum peak intensity at ∼605 nm, while the corresponding peak maxima for green and blue components were observed at around 510 nm and 400 nm, respectively. We also measured the excitation dependent fluorescence from each of these components. Fig. 2e showed that in the case of the red component, no emission was visible below 500 nm, rather very strong excitation independent emission with an intensity maximum at around 605 nm along with two progression peaks at around 650 nm and 710 nm were observed (Fig. 2e). Interestingly, similar to the unpurified sample, the red component emission spectrum showed a mirror symmetry with the corresponding absorption spectrum. Moreover, the measured excitation spectra at the corresponding emission maxima for the red component perfectly matches with the absorption spectrum (Fig. S1†). All the above-mentioned observations pose a question on whether the red component is a real CND. If so, then how is excitation independent emission possible? Nevertheless, this could be possible by considering the red emissive component as one of the following: (1) a CND like structure with a perfect homogeneous size distribution, (2) CNDs with a large number of homogeneous surface states/molecular states or (3) an independent molecular fluorophore.
The blue component, on the other hand, showed both excitation dependent (below 400 nm) and independent emission spectra with an intensity maximum at around 450 nm (Fig. 2g). It should be noted that the red component showed the maximum intensity in emission spectra when excited at the molecular state absorption (580 nm), while the blue component showed the maximum intensity when excited at surface state transition (∼350 nm). In addition, when excited within 240 to 280 nm, the blue component showed both excitation dependent and independent emission. Interestingly, when the blue component was excited after 300 nm, only excitation independent emission was observed. Since the transition in the absorption spectrum between 240 and 280 nm was assigned to the CC and CN of the core state, it could be concluded that the excitation dependent emission arises from the core along with different surface states. Interestingly, the homogeneous size distribution of the blue component as observed under a TEM (Fig. S2†) ruled out the possibility of size dependent multicolor emission.41 This data suggested that the excitation independent emission is actually due to the molecular fluorophore attached to the surface of the core structure (quasi CND like structure) as shown in Fig. 2d. Similar to the blue component, the green component also showed both excitation dependent emission below 400 nm and excitation independent emission spectra with an intensity maximum at around 510 nm (Fig. 2f). A comparison of the emission spectra in Fig. 2f and g suggests that the extent of excitation dependent emission as compared to the independent emission is more in the blue component than the green component. This suggests that the amount of core state formation is less in the green component as compared to the blue component. The detailed analysis and assignments of both the absorption and emission spectra for all three components directed us to conclude that the red emissive component is an independent molecular fluorophore formed in the mixture, while the blue and green components might have a quasi CND structure with a variable amount of core and surface structures. Fig. 2d and 3 show their detailed structural and optical transitions.
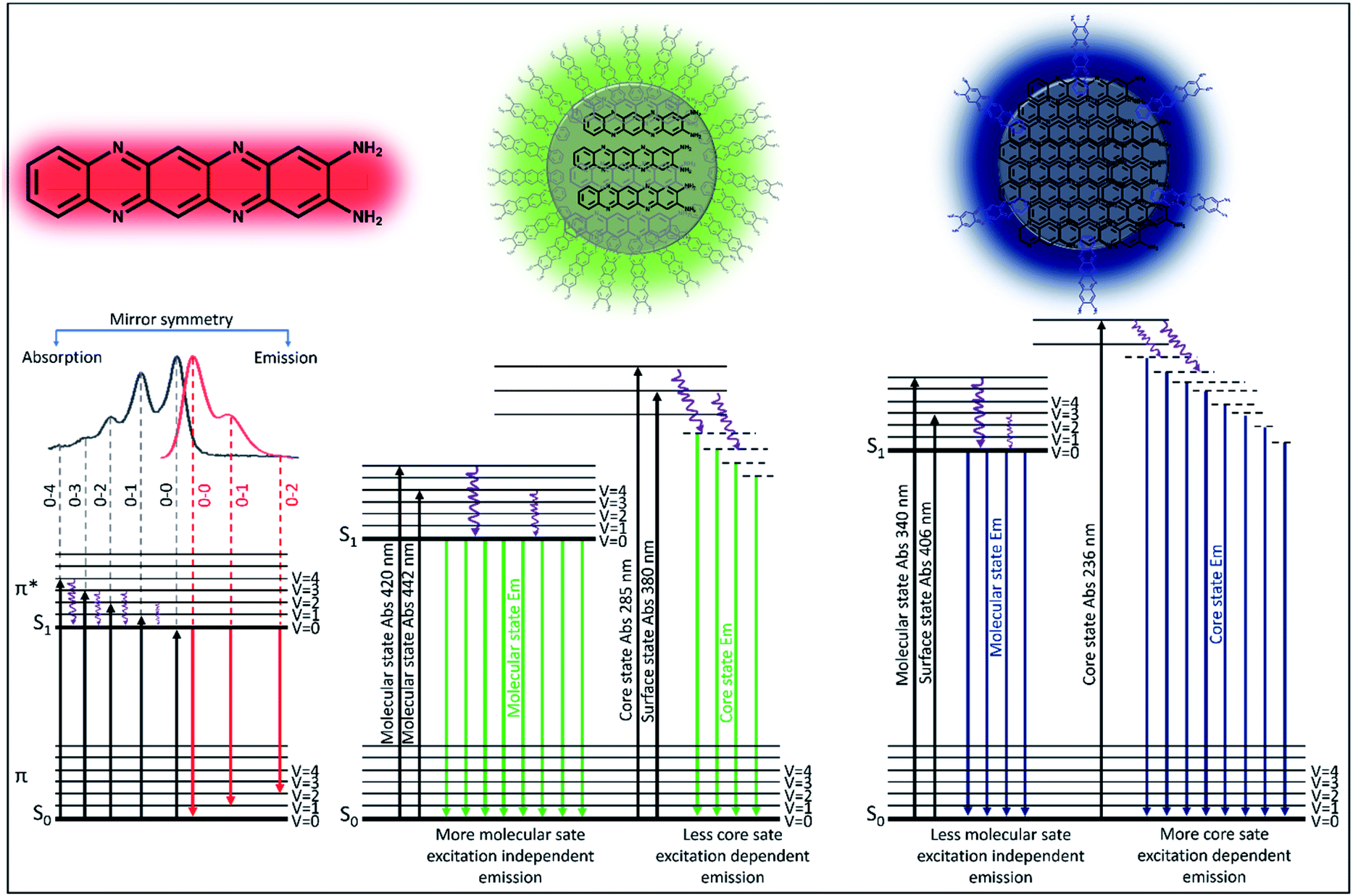 | ||
| Fig. 3 Schematic diagram of the real experimental absorption and emission of light by the red, green and blue emissive components. The red component shows only molecular fluorophore like transition with nicely arranged vibrational overtone bands observed both in absorption and emission spectra. The green and blue components show molecular state, surface state and core state absorption and emission, while the surface and core states are stronger in blue emission. | ||
To validate our hypothesis on the structural assignments of all these components, we carried out several experiments. To our curiosity, first, we performed fluorescence correlation spectroscopy (FCS) measurements, which is one of the best techniques to deduce the size of a fluorescence emitter.42,43 It is a single molecule fluorescence fluctuation technique that directly probes the emitter diffusion with high accuracy by measuring the fluorescence fluctuation within a confocal volume. By fitting the above fluorescence fluctuation to the analytical autocorrelation function one can obtain the diffusion coefficient and later the hydrodynamic diameter of the fluorescent species. Before carrying out the real measurements, the instrument was calibrated with a standard dye (Atto 647). The normalized autocorrelation curve of the red emissive component as shown in Fig. 4a provided a hydrodynamic diameter of 0.56 nm. Such a small size could only be assigned to the size of a molecular fluorophore. Next, we performed dialysis of the red component using a ∼1 kDa cut off membrane. Interestingly, the red component easily came out from the dialysis bag and showed absorption and emission spectra (Fig. S3a†) similar to that shown in Fig. 2d. The measured FTIR data as presented in Fig. 4b confirm the presence of N–H (3407 cm−1), C–H (2920 cm−1), CN (1620 cm−1), and C–N (1350 cm−1) stretching vibrations of the red emissive component. The signature D (1355 cm−1) and G (1590 cm−1) bands, which signify the defect and graphitic nature of carbon based materials, didn't appear in the Raman spectra as shown in Fig. 4c. Rather, a few sharp transitions at 1230 cm−1, 1369 cm−1 and 1480 cm−1 attributed to N–H bending, C–N and CN stretching, respectively, were observed. The Raman data disproves any formation of a CND like graphitic core structure in the red component. In addition, instead of a broad band at 2 theta, several sharp peaks were obtained in the XRD data (Fig. S3b†). More interestingly, a complete mass loss of the red component within 200 °C was observed in the TGA data, which exclusively suggests the highly volatile organic nature of the red emissive component (Fig. 4d). We have carried out a mass analysis of the red emissive component. The actual mass of the red emissive component, which was denoted as QXPDA (C18H12N6), is m/z = 312. The mass spectrum as presented in Fig. S4† shows weak adduct ions with m/z = 313 (C18H12N6 + H)+ and strong peaks at m/z = 318, 335 and 353, which correspond to [318 (C18H12N6 + Li)+], [335 (C18H12N6 + Na)+] and [353 (C18H12N6 + K + 2H)+], respectively. Further, we carried out energy dispersive X-ray analysis (EDX) elemental analysis, which showed that QXPDA comprised carbon and nitrogen only with a dominant amount of carbon (Fig. S5†). Finally, we recorded the 1H-NMR spectrum to shed light on the chemical structure of the red emissive component (Fig. S6†). The 1H-NMR data of the red component are as follows (500 MHz, DMSO-d6): δ 8.58 (q, J = 3.45 Hz, 1H), 8.15 (q, J = 3.45 Hz, 1H), 7.64 (b, 1H), 7.42–7.32 (m, 1H), 7.25 (d, J = 10.95 Hz, 1H), 7.17 (b, 1H), 6.88 (s, 1H), 6.71 (s, 1H), 6.33 (s, 1H), 4.37 (b, 1H), 4.11 (s, 1H), 4.02 (dd, J1 = 6.85 Hz, J2 = 7.55 Hz, 1H), 3.56 (s, 1H), 3.44 (dd, J1 = 6.2 Hz, J2 = 6.85 Hz, 1H), 3.36 (s, 1H), 3.16 (s, 1H), 1.98 (s, 2H), 1.17 (t, J = 6.85 Hz, 2H), 1.05 (t, J = 6.85 Hz, 3H). The above proton NMR data suggested the tentative structure of the molecular fluorophore to be QXPDA (Fig. 2 and 3). The formation of QXPDA is quite obvious by following the oxidation reaction scheme of OPDA in the presence of oxygen in a hydrothermal reaction (Fig. S7†). Although at the initial stage there is a chance of production of phenazine-2,3-diamine, the long-term reaction finally produced only QXPDA.44 This was verified by the measured absorption and emission data of the commercially available phenazine-2,3-diamine (Fig. S8†). Interestingly, the hydrodynamic diameter obtained from the FCS results for the blue emissive species was much larger (6 nm), which suggests the CND like structure (Fig. 4a). In addition, the TGA data showed a mass loss only after 550 °C, thus confirming the bigger size and the thermally stable dot-like structure. However, the similarity in the Raman spectra of QXPDA and the blue component dot structure suggests the formation of the QXPDA aggregated structure rather than a pure graphitic core structure in the blue emissive species.
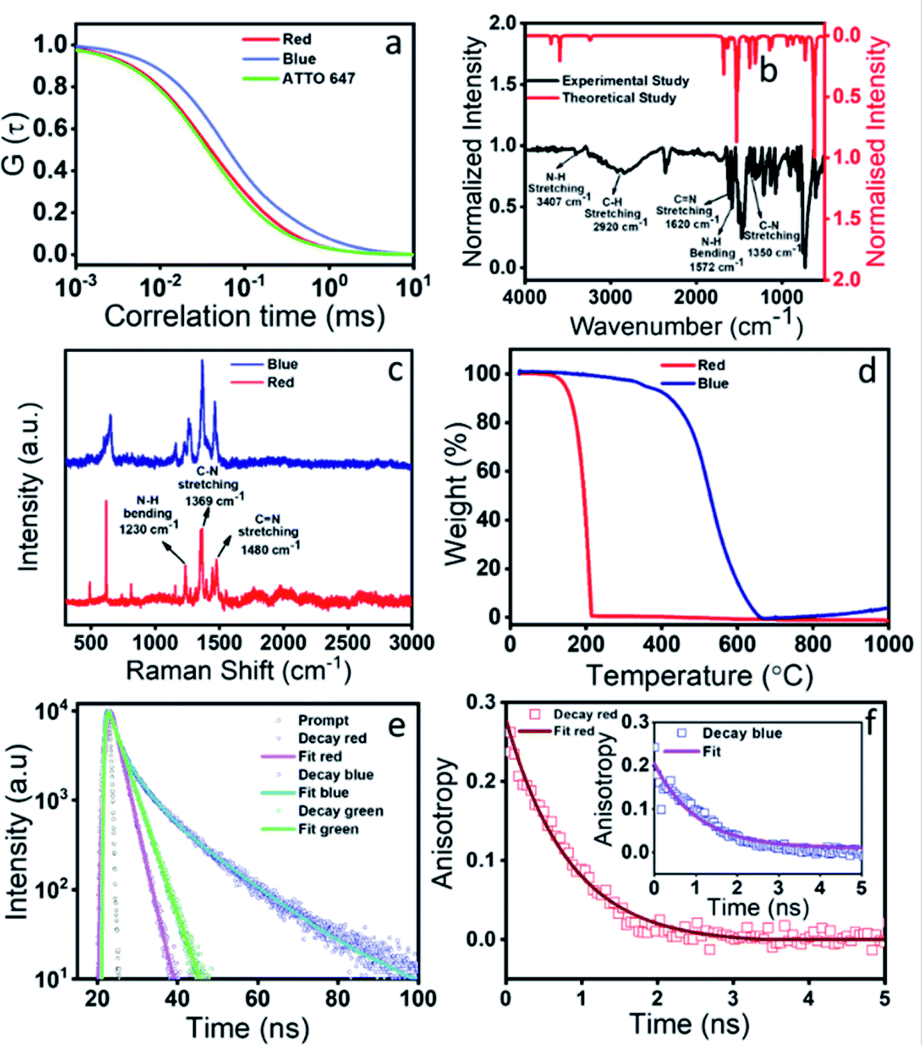 | ||
| Fig. 4 Detailed characterization of red and blue components. (a) FCS data show that the autocorrelation curve of the red emissive component is very similar to that of the ATTO 647 dye. This suggests a molecular fluorophore like signature of the red emissive component with a size of 0.6 nm. The blue emissive component has a much slower autocorrelation decay curve that corresponds to a size of ∼6 nm. (b) Experimental and theoretical simulated FTIR spectra of the red component. The fingerprint region shows similarity in the vibrational bands, especially the highly intense CN, N–H, and C–N stretching modes. (c) Raman spectra of both blue and red components show very high similarity of the vibrational bands. No signature of D and G bands for the graphitic material is observed. (d) TGA data show that the red component loses its mass within 200 °C, while the blue component shows thermal degradation only after 500 °C. (e) Lifetime data of both blue and red components. The blue component has a much higher lifetime in comparison to the red component, thus suggesting higher radiative decay for the blue component. (f) Anisotropy decay of the red emissive component and (inset) blue component. The rotational correlation time was found to be much larger in the blue component suggesting its larger size in comparison to the red component. | ||
To correlate the structural transformation of these components, we calculated the absolute quantum yield (QY) and zeta potential. The absolute QY was measured by the integrating sphere method. Interestingly, the QY of the blue component was found to be as high as 54.7% and for the red component it was the lowest i.e., 9.1%, while the QY of the green component was obtained as 18.8%. The increased QY of the blue emission could be attributed to the nitrogen doped core structure and the aggregation induced rigidity of the molecular state.45,46 The increased rate of radiative relaxation is a very common phenomenon in aggregation induced emission.47 The increase in the lifetime value from the red component (2.15 ns) to the blue component (5.46 ns) indeed supports the increase in the QY of the blue component (Fig. 4e). Such an observation of the increased QY along with spectral blue shift which occurred due to the reduction of polarity is a very well-known phenomenon in aggregation induced rigidity of the structure. The structural rigidity of the molecule prevents energy loss through non radiative rotational and vibrational relaxation processes, which occur in less rigid or freely rotating functional groups.48–50 The nitrogen-based heterocycles or amine functionalized compounds undergo extensive intersystem crossing, excited state predissociation and internal conversion, which reduce the fluorescence intensity.51,52 Upon aggregation, such processes are greatly hindered and increase the fluorescence intensity. Fig. 2b clearly shows that the blue emissive CNDs are eluted out in a less polar solvent than the red emissive species. This observation suggests a less polar nature of the blue emissive CNDs. As a result, the emission in blue CNDs occurs from the high-lying excited state as compared to the red emissive QXPDA. The hydrodynamic diameter of ∼6 nm for the blue component as supported by the FCS data, supports the CND like size. In addition, the increased anisotropy in the case of the blue component supports a more rigid structure of the blue component than the red component (Fig. 4f). The measured zeta potential also supports our hypothesis. A higher zeta potential value of 24 mV was observed for the red emissive species, while it was reduced to 7 mV for the blue component (Fig. S9†). The substantial decrease in zeta potential in the blue component suggests a considerable reduction of the molecular state (fluorophore) and the formation of the aggregated core structure. Recently, self-assembly of a molecular fluorophore, namely 5-oxo-1,2,3,5-tetrahydroimidazo-[1,2-α]-pyridine-7-carboxylic acid (IPCA), has been shown to form an organized stacked blue emissive CND like structure.53 It is interesting to point out here that, although the QY of red emission is much lower (∼9.1%) than the blue emission (∼54.7%), the emission spectral behavior and the QY of the as-synthesized crude sample is largely dictated by the large amount of the red emissive fluorophore (∼80%) present in the synthesis mixture in comparison to the very small amount of the blue component (∼7%).
Theoretical interpretation of photoluminescence of the molecular fluorophore
In order to obtain details of the origin of the absorptions, emissions, and corresponding vibronic progressions of the red component, we turn to theoretical spectroscopic calculations at the TD-DFT level using three different density functionals, namely CAM-B3LYP, B3LYP, and M06. Details of the theoretical calculations are documented in the ESI.† Among the three functionals, only CAM-B3LYP could produce spectra closest to the experimental ones with a maximum spectral shift of ∼40 nm to the peak positions, which is within the uncertainly limit of our TD-DFT calculations (Fig. 5b). We analyzed three molecular species, phenazine-2,3-diamine (PDA), QXPDA and pyrazino[2,3-b:5,6-b′]diphenazine-2,3-diamine (PZDDA), as shown in Fig. S7,† which could potentially form during the synthesis of CNDs. However, according to our best estimated CAM-B3LYP results, only the absorption and emission spectra of QXPDA match with the experimentally recorded red component spectra, whereas the calculated spectra for PDA and PZDDA are far off from the experimental ones (Fig. S10†). In addition to the peak positions, the vibronic overtone bands with descending energy difference, 0–0 → 0–1 = 46 nm, 0–1 → 0–2 = 40 nm, 0–2 → 0–3 = 35 nm in the absorption and ascending energy difference 0–0 → 0–1 = 63 nm, 0–1 → 0–2 = 69 nm in the emission closely match with the experimental results (Fig. 5a). This provides conclusive evidence that QXPDA featuring a five-membered ring is responsible for the red component emission.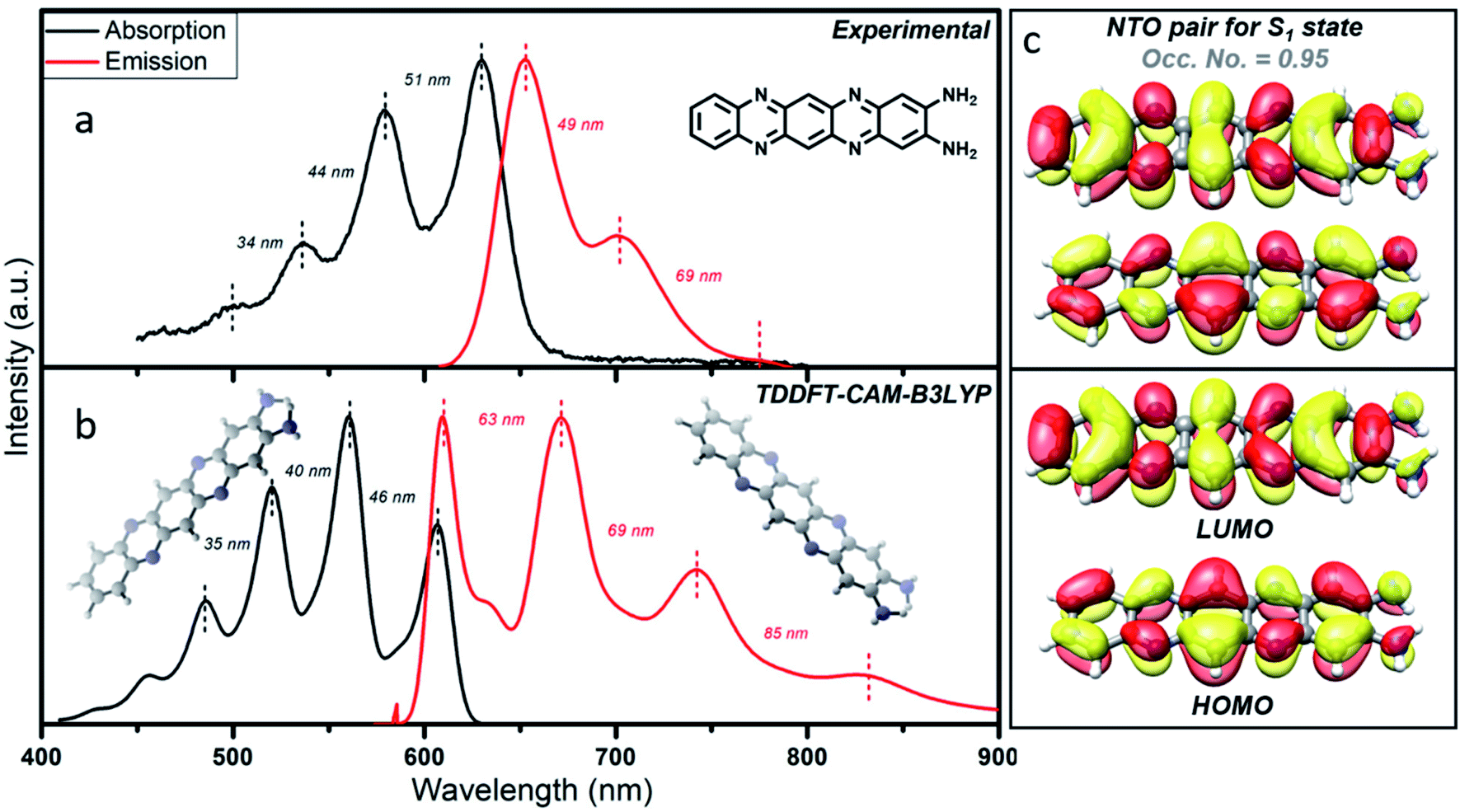 | ||
| Fig. 5 Theoretical interpretation of photoluminescence of the molecular fluorophore. (a) Experimental and (b) TD-DFT CAM-B3LYP computed absorption and emission spectra along with the vibronic overtones. (c) Natural transition orbitals (NTOs) and canonical molecular orbitals (HOMO and LUMO) of the organic fluorophore, QXPDA, responsible for the red component. | ||
To obtain a detailed picture of the electronic nature of the optical transitions, we analyzed the three lowest singlet excited states, S1, S2, and S3, of QXPDA. As predicted by the TD-DFT method, the S1 state is dominated by a HOMO → LUMO transition (94%) of a π → π* nature (Fig. 5c), whereas the closely lying S2 and S3 states are dominated by (HOMO − 3) → LUMO (84%) and (HOMO − 4) → LUMO (84%) transitions, respectively, which are of an n → π* nature. The S1 state is central to our discussion as it plays a key role in the absorption and emission behavior of QXPDA. To gain a more qualitative description of the electronic excitation, we investigated the natural transition orbital (NTO) pairs that constitute the S1 state. As shown in Fig. 5c, the S1 state is solely comprised of one NTO pair with an occupation number close to one, 0.95. Notably, the NTOs are of a similar electronic nature to that of the HOMO and LUMO.
TEM and SEM imaging of the components and their interpretation
For in-depth structural characterization, we performed TEM analysis of all three components and SEM of the red component (Fig. 6). The blue component depicted a clear aggregated core structure with a d-spacing of 0.21 nm (Fig. 6a and b). The green component also showed a dot-like ∼3 nm amorphous structure (Fig. 6c and d). The blue component can thus be considered a CND like material, while the green component is supposed to be an aggregated fluorophore structure. Interestingly, distinct morphologies were observed for the red component under different environmental conditions. For example, it showed either small dots, large dots or ribbon-like structures under a TEM. These suggested the molecular fluorophore behavior of the red component, which can undergo different types of aggregation processes under different conditions (Fig. 6e–h). The morphology of the red component is further confirmed using a scanning electron microscope (SEM), which clearly shows thread, ribbon, or flower-like structures (Fig. 6i–k). The elemental composition obtained from EDX confirmed that these threads, which are made up of the red component, were entirely comprised of carbon and nitrogen only (Fig. S11†). | ||
| Fig. 6 TEM image of the (a and b) blue and (c and d) green component. (e–h) TEM image of the aggregated structure of the red component under different experimental conditions. (i–k) SEM image of the red component shows ribbon, flower and seed like structures. | ||
Red CNDs synthesized under different conditions
We have synthesized the red emissive CNDs by changing the precursor molecules as reported in the literature and also by changing the temperature of the reaction.26,39 Surprisingly, in all the cases, the observations were closely similar as discussed above, albeit with minor changes in the optical properties. For example, red CNDs synthesized from (1) the OPDA and dopamine (DOP) mixture and (2) OPDA and catechol (CAT) in a hydrothermal synthesis showed an extensive amount of red emission along with blue emissive components (Fig. 7). Interestingly, when we performed the chromatographic separation, all the three-color emissive components, as reported in this work, were observed. The red components, in both cases, showed completely excitation independent emission. In addition, the FCS measurements confirmed the size of these components within the range of 0.5–0.7 nm (Fig. S12†). A slight difference in the emission spectra, especially in the green component, suggests the different extent of aggregation and core structure formation. In all these cases the red emissive component was present in a substantial amount and thus decided the color of the as-synthesized solution.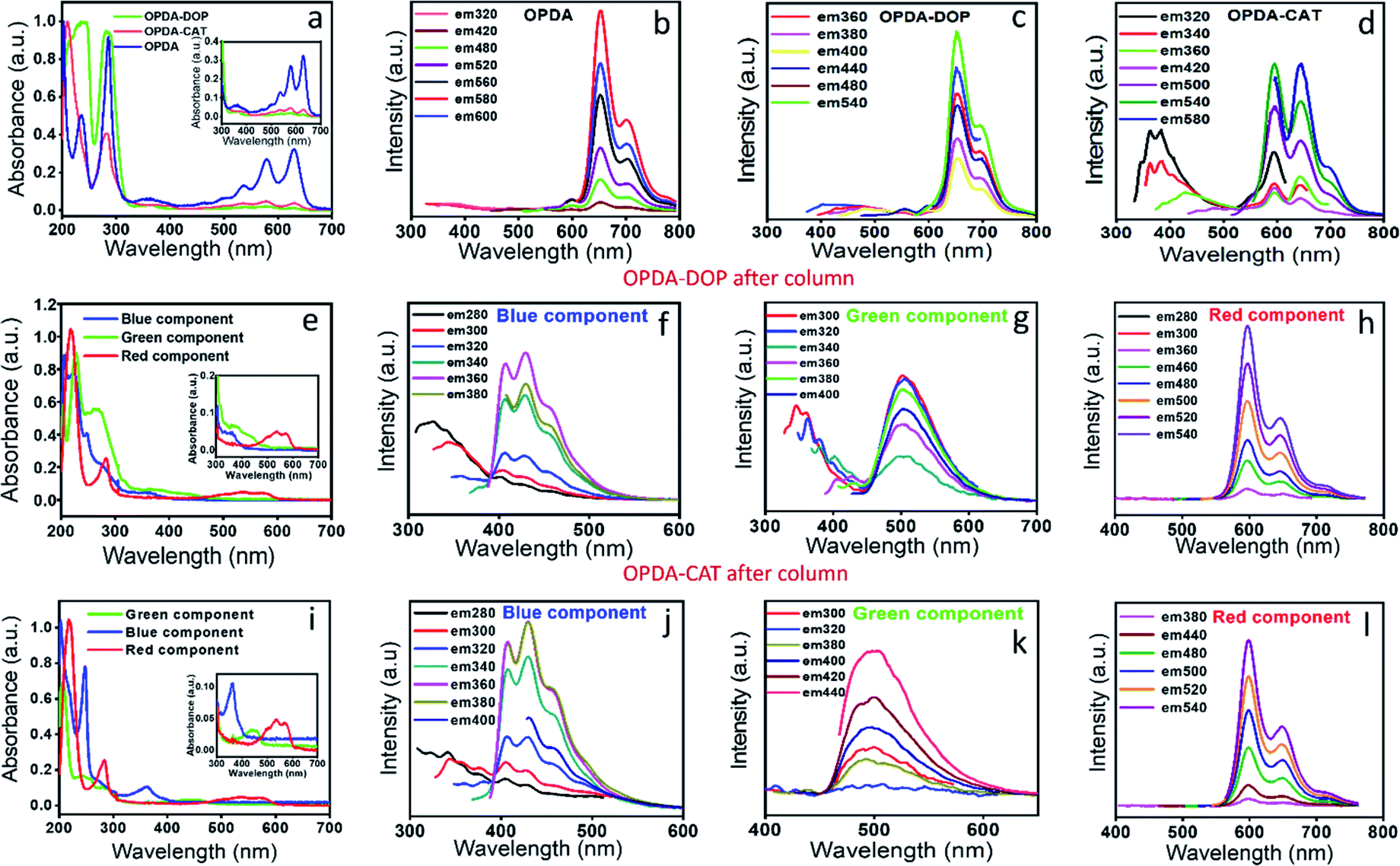 | ||
| Fig. 7 (a) Absorption of the red emissive CNDs synthesized using different precursor molecules. (Blue color) CNDs synthesized using OPDA as the only precursor molecule, (green color) CNDs synthesized from OPDA and dopamine (DOP) and (red color) CNDs synthesized from OPDA and catechol (CAT). (b–d) Emission spectra for the CNDs synthesized from OPDA, OPDA–DOP and OPDA–CAT. It is seen that the emission intensity or the extent of the blue component depends on the precursor molecules. (e & i) Absorption spectra of column separated red, green and blue components for OPDA–DOP and OPDA–CAT. The observed spectral features have similarities up to a large extent. (f–h) The emission spectra with the change in excitation wavelength for the red, green and blue components for OPDA–DOP and for (j–l) OPDA–CAT. Interestingly, the red emission spectral features in all the three cases bear extreme similarities, suggesting the origin from a similar type of chemical structure. The excitation independent part in the blue and green component also bears similarities, while the excitation dependent part has a slight difference, suggesting the slight difference in the extent of core structure formation. | ||
We also varied the reaction time and observed that with the increase in reaction time, the amount of the green component and finally the blue component in the reaction mixture increases substantially (Fig. S13†). Finally, to verify the role of HCl in the red emissive nature of the synthesized CND solution, we carried out systematic CND synthesis by varying the percentage concentration of HCl. Fig. S14† shows the presence of only red emissive species when a lower concentration of HCl (1% and 5%) was used, while a further increase of the HCl concentration to 9% leads to the appearance of the blue emissive component. It could be proposed that the amount of the red emissive molecular fluorophore is optimum up to a 5% HCl concentration. A further increase in HCl concentration may trigger the formation of the rigid blue emissive nature of CNDs. This conclusion is in the line with the long-term reaction conditions required for the synthesis of CNDs, as reported by Ehrat et al.54 or the increase in temperature as shown in Fig. S13.†22
Cell imaging by the red and blue emissive species
Finally, we checked the cytotoxicity assay of the as-synthesized CNDs and each of the well-separated components to check their biocompatibility for the bioimaging application. Fig. 8i shows that while the as-synthesized samples showed a little toxicity towards MCF-7 cells at higher concentration, the individual separated components are almost nontoxic. Finally, we checked the bioimaging of the as-synthesized and the highly purified red component. Confocal microscopy images show that the cells incubated with the as-synthesized material have the capability of multicolor imaging from blue to green to red (Fig. 8b–d), while the pure red component appears only in the red channel (Fig. 8f–h). Lastly, we carried out fluorescence lifetime imaging (FLIM) to check whether it could be used as a probe in metal induced energy transfer (MIET), one of the newly developed techniques for cellular dynamic study. Interestingly, we could see that the red component provides almost homogeneous lifetime distribution throughout the cytoplasm and nucleus with a full width at half maximum (FWHM) of 0.40 ns (Fig. 8j and k). All the data suggested that the newly synthesized red component can find potential bioimaging applications with very high efficiency.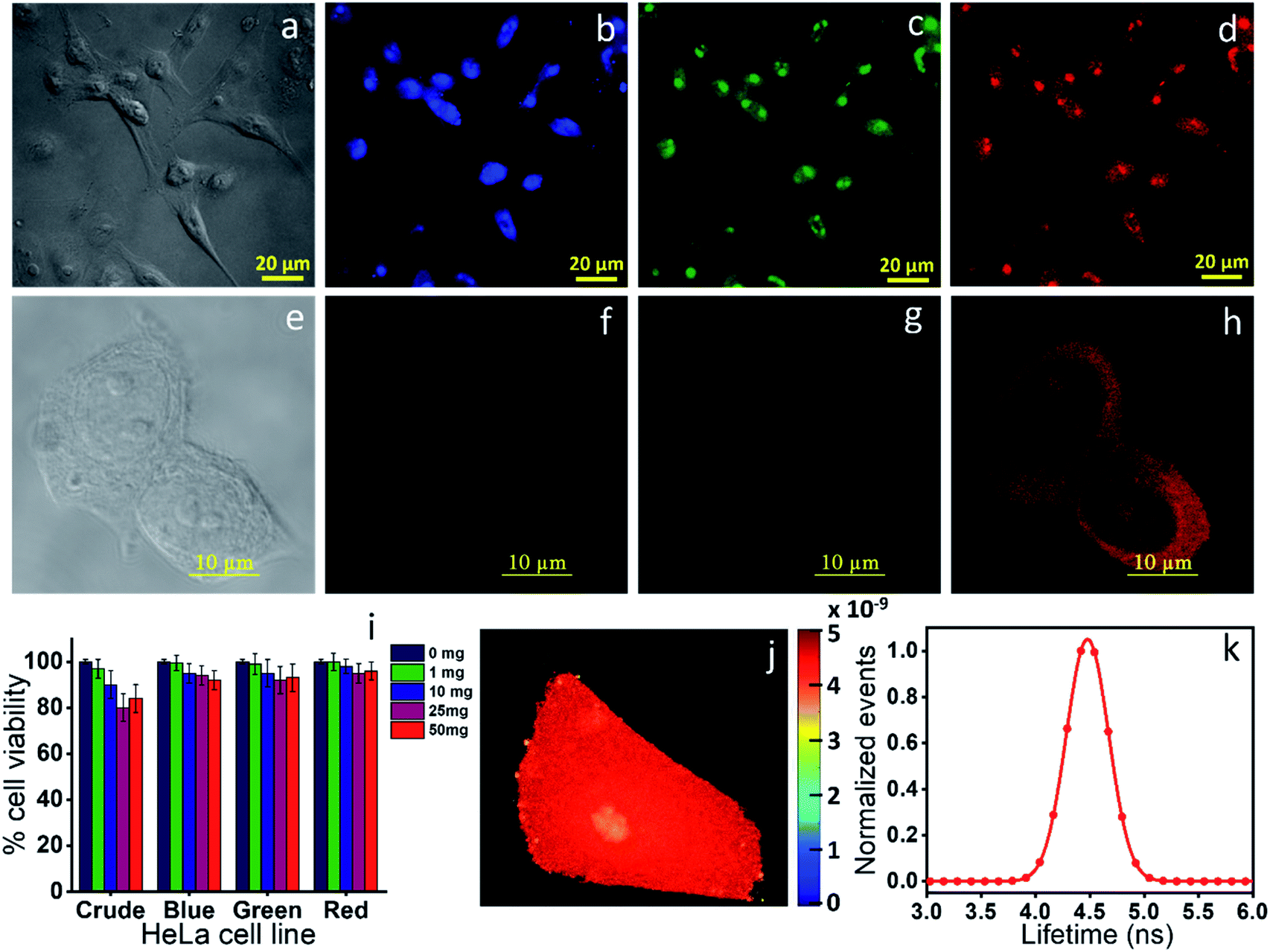 | ||
| Fig. 8 Bioimaging of the (a–d) crude red emissive CNDs obtained from OPDA and (e–h) the column purified red component. All the three color images (blue, green and red) were observed in all the three channels (blue, green and red) for the crude material, while only red color was observed for the red component. (i) Cytotoxicity data of the crude and all three column separated components, suggesting the non-toxic nature of the materials. (j & k) Fluorescence lifetime imaging and the corresponding distribution. The data show a homogeneous lifetime of the red component throughout the cell with a FWHM of 0.4 ns. | ||
Conclusions
In conclusion, for the first time, we have revealed the actual origin of the absorption and emission of light in red emissive CNDs, which were synthesized using OPDA as the precursor molecule. We showed that the emission in red emissive CNDs actually originates from a molecular fluorophore, QXPDA, which is largely present in the synthesis mixture as a by-product. The results from the theoretical calculations at the TD-DFT level further strengthen the origin of the red emission from QXPDA. The real CND like properties are observed from the blue emissive component, which is present in a very small amount in the solution. The red emissive fluorophore showed only ∼9% absolute QY with complete excitation independent emission spectra. On the other hand, the blue emissive species show a very high QY of ∼55% with both excitation dependent and independent spectra. The excitation dependent emission originated from the core and different surface states, whereas the excitation independent emission originated from the attached fluorophore in the blue emissive CNDs. We further showed that the formation of QXPDA, in terms of the optical signature, most likely is a general phenomenon in the synthesis of red emissive CNDs, irrespective of any other secondary molecule used along with OPDA. These results suggest that the CNDs produced by a hydrothermal method should be analyzed with proper consideration and extensive purification techniques should be employed before concluding the nature of the emission in CNDs.Author contributions
Neeraj Soni and Shivendra Singh performed all the synthesis, purification, and characterization of the materials. Shubham Sharma performed the theoretical calculation under the guidance of Bhaskar Mondal. Gayatri Batra helped with some optical measurements. Kush Kaushik performed FCS measurements. Chethana Rao performed all the TEM measurements. Navneet C. Verma performed the bioimaging and helped with analyzing the optical data. Aditya Yadav and Neeraj Soni performed all the experiments and helped in designing several experiments and Chayan K. Nandi guided the complete project and wrote the manuscript with the help of Aditya Yadav, Neeraj Soni and Bhaskar Mondal.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Chayan K. Nandi acknowledges the facilities of the Advanced Materials Research Centre (AMRC) and BioX center of IIT Mandi, India. Bhaskar Mondal acknowledges the high-performance computing (HPC) facility at IIT Mandi for providing the computational resources. Aditya Yadav thanks the Council of Scientific and Industrial Research, India [CSIR JRF:09/1058(0014)/2019-EMR-I]. Shubham Sharma thanks the University Grants Commission, India [16-9(June 2019)/2019(NET/CSIR)]. Navneet C. Verma thanks the Council of Scientific and Industrial Research, India [CSIR SRF:9/1058(07)/2017-EMR-I].References
- S.-T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y.-P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS.
- S. Paulo, E. Palomares and E. Martinez-Ferrero, Nanomaterials, 2016, 6, 157 CrossRef.
- T. Feng, X. Ai, G. An, P. Yang and Y. Zhao, ACS Nano, 2016, 10, 4410–4420 CrossRef CAS.
- X. Li, M. Rui, J. Song, Z. Shen and H. Zeng, Adv. Funct. Mater., 2015, 25, 4929–4947 CrossRef CAS.
- L. Pan, S. Sun, A. Zhang, K. Jiang, L. Zhang, C. Dong, Q. Huang, A. Wu and H. Lin, Adv. Mater., 2015, 27, 7782–7787 CrossRef CAS.
- B. van Dam, H. Nie, B. Ju, E. Marino, J. M. J. Paulusse, P. Schall, M. Li and K. Dohnalová, Small, 2017, 13, 1702098 CrossRef.
- A. Sharma, T. Gadly, A. Gupta, A. Ballal, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2016, 7, 3695–3702 CrossRef CAS.
- K. Mishra, S. Koley and S. Ghosh, J. Phys. Chem. Lett., 2019, 10, 335–345 CrossRef.
- J. B. Essner, J. A. Kist, L. Polo-Parada and G. A. Baker, Chem. Mater., 2018, 30, 1878–1887 CrossRef CAS.
- N. C. Verma, C. Rao, A. Singh, N. Garg and C. K. Nandi, Nanoscale, 2019, 11, 6561–6565 RSC.
- S. Zhu, Y. Song, X. Zhao, J. Shao, J. Zhang and B. Yang, Nano Res., 2015, 8, 355–381 CrossRef CAS.
- N. C. Verma, A. Yadav and C. K. Nandi, Nat. Commun., 2019, 10, 2391 CrossRef.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS.
- C. J. Reckmeier, J. Schneider, Y. Xiong, J. Häusler, P. Kasák, W. Schnick and A. L. Rogach, Chem. Mater., 2017, 29, 10352–10361 CrossRef CAS.
- S. K. Das, Y. Liu, S. Yeom, D. Y. Kim and C. I. Richards, Nano Lett., 2014, 14, 620–625 CrossRef CAS.
- D. Kozawa, Y. Miyauchi, S. Mouri and K. Matsuda, J. Phys. Chem. Lett., 2013, 4, 2035–2040 CrossRef CAS.
- V. Gude, A. Das, T. Chatterjee and P. K. Mandal, Phys. Chem. Chem. Phys., 2016, 18, 28274–28280 RSC.
- W. Wang, B. Wang, H. Embrechts, C. Damm, A. Cadranel, V. Strauss, M. Distaso, V. Hinterberger, D. M. Guldi and W. Peukert, RSC Adv., 2017, 7, 24771–24780 RSC.
- W. Kasprzyk, T. Świergosz, S. Bednarz, K. Walas, N. V Bashmakova and D. Bogdał, Nanoscale, 2018, 10, 13889–13894 RSC.
- L. Shi, J. H. Yang, H. B. Zeng, Y. M. Chen, S. C. Yang, C. Wu, H. Zeng, O. Yoshihito and Q. Zhang, Nanoscale, 2016, 8, 14374–14378 RSC.
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC.
- M. J. Krysmann, A. Kelarakis, P. Dallas and E. P. Giannelis, J. Am. Chem. Soc., 2012, 134, 747–750 CrossRef CAS.
- X. Meng, Q. Chang, C. Xue, J. Yang and S. Hu, Chem. Commun., 2017, 53, 3074–3077 RSC.
- S. Khan, A. Sharma, S. Ghoshal, S. Jain, M. K. Hazra and C. K. Nandi, Chem. Sci., 2018, 9, 175–180 RSC.
- J. Schneider, C. J. Reckmeier, Y. Xiong, M. von Seckendorff, A. S. Susha, P. Kasák and A. L. Rogach, J. Phys. Chem. C, 2017, 121, 2014–2022 CrossRef CAS.
- S. Lu, L. Sui, J. Liu, S. Zhu, A. Chen, M. Jin and B. Yang, Adv. Mater., 2017, 29, 1603443 CrossRef.
- H. Ding, J.-S. Wei, P. Zhang, Z.-Y. Zhou, Q.-Y. Gao and H.-M. Xiong, Small, 2018, 14, 1800612 CrossRef.
- D. Chao, W. Lyu, Y. Liu, L. Zhou, Q. Zhang, R. Deng and H. Zhang, J. Mater. Chem. C, 2018, 6, 7527–7532 RSC.
- T. Zhang, J. Zhu, Y. Zhai, H. Wang, X. Bai, B. Dong, H. Wang and H. Song, Nanoscale, 2017, 9, 13042–13051 RSC.
- K. Jiang, S. Sun, L. Zhang, Y. Lu, A. Wu, C. Cai and H. Lin, Angew. Chem., Int. Ed., 2015, 54, 5360–5363 CrossRef CAS.
- M. Shamsipur, A. Barati, A. A. Taherpour and M. Jamshidi, J. Phys. Chem. Lett., 2018, 9, 4189–4198 CrossRef CAS.
- A. Sharma, T. Gadly, S. Neogy, S. K. Ghosh and M. Kumbhakar, J. Phys. Chem. Lett., 2017, 8, 5861–5864 CrossRef CAS.
- P. Zhu, K. Tan, Q. Chen, J. Xiong and L. Gao, Chem. Mater., 2019, 31, 4732–4742 CrossRef CAS.
- M. Niu, F. Zheng, X. Yang, P. Bi, L. Feng and X. Hao, Org. Electron., 2017, 49, 340–346 CrossRef CAS.
- L. Ma, K. Zhang, C. Kloc, H. Sun, M. E. Michel-Beyerle and G. G. Gurzadyan, Phys. Chem. Chem. Phys., 2012, 14, 8307–8312 RSC.
- J. J. Burdett, A. M. Müller, D. Gosztola and C. J. Bardeen, J. Chem. Phys., 2010, 133, 144506 CrossRef.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- P. Yu, X. Wen, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 25552–25557 CrossRef CAS.
- Q. Guan, R. Su, M. Zhang, R. Zhang, W. Li, D. Wang, M. Xu, L. Fei and Q. Xu, New J. Chem., 2019, 43, 3050–3058 RSC.
- M. Zhang, R. Su, J. Zhong, L. Fei, W. Cai, Q. Guan, W. Li, N. Li, Y. Chen, L. Cai and others, Nano Res., 2019, 12, 815–821 CrossRef CAS.
- S. Khan, A. Gupta, N. C. Verma and C. K. Nandi, Nano Lett., 2015, 15, 8300–8305 CrossRef CAS.
- N. Pal, S. D. Verma, M. K. Singh and S. Sen, Anal. Chem., 2011, 83, 7736–7744 CrossRef CAS.
- S. A. Sanchez, E. Gratton, A. L. Zanocco, E. Lemp and G. Gunther, PLoS One, 2011, 6, e29278 CrossRef CAS.
- D. Rodríguez-Padrón, A. D. Jodlowski, G. de Miguel, A. R. Puente-Santiago, A. M. Balu and R. Luque, Green Chem., 2018, 20, 225–229 RSC.
- J. Manioudakis, F. Victoria, C. A. Thompson, L. Brown, M. Movsum, R. Lucifero and R. Naccache, J. Mater. Chem. C, 2019, 7, 853–862 RSC.
- X. Zhou, G. Zhao, X. Tan, X. Qian, T. Zhang, J. Gui, L. Yang and X. Xie, Microchim. Acta, 2019, 186, 67 CrossRef.
- P. Xu, F. Kang, W. Yang, M. Zhang, R. Dang, P. Jiang and J. Wang, Nanoscale, 2020, 12, 5084–5090 RSC.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald Jr, J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS.
- H. Liu, Y. Gu, Y. Dai, K. Wang, S. Zhang, G. Chen, B. Zou and B. Yang, J. Am. Chem. Soc., 2020, 142, 1153–1158 CrossRef CAS.
- C. M. Legaspi, R. E. Stubbs, M. Wahadoszaman, D. J. Yaron, L. A. Peteanu, A. Kemboi, E. Fossum, Y. Lu, Q. Zheng and L. J. Rothberg, J. Phys. Chem. C, 2018, 122, 11961–11972 CrossRef CAS.
- C. G. Freeman, M. J. McEwan, R. F. C. Claridge and L. F. Phillips, Chem. Phys. Lett., 1971, 8, 77–78 CrossRef CAS.
- M. Sun, C.-Y. Hong and C.-Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS.
- M. Langer, M. Paloncýová, M. Medved and M. Otyepka, J. Phys. Chem. Lett., 2020, 11, 8252–8258 CrossRef CAS.
- F. Ehrat, S. Bhattacharyya, J. Schneider, A. Löf, R. Wyrwich, A. L. Rogach, J. K. Stolarczyk, A. S. Urban and J. Feldmann, Nano Lett., 2017, 17, 7710–7716 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05879c |
| ‡ Neeraj and Shivendra equally contributed to this work. |
| § Department of Nanoscience and Nanotechnology Technion, Israel Institute of Technology Haifa, 3200003, Israel. |
| ¶ Discipline of Chemistry, Indian Institute of Technology Indore, M.P., 453552, India. |
| || Deutsches Elektronen-Synchrotron (DESY), Notkestraße 85, 22607, Hamburg, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |