
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational strategy for intrinsically disordered protein ligand design leads to the discovery of p53 transactivation domain I binding compounds that activate the p53 pathway†
Hao
Ruan
a,
Chen
Yu
a,
Xiaogang
Niu
bc,
Weilin
Zhang
a,
Hanzhong
Liu
d,
Limin
Chen
e,
Ruoyao
Xiong
a,
Qi
Sun
a,
Changwen
Jin
bc,
Ying
Liu
*ad and
Luhua
Lai
 *ade
*ade
aBNLMS, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: lhlai@pku.edu.cn; Tel: +861062757486
bCollege of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China
cBeijing Nuclear Magnetic Resonance Center, Peking University, Beijing 100871, China
dCenter for Quantitative Biology, Academy of Advanced Interdisciplinary Studies, Peking University, Beijing 100871, China. E-mail: liuying@pku.edu.cn; Tel: +861062751490
ePeking-Tsinghua Center for Life Sciences, Peking University, Beijing 100871, China
First published on 28th December 2020
Abstract
Intrinsically disordered proteins or intrinsically disordered regions (IDPs) have gained much attention in recent years due to their vital roles in biology and prevalence in various human diseases. Although IDPs are perceived as attractive therapeutic targets, rational drug design targeting IDPs remains challenging because of their conformational heterogeneity. Here, we propose a hierarchical computational strategy for IDP drug virtual screening (IDPDVS) and applied it in the discovery of p53 transactivation domain I (TAD1) binding compounds. IDPDVS starts from conformation sampling of the IDP target, then it combines stepwise conformational clustering with druggability evaluation to identify potential ligand binding pockets, followed by multiple docking screening runs and selection of compounds that can bind multi-conformations. p53 is an important tumor suppressor and restoration of its function provides an opportunity to inhibit cancer cell growth. TAD1 locates at the N-terminus of p53 and plays key roles in regulating p53 function. No compounds that directly bind to TAD1 have been reported due to its highly disordered structure. We successfully used IDPDVS to identify two compounds that bind p53 TAD1 and restore wild-type p53 function in cancer cells. Our study demonstrates that IDPDVS is an efficient strategy for IDP drug discovery and p53 TAD1 can be directly targeted by small molecules.
Introduction
Intrinsically disordered proteins or intrinsically disordered regions (IDPs) play central roles in transcription, signaling, important cellular processes and cellular assembly.1 Given their prevalence in many diseases, IDPs are considered as potential drug targets.2,3 However, designing small molecules by directly targeting IDPs is still in its infancy.4–7 Despite difficulties in ligand discovery for directly targeting IDPs, a number of compounds binding to IDPs were reported. For example, compound YK-4-479 was reported to inhibit EWS-FLI1 and RNA helicase interaction by binding to the disordered oncogenic fusion protein EWS-FLI1, thus leading to tumor cell growth inhibition.8 Based on NMR screening, several compounds were discovered to bind to the p27 kinase inhibitory domain.9 A recent study screened FDA-approved drugs using the fluorescence thermal denaturation strategy and several small molecule drugs were observed to bind to nuclear protein 1.10 In addition, other small molecule ligands targeting disordered proteins, including c-Myc, AF9, α-synuclein, tau, androgen receptor and MBD2 have been reported.11–18 In these investigations, rational drug design and discovery for direct IDP targeting are emerging.Tóth et al. utilized a fragment-based mapping strategy to identify potential binding pockets and found an active compound (ELN484228) targeting α-synuclein.13,19 Baggett et al. discovered small molecules targeting tau by coupling computational docking and machine learning-based screening.17 Recently, we used molecular docking to screen cMyc370–409 binding compounds based on NMR derived multiple conformations and found several active compounds.15 We found that these active compounds bound to different conformations with similar affinity, while the non-active compounds only bound to a small fraction of conformations, implying that IDP drugs should simultaneously bind to multiple conformations of the IDP.
The tumor suppressor p53 is a transcription factor that induces cell cycle arrest, senescence or apoptosis in response to cellular stresses.20 For approximately 50% human tumors, the TP53 gene is disabled by mutation or deletion.21,22 A recent study suggests that TP53 gene missense mutations do not lead to gain of function in myeloid malignancies but instead exert a dominant-negative effect, leading to impairment of the remaining wild-type allele.23 Wild-type p53 inactivation mostly results from elevated expression of its cellular inhibitors, MDM2 and/or MDMX, which has motivated extensive efforts to develop drugs to activate p53 function.24–30 Several MDM2 inhibitors are now under clinical evaluation. However, as MDM2 is involved in normal hematopoiesis, MDM2 inhibitors cause dose dependent adverse events including gastrointestinal and hematological adverse events that cannot be well controlled by simple adjustment of schedule treatment.31,32 Therefore, the quest for new pharmacological strategies to target the MDM2/X-p53 axis remains one of the main challenges in the MDM2/X-p53 axis drug discovery. Instead of targeting MDM2/X, we intended to take an alternative approach—directly targeting p53 to release it from MDM2/X—to reactivate p53 function.
The full-length p53 comprises an N-terminal transactivation domain (TAD), followed by the central DNA-binding core domain (DBD), the tetramerization domain (TET) and the C-terminal regulatory domain. The free TAD is disordered and consists of two subdomains TAD1 (residues 1–40) and TAD2 (residues 40–61).33,34 Both MDM2 and MDMX inhibit p53 transactivation function by engaging with its TAD1.35–37 We hypothesize that p53 TAD1 ligands that disrupt p53 binding to both MDM2 and MDMX should be able to restore wild-type p53 activity. In fact, activating wild-type p53 by directly targeting p53 has been shown challenging. Although compound RITA has been reported to activate p53 function in tumors by targeting p53, there is no strong evidence supporting the direct binding between RITA and the N-terminus of p53.38,39 Therefore, it remains an open question whether compounds directly binding to the N-terminus of p53 can be found.
Here we developed a hierarchical computational strategy for IDP drug virtual screening (IDPDVS) and applied it to discover ligands that can bind to p53 TAD1 to inhibit p53-MDM2/X interaction. This led to the identification of several small molecules that can bind to p53 TAD1 and activate wild-type p53 function in living cells, suggesting the feasibility of IDPDVS and the new pharmacological strategy of targeting p53 to inhibit p53-MDM2/X interaction.
Results
The virtual screening strategy for IDP drug discovery
Due to the highly flexible nature of IDPs, generally applicable rational design methods for IDP drug discovery are lacking. We developed a virtual screening strategy for IDP drug discovery (IDPDVS) and tested it in p53 TAD1 binding compound discovery (Fig. 1). IDPDVS contains four major steps. In the first step, conformation sampling is performed to generate possible conformations for the IDP target. In the second step, a stepwise conformation clustering procedure is performed, which includes local clustering based on the knowledge of the functional region within the IDP and subsequent global clustering. In the third step, representative druggable conformations are selected according to the availability of druggable binding sites. In the fourth step, multiple virtual screening runs are performed for all the druggable conformations, and compounds that can bind to multiple conformations with high docking scores are selected for further experimental validation.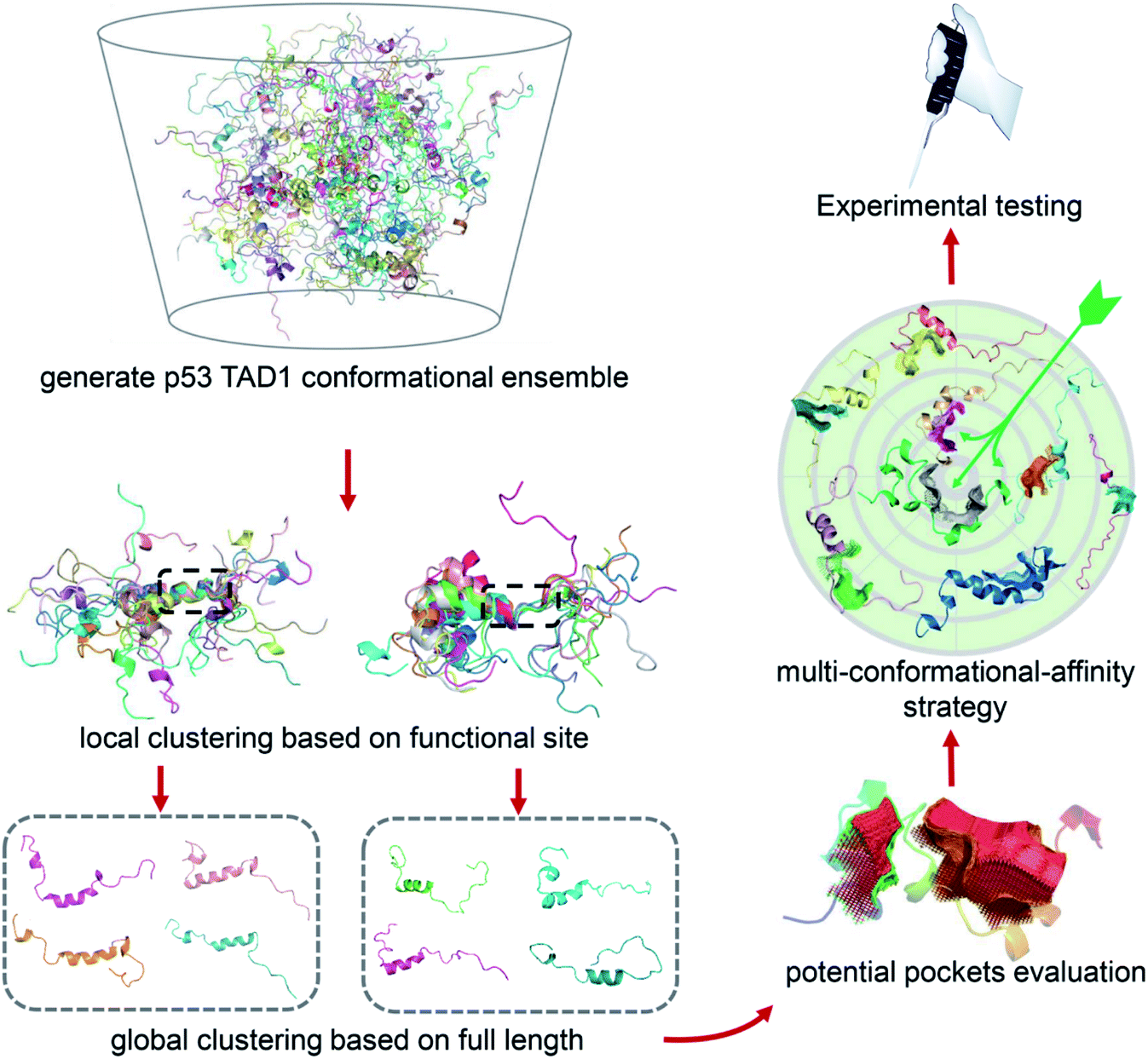 | ||
| Fig. 1 Illustration of the virtual screening strategy for IDP drug discovery (IDPDVS). The p53 TAD1 system was used as an example. Conformational ensemble should be generated first using MD simulations. After performing local and global clustering and ligand binding site detection, multiple docking runs will be performed for all the druggable conformations. Top-ranking compounds that can bind to multiple conformations are selected for further experimental testing. | ||
Conformation analysis of p53 TAD1 and virtual screening of binding compounds
Since IDPs generally have lower energy barriers, conformation sampling of IDPs remains challenging.40,41 For p53 TAD1, we performed two types of MD simulations, including (i) classical all-atom molecular dynamics (cMD) simulations in an explicit solvent with initial structure from the p53 TAD1-MDMX complex structure,42 (ii) replica exchange molecular dynamics (REMD) simulations using an implicit water model with the random coil conformation as the initial state. For cMD and REMD, simulations were performed for 900 ns and 8.5 μs, respectively. The trajectory at 298 K was used in the further analysis for REMD. The two independent simulations starting from different initial structures, either the highly helical structure extracted from the p53 TAD1-MDMX complex or the highly extended random coil structure gave conformation ensembles with similar secondary structure content, indicating the convergence of simulations. In addition, residues 19–25 take interchangeable disordered and partially helical conformations, which is consistent with the NMR results (Fig. S1A and B†).43These trajectories were then analyzed to identify druggable conformations and binding sites. Locating potential small molecule binding sites in IDPs is difficult due to their high flexibility. Direct conformational clustering based on backbone RMSD values often gives too many small clusters, which makes subsequent ligand discovery study unapproachable. Given that residues 19–26 play essential roles in p53 TAD1-MDM2 binding,35,44 we first performed a local clustering based on this functional region of p53 TAD1. We then carried out the second-round clustering for each cluster from the primary clustering results with the full-length TAD1 backbone RMSD values as clustering metrics. This procedure gave 215 and 1091 clusters for cMD and REMD trajectories, respectively (Fig. S1C and D†). The representative structure, which has the minimal sum of the squared displacements with other structures in the cluster, was selected from each cluster. To detect potential binding sites for all the representative structures, the ligand-binding site detection program, CAVITY,45,46 was applied. Subsequently, structures containing pockets that include the functional region, have large cluster population and a high CAVITY druggability score (average pKd > 6, volume > 300 Å3) were considered as potential druggable conformations. Accordingly, eight potential druggable conformations of p53 TAD1 were identified from this analysis for further docking study (Fig. S1E†).
We then used all the eight conformations to carry out eight parallel virtual screening runs against the SPECS library containing ∼20,000 compounds (see Methods). To balance computational cost and docking precision, we applied a two-step dock protocol. As docking scores depend on the sizes and chemical features of the binding pockets, the distributions of the docking scores of the eight conformations are quite different (Fig. S2†). Thus we compared the docking score rankings rather than the absolute docking scores to select possible binding compounds. Specifically, all molecules were docked and scored using the Glide HTVS mode first; then the top 50% molecules from the HTVS step were redocked and scored using the Glide SP mode. After performing the two-step docking procedure, we selected the 1% top-ranking compounds in the second step (the 0.5% top-ranking compounds of all the compounds) from each of the eight conformations. We then analyzed and compared all the top-ranking compounds from the eight docking runs and selected molecules that are among the top 1% in at least three different conformations as potential p53 TAD1 binding compounds. These potential p53 TAD1 binding compounds were then visually inspected, and 244 compounds were selected for further experimental testing.
p53 TAD1 binding and cell-growth inhibition activities of the selected compounds
We purchased the 244 selected molecules and tested their binding ability with the p53 TAD1 peptide using surface plasmon resonance (SPR). For primary screening, the N-terminal biotinylated p53 TAD1 peptide was immobilized onto an SA chip and molecules were injected at 50 and 100 μM concentrations onto the surface of the chip. Molecules with dose-dependent responses above 0.5 RU were selected. This initial screening identified 10 binding compounds (Table S1†). We then evaluated their effects on cancer cell viability using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay in the p53 wild-type MCF-7 cell line. All the 10 compounds showed modest activities in the cell viability assay (Table S1†). The most potent compound, PKUMDL-RH-1047 (1047), demonstrated an IC50 (the concentration that inhibits the survival of cells by 50%) of 36.8 ± 13.1 μM (Fig. S3A†). We then measured the binding strength of 1047 to the p53 TAD1 peptide using SPR, and the equilibrium dissociation constant (KD) was 13.8 ± 4.8 μM (Fig. 2A, B and S4A†).We further tested whether 1047 can inhibit p53-MDM2 interaction after binding to p53 TAD1. To this end, an SPR competitive binding assay was conducted, where the biotin–p53 TAD1 peptide was immobilized onto an SA chip and serial concentrations of 1047 pre-incubated with MDM2 were injected onto the surface of the chip. The competition assay data showed that 1047 reduced MDM2 binding to p53 TAD1 peptide in a concentration-dependent manner (Fig. 2C). To verify the selectivity of 1047, we tested its activity using cancer cell lines with different p53 genetic backgrounds, including cell lines with wild type p53 (MCF-7, overexpressing MDMX; HCT116, overexpressing MDM2), mutant p53 (MDA-MB-231), and deficient p53 (H1299). Treatment with 1047 led to dose-dependent growth inhibition of HCT116 and MCF-7, with IC50 values of 23.6 ± 1.8 μM and 36.8 ± 13.1 μM, whereas IC50 values were beyond 50 μM in H1299 and MDA-MB-231 cell lines (Fig. S3A†). In comparison, 1047 caused less toxicity in p53 mutation/deficiency cell lines, demonstrating selectivity toward cell lines with wild-type p53 to a certain extent.
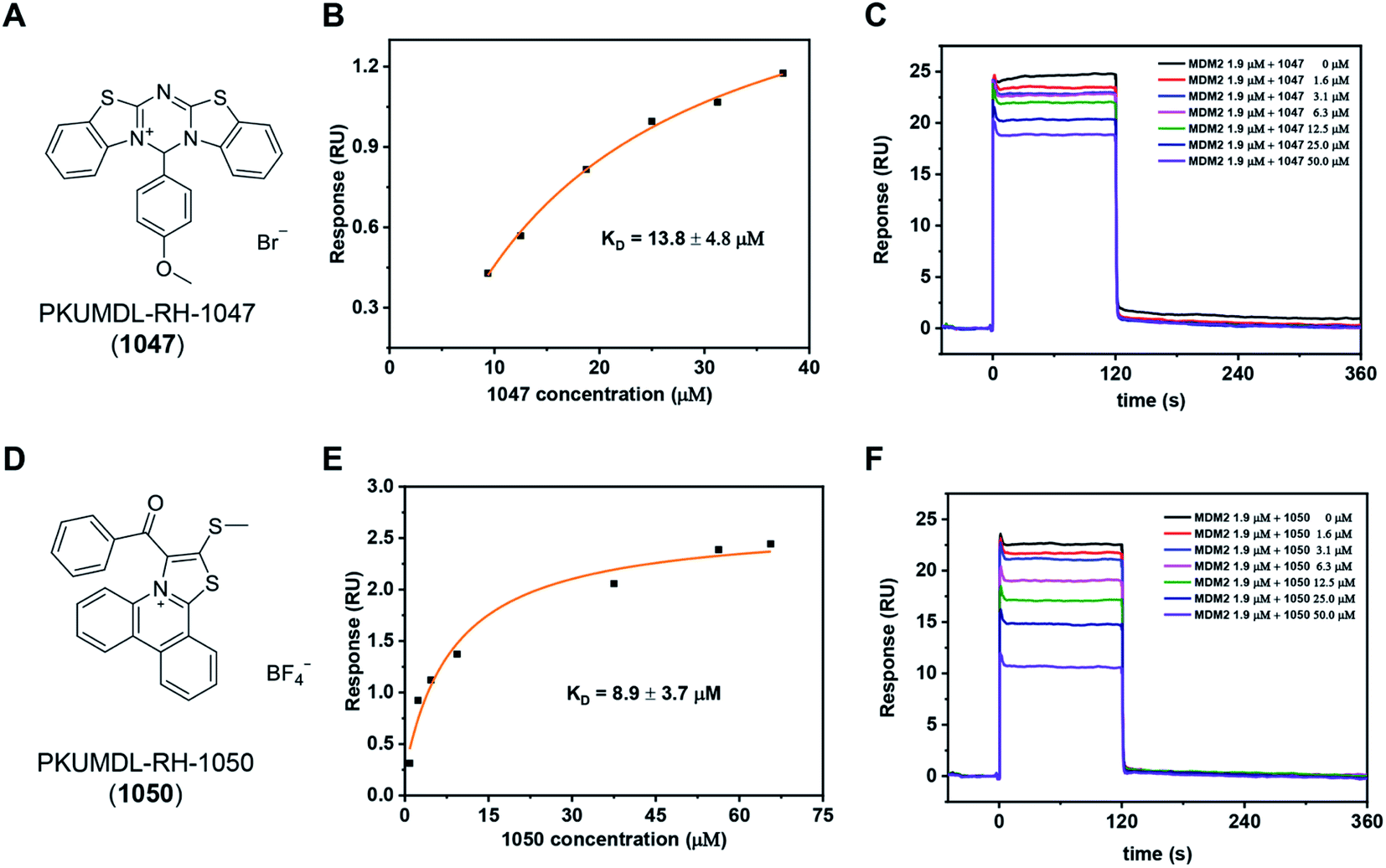 | ||
| Fig. 2 Discovery of p53 TAD1 binding compounds 1047 and 1050. (A and D) Chemical structure of the compound 1047 and 1050. (B and E) SPR results of the binding affinity measurements between p53 TAD1 ligands and the p53 TAD1 peptide on an SA chip. (C and F) Competitive binding assay results by SPR. | ||
We then performed a two-dimensional chemical similarity search using binary fingerprint and selected 70 analogs of 1047 for experimental testing. Similar to the first-round experimental studies, we tested their p53 TAD1 binding using SPR and their effects on cancer cell viability. Five compounds were active in p53WT cell lines with IC50 values less than 50 μM (Table S2†). Among them, compound PKUMDL-RH-1050 (1050) showed selectivity toward p53WT cell lines (Fig. S3B†). We then determined the binding strength of 1050 to p53 TAD1 (KD = 8.9 ± 3.7 μM) by SPR (Fig. 2D, E and S4B†) and tested its ability to break p53 TAD1 and MDM2 binding using the SPR competitive binding assay as mentioned above. Compound 1050 significantly reduced the binding of MDM2 to the p53 TAD1 peptide (Fig. 2F). To rule out the possibility that p53 TAD1 ligands might bind to MDM2, we immobilized MDM2 to a CM5 sensor chip and planned to measure compound binding by SPR. However, as the MDM2 protein immobilized on the CM5 sensor chip via amine coupling could not bind to the p53 TAD1 peptide (data not shown), we switched to using microscale thermophoresis (MST) to measure binding. The MST experiments showed that the p53 TAD1 peptide strongly bound to MDM2, consistent with previous reports.43,47 In contrast, 1047 and 1050 showed no detectable binding to MDM2 with concentrations up to 200 μM (Fig. S6†), thus ruling out the possibility that these compounds might also bind to MDM2.
p53 TAD1 binding compounds activate the p53 pathway in cells
We used a cellular protein interaction assay, termed nanoBRET (bioluminescence resonance energy transfer), to verify whether our p53 TAD1 binding compounds can disrupt p53 and MDM2 interaction in living cells. In this assay, MDM2 was designed as an energy donor, expressed as a fusion to NanoLuc luciferase, and p53 as an energy acceptor, expressed and fluorescently labelled as a fusion to HaloTag. As shown in Fig. 3A, treatment of cells with p53 TAD1 ligands 1047 or 1050 results in specific reduction of the BRET signal, indicating reduced interaction between p53 and MDM2.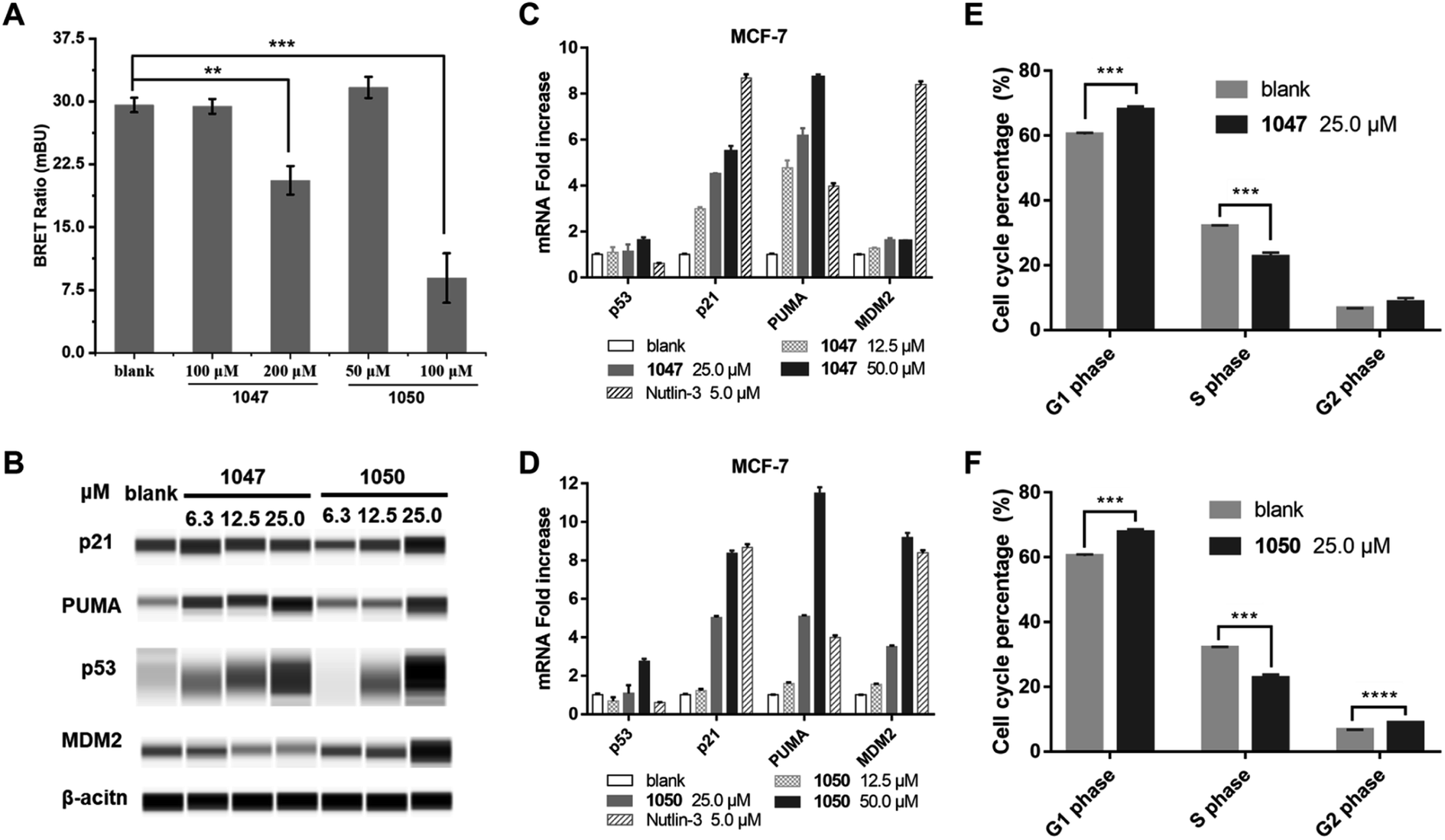 | ||
| Fig. 3 p53 TAD1 binders inhibit p53-MDM2 interaction in cells and activate the p53 pathway in cancer cells in MCF-7. (A) Compounds 1047 and 1050 inhibit p53-MDM2 interaction in the nanoBRET cellular assay. Data presented as mean (n = 3). *P < 0.03, **P < 0.002, ***P < 0.0002 (unpaired two-tailed t-test). (B) Compound 1050 stabilizes p53 and elevates protein levels of p53 target p21 and puma. (C and D) Compound 1047 and 1050 treatment induces dose-dependent expression of p53 target genes in MCF-7 cell line by quantitative PCR. (E and F) Compounds 1047 and 1050 induce cell cycle arrest in MCF-7 cells. Data presented as mean (n = 3). ***P < 0.0002, ****P < 0.0001 (unpaired two-tailed t-test). | ||
According to the model of p53 regulation by its negative regulator MDM2 and MDMX, inhibition of p53-MDM2/MDMX interaction would activate the p53 pathway in cells with wild-type p53.33,35 The on-target effect of p53 TAD1 ligands in a biological context of a living cell was evaluated by assessing the expression level of p53 and other p53-regulating genes. As shown in Fig. 3B–D, treatment with 1047 or 1050 induced accumulations of the p53 protein, whereas transcription of the p53 gene was unaffected, suggesting that treatment of p53 TAD1 ligands elevated the p53 protein levels by reducing its degradation by MDM2. Furthermore, accumulation of the p53 protein induced the transcription of the p53 target genes p21, puma and MDM2 in a dose-dependent manner (Fig. 3C and D). Up-regulation of p21, puma, and MDM2 proteins was also observed with 1050 treatment (Fig. 3B). We note that 1047 treatment did not induce significant accumulations of p21 and MDM2, which may result from other unknown effects.
Accumulation of p21 upon activation of p53 is expected to trigger cell cycle arrest in p53 wild-type cells. We tested the effect of 1047 and 1050 treatment on the cell cycle progression in MCF-7 cells. Both molecules showed effective cell-cycle arrest in G1 and G2/M phases, which led to depletion of the S phase compartment (Fig. 3E and F). Taken together, these data indicated that 1047 and 1050 treatment activated the p53 pathway in cancer cells with wild-type p53.
Binding mode analysis of the p53 TAD1 ligands
To locate the primary binding site within p53 TAD1, we tested the interaction between p53 TAD1 ligands and the p53 TAD1 peptide using NMR spectroscopy. We recorded two-dimensional 1H–1H total correlation spectroscopy (TOCSY) of p53 TAD1 and p53 TAD1 with active ligands. Significant chemical shift perturbations (CSPs) were observed for aromatic hydrogen atoms in TOCSY spectra of the p53 TAD1 peptide after addition of 1047 and 1050 (Fig. 4A). After resonance assignment with COSY and NOESY spectra, residue W23 was identified with large CSPs (Fig. 4A). To further analyze the specificity of binding between the active ligands and the p53 TAD1 peptide, we shuffled the p53 TAD1 peptide sequence and then acquired TOCSY spectra under the same experimental conditions. For wild-type p53 TAD1 peptide, p53 TAD1 ligands 1047 and 1050 treatment induced 0.011 ppm and 0.015 ppm (F1) CSPs, respectively, while only 0.005 ppm and 0.008 ppm (F1) CSPs were observed for the shuffled p53 TAD1 peptide (Fig. 4B, S7 and Table S3†). Collectively, these results demonstrated that the interaction between the p53 TAD1 ligands and p53 TAD1 was specific and sequence-dependent.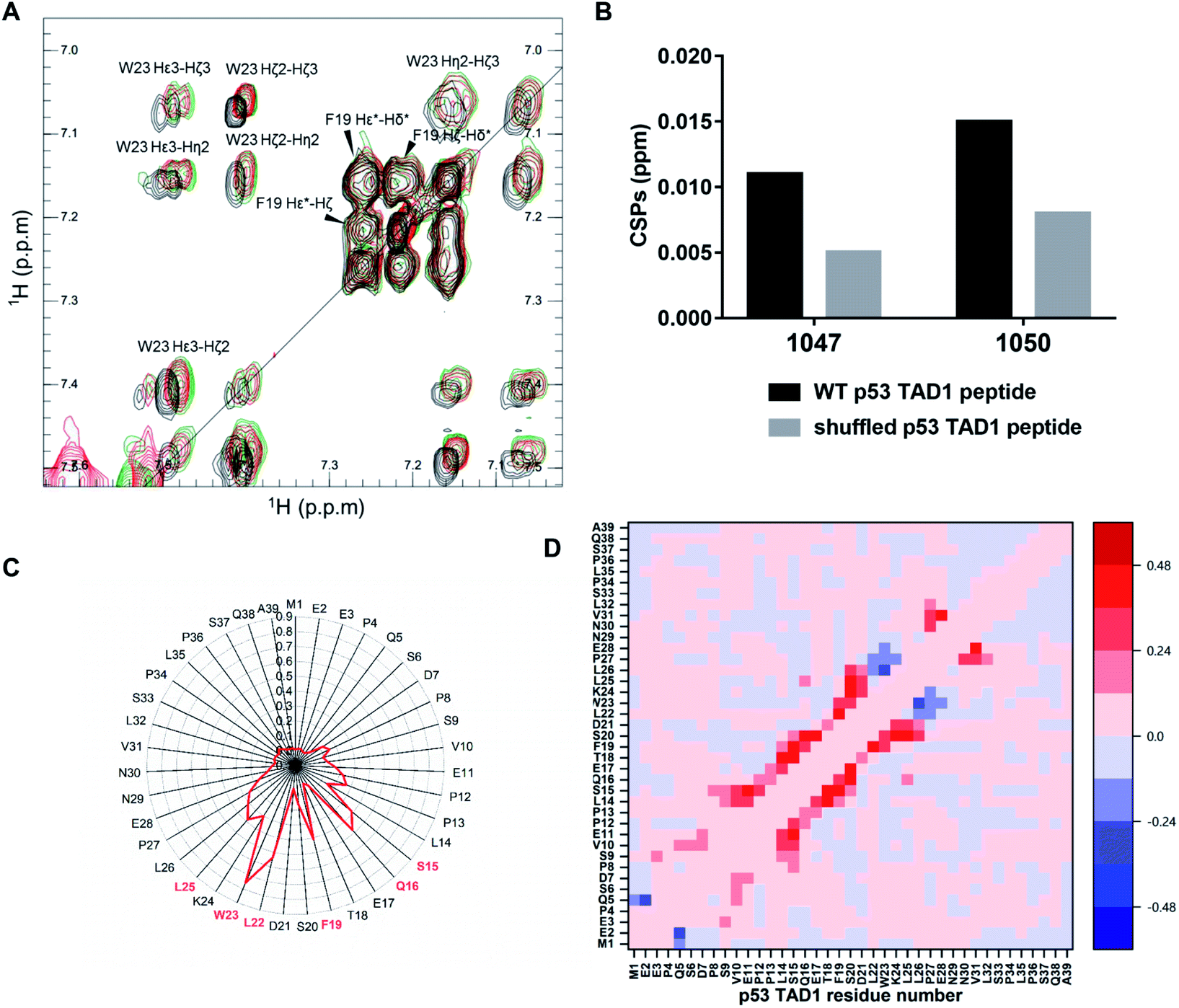 | ||
| Fig. 4 p53 TAD1 ligands bind to the p53 TAD1 peptide specifically. (A) Overlay of TOCSY spectra of free p53 TAD1 peptide (black, 100 μM) and the p53 TAD1 peptide–p53 TAD1 ligand complex (1047 red, 1050 green, 200 μM). (B) CSPs induced by 1047 and 1050 treatment for the p53 TAD1 peptide and the shuffled p53 TAD1 peptide. (C) Contact probabilities between p53 TAD1 and 1047. (D) Contact probability differences between p53 TAD1 residues for apo (free p53 TAD1 state) and holo (binding with active compound 1047) p53 TAD1 ensemble. Red/blue indicates increased contact probability and reduced contact probability after binding to 1047, respectively. | ||
We further performed MD simulations to gain insight into the binding features of p53 TAD1 and 1047. Given that force fields originally developed for ordered proteins are insufficient in sampling IDPs,48,49 we firstly tested the newly developed force fields and water models parameterized for IDPs to better sample the disordered p53 TAD1 ensemble. RSFF2 was developed by modifying the Amber ff99SB force field based on conformational free-energy distribution of the 20 amino acid residues from a protein coil library.50 The new water model TIP4P-D corrects deficiencies in modeling water dispersion interactions, thus it greatly improves the description of disordered protein states.51 The previous study illustrates that combination of RSFF2 and TIP4P-D turns out to achieve better balance between different protein secondary structures.52 Therefore, RSFF2/TIP4P-D combination was employed for p53 TAD1 ensemble sampling. Strikingly, this combination led to a more extended protein structure and more reasonable helix content in p53 TAD1 ensemble trajectories compared to NMR experiment results (Fig. S8†).43
According to our docking results from the first-round virtual screening, 1047 was predicted to bind to four conformations among the eight representative ones (Fig. S9, Table S4†). The four starting complex structures were subject to four 300 nanosecond MD simulations, respectively. To evaluate simulation convergence of p53 TAD1-ligand complexes, we analyzed time-dependent changes of secondary structure content with a time interval of 50 ns starting from 0 ns, as shown in Fig. S10.† In the late stages of simulations (from ∼200 ns), secondary structure content tends to be stable, indicating convergence of simulations. To characterize the binding site, the interactions between hydrogen atoms in 1047 and different protein residues (d < 4.5 Å) were analyzed. Compound 1047 interacted with the p53 TAD1 peptide throughout three independent 300 nanosecond trajectories except the one in which no interactions were observed between 261 and 300 ns (Fig. S11†). Subsequently, detailed analyses were performed based on the three independent 300 nanosecond trajectories. Compound 1047 binding reduced the radius of gyration (Rg) and increased the helix content (Fig. S12†). It seems that 1047 binding narrows the radius of gyration distribution of p53 TAD1. To characterize the binding features between p53 TAD1 and 1047, we calculated the contact probability of p53 TAD1 and 1047. Fig. 4C shows that 1047 comes into contact primarily with the p53 TAD1 functional region, especially W23, which is consistent with the TOCSY spectra results. Difference in contact probability between protein residues revealed the impact of 1047 binding on p53 TAD1 conformation ensemble. As in Fig. 4D, 1047 binding increased helical contacts in residues 15 to 26 and decreased helical contacts in residues 27 to 29.
Discussion
There is an increasing interest in discovering IDP binding compounds using rational drug design approaches. However, due to the heterogeneous nature of the IDP conformation ensemble, conventional structure-based drug design methods cannot be applied directly. We developed a novel virtual screening strategy for IDP drug discovery (IDPDVS) and successfully used it to discover two compounds that bind to p53 TAD1, break p53-MDM2 binding and activate p53 in living cells. IDPDVS should be generally applicable in IDP drug discovery, which can be further optimized based on more application cases.Insights into the dynamic and ensemble nature of the IDP structure have revealed unprecedented challenges for drug discovery targeting IDPs via rational drug design approaches. To surmount these challenges, several key issues need to be solved. First, a comprehensive conformational library has to be generated. We used classical MD and enhanced sampling REMD with different initial structures, and then combined with the latest force field, ff14SB, to acquire conformation space.53 After identifying the active compound 1047, we further used the newly developed force field RSFF2 and water model TIP4P-D parameterized for IDPs to investigate the interaction between p53 TAD1 and 1047. Although several force fields have been released to model IDPs,50,54–57 they should be assessed on the protein of interest and compared with experimental results. Our data showed that the p53 TAD1 ensemble-structures generated by applying the newly developed force field RSFF2 combined with the TIP4P-D water model are consistent with NMR experimental results. In addition, drug design targeting IDP would benefit from extensive simulations. For example, high throughput MD simulations coupled with Markov State Models towards p53 TAD1 revealed the macrostate interconversion,58 which is helpful for designing small molecules that more efficiently trap IDP in nonfunctional conformations.
Second, potential binding sites need to be predicted. Delicate conformation analyses are needed because interactions between IDPs and their ligands are also dynamic, which leads to dynamic binding sites within IDPs.59–61 We used the local segment clustering method followed by full-length clustering to group conformations. The pocket detection and analysis program CAVITY was used to predict potential binding pockets and assess their druggability. Coincidentally, Baggett et al. developed a segmented clustering method to identify well-populated local conformations as potential binding sites.17 Third, ensemble docking has to be used to consider the flexibility of IDPs. Stelzer et al. successfully identified experimentally validated RNA-targeting compounds via ensemble docking.62
Furthermore, due to the dynamic binding modes between IDPs and their ligands, we prefer molecules being able to bind to multiple conformations according to docking scores. Our previous studies in c-Myc ligand discovery revealed that active compounds tend to bind multiple conformations simultaneously and compounds that only strongly bind to one conformation are usually not active. Therefore, we selected eight conformations for p53 TAD1 and applied the “multi-conformational-affinity” strategy in compound selection. The novel IDP drug discovery strategy developed here provides a general approach for IDP ligand discovery, especially for IDPs without known active small-molecule ligands.
Our multi-conformational-affinity compound selection rule reflects the dynamic interaction nature of IDPs with their ligands. It has been found that IDPs remain disordered even after ligand binding and ligands bind to IDPs at multiple binding sites, exhibiting “ligand cloud” or “specific diffuse” binding mechanisms.7,59–61,63 To further test our computational strategy, we performed negative control experiments. We purchased ten single conformation binding compounds and tested their binding strength with p53 TAD1. Each of these ten compounds only binds one p53 TAD1 conformation with docking score among top 1% and other conformations with low scores that rank outside of top 1% (Table S4†). In the SPR binding test, none of them showed binding to p53 TAD1, further validating the rationale of our screening strategy (Table S5†). All the active and the negative control compounds have distinct chemical structures with Tanimoto coefficients less than 0.25 (Tables S6 and S7†).
Our NMR studies showed that the two active compounds we identified interacted with the functional site of p53 TAD1. MD simulations revealed that 1047 made dynamic and extensive hydrophobic interactions with p53 TAD1. Furthermore, CSPs of WT p53 TAD1 induced by p53 TAD1 ligand treatment were significantly larger than the shuffled p53 TAD1 control peptide, demonstrating the specificity and sequence dependence of the interaction between p53 TAD1 and small-molecule ligands. We also plotted the hydropathy of p53 TAD1 along with residue number, calculated according to the hydrophilicity scale of Kyte–Doolittle.64 As can be seen from Fig. S13,† the binding site in p53 TAD1 not only displays high hydrophobicity, but also contains aromatic residues. Together, these findings highlight that hydrophobic contacts and π–π interactions play critical roles in p53 TAD1–ligand interaction. Furthermore, by comparison of the physicochemical properties of all the active and the negative control compounds, we found that the active compounds tend to possess higher molecular weight and more ring structures compared to the negative control compounds (Fig. S14†), which is consistent with our analysis that hydrophobic interactions play important roles between p53 TAD1 and the active compounds. We suggest that hydrophobic residue clusters containing aromatic residues can be used as an indicator of potential IDP-ligand binding site.
To eliminate possible assay interference of the compounds, we analyzed the two active compounds using the PAINS server65 and searched the literature for their reported biological activities. Both compounds 1047 and 1050 do not belong to PAINS and none of them has reported biological activity so far. We also performed the Similarity Ensemble Approach (SEA) analysis for these two compounds to evaluate off-target probabilities.66 Only one protein (MYLK_human, Myosin light chain kinase) was marginally predicted as possible 1047 target and none was predicted for 1050, suggesting low off-target possibilities of the active ligands 1047 and 1050 (Fig. S15†). As these two compounds activate the p53 pathway by directly binding to p53 rather than MDM2, they should be able to avoid the gastrointestinal and hematological adverse events caused by MDM2 inhibitors. These compounds can be further optimized in future studies.
IDPs are closely associated with many diseases. Recent advances have shown that IDPs are key drivers of membraneless bodies formed by phase separation.67–69 Targeting IDPs to regulate liquid–liquid phase separation will open up new avenues for therapeutic intervention.70,71 Given that virtual screening can be scaled up to include millions of compounds, the IDPDVS strategy that we developed can be applied to screen compounds to modulate IDP-mediated pathologies and diseases. Although our strategy is generally applicable, special concerns should be considered before applying it. Since IDPs are typically rich in polar and charged amino acid residues and lack bulk hydrophobic residues, druggability of the IDP of interest should be evaluated before using the IDPDVS strategy. We suggest that the existence of a high average hydrophobicity region enriched with aromatic residues is one good indicator of druggability. In addition, some IDPs have significant long-range intrachain contacts, and active small molecules may function by inhibiting the long-range intrachain contact. In this case, although conformations with non-long range intrachain contacts have low population in molecular simulations of the free state, these conformations may be highly druggable. With the improvement of IDP conformation ensemble characterization methods and our understanding of IDP and ligand interactions, we are expecting that better strategies of rational drug design targeting IDP emerge ahead.
Materials and methods
MD simulations and analysis
To sample p53 TAD1 conformations, classical all-atom MD and REMD simulations were performed with AMBER14SB force fields and AMBER14 software package.53,72 For classical all-atom MD simulations, the conformation of the p531–39 peptide (p53 TAD1) within the p300 Taz1–p53 TAD1 structure (PBD ID, 2K8F) was used as the starting structure and the peptide termini were amidated.42 The TIP4P-EW water molecules were added as the solvent and the solute atoms were at least 15 Å away from the boundary of the cubic box. The net charge of the system was neutralized with counterions. The system was minimized using the steepest descent approaches, followed by conjugate gradient minimization. Heat and density equilibration were performed on the system for 100 ps. Three 300 ns simulations were conducted in the NPT (P = 1 atm and T = 298 K) ensemble. The particle mesh Ewald (PME) algorithm was used to handle long-range electrostatic interactions and the nonbonded cutoff was set to 8 Å. The SHAKE algorithm was used to restrict the covalent bonds between heavy atoms and hydrogen atoms. For the REMD simulation, the initial structure was random coil and the peptide termini were also amidated. A total of 36 replicas were adopted with temperature ranging from 279 K to 600 K. Replica exchanges were attempted every 10 ps. Each replica started from the same structure but with different random number seeds for the initial velocity assignment and at different temperatures. The REMD was performed with a generalized Born/surface area (GB/SA) implicit solvent model. The cutoff for non-bonded interactions and for the GB pairwise summations involved in calculating Born radii was 999 Å. Each replica was simulated for 238 ns.To explore suitable force fields for p53 TAD1, all-atom MD simulations were conducted with the GROMCS 5.0.7 package.73,74 The RSFF2 force field with the TIP4P-D water model was used.50,51 The initial structure was also from the p300 Taz1–p53 TAD1 structure with termini amidated. Electrostatics was treated using the PME method, and Van der Waals interactions were cut off at 10 Å. All bonds involving hydrogen were restrained using LINCS. For molecular dynamics simulations of p53 TAD1 complexes with compounds, the RSFF2 force field and the general amber force field (gaff) were used for p53 peptide and ligands, respectively.
Druggable conformation selection and virtual screening
Structural clustering was conducted with AmberTools14 using the average-linkage algorithm. Preliminary clustering was performed on the p53 backbone atoms of residues 19–26, stopping when minimum distance between clusters is 1.5 Å, followed by secondary clustering on the p53 backbone atoms of residues 1–39 for every cluster from preliminary clustering, stopping when the minimum distance between clusters is 6 Å. Binding site detection and druggability prediction of various conformations were performed using the CAVITY program.45,46 We selected structures satisfying the following three rules: (i) pockets are located in the functional site; (ii) high druggability score from CAVITY (average pKd > 6, volume > 300 Å3); (iii) large cluster population. Finally, eight conformations were selected as potential druggable conformations.High-throughput docking was performed using Glide.75,76 We chose two docking modes with different precisions from Glide: high throughput virtual screening (HTVS) and standard precision (SP), to screen SPECS libraries. Top 50% molecules with the best scoring function from the HTVS mode were redocked using the SP mode to obtain top 1% molecules (the 0.5% top-ranking compounds for all compounds). Ensemble-based docking was conducted using the selected eight conformations.
MDM2 expression and purification
Samples of human MDM2, corresponding to residues 17–125, were expressed in Escherichia coli strain BL21 (DE3), using recombinant plasmid pGEX6P2-MDM2 (17–125) (Addgene plasmid #62063).43 Recombinant cells were cultivated at 37 °C in LB medium. At an OD600 of 0.6–0.8, expression was induced by the addition of 0.5 mM IPTG and cultures were grown at 25 °C for 6 h. E. coli cells were resuspended in binding buffer (50 mM Tris–HCl, 300 mM NaCl, 2.5 mM EDTA, 2 mM DTT, pH 7.4), lysed by sonication supplemented with 1 mM PMSF and sedimented at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min. The supernatant lysates were chromatographed on a GSTrap column and the MDM2 was cleaved from the column using 1:100 ratio of HRV3C protease with the cleavage buffer containing 50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.3. Protein fractions were eluted with the cleavage buffer with three column volumes and then were buffer exchanged into 50 mM sodium phosphate, pH 6.6. Fractions were concentrated and further purified on a 5 ml HiTrap SP HP column. Proteins were washed with buffer 50 mM sodium phosphate, 500 mM NaCl, pH 6.6 and the protein peaks were collected and concentrated. The Sephacryl S-200 gel-filtration column was applied for final purification with buffer containing 50 mM sodium phosphate, and 300 mM NaCl, at pH 7.0 (Fig. S5†).
000 rpm for 30 min. The supernatant lysates were chromatographed on a GSTrap column and the MDM2 was cleaved from the column using 1:100 ratio of HRV3C protease with the cleavage buffer containing 50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA, and 1 mM DTT, pH 7.3. Protein fractions were eluted with the cleavage buffer with three column volumes and then were buffer exchanged into 50 mM sodium phosphate, pH 6.6. Fractions were concentrated and further purified on a 5 ml HiTrap SP HP column. Proteins were washed with buffer 50 mM sodium phosphate, 500 mM NaCl, pH 6.6 and the protein peaks were collected and concentrated. The Sephacryl S-200 gel-filtration column was applied for final purification with buffer containing 50 mM sodium phosphate, and 300 mM NaCl, at pH 7.0 (Fig. S5†).
Surface plasmon resonance (SPR)
A Biacore T200 (GE Healthcare) system was used for all experiments. For preliminary compound screening, N-terminal biotinylated p531–39 peptide (synthesized by GL Biochem Ltd, purity ≥ 95%) was captured on a Sensor Chip SA (GE Healthcare) at a density of about 500 RU. We performed initial screening of compounds at 50 μM and 100 μM in 1× PBS-P (GE Healthcare) with 5% DMSO. Samples were injected for 120 s at a flow rate of 10 μL min−1 and dissociation time was 240 s. In the direct binding assay, serial dilution of molecules was made in the running buffer. For competitive binding assay, serial dilutions of molecules with the MDM2 protein at a 1.9 μM concentration were injected. Regeneration was achieved with 10 mM glycine-HCl, pH 3.0 for 5 s after the disassociation stage to remove residual MDM2 proteins on the chip. Biacore T200 Evaluation was used for data analysis.Microscale thermophoresis (MST) measurements
MDM2 was labeled using the RED-NHS Labeling Kit (NanoTemper Technologies). The labeling reaction was performed at RT for 30 min and then the unreacted dye was removed with the supplied dye removal columns equilibrated with buffer (50 mM PB, 150 mM NaCl, pH 6.8). A volume of 10 μL of labeled protein was mixed with 10 μL of ligand in 50 mM PB, 150 mM NaCl, pH 6.8, 0.1% Tween-20, 2 mg ml−1 BSA, 5% DMSO. Thermophoresis was measured using a Monolith NT.115 instrument and instrument parameters were adjusted to 50% LED power and medium MST power. The thermophoresis + T-Jump signal was employed for data analysis.NMR experiments
NMR experiments were performed at 25 °C using a Bruker Advanced 600 MHz spectrometer equipped with cryo probes. For 2D 1H–1H TOCSY, COSY and NOESY spectra, NMR samples contained 100 μM p53 TAD1 peptide in 5 mM Na phosphate, pH 6.3, 200 mM NaCl, 5% DMSO-D6, and 95% D2O. Samples of 200 μM p53 TAD1 binder treatment for 100 μM p53 TAD1 peptide, shuffled peptide and amino acid L-tryptophan in 5 mM Na phosphate, pH 6.3, 200 mM NaCl, 5% DMSO-D6, and 95% D2O were prepared for 2D 1H–1H TOCSY spectra, respectively. Shuffled p53 TAD1 peptide sequence: MSPPSEDNWE SKSQPQELVP PVDSLQLESL PFTLNEPLA. TOCSY spectra were acquired with a relaxation delay between scans of 1.5 s, 60 ms TOCSY mixing time. NOESY spectra were acquired with a relaxation delay between scans of 2 s, 150 ms NOESY mixing time. All experiments used shaped pulses for water suppression, and the 2D spectra were processed using Bruker® Topspin software to 2048 × 1024 points and analyzed using CcpNmr software.77 The wild type and shuffled p53 TAD1 peptide were synthesized by GL Biochem Ltd (purity ≥ 95%).Cell culture and MTT assay
MCF-7 and HCT116 from the China Infrastructure of Cell Line Resources were cultured in DMEM and IMDM (Gibco), respectively. H1299 from the American Type Culture Collection (ATCC) was cultured in RMPI-1640 culture medium (Gibco). All cell culture media contained 10% PBS, 100 U ml−1 penicillin, and 100 μg ml−1 streptomycin. Cells in exponential growth were seeded at 2000–3000 cells per well in 96-well culture plates and allowed to grow for 24 h before treatment. Culture media were added to stock molecular solutions and final molecular formulations contained 0.2% DMSO (v/v) and 0.01% Tween 80 (v/v). Spectroscopic reading was taken 72 h after treatment, and the cell survival percentage was calculated as the percentage absorbance of treated wells versus reference wells.Bioluminescence resonance energy transfer (BRET) assays
HEK293T cells (800000 cells per well) were plated in a 6-well plate 4–6 h prior to treatment and co-transfected with the NanoLuc-MDM2 fusion vector and p53-HaloTag fusion vector using FuGENE HD transfection reagent (PROMEGA). After a 20 h overnight incubation period post-transfection cells were collected, washed with PBS, and exchanged into DMEM media with 4% FBS. Cell density was adjusted to 20000 cells per ml and then divided into two pools where either HaloTag NanoBRET 618 ligand was added or DMSO. Subsequently, cells were re-plated in a 96-well assay white plate and p53 TAD1 ligands were then added directly to media. After incubation for 4–6 h, Nano-Glo substrate was added to each control and experimental sample. The donor emission (460 nm) and acceptor emission (618 nm) were measured within 10 minutes of substrate addition using the GloMax Discover System with preloaded filters for donor 450 nm/8 nm bandpass and acceptor 600 nm long pass filters.
Western-blot analysis
Cells were exposed to various concentrations of testing compounds for 24 h, and then lysed using RIPA buffer supplemented with cocktails of protease inhibitors. The following primary antibodies were used in this study: p53 (#Sc-126, Santa Cruz Biotechnology, 1:10), PUMA (#12450, Cell Signaling Technology, 1:10), MDM2 (#Sc-5304, Santa Cruz Biotechnology, 1:10), β-actin (#3700S, Cell Signaling Technology, 1:100). Protein separation and detection were performed using an automated capillary electrophoresis system (Simple Western system and Compass software; Protein Simple). In short, after performing capillary electrophoresis in a Wes Simple Western System, sample proteins were analyzed based on the chemiluminescence signal peaks generated, which were transformed into digital images depicting bands observed in the western blot analysis. Western-blot results were analyzed using the Protein Simple software Compass.
Quantitative real-time PCR
Cells were seeded in 6 well plates (30000 cells per well) 24 hours prior to treatment. Cells were treated with testing compounds for 12 h and control cells were treated with an equivalent concentration of the solvent DMSO. Total RNA was extracted from culture cells using the Eastep Super Total RNA Extraction Kit (Promega). After converting total RNA to cDNA using the PrimeScript RT reagent Kit (TaKaRa), the relative quantity of p53, p21, puma and MDM2 transcripts was determined using SYBR Select Master Mix (Life Technologies) and β-actin as a normalization control. The sequence of the primers was as follows: p53 (F: CTGGGACGGAACAGCTTTGA; R: CCTTTCTTGCGGAGATTCTCTTC), p21 (F: CTGGAGACTCTCAGGGTCGAA; R: CGGCGTTTGGAGTGGTAGAA), PUMA (F: GACCTCAACGCACAGTACGAG; R: AGGAGTCCCATGATGAGATTGT), MDM2 (F: AATCATCGGACTCAGGTACAT C; R: CTGTCTCACTAATTGCTCTCCT).
Flow cytometric analysis of the cell cycle
HCT116 and MCF-7 cells in exponential growth were seeded at 300000 cells per well in 6-well plates and incubated for 36 h before treatment. After being incubated with testing compounds for 24 h, cells were detached with trypsin and collected by centrifugation at 1000g for 5 min. Then cells were washed twice with PBS and fixed in 70% (v/v) ethanol at 4 °C for 2 h, followed by incubation with RNase and staining with propidium iodide at 37 °C for 30 min. The suspensions were then analyzed using a Becton Dichinson FACScan (BD Biosciences, San Jose, CA).
Author contributions
L. L. developed the concept of this study. H. R. designed and performed experiments, analyzed data and prepared the draft manuscript. C. Y., X. N., W. Z., H. L., L. C., R. X., and Q. S. performed experiments and analyzed data. C. J, Y. L. and L. L. analyzed data. Y. L. and L. L. supervised the work and edited the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors thank Prof. Zhirong Liu and Prof. Chun Tang for helpful discussions. This work was supported in part by the Ministry of Science and Technology of China (2016YFA0502303) and the National Natural Science Foundation of China (Grant No. 21633001, 21877003). Some of the simulations and analysis were performed on the High Performance Computing Platform of the Peking-Tsinghua Center for Life Sciences at Peking University. All NMR experiments were carried out at the Beijing NMR Center and the NMR facility of National Center for Protein Sciences at Peking University.References
- P. E. Wright and H. J. Dyson, Intrinsically disordered proteins in cellular signalling and regulation, Nat. Rev. Mol. Cell Biol., 2015, 16, 18–29 CrossRef CAS.
- V. N. Uversky, C. J. Oldfield and A. K. Dunker, Intrinsically disordered proteins in human diseases: introducing the D2 concept, Annu. Rev. Biophys., 2008, 37, 215–246 CrossRef CAS.
- S. J. Metallo, Intrinsically disordered proteins are potential drug targets, Curr. Opin. Chem. Biol., 2010, 14, 481–488 CrossRef CAS.
- Y. Cheng, T. LeGall, C. J. Oldfield, J. P. Mueller, Y. Y. Van, P. Romero, M. S. Cortese, V. N. Uversky and A. K. Dunker, Rational drug design via intrinsically disordered protein, Trends Biotechnol., 2006, 24, 435–442 CrossRef CAS.
- A. K. Dunker and V. N. Uversky, Drugs for 'protein clouds': targeting intrinsically disordered transcription factors, Curr. Opin. Pharmacol., 2010, 10, 782–788 CrossRef CAS.
- V. N. Uversky, Intrinsically disordered proteins and novel strategies for drug discovery, Expert Opin. Drug Dis., 2012, 7, 475–488 CrossRef CAS.
- H. Ruan, Q. Sun, W. Zhang, Y. Liu and L. Lai, Targeting intrinsically disordered proteins at the edge of chaos, Drug Discov. Today, 2019, 24, 217–227 CrossRef CAS.
- H. V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. Barber-Rotenberg, L. Yuan, O. D. Abaan, T. H. Chou, S. Dakshanamurthy, M. L. Brown, A. Uren and J. A. Toretsky, A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma, Nat. Med., 2009, 15, 750–756 CrossRef CAS.
- L. I. Iconaru, D. Ban, K. Bharatham, A. Ramanathan, W. Zhang, A. A. Shelat, J. Zuo and R. W. Kriwacki, Discovery of small molecules that inhibit the disordered protein, p27(Kip1), Sci. Rep., 2015, 5, 15686 CrossRef CAS.
- J. L. Neira, J. Bintz, M. Arruebo, B. Rizzuti, T. Bonacci, S. Vega, A. Lanas, A. Velazquez-Campoy, J. L. Iovanna and O. Abian, Identification of a drug targeting an intrinsically disordered protein involved in pancreatic adenocarcinoma, Sci. Rep., 2017, 7, 39732 CrossRef CAS.
- X. Yin, C. Giap, J. S. Lazo and E. V. Prochownik, Low molecular weight inhibitors of Myc-Max interaction and function, Oncogene, 2003, 22, 6151–6159 CrossRef CAS.
- R. S. Srinivasan, J. B. Nesbit, L. Marrero, F. Erfurth, V. F. LaRussa and C. S. Hemenway, The synthetic peptide PFWT disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells, Leukemia, 2004, 18, 1364–1372 CrossRef CAS.
- G. Toth, S. J. Gardai, W. Zago, C. W. Bertoncini, N. Cremades, S. L. Roy, M. A. Tambe, J. C. Rochet, C. Galvagnion, G. Skibinski, S. Finkbeiner, M. Bova, K. Regnstrom, S. S. Chiou, J. Johnston, K. Callaway, J. P. Anderson, M. F. Jobling, A. K. Buell, T. A. Yednock, T. P. Knowles, M. Vendruscolo, J. Christodoulou, C. M. Dobson, D. Schenk and L. McConlogue, Targeting the intrinsically disordered structural ensemble of α -synuclein by small molecules as a potential therapeutic strategy for Parkinson's disease, PloS one, 2014, 9, e87133 CrossRef.
- E. De Mol, R. B. Fenwick, C. T. Phang, V. Buzon, E. Szulc, A. de la Fuente, A. Escobedo, J. Garcia, C. W. Bertoncini, E. Estebanez-Perpina, I. J. McEwan, A. Riera and X. Salvatella, EPI-001, a compound active against castration-resistant prostate cancer, targets transactivation unit 5 of the androgen receptor, ACS Chem. Bio., 2016, 11, 2499–2505 CrossRef CAS.
- C. Yu, X. Niu, F. Jin, Z. Liu, C. Jin and L. Lai, Structure-based inhibitor design for the intrinsically disordered protein c-Myc, Sci. Rep., 2016, 6, 22298 CrossRef CAS.
- S. H. Choi, M. Mahankali, S. J. Lee, M. Hull, H. M. Petrassi, A. K. Chatterjee, P. G. Schultz, K. A. Jones and W. Shen, Targeted disruption of Myc-Max oncoprotein complex by a small molecule, ACS Chem. Biol., 2017, 12, 2715–2719 CrossRef CAS.
- D. W. Baggett and A. Nath, The rational discovery of a tau aggregation inhibitor, Biochemistry, 2018, 57, 6099–6107 CrossRef CAS.
- M. Y. Kim, I. Na, J. S. Kim, S. H. Son, S. Choi, S. E. Lee, J. H. Kim, K. Jang, G. Alterovitz, Y. Chen, A. van der Vaart, H. S. Won, V. N. Uversky and C. G. Kim, Rational discovery of antimetastatic agents targeting the intrinsically disordered region of MBD2, Sci. Adv., 2019, 5, eaav9810 CrossRef CAS.
- M. Zhu, A. De Simone, D. Schenk, G. Toth, C. M. Dobson and M. Vendruscolo, Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide, J. Chem. Phys., 2013, 139, 035101 CrossRef.
- B. Vogelstein, D. Lane and A. J. Levine, Surfing the p53 network, Nature, 2000, 408, 307–310 CrossRef CAS.
- A. Feki and I. Irminger-Finger, Mutational spectrum of p53 mutations in primary breast and ovarian tumors, Crit Rev. Oncol. Hematol., 2004, 52, 103–116 CrossRef.
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Awakening guardian angels: drugging the p53 pathway, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS.
- S. Boettcher, P. G. Miller, R. Sharma, M. McConkey, M. Leventhal, A. V. Krivtsov, A. O. Giacomelli, W. Wong, J. Kim, S. Chao, K. J. Kurppa, X. Yang, K. Milenkowic, F. Piccioni, D. E. Root, F. G. Rucker, Y. Flamand, D. Neuberg, R. C. Lindsley, P. A. Janne, W. C. Hahn, T. Jacks, H. Dohner, S. A. Armstrong and B. L. Ebert, A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies, Science, 2019, 365, 599–604 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science, 2004, 303, 844–848 CrossRef CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, Structure-based design of potent non-peptide MDM2 inhibitors, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS.
- M. Liu, C. Li, M. Pazgier, C. Li, Y. Mao, Y. Lv, B. Gu, G. Wei, W. Yuan, C. Zhan, W. Y. Lu and W. Lu, D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321–14326 CrossRef CAS.
- Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K. H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS.
- M. Wade, Y. C. Li and G. M. Wahl, MDM2, MDMX and p53 in oncogenesis and cancer therapy, Nat. Rev. Cancer, 2013, 13, 83–96 CrossRef CAS.
- W. Wang, J. J. Qin, S. Voruganti, K. S. Srivenugopal, S. Nag, S. Patil, H. Sharma, M. H. Wang, H. Wang, J. K. Buolamwini and R. W. Zhang, The pyrido[b]indole MDM2 inhibitor SP-141 exerts potent therapeutic effects in breast cancer models, Nat. Commun., 2014, 5, 5086 CrossRef CAS.
- A. Burgess, K. M. Chia, S. Haupt, D. Thomas, Y. Haupt and E. Lim, Clinical overview of MDM2/X-targeted therapies, Front. Oncol., 2016, 6, 7 Search PubMed.
- V. Tisato, R. Voltan, A. Gonelli, P. Secchiero and G. Zauli, MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer, J. Hematol. Oncol., 2017, 10, 133 CrossRef.
- A. C. Joerger and A. R. Fersht, Structural biology of the tumor suppressor p53, Annu. Rev. Biochem., 2008, 77, 557–582 CrossRef CAS.
- M. Wells, H. Tidow, T. J. Rutherford, P. Markwick, M. R. Jensen, E. Mylonas, D. I. Svergun, M. Blackledge and A. R. Fersht, Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5762–5767 CrossRef CAS.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain, Science, 1996, 274, 948–953 CrossRef CAS.
- N. A. Laurie, S. L. Donovan, C. S. Shih, J. Zhang, N. Mills, C. Fuller, A. Teunisse, S. Lam, Y. Ramos, A. Mohan, D. Johnson, M. Wilson, C. Rodriguez-Galindo, M. Quarto, S. Francoz, S. M. Mendrysa, R. K. Guy, J. C. Marine, A. G. Jochemsen and M. A. Dyer, Inactivation of the p53 pathway in retinoblastoma, Nature, 2006, 444, 61–66 CrossRef CAS.
- C. L. Brooks and W. Gu, p53 ubiquitination: Mdm2 and beyond, Mol. Cell, 2006, 21, 307–315 CrossRef CAS.
- V. Grinkevich, N. Issaeva, S. Hossain, A. Pramanik and G. Selivanova, NMR indicates that the small molecule RITA does not block p53-MDM2 binding in vitro - Reply, Nat. Med., 2005, 11, 1136–1137 CrossRef CAS.
- N. Issaeva, P. Bozko, M. Enge, M. Protopopova, L. G. G. C. Verhoef, M. Masucci, A. Pramanik and G. Selivanova, Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors, Nat. Med., 2004, 10, 1321–1328 CrossRef CAS.
- C. K. Fisher and C. M. Stultz, Constructing ensembles for intrinsically disordered proteins, Curr. Opin. Struct. Biol., 2011, 21, 426–431 CrossRef CAS.
- G. Wei, W. Xi, R. Nussinov and B. Ma, Protein ensembles: How does nature harness thermodynamic fluctuations for Life? The diverse functional roles of conformational ensembles in the cell, Chem. Rev., 2016, 116, 6516–6551 CrossRef CAS.
- H. Q. Feng, L. M. M. Jenkins, S. R. Durell, R. Hayashi, S. J. Mazur, S. Cherry, J. E. Tropea, M. Miller, A. Wlodawer, E. Appella and Y. Bai, Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation, Structure, 2009, 17, 202–210 CrossRef CAS.
- W. Borcherds, F. X. Theillet, A. Katzer, A. Finzel, K. M. Mishall, A. T. Powell, H. Wu, W. Manieri, C. Dieterich, P. Selenko, A. Loewer and G. W. Daughdrill, Disorder and residual helicity alter p53-Mdm2 binding affinity and signaling in cells, Nat. Chem. Biol., 2014, 10, 1000–1002 CrossRef CAS.
- H. F. Chen and R. Luo, Binding induced folding in p53-MDM2 complex, J. Am. Chem. Soc., 2007, 129, 2930–2937 CrossRef CAS.
- Y. Xu, S. Wang, Q. Hu, S. Gao, X. Ma, W. Zhang, Y. Shen, F. Chen, L. Lai and J. Pei, CavityPlus: a web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction, Nucleic Acids Res., 2018, 46, W374–W379 CrossRef CAS.
- Y. Yuan, J. Pei and L. Lai, Binding site detection and druggability prediction of protein targets for structure-based drug design, Curr. Pharm. Des., 2013, 19, 2326–2333 CrossRef CAS.
- E. Aberg, O. A. Karlsson, E. Andersson and P. Jemth, Binding kinetics of the intrinsically disordered p53 family transactivation domains and MDM2, J. Phys. Chem. B, 2018, 122, 6899–6905 CrossRef.
- K. Kasahara, H. Terazawa, T. Takahashi and J. Higo, Studies on molecular dynamics of intrinsically disordered proteins and their fuzzy complexes: A mini-review, Comput. Struct. Biotechnol J., 2019, 17, 712–720 CrossRef CAS.
- S. H. Chong, P. Chatterjee and S. Ham, Computer simulations of intrinsically disordered proteins, Annu. Rev. Phys. Chem., 2017, 68, 117–134 CrossRef CAS.
- C. Y. Zhou, F. Jiang and Y. D. Wu, Residue-specific force field based on protein coil library. RSFF2: modification of AMBER ff99SB, J. Phys. Chem. B, 2015, 119, 1035–1047 CrossRef CAS.
- S. Piana, A. G. Donchev, P. Robustelli and D. E. Shaw, Water dispersion interactions strongly influence simulated structural properties of disordered protein states, J. Phys. Chem. B, 2015, 119, 5113–5123 CrossRef CAS.
- H. N. Wu, F. Jiang and Y. D. Wu, Significantly improved protein folding thermodynamics using a dispersion-corrected water model and a new residue-specific force field, J. Phys. Chem. Lett., 2017, 3199–3205 CrossRef CAS.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS.
- D. Song, W. Wang, W. Ye, D. Ji, R. Luo and H. F. Chen, ff14IDPs force field improving the conformation sampling of intrinsically disordered proteins, Chem. Biol. Drug Des., 2017, 89, 5–15 CrossRef CAS.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. de Groot, H. Grubmuller and A. D. MacKerell Jr, CHARMM36m: an improved force field for folded and intrinsically disordered proteins, Nat. Methods, 2017, 14, 71–73 CrossRef CAS.
- P. Robustelli, S. Piana and D. E. Shaw, Developing a molecular dynamics force field for both folded and disordered protein states, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4758–E4766 CrossRef CAS.
- H. Liu, D. Song, H. Lu, R. Luo and H. F. Chen, Intrinsically disordered protein-specific force field CHARMM36IDPSFF, Chem. Biol. Drug Des., 2018, 92, 1722–1735 CrossRef CAS.
- P. Herrera-Nieto, A. Perez and G. De Fabritiis, Characterization of partially ordered states in the intrinsically disordered N-terminal domain of p53 using millisecond molecular dynamics simulations, Sci. Rep., 2020, 10, 12402 CrossRef CAS.
- F. Jin, C. Yu, L. Lai and Z. Liu, Ligand clouds around protein clouds: a scenario of ligand binding with intrinsically disordered proteins, PLoS Comput. Biol., 2013, 9, e1003249 CrossRef.
- D. Ban, L. I. Iconaru, A. Ramanathan, J. Zuo and R. W. Kriwacki, A small molecule causes a population shift in the conformational landscape of an intrinsically disordered protein, J. Am. Chem. Soc., 2017, 139, 13692–13700 CrossRef CAS.
- G. T. Heller, F. A. Aprile, M. Bonomi, C. Camilloni, A. De Simone and M. Vendruscolo, Sequence specificity in the entropy-driven binding of a small molecule and a disordered peptide, J. Mol. Biol., 2017, 429, 2772–2779 CrossRef CAS.
- A. C. Stelzer, A. T. Frank, J. D. Kratz, M. D. Swanson, M. J. Gonzalez-Hernandez, J. Lee, I. Andricioaei, D. M. Markovitz and H. M. Al-Hashimi, Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble, Nat. Chem. Biol., 2011, 7, 553–559 CrossRef CAS.
- J. Michel and R. Cuchillo, The impact of small molecule binding on the energy landscape of the intrinsically disordered protein c-Myc, PloS one, 2012, 7, e41070 CrossRef CAS.
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157, 105–132 CrossRef CAS.
- J. B. Baell and G. A. Holloway, New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS.
- M. J. Keiser, B. L. Roth, B. N. Armbruster, P. Ernsberger, J. J. Irwin and B. K. Shoichet, Relating protein pharmacology by ligand chemistry, Nat. Biotechnol., 2007, 25, 197–206 CrossRef CAS.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Biomolecular condensates: organizers of cellular biochemistry, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS.
- Y. Shin and C. P. Brangwynne, Liquid phase condensation in cell physiology and disease, Science, 2017, 357, eaaf4382 CrossRef.
- S. Alberti, A. Gladfelter and T. Mittag, Considerations and challenges in studying liquid-liquid phase separation and biomolecular condensates, Cell, 2019, 176, 419–434 CrossRef CAS.
- K. Cermakova and H. C. Hodges, Next-generation drugs and probes for chromatin biology: From targeted protein degradation to phase separation, Molecules, 2018, 23, 1958 CrossRef.
- D. G. Brown, J. Shorter and H. J. Wobst, Emerging small-molecule therapeutic approaches for amyotrophic lateral sclerosis and frontotemporal dementia, Bioorg. Med. Chem. Lett., 2020, 30, 126942 CrossRef CAS.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, The Amber biomolecular simulation programs, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- W. F. Vranken, W. Boucher, T. J. Stevens, R. H. Fogh, A. Pajon, M. Llinas, E. L. Ulrich, J. L. Markley, J. Ionides and E. D. Laue, The CCPN data model for NMR spectroscopy: development of a software pipeline, Proteins, 2005, 59, 687–696 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04670a |
| This journal is © The Royal Society of Chemistry 2021 |