
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe video-rate imaging of sub-10 nm plasmonic nanoparticles in a cellular medium free of background scattering†
He
Gao
,
Pei
Wu
,
Pei
Song
,
Bin
Kang
*,
Jing-Juan
Xu
 * and
Hong-Yuan
Chen
* and
Hong-Yuan
Chen
State Key Laboratory of Analytical Chemistry for Life Science, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: binkang@nju.edu.cn; xujj@nju.edu.cn
First published on 4th January 2021
Abstract
Plasmonic nanoparticles (e.g., gold, silver) have attracted much attention for biological sensing and imaging as promising nanoprobes. Practical biomedical applications demand small gold nanoparticles (Au NPs) with a comparable size to quantum dots and fluorescent proteins. Very small nanoparticles with a size below the Rayleigh limit (usually <30–40 nm) are hard to see by light scattering using a dark-field microscope, especially within a cellular medium. A photothermal microscope is able to detect very small nanoparticles, down to a few nanometers, but the imaging speed is usually too slow (minutes to hours) to image living cell processes. Here an absorption modulated scattering microscopy (AMSM) method is presented, which allows for the imaging of sub-10 nm Au NPs within a cellular medium. The unique physical mechanism of AMSM offers the remarkable ability to remove the light scattering background of the cellular component. In addition to having a sensitivity comparable to that of photothermal microscopy, AMSM has a much higher imaging speed, close to the video rate (20 fps), which allows for the dynamic tracking of small nanoparticles in living cells. This AMSM method might be a valuable tool for living cell imaging, using sub-10 nm Au NPs as biological probes, and thereby unlocking many new applications, such as single molecule labeling and the dynamic tracking of molecular interactions.
Introduction
Applications using advanced probes play an important role in the development of modern biomedicine. Noble metal nanoparticles, described as plasmonic nanoparticles, scatter very bright and stable light signals (i.e., Mie scattering) because of their unique plasmonic resonance properties.1,2 This scattering signal could only slightly undergo optical quenching or photobleaching like fluorescence does, therefore the plasmonic nanoparticles can withstand strong light excitation.3 Usually, the noble metals such as gold have stable chemical properties in biological systems and have little risk of releasing heavy metal ions when comparing to some quantum dots.4–6 Because of these advantages, plasmonic nanoparticles are especially suitable for the study of long-term biological processes continuing from hours up to days, such as transmembrane transport,7,8 intracellular delivery,9 cell migration,10 cell division and so on.11,12 Moreover, the dispersed or aggregated states of nanoparticles within cells, or interactions between nanoparticles and surrounding molecules, could be distinguished by the color or spectral changes of light scattering, which allows for the study of many intracellular molecular processes.13–16Plasmonic nanoprobes have many advantages and have been extensively researched for use as new biological probes in the past decades, and various cell analysis methodology were demonstrated. However, thus far, they have not been widely accepted by biologists for use in cell imaging as fluorescent dyes and proteins, as well as quantum dots, have been.17–19 One of the main reasons for this is that the nanoparticles used in most conceptual studies are too large, typically in the range of 30–50 nm. Nanoparticles of this size are much larger than most of the biological molecules in cells, and even comparable to the size of some organelles (i.e., early endosomes and lysosomes). From a biological point of view, such large probes may modify the native behavior of biomolecules. If the size of the plasmonic probes could be as small as a few nanometers, which is equivalent to the size of fluorescent proteins (4–6 nm) and quantum dots (<10 nm), it will be easier for them to be commonly accepted in practical applications.19–21 From the material perspective, noble metal nanoparticles with sizes down to several nanometers were still stable and not hard to synthesize.22 The real challenge is how to see such small nanoparticles within cells. Obviously, regular dark field microscopy based on plasmonic scattering is not able to detect such small nanoparticles with sizes far beyond the Rayleigh limit (∼40 nm) because the scattering cross-section of the nanoparticles are scaled down by almost six powers of their size.23,24 Worse, the cellular medium is a heterogeneous environment with many strong scattering organelles, resulting in a strong scattering background. The weak light scattering signal, could be somewhat compensated for by using a more sensitive camera. However, the light scattering background from cellular components is hard to fully eliminate because the Rayleigh scattering from subcellular components usually have a wide spectrum. Even some background scavenging reagents, background smoothing algorithms, and so on, have been tried to reduce the intracellular scattering background, however, the problem has not yet been fully solved.25–28
To detect small plasmonic nanoparticles, several types of photothermal imaging methods were developed which were based on plasmonic resonance absorption instead of scattering, because the absorption cross section of the nanoparticles was scaled down by three powers of the particle size (slower than scattering). Laser scanning photothermal imaging is able to detect nanoparticles down to a size of a few nanometers, and this enabled high resolution imaging of the cellular structure. In this type of imaging, the photothermal signals, defined by the relative change of the probe beam with and without heat beam, were still scaled down because of the absorption cross-section.29–31 Thus, small nanoparticles below 10 nm require a tightly focused and high frequency modulated heat beam and a lock-in amplifier to extract the weak photothermal signal.32 For imaging of living cells, such a strong and focused laser beam might cause irreversible cell damage.33 Also, this type of laser scanning based imaging method usually needs minutes to hours to obtain a frame of the image, which seems too slow for living cell imaging because many dynamic biological events occur in seconds or even faster. In addition to laser scanning photothermal imaging methodology, photothermal imaging could also be realized based on surface plasmon resonance (SPR).34 The SPR imaging enabled the detection of a photothermal signal at a very fast speed, however, it seemed to only just produce an image of the nanoparticles inside the cells because the SPR effect relies greatly on the interface of the gold film.35 Recently, there have also been some new photothermal imaging methods, such as widefield photothermal sensing (WPS), which aimed to break the obstacle of imaging speed.36 Such a photothermal method is still based on the absorption characteristics of the objects. In addition to an absorption-based principle, interferometric optical detection can also be used to image 2–5 nm Au NPs based on their scattering properties.37 In cells, it usually requires larger nanoparticles to achieve a good image quality because the cellular environment contains a lot of scattering objects and this contributes to a strong scattering background.38
Here, a method, called absorption modulated scattering microscopy (AMSM) is demonstrated. This AMSM imaging method applied to both the resonance absorption and the scattering properties of plasmonic nanoparticles, rather than them each individually. Thus, the AMSM method exhibited a remarkable ability for the removal of the scattering background. Compared to regular dark-field microscopy that can typically only detect 30–50 nm Au NPs, the AMSM method was able to detect much smaller nanoparticles with a size down to ∼9 nm. Moreover, the imaging speed of the AMSM method is much faster than regular laser-scanning photothermal microscopy, and this allows for real-time video rate (20 fps) imaging of sub-10 nm nanoparticles in living cells. This AMSM method might encourage the study of biological processes within living cells using small Au NPs as biological probes and dynamic optical tracking.
Results and discussion
The AMSM is based on a pump-probe detection technique,39 which is realized using a self-built setup (Fig. 1 and S1, ESI†). A 532 nm pulse laser (∼5 ns) was used as a pump beam to heat the Au NPs, and pulsed white light (∼6 μs) was used as a probe beam to detect the light scattering signal from the Au NPs (Fig. 1a and S2, ESI†). Without a pump beam, the temperature of the liquid medium around the Au NPs was uniform and the intensity of the light scattering was defined as IOFF. Once the nanoparticle was irradiated by the pump beam, it was heated to a ‘hot’ state in hundreds of picoseconds by photon–phonon interaction.40–42 After a very short time (∼ns), the nanoparticle transfers a part of its heat to the surrounding medium, which resulted in a localized thermal field in the medium. As the refractive index of the medium depends on temperature, a local refractive index change was formed in the medium surrounding the nanoparticles, which is often called a “nano-thermal lens”.31,32 Assisted by the effect of the nano-lens, more light scattered from the Au NPs was captured by the imaging unit. Under these conditions, the light scattering intensity is defined as ION. Hence, the absorption modulated scattering signal Φ was defined as Φ = (ION − IOFF)/IOFF, which represented the change of the light scattering of the nanoparticles caused by absorption of the pump energy. After stopping the pump, the nanoparticles were cooling down and the “nano-thermal lens” disappeared in a few microseconds.43 Then, without pumping the system returned to the initial state and the light scattering intensity returned to IOFF until the next time heat was applied.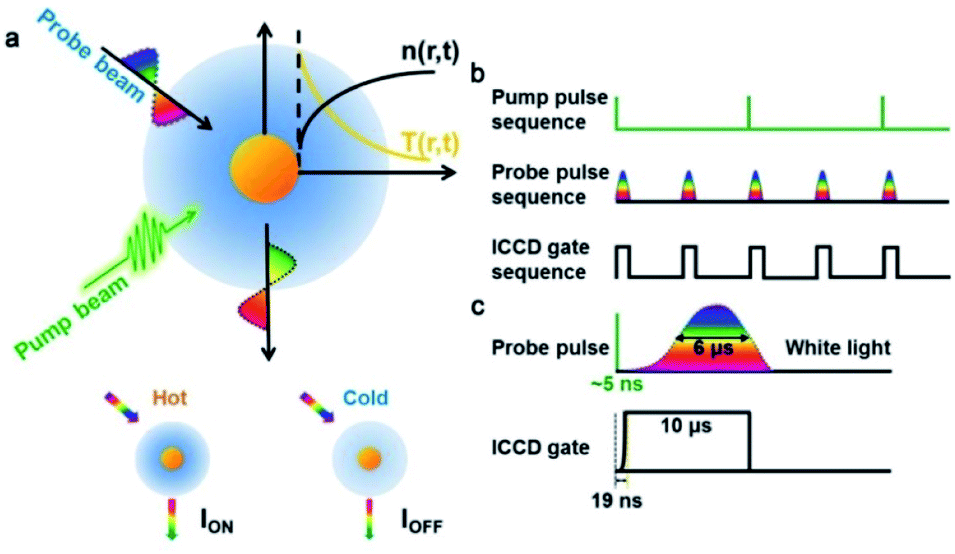 | ||
| Fig. 1 Schematic diagrams of absorption modulating scattering microscopy (AMSM). (a) The principle of the pump-probe imaging of a single gold nanoparticle. (b) The time sequence of the pump pulse (20 Hz), probe pulse, and camera gate (40 Hz). (c) A schematic diagram of the working time window of the probe pulse and camera gate in one time period of imaging. | ||
To achieve video rate imaging, time sequence control was introduced in to the previously described system. The pump laser worked at 20 Hz and a delay generator was used to synchronize frequency and generate a transistor–transistor logic (TTL) signal sequence at 40 Hz, which acted on the probe beam source and imaging module (Intensified Camera, ICCD) to collect the light scattering signal (Fig. 1b). The time interval of the probe sequences was 25 ms, and in one cycle of the pump, the scattering intensity was recorded as ION, and 25 ms later, the scattering intensity was recorded as IOFF after the nanoparticle was fully cooled. By subtracting IOFF from ION in chronological order, the absorption modulated scattering signal Φ, varying with time was be obtained. As illustrated in Fig. 1c, about 6 μs was needed for the probe beam (pulse xenon lamp) to reach maximum brightness and the half-width of time duration was also about 6 μs. Thus, the ICCD gate time was set to a duration of 10 μs to ensure that most of the scattered light was captured (Fig. 1c) with a very short start time of 19 ns.
As mentioned previously, the signal Φ originated from the change of refractive index, caused by light absorption, in the medium surrounding the AU NPs. Thus, the Φ signal value can be ultimately expressed as (see ESI† for details):44,45
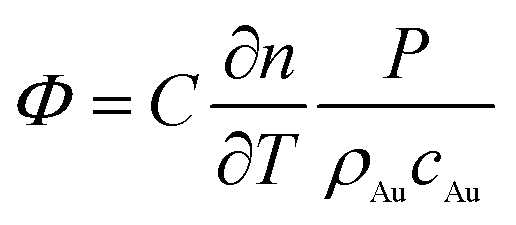 | (1) |
The feasibility of this imaging method was first demonstrated in vitro (Fig. 2). The Au NPs coated with polyethylene glycol (Au-PEG) with a size of 80 nm were immersed into glycerol (GC) medium to simulate the environment of the cytoplasm with a similar refractive index. Fig. 2a shows an example of a raw image captured by ICCD, and its corresponding Φ signal image is shown in Fig. 2b. All the Au NPs in Fig. 2a were found in Fig. 2b and the only exception was the super bright dust particle. Dust particles have a much larger size (μm) and a huge scattering cross section, thus they scatter much more light than the Au NPs. For scattering imaging, the background scattering from this type of super bright objects must be considered, especially in a complex environment such as cells. Therefore, the capability of the AMSM method for eliminating such strong scattering background signals, was then investigated using dust particles and polystyrene spheres as models (Fig. S3, ESI†). As expected, because the absorption characteristics of dust particles or polystyrene spheres were significantly different to Au NPs, then they could not be heated by the pump laser and therefore cannot be detected in the final AMSM image. These results suggested that the signal extraction capabilities of this system occur from the complex scattering background.29
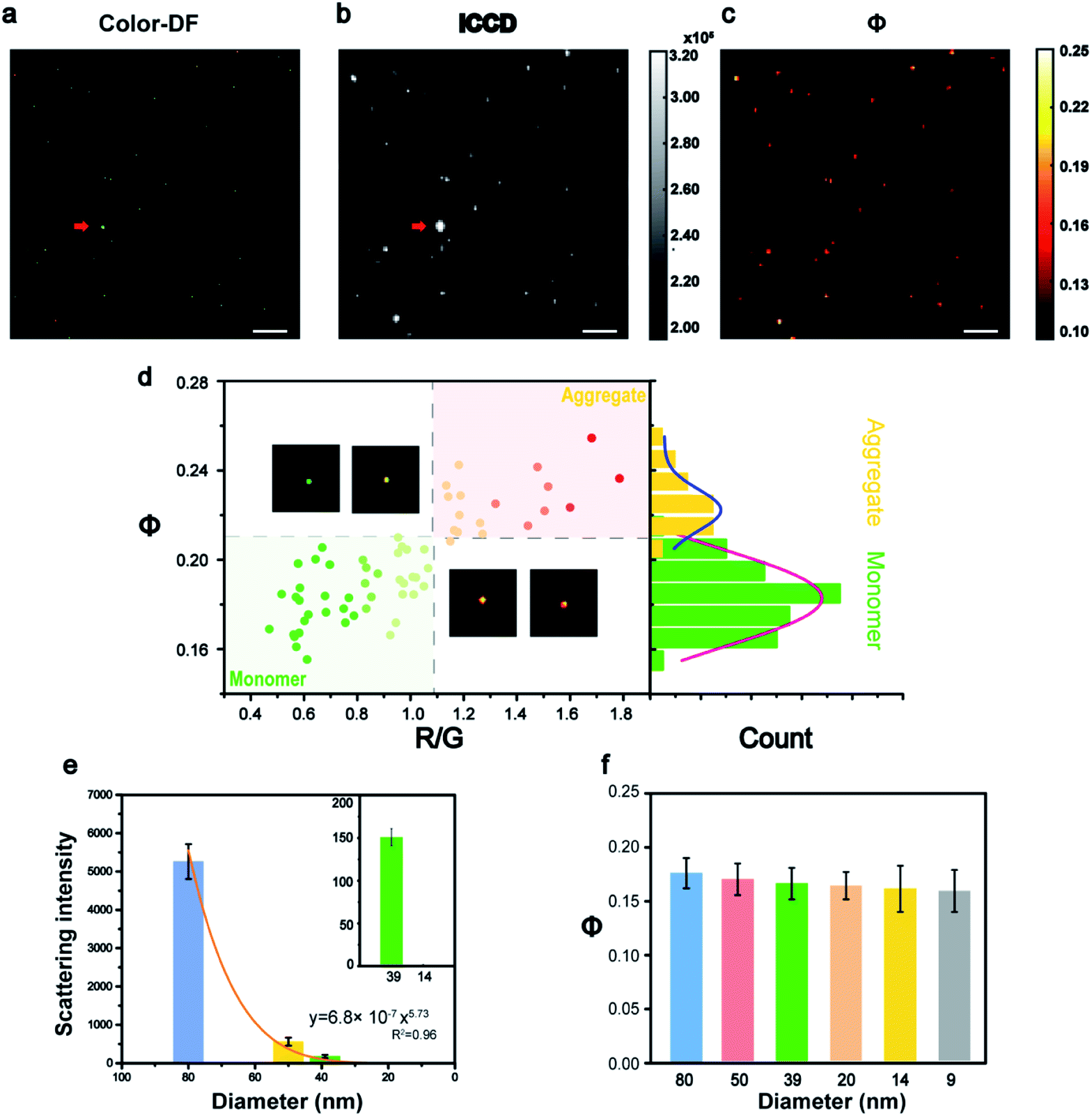 | ||
| Fig. 2 Colocalization imaging analysis of 80 nm Au NPs: (a) a color dark field image, (b) an ICCD image, and (c) a Φ signal image of nanoparticles on a glass substrate (scale bars: 10 μm). (d) A scatter plot of the Φ values versus R/G values of each nanoparticle and statistical histograms of the Φ distribution of monomeric (green) and aggregated (yellow) particles. (e) Light scattering intensity, and (f) Φ signal intensity of different sized nanoparticles. The red arrows in (a) and (b) indicate dust particles. | ||
In cell imaging using Au NPs as a probe, particle aggregation is almost inevitable. The states of the particle monomers and aggregates could usually be distinguished in true color dark field images, because aggregates show a different color to monomers (Fig. 2d). The RGB channels of the color dark field image were then split, and the red/green (R/G) value of each of the nanoparticles were calculated. Then the R/G values and the corresponding Φ values of a large number of nanoparticles in many frames of images were plotted and are shown in Fig. 2d and S4 (ESI†). The populations of monomers and aggregates were clearly distinguished according to either the R/G values or the Φ signal intensity, because the particle aggregates exhibited a red-shift color and a higher Φ signal. Then the Φ signal values of these two populations were analyzed statistically and the results are shown in the right histogram. This result verified the ability of this method to identity the monomeric and aggregated state of nanoparticles, which is an important issue in the imaging of nanoparticles within cells. The detailed procedure for data processing is presented in Fig. S5 (ESI†).
According to eqn (1), the Φ signal is independent of particle size in theory. To prove this concept, the intensity of the light scattering and the Φ signal of nanoparticles with a size range from 80 to 9 nm were measured (Fig. 2e, f and S6–S10, ESI†). From the dark field images, it can be seen that most of the particles were dispersed monomers on glass, because the surface of the Au NPs is wrapped by PEG. The intensity of the scattering signal dropped sharply with the particle size, which was consistent with the Mie theory (Fig. 2e). With the decrease of particle size, the scattering signal decreased sharply by a power of 5.73, which was close to the Mie theory where the scattering cross section dropped with particle size by a power of 6 (Fig. S10, ESI†). Whereas the Φ signals of different sized nanoparticles remained almost in the same range, with only with a slight drop (∼8%), together with a size decrease from 80 nm to 9 nm (Fig. 2f). This tendency was basically consistent with the theory of eqn (1), where the slight drop of Φ signals of very small nanoparticles could be attributed to the linearity response of the camera. The influence of the pump energy, surface coating, and surrounding medium on the intensity of the Φ signal was investigated further (Fig. S11†). For a given medium, the Φ signal intensity was proportional to the pump laser power. However, different surface modifications on the nanoparticles did not alter the intensity of their Φ signals.
The AMSM imaging of nanoparticles within cells was then demonstrated. For the convenience of observation and also to minimize the adverse effect on cells, the incubation concentration of Au NPs was controlled to ensure that only a small number of nanoparticles were assimilated into the cells. According to the dark field images of the cells, the numbers of nanoparticles within each cell were around ∼102. The Au NPs incubated with HeLa cells were captured in a colored dark field image and in the Φ signal image (Fig. 3a–c). Looking at the population in the scatter plot of R/G ratio and Φ signal intensity, the monomeric and aggregated gold nanoparticles could be differentiated (Fig. 3d). Thus, the positions of each nanoparticle could be located within the cells (Fig. 3e). The assignment of monomeric or aggregated nanoparticles matched well with the assignment from the colored dark field image. The HeLa cell which had not been incubated with Au NPs was also checked (Fig. S12, ESI†). Compared to the dark field image (Fig. 3a), the Φ signal image (Fig. 3c) showed very little background scattering from cellular components, because the cellular components did not have as strong a resonance absorption as the Au NPs.
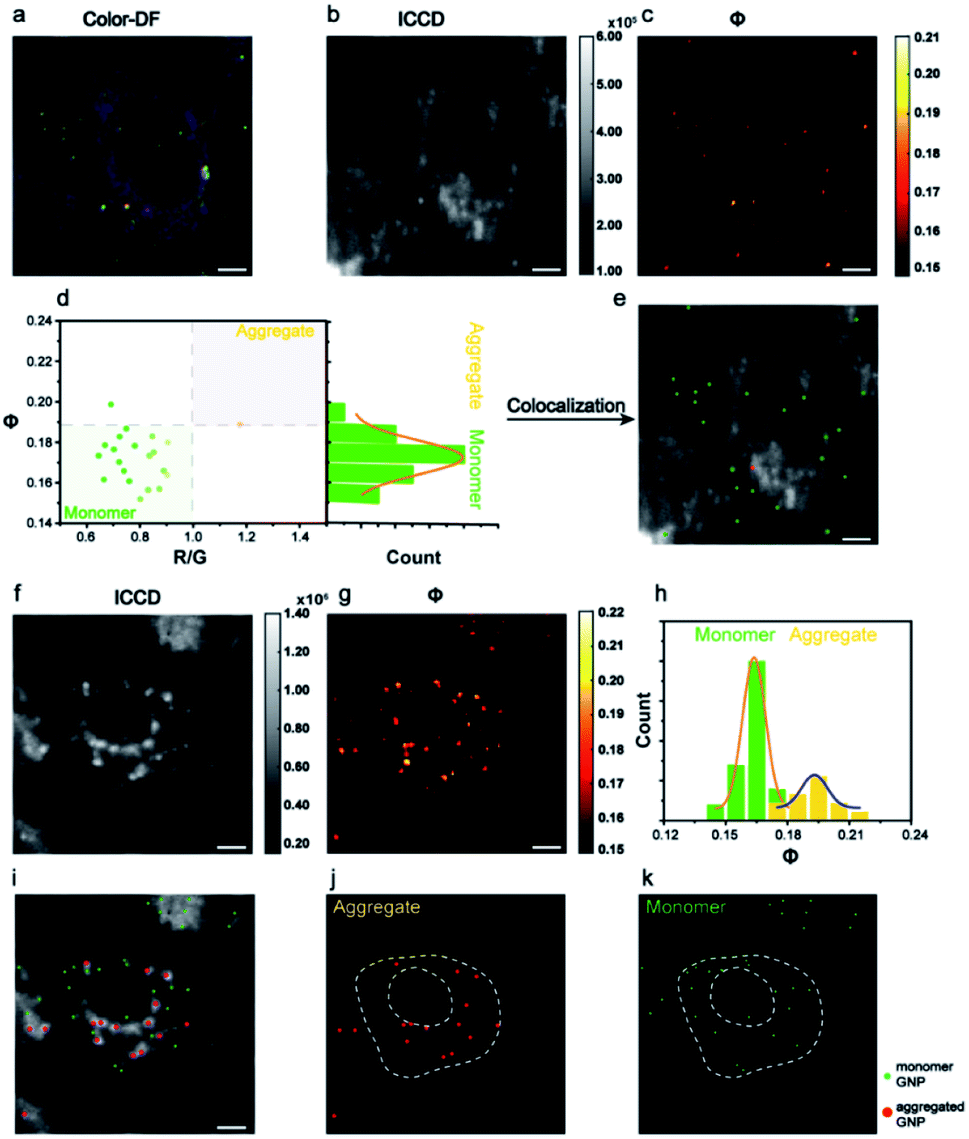 | ||
| Fig. 3 Colocalization imaging of Au NPs in a single cell. (a) A color dark field image, (b) an ICCD image, and (c) a Φ signal image of HeLa cells incubated with 80 nm Au NPs. (d) A scatter plot of the Φ signal versus R/G ratio and a Φ distribution histogram of monomeric (green) and aggregated (yellow) nanoparticles. (e) The localization of monomeric (green) and aggregated (red) nanoparticles in HeLa cells. (f) An ICCD dark field image and (g) a Φ image of 39 nm Au NPs in HeLa cells. (h) The corresponding Φ distribution histogram of monomeric and aggregated nanoparticles. (i) The localization of monomeric (green) and aggregated (red) 39 nm Au NPs in HeLa cells. (j and k) Splitting localization images of monomeric (j) and aggregated (k) nanoparticles. Scale bars: 5 μm. | ||
Then the same imaging procedure was applied by using 39 nm Au NPs (Fig. 3f and g). After signal extracting and particle counting, the Φ distribution histogram of monomeric and aggregated nanoparticles are shown in Fig. 3h. Compared to the 80 nm nanoparticles, more aggregated particles were observed with the 39 nm nanoparticles. The position of all monomeric and aggregated Au NPs in cells were recognized and marked (Fig. 3i–k). Usually, nanoparticles with a size below 40 nm were hard to capture via regular dark field imaging. Here, the Φ value intensity could still be used to identify the status of the Au NPs. A complete set of signal analysis processes for cell imaging is described in detail in Fig. S13 and S14 (ESI†).
Next, the size of the Au NPs was gradually reduced to explore the limit of the laboratory-built AMSM system. The 20 nm nanoparticles still can appear through the scattering background cell in the ICCD image (Fig. 4a), but the 14 nm and 9 nm nanoparticles were totally hidden in the background scattering (Fig. 4b and c). Fortunately, small nanoparticles with a size down to 9 nm could still be clearly imaged using the AMSM equipment. Some strong scattering light from dust particles or cellular components were successfully filtered.
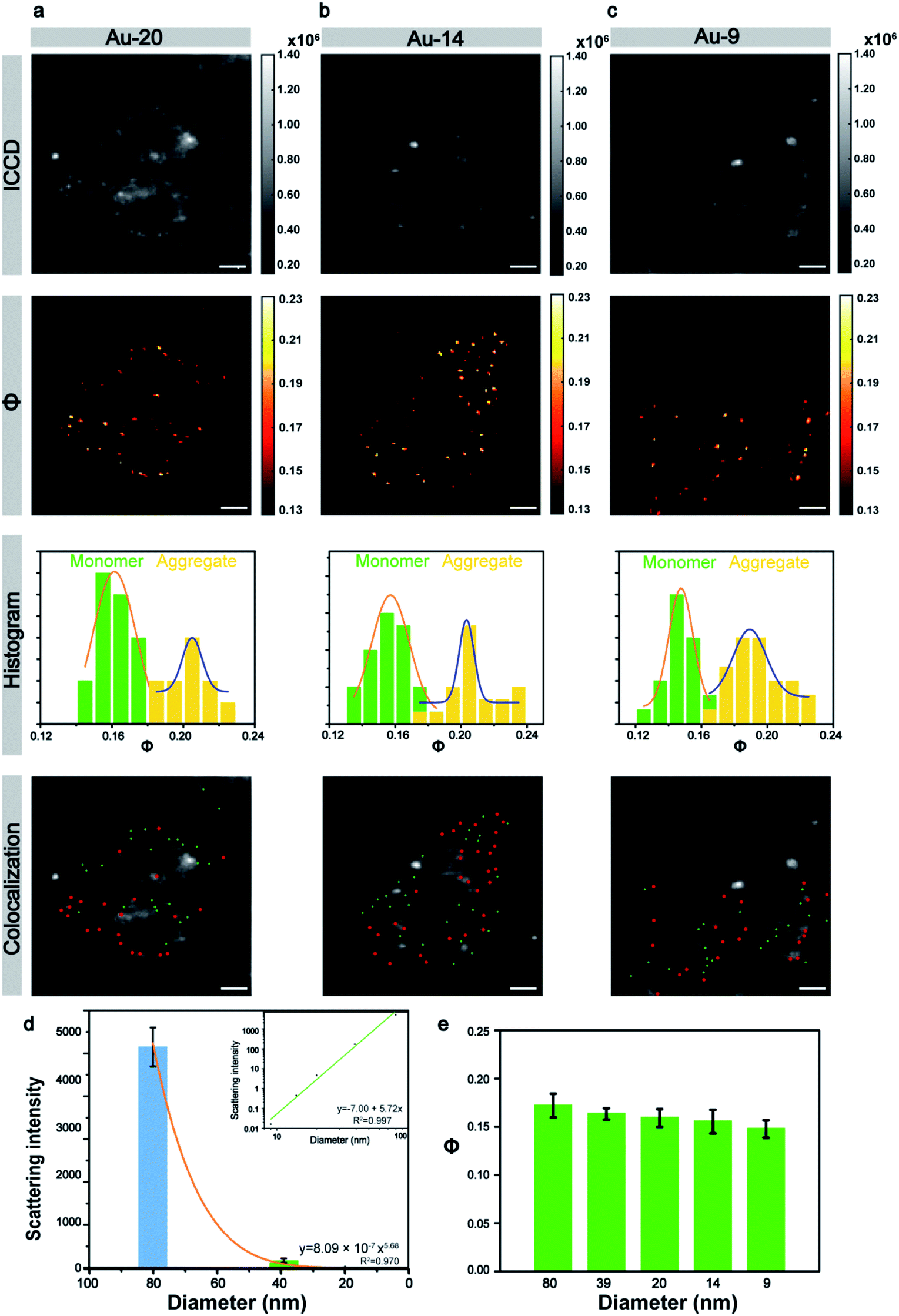 | ||
| Fig. 4 The imaging of small Au NPs within cells. (a–c) ICCD images, corresponding Φ images, Φ distribution histograms, and the spatial positions of the monomeric and aggregated Au NPs with sizes of (a) 20 nm, (b) 14 nm, and (c) 9 nm. (d) Light scattering intensity of nanoparticles with different sizes in HeLa cells; inset: the linear fitting of the logarithm of light scattering intensity versus particle size. (e) The Φ signal value of Au NPs with different sizes in a HeLa cell (scale bars: 5 μm). | ||
In a similar way to the large size particles, the Φ signals of the small size Au NPs also had two distinct distributions. The particle group with relatively small Φ values had monomeric particles and the group with relatively large Φ values had particle aggregation. When the intracellular monomeric and aggregated gold nanoparticles were separated and then using a similar overlay image as before, the position of the Au NPs could be accurately distinguished. The scattering intensity of the different sized nanoparticles with in a cellular medium, was analyzed statistically and the scattering signal dropped sharply with the decrease of particle size by a power of 5.68 (Fig. 4d), which approximately complied with the Mie theory. However, the intensity of the Φ signal decreased by only 14% although the particle size was reduced to more than one-eighth of the initial particle size (Fig. 4e). This tendency was also consistent with the theory of AMSM. The universality of AMSM was also verified by the image of small sized Au NPs in MCF-7 cells (Fig. S15, ESI†).
Dynamic tracking technology is needed urgently in cell imaging to reveal intracellular events in real time. The benefit from the principle of the time sequence control of the AMSM method, is that video-rate or even fast imaging is feasible (see video in the ESI†). To achieve this, 40 frames of HeLa cells with 9 nm Au NPs were recorded (Fig. 5a) in one second, and finally 20 frames of Φ signal images were obtained with a frame rate of 20 fps (Fig. 5b). Four particles were extracted as examples and their Φ signals over time are shown in Fig. 5c. Particles 1 and 2 show relatively stable Φ values of about 0.15, and thus they were monomeric Au NPs according to the previous discussion. Particles 3 and 4 were identified as aggregates from the Φ signal, which showed a stronger intensity and a wider variation range than that of the monomers. Next the position information of these four nanoparticles at each frame was extracted to track their states of motion (Fig. 5d). The two monomeric Au NPs 1 and 2 exhibited fast movement with a long journey of 1736 nm and 1438 nm. However, the aggregated particles 3 and 4 showed a slower movement and a shorter journey within 500 nm. Apparently, the moving speed of the aggregates within the cellular medium was much slower than the movement of the monomers. Here the dynamic imaging ability of method, is just showing that the moving states of the Au NPs within the cells could be tracked at video rate (20 fps) for a long time if necessary.
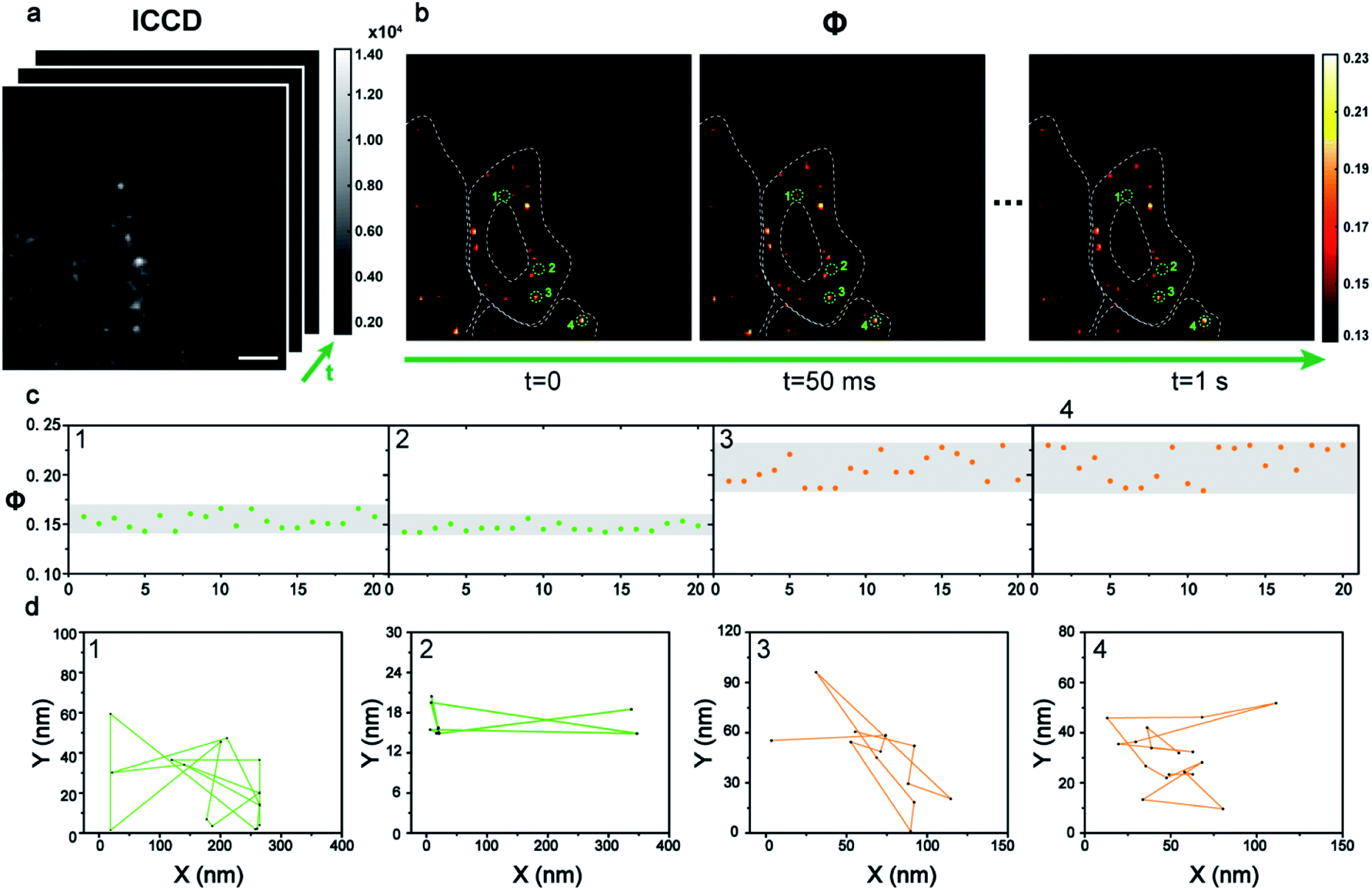 | ||
| Fig. 5 The dynamic tracking of 9 nm Au NPs in cells. (a) A series of dark field images of a HeLa cell incubated with Au NPs captured by ICCD in sequence (scale bars: 5 μm). (b) The corresponding Φ signal images of intracellular Au NPs in 1 s with a time interval of 50 ms. (c) The variation of the Φ signal of four typical particles extracted from the dynamic Φ signal image. (d) The movement tracking of the four particles in 1 s. A video of the Au NP tracking is shown in the ESI.† | ||
Conclusions
Absorption modulated scattering microscopy (AMSM) was demonstrated for the imaging of small gold nanoparticles within a cellular scattering medium. Because AMSM utilized both absorption and scattering rather than just a single component, only subjects with characteristic features of resonance absorption and scattering were detected, and the scattering background from the cellular component could be almost fully removed. The AMSM signal was very sensitive to the nanoparticle state, which makes it possible to distinguish monomers or aggregates in the cellular environment. Compared to regular darkfield microscopy, AMSM could detect much smaller nanoparticles with a size far beyond the Rayleigh limit, with a sensitivity comparable to that of photothermal microscopy. The imaging speed of AMSM could be close to the rate of a video or, in principle, even faster if used with a high-frequency pulsed laser and a fast camera. Because of the stable signal intensity, fast imaging speed, background removal ability, and lack of photobleaching, the AMSM imaging method has great potential for real-time cell imaging using small sized gold nanoprobes to determine the dynamics of molecular processes within living cells.Experimental
Reagents
The O-[2-(3-mercaptopropionylamino)ethyl]-O′-methylpolyethylene glycol (Mw = 5000, SH-PEG), and glycerol (GC, ≥99.5%) were obtained from Sigma-Aldrich Co. (USA). Human cervical cancer (HeLa) cells, Michigan Cancer Foundation-7 (MCF-7) cells, paraformaldehyde (4%), phosphate buffer solution (PBS, 10 mM, pH = 7.4) were provided were obtained from KeyGEN Biotech. Co. (Nanjing, China). The polystyrene spheres were purchased from the Zhejiang Tianke Hi-Tech Development Co., Ltd (Zhejiang, China). Gold nanoparticles (Au NPs) with an average diameter of 9 nm, 14 nm, 20 nm, 39 nm and 80 nm were purchased from Ted Pella, Inc. (USA) (Fig. S16, ESI†). Ultra-pure water from Millipore Milli-Q (18 MΩ cm−1) was used in the experiments.Samples
The preparation processes of the Au-PEG particles with different particle sizes were essentially the same. The Au NPs of different sizes [9 nm (9.5 nM), 14 nm (2.3 nM), 20 nm (1.2 nM), and 40 nm (150 pM)] were mixed with the same volume of 1 mmol L−1 SH-PEG to prepare Au-PEG particles of different sizes. At the same time, the desired Au-PEG was obtained by mixing the 80 nm Au NPs (18 pM) with an equal volume of 0.1 mmol L−1 PEG solution. The mixture was maintained at 37 °C and stirred continuously for 24 h. The resulting Au-PEG were then centrifuged at an appropriate speed, and then washed several times.The Au NPs, Au-PEG or Au-DNA were absorbed on positively charged glass slides for 30 min. Then the Au- or Au-complex coated glass slides were washed three times and placed on a microscope objective for examination. Moreover, water or glycerol was dropped on each glass slide to acted as a surrounding medium.
The HeLa and MCF-7 cells were routinely cultured in DMEM medium with 10% fetal bovine serum, 2% antibiotics, and 5% CO2 in a 37 °C incubator. When making the samples, the HeLa and MCF-7 cells were seeded on a glass coverslip. After culturing for 24 h, normal Au-PEG solution was added and then the solution containing the cells was incubated. After 5 h, the HeLa, and MCF-7 cells coated glass coverslips were washed several times with PBS to remove the excess nanoparticles and were ready for the detection of living cells. The fixed cell samples were prepared by incubating them with 4% paraformaldehyde for an additional 15 min, and then the coverslips coated with the fixed cells were washed and soaked in PBS solution before examining them for the presence of living cells. The temperature of the glass slides and glass coverslips were controlled by a thermal platform equipped with a sensitive temperature control system. For all the cell experiments on microscope, at least three independent trials of the experiments were performed and 10–20 cells were randomly selected each time for measurement. The results of the MTT assay confirmed that the cell activity of the cells under different experimental conditions was greater than 96%, indicating that the AMSM imaging method caused no photodamage of the cells (Fig. S17, ESI†).
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was mainly supported by the National Natural Science Foundation of China (21675081 and 22034003), the Excellent Research Program of Nanjing University (ZYJH004), and the State Key Laboratory of Analytical Chemistry for Life Sciences (5431ZZXM2002).References
- H. C. Hulst and H. C. van de Hulst, Light scattering by small particles, Courier Corporation, 1981 Search PubMed.
- G. Mie, Ann. Phys., 1908, 330, 377–445 CrossRef.
- S. Schultz, D. R. Smith, J. J. Mock and D. A. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 996 CrossRef CAS.
- W. Eck, G. Craig, A. Sigdel, G. Ritter, L. J. Old, L. Tang, M. F. Brennan, P. J. Allen and M. D. Mason, ACS Nano, 2008, 2, 2263–2272 CrossRef CAS.
- E. Oh, R. Liu, A. Nel, K. B. Gemill, M. Bilal, Y. Cohen and I. L. Medintz, Nat. Nanotechnol., 2016, 11, 479–486 CrossRef CAS.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS.
- C. Rosman, S. Pierrat, A. Henkel, M. Tarantola, D. Schneider, E. Sunnick, A. Janshoff and C. Sönnichsen, Small, 2012, 8, 3683–3690 CrossRef CAS.
- X. Nan, P. A. Sims and X. S. Xie, ChemPhysChem, 2008, 9, 707–712 CrossRef CAS.
- I. H. El-Sayed, X. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS.
- J. A. Yang, H. T. Phan, S. Vaidya and C. J. Murphy, Nano Lett., 2013, 13, 2295–2302 CrossRef CAS.
- G. A. Craig, P. J. Allen and M. D. Mason, in Cancer Nanotechnology: Methods and Protocols, ed. S. R. Grobmyer and B. M. Moudgil, Humana Press, Totowa, NJ, 2010, pp. 177–193, DOI:10.1007/978-1-60761-609-2_12.
- W. Qian, X. Huang, B. Kang and M. El-Sayed, J. Biomed. Opt., 2010, 15, 046025 CrossRef.
- C. Sönnichsen, B. M. Reinhard, J. Liphardt and A. P. Alivisatos, Nat. Biotechnol., 2005, 23, 741–745 CrossRef.
- L. Zhang, Y. Li, D.-W. Li, C. Jing, X. Chen, M. Lv, Q. Huang, Y.-T. Long and I. Willner, Angew. Chem., Int. Ed., 2011, 50, 6789–6792 CrossRef CAS.
- K. Lee, Y. Cui, L. P. Lee and J. Irudayaraj, Nat. Nanotechnol., 2014, 9, 474–480 CrossRef CAS.
- M.-X. Li, C.-H. Xu, N. Zhang, G.-S. Qian, W. Zhao, J.-J. Xu and H.-Y. Chen, ACS Nano, 2018, 12, 3341–3350 CrossRef CAS.
- F. Wang, Y. Zhu, L. Zhou, L. Pan, Z. Cui, Q. Fei, S. Luo, D. Pan, Q. Huang, R. Wang, C. Zhao, H. Tian and C. Fan, Angew. Chem., Int. Ed., 2015, 54, 7349–7353 CrossRef CAS.
- R. Tang, J. Xue, B. Xu, D. Shen, G. P. Sudlow and S. Achilefu, ACS Nano, 2015, 9, 220–230 CrossRef CAS.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Physiol. Rev., 2010, 90, 1103–1163 CrossRef CAS.
- B. Dubertret, P. Skourides, D. J. Norris, V. Noireaux, A. H. Brivanlou and A. Libchaber, Science, 2002, 298, 1759 CrossRef CAS.
- B. Zhang, F. Zhang, P. Zhang, D. Shen, X. Gao and G. Zou, Anal. Chem., 2019, 91, 3754–3758 CrossRef CAS.
- B. H. Kim, M. J. Hackett, J. Park and T. Hyeon, Chem. Mater., 2014, 26, 59–71 CrossRef CAS.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 7238–7248 CrossRef CAS.
- M. A. van Dijk, A. L. Tchebotareva, M. Orrit, M. Lippitz, S. Berciaud, D. Lasne, L. Cognet and B. Lounis, Phys. Chem. Chem. Phys., 2006, 8, 3486–3495 RSC.
- H. Hama, H. Kurokawa, H. Kawano, R. Ando, T. Shimogori, H. Noda, K. Fukami, A. Sakaue-Sawano and A. Miyawaki, Nat. Neurosci., 2011, 14, 1481–1488 CrossRef CAS.
- D. Sun, J. Fan, C. Liu, Y. Liu, Y. Bu, C. J. Lyon and Y. Hu, Anal. Chem., 2016, 88, 12001–12005 CrossRef CAS.
- Y. Cui, X. Wang, W. Ren, J. Liu and J. Irudayaraj, ACS Nano, 2016, 10, 3132–3143 CrossRef CAS.
- Z. Ye, L. Wei, X. Zeng, R. Weng, X. Shi, N. Wang, L. Chen and L. Xiao, Anal. Chem., 2018, 90, 1177–1185 CrossRef CAS.
- D. Boyer, P. Tamarat, A. Maali, B. Lounis and M. Orrit, Science, 2002, 297, 1160 CrossRef CAS.
- L. Cognet, C. Tardin, D. Boyer, D. Choquet, P. Tamarat and B. Lounis, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11350 CrossRef CAS.
- M. Selmke, M. Braun and F. Cichos, ACS Nano, 2012, 6, 2741–2749 CrossRef CAS.
- A. Gaiduk, P. V. Ruijgrok, M. Yorulmaz and M. Orrit, Chem. Sci., 2010, 1, 343–350 RSC.
- H. Zhang, W. Gao, Y. Liu, Y. Sun, Y. Jiang and S. Zhang, Anal. Chem., 2019, 91, 12581–12586 CrossRef CAS.
- Z. Chen, X. Shan, Y. Guan, S. Wang, J.-J. Zhu and N. Tao, ACS Nano, 2015, 9, 11574–11581 CrossRef CAS.
- W. Jing, Y. Wang, Y. Yang, Y. Wang, G. Ma, S. Wang and N. Tao, ACS Nano, 2019, 13, 8609–8617 CrossRef CAS.
- Y. Bai, D. Zhang, L. Lan, Y. Huang, K. Maize, A. Shakouri and J.-X. Cheng, Sci. Adv., 2019, 5, eaav7127 CrossRef CAS.
- V. Jacobsen, P. Stoller, C. Brunner, V. Vogel and V. Sandoghdar, Opt. Express, 2006, 14, 405–414 CrossRef CAS.
- R. W. Taylor, R. G. Mahmoodabadi, V. Rauschenberger, A. Giessl, A. Schambony and V. Sandoghdar, Nat. Photonics, 2019, 13, 480–487 CrossRef CAS.
- A. Schmidt, M. Chiesa, X. Chen and G. Chen, Rev. Sci. Instrum., 2008, 79, 064902 CrossRef.
- J. H. Hodak, A. Henglein and G. V. Hartland, J. Phys. Chem. B, 2000, 104, 9954–9965 CrossRef CAS.
- M. Rashidi-Huyeh and B. Palpant, J. Appl. Phys., 2004, 96, 4475–4482 CrossRef CAS.
- Z. Qin and J. C. Bischof, Chem. Soc. Rev., 2012, 41, 1191–1217 RSC.
- N. N. Nedyalkov, S. E. Imamova, P. A. Atanasov, R. A. Toshkova, E. G. Gardeva, L. S. Yossifova, M. T. Alexandrov and M. Obara, Appl. Surf. Sci., 2011, 257, 5456–5459 CrossRef CAS.
- T. L. Bergman, F. P. Incropera, D. P. DeWitt and A. S. Lavine, Fundamentals of heat and mass transfer, John Wiley & Sons, 2011 Search PubMed.
- C. D. S. Brites, M. C. Fuertes, P. C. Angelomé, E. D. Martínez, P. P. Lima, G. J. A. A. Soler-Illia and L. D. Carlos, Nano Lett., 2017, 17, 4746–4752 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic route, additional figures and table. See DOI: 10.1039/d0sc04764c |
| This journal is © The Royal Society of Chemistry 2021 |