
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCooperative catalytically active sites for methanol activation by single metal ion-doped H-ZSM-5†
Wei-Che
Lin‡
a,
Simson
Wu‡
a,
Guangchao
Li
a,
Ping-Luen
Ho
a,
Yichen
Ye
a,
Pu
Zhao
a,
Sarah
Day
b,
Chiu
Tang
b,
Wei
Chen
 c,
Anmin
Zheng
c,
Benedict T. W.
Lo
*d and
Shik Chi
Edman Tsang
*a
c,
Anmin
Zheng
c,
Benedict T. W.
Lo
*d and
Shik Chi
Edman Tsang
*a
aWolfson Catalysis Centre, Department of Chemistry, University of Oxford, Oxford, OX1 3QR, UK. E-mail: edman.tsang@chem.ox.ac.uk
bDiamond Light Source Ltd, Harwell Science and Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
cWuhan Institute of Physics and Mathematics, CAS, Wuhan 430071, China
dDepartment of Applied Biology and Chemical Technology, Hong Kong Polytechnic University, Hong Kong, China
First published on 21st October 2020
Abstract
Catalytic conversion of methanol to aromatics and hydrocarbons is regarded as a key alternative technology to oil processing. Although the inclusion of foreign metal species in H-ZSM-5 containing Brønsted acid site (BAS) is commonly found to enhance product yields, the nature of catalytically active sites and the rationalization for catalytic performance still remain obscure. Herein, by acquiring comparable structural parameters by both X-ray and neutron powder diffractions over a number of metal-modified ZSM-5 zeolites, it is demonstrated for the first time that active pairs of metal site-BAS within molecular distance is created when single and isolated transition metal cation is ion-exchanged with the zeolites. According to our DFT model, this could lead to the initial heterolytic cleavage of small molecules such as water and methanol by the pair with subsequent reactions to form products at high selectivity as that observed experimentally. It may account for their active and selective catalytic routes of small molecule activations.
Introduction
Recently, the efficient provision of energy and fine chemical has been a critical consideration in the formulation of many global and regional policies. It relies heavily on the use of heterogeneous catalysts, thanks to the ease of catalyst-product separation and their high thermal/chemical stability. Two common types of heterogeneous catalysts are nanomaterials and porous materials. Both materials possess high surface area and high ratio of active sites. To the greatest extent, this can be maximized by establishing ‘single-site’. Akin to the homogeneous and biological enzyme analogues, isolated metal species can offer great possibilities in rationally tuning the catalytic properties by tailoring the geometric and electronic properties. Recent research has blossomed in the preparation of isolated metal species supported on solid-state materials. Notably, single-site Pt/FeOx has been first acquainted.1 A series of related materials have been subsequently reported, including Ir/FeOx,2 Pd/CeO2,3 Pt/graphene,4 and so forth. Although the argument for high activity of a single metal site is well received, many catalytic reactions show superior activity or selectivity dependent on the support solid or surface structures of the hosts, indicative of more than one catalytically sites involved. In a similar line, microporous zeolites have been well-identified for many decades that can host isolated metal species through ion-exchange with their Brønsted acid sites (BASs).5 Also, isolated metal species on non-acidic support materials do not offer comparable catalytic performance, which suggests their cooperative catalytic roles with the BAS in proximity. Metal-modified zeolites are shown to catalyze some diverse but important reactions in petrochemical processing.Recently, catalytic conversion of methanol to useful products such as aromatics and hydrocarbons is regarded as a key alternative technology to oil processing. For example, Ga- and Zn-modified ZSM-5 (MFI framework type) zeolites are used for the production of valuable aromatic compounds at higher yields from methanol feedstock,6 where methanol can be readily obtained via syngas from cleaner sources such as natural gas or biomass.7 Similarly, methanol can also be converted into related hydrocarbon products, such as to olefins8 and gasoline by using zeolites of different pore structures.9 However, the mechanism of how methanol is first activated and the fates of intermediates over acidic zeolites are still not fully understood. It is generally believed that methanol is first protonated at a Brønsted acid site (BAS), followed by subsequent reactions via dimethyl ether (DME).10–14 The incorporation of transition-metal species into the acidic zeolites is a typical way to improve the catalytic performance; however, the role of metal ions in synergy with BAS to improve catalytic performance remains obscure.
Acid–base interactions generally play a significant role in the activation of small molecules in catalysis. Different from conventional homogeneous frustrated Lewis pair (FLP) catalysts, surface atoms of heterogeneous catalysts are firmly held within rigid 2-D or 3-D lattice matrices.15 Spatial constraints for the formation of classic Lewis acid–base adducts in transition metal ion-doped zeolites could equally favour the creation of a similar ion pair in the ‘rigid’ framework. The main problem is the difficulty in confirming this assertion by high quality-single crystals of modified zeolites to correlate the structure–activity relationship. Our group has recently developed a reliable methodology that can offer extensive information about the active sites within microporous materials. By combining synchrotron X-ray powder diffraction (SXRD)-neutron powder diffraction (NPD)-Rietveld refinement and extended X-ray absorption fine structure (EXAFS), even the small adsorbed organic or inorganic molecules on the active sites can be determined at high precision.16 It has enabled us a facile method in the rational design of new and more effective catalytic systems. Recent examples include the incorporation of Zn2+ centres onto the H-ZSM-5 framework for acid site catalyzed biomass activation17 and ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) W
W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O sites for metathesis reactions.18
O sites for metathesis reactions.18
This work first reveals the crystal structures of three specifically selected metal modified (Fe, Zn and Ag) ions doped H-ZSM-5 catalysts and the respective coordination environments of these metal ion species to form constrained metal–BAS ion pair via the combined use of advanced characterization techniques. The structure–activity relationships with respect to their small molecules (namely methanol and water) activations are discussed. It is emphasized that we here aim to provide rationale and insights for the initial chemical bond activation of small molecules by the metal–BAS ion pair without providing the comprehensive account for subsequent reactions to products in the zeolite cage.
Results and discussion
The catalysts were prepared via typical ion-exchange, followed by calcination at 400 °C (see ESI†). The H-ZSM-5 zeolites (space group of Pnma) were specifically chosen with high crystallinity and optimal acidity for versatile crystallographic investigation. As characterized in our previous work,17 the structural formula of the H-ZSM-5 is H4.48Al4.48Si43.52O96 (formula mass = 2883.6) which renders about one Al site per asymmetric unit (acid site) enabling the structural study with the metal ion. Similar content of metal species was thus incorporated to ensure a fair and well-controlled study. The content of Fe, Cu, Zn, and Ag per unit cell of H-ZSM-5 are 1.43(2), 1.89(3), 1.97(4), and 1.67(3), respectively (as determined by the Rietveld refinement of the high-resolution SXRD data). The extents of metal incorporation are comparable to each other. The zeolite catalysts were tested in a laboratory reactor at Oxford (Fig. S1†). Typically, for methanol conversion, the catalytic performance was compared systemically at the optimized reaction condition of 397 °C and 10 bars for 24 h. The methanol flow rate of 0.016 mL min−1 was pumped by N2 (3 mL min−1) into the fixed-bed stainless steel reactor. 0.5 g of zeolite catalysts were sandwiched evenly by quartz wool plugs. The catalytic results of the MTA reaction over Fe-ZSM-5, Cu-ZSM-5, Zn-ZSM-5 and Ag-ZSM-5 are presented in Fig. 1. To ensure high reliability of the catalysis data, the mass balance was carefully monitored and achieved around 95%.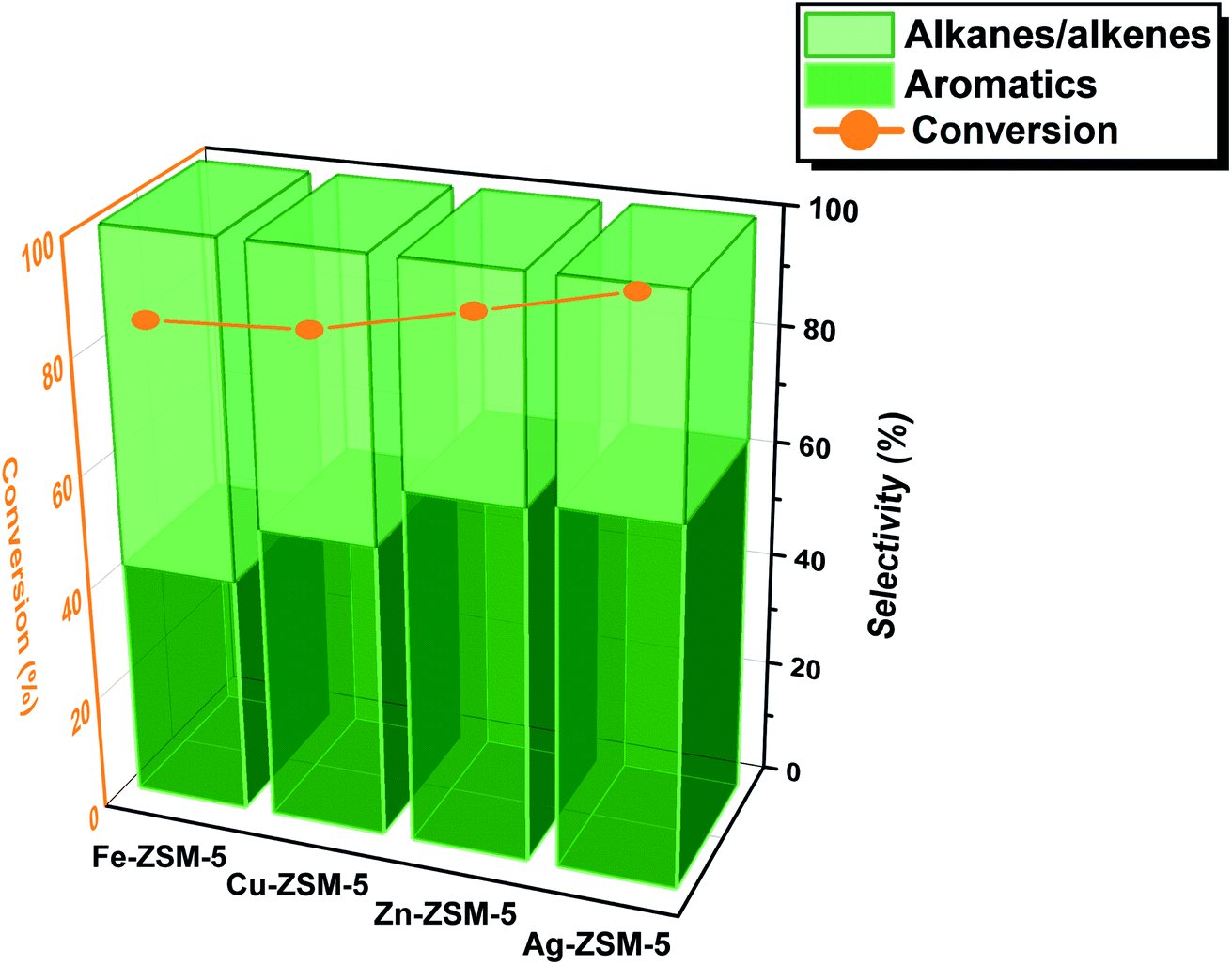 | ||
| Fig. 1 MTA catalytic results for H-ZSM-5 exchanged with metal ions including Fe, Cu, Zn and Ag. Liquid methanol with a flow rate of 0.016 mL min−1 was pumped into a fixed bed stainless steel reactor that hosted 0.5 g of the ion-exchanged catalysts sandwiched by quartz wool plugs. The carrier gas was N2 with a feed rate of 3 mL min−1 at reaction conditions of 397 °C and 10 bar. The conversion and selectivity are taken at steady-state, and the total selectivity with respect to converted methanol to all these products is 100%; measurement errors by gas chromatography are within 10%. | ||
As seen in Fig. 1, all four metal ion-doped H-ZSM-5 catalysts present with progressively increasing conversion of over 90%. Interestingly, the H-ZSM-5 sample exchanged with Fe ions gives only 40% selectivity to total aromatics (BTX and other substituted aromatics) but with 60% selectivity to alkanes/alkenes (see typical product distribution in Tables S1–S3 and Fig. S2, S3†). Across the first-row transition metal ions, there is an apparent increase in the formation of desirable aromatic products from methanol at the expense of alkanes/alkenes. The H-ZSM-5 sample exchanged with Zn ions gives 60% selectivity to total aromatics. This result matches with the earlier claims that late transition metals in the periodic table (group IB) such as Zn and Ga- ZSM-5 show higher selectivity for aromatics formation.6 Ag ions also show a similar or slightly higher selectivity to that of Zn ions for the synthesis of aromatics but at a virtual completion of methanol conversion. The catalytic methanol conversions to aromatics depend on the effective nuclear charge (Zeff) of the exchanged metal ions with the lowest value at Fe and the highest at Ag. We are interested in further investigating the structure–activity relationship in order to address why the late transition metal ions work well in synergy with the acidic zeolite for the methanol conversions to aromatics particularly how the methanol is activated in the first place.
By making use of unit cells with perfect long-range order as the templates, the use of SXRD, NPD combined with Rietveld refinement can elucidate the positions of extra-framework modifiers in zeolites. The alteration in the scattering parameters by foreign ions/molecules enables the probing of their adsorption geometries and interactions with the BASs in terms of atomic distances and angles.18,19 From EXAFS (Fig. S4–S9 and Tables S4, S6†) and microscopy (Fig. S10 and S11†), it is clear that the exchanged metal ions do not seem to form aggregates/nanoparticles but are fully dispersed as single entities in the ZSM-5. There are more than one forms of metal ion species found in the internal cavity of the zeolites. The Rietveld refinements of the SXRD data collected on Fe-ZSM-5, Zn-ZSM-5 and Ag-ZSM-5 at 25 °C revealed at least two types of sites, which are presented in Fig. 2. Type 1 is located close to the straight channels. For example, Fe(H2O)63+, Zn(H2O)62+ and Ag(H2O)2+ cationic species are found, respectively in the larger straight channel of the negatively charged frameworks. Thus, these original hydrated cations from corresponding precursor salts must have penetrated and stabilized in the large pore of the zeolite structure during the ion-exchange with optimized enthalpy and entropy. We cannot resolve the positions of the counter-ions, which may suggest their random distribution on the extra-frameworks as previously discussed.20
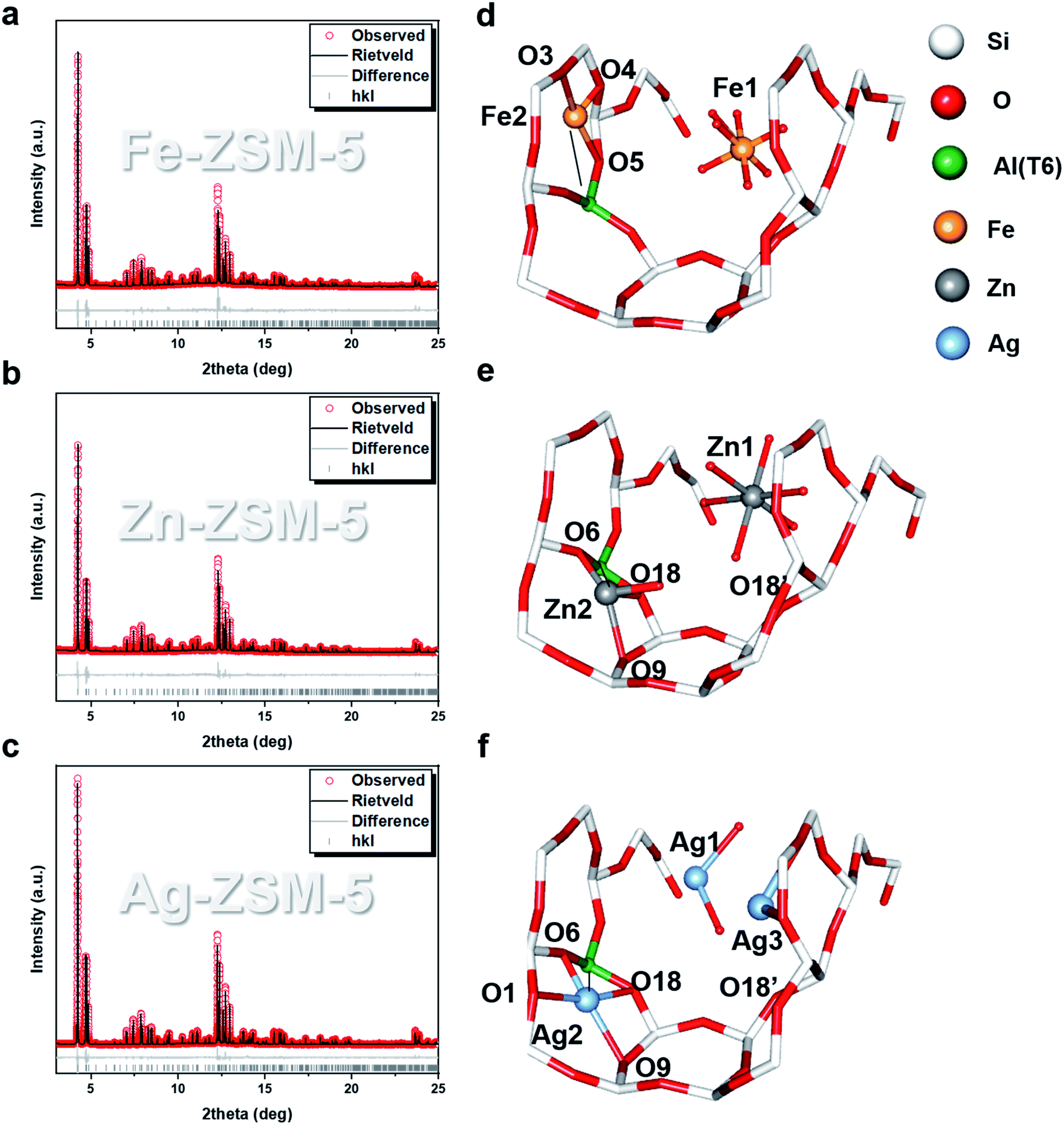 | ||
| Fig. 2 SXRD and Rietveld refinement measured at 25 °C of (a) Fe-ZSM-5, (Rwp = 7.956%, χ2 = 1.581), (b) Zn-ZSM-5, (Rwp = 7.896%, χ2 = 2.237) and (c) Ag-ZSM-5, (Rwp = 7.212%, χ2 = 2.181). Their corresponding Rietveld refined crystal structures are shown in (d)–(f). The mirror symmetry of the extra-framework species is disregarded for clarity. See main text for the detailed bond distance descriptions. The atomic and crystallographic parameters are summarised in Tables S7–S10.† Atoms are represented in ball/sticks. | ||
Apart from these unmodified/non-interactive metal ion species, type 2 species is also found in the same zeolite but located in much closer proximity to the earlier identified BASs (T6, at the straight-sinusoidal channel crossing) of the zeolitic framework than that of the type 1 species in the larger straight channel. It is thought that such metal ion species are resulted from the direct ion-exchange with the only protonic BASs in H-ZSM-5 despite the decrease in entropy in the smaller cavity. By analyzing the M–Oframework bond distances (which will be later discussed), these metal ion species are anchored on the oxygen-wall of the MFI framework by coordinating with one or more of the proton-depleted Oframework atoms 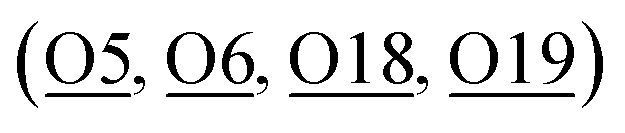 adjacent to the Al T6 site, as well as with the nearby surface oxygens for the displacement of hydrated water molecules. For Fe-ZSM-5, 0.96(2) occupancy of Fe-2 site is coordinated with three framework oxygens
adjacent to the Al T6 site, as well as with the nearby surface oxygens for the displacement of hydrated water molecules. For Fe-ZSM-5, 0.96(2) occupancy of Fe-2 site is coordinated with three framework oxygens 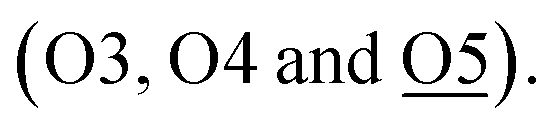 For Zn-ZSM-5, although the Zn (Zn-2) species do not take residence at the same site (depending on the charge/radius ratio and covalency of the metal ion) an occupancy of 0.14(1) of Zn-2 is found coordinated by three framework oxygen atoms
For Zn-ZSM-5, although the Zn (Zn-2) species do not take residence at the same site (depending on the charge/radius ratio and covalency of the metal ion) an occupancy of 0.14(1) of Zn-2 is found coordinated by three framework oxygen atoms 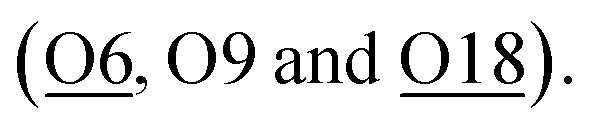 By taking account of the average coordination number of 5-CN derived from EXAFS as well as relative site occupancy from the site 1 (6-CN) and site 2 (4-CN) species from SXRD refinement (see Table 1), it reveals that a remaining fourth non-framework coordination with terminal oxygen (either water or hydroxyl molecule) associated the tetrahedral site 2 metal ion species as an anchored species.
By taking account of the average coordination number of 5-CN derived from EXAFS as well as relative site occupancy from the site 1 (6-CN) and site 2 (4-CN) species from SXRD refinement (see Table 1), it reveals that a remaining fourth non-framework coordination with terminal oxygen (either water or hydroxyl molecule) associated the tetrahedral site 2 metal ion species as an anchored species.
| Catalyst | CNSXRD | CNEXAFS | R SXRD [Å] | R EXAFS [Å] | E not [eV] |
|---|---|---|---|---|---|
| a Estimated standard deviation (ESD) are presented in brackets; CN stands for coordination number; R represents derived atomic distance (see ESI). b The calculation of CNSXRD is based on averaging the total coordination number of each transition metal site multiple by their occupancy. c R SXRD is the occupancy weighted bond length of each M–O. | |||||
| Fe-ZSM-5 | 4.7(5) | 5.3(6) | 1.71(5) | 1.79(3) | 0.7 |
| Zn-ZSM-5 | 4.8(2) | 5.2(2) | 2.11(2) | 2.07(1) | 3.4 |
| Ag-ZSM-5 | 2.7(2) | 2.7(3) | 2.19(3) | 2.33(3) | 0.1 |
Similarly, according to the Rietveld refinement of the SXRD data collection on Ag-ZSM-5, site 1 is also occupied by a slightly bent di-aqua Ag-1 species near the straight channel (occupancy 0.13(1)). Agreeably, there is Ag-2 site (occupancy 0.05(1)) which is located on another 10-membered ring adjacent to the Al T6 (BAS). Ag-2 appears to be coordinated with four nearby framework oxygen atoms presumably due to the smallest size of the cations to gain the highest surface oxygen coordination: 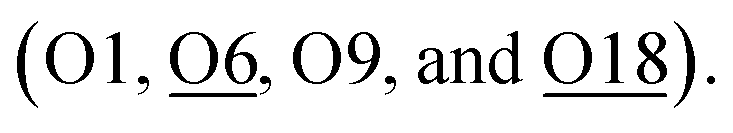 Notice that Ag-2–O6 is notably shorter than the average Ag–O value of 2.33 Å (deduced from EXAFS), which may have caused by electrostatic interaction due to the ion-exchange of protons with an additional contribution of covalency between Ag and O moieties. Unlike the Fe-2 and Zn-2 sites, the corresponding Ag-2 site does not seem to bond with any non-framework oxygen (no hydrated water molecule or hydroxyl species). The hybridization of dz2-s orbitals in Ag+ does not support higher coordination; the surface oxygens are hence sufficient to coordinate the Ag ion without major distortion. Interesting, we also note a third Ag+ site, namely Ag-3 site (occupancy 0.09(1)), which has longer bond distances from the generally strong anchored BASs. It is found coordinated with three framework oxygen atoms but at longer Ag–O distances (O2, O3 and O20). Notice that the average CN and bond distances of these metal ions derived from the SXRD and EXAFS measurements are consistent with each other. The associated XANES (Tables S4–S6†) matched with the form charges of Ag+, Fe3+, Zn2+, respectively, replacing the corresponding number(s) of H+ from BAS sites.
Notice that Ag-2–O6 is notably shorter than the average Ag–O value of 2.33 Å (deduced from EXAFS), which may have caused by electrostatic interaction due to the ion-exchange of protons with an additional contribution of covalency between Ag and O moieties. Unlike the Fe-2 and Zn-2 sites, the corresponding Ag-2 site does not seem to bond with any non-framework oxygen (no hydrated water molecule or hydroxyl species). The hybridization of dz2-s orbitals in Ag+ does not support higher coordination; the surface oxygens are hence sufficient to coordinate the Ag ion without major distortion. Interesting, we also note a third Ag+ site, namely Ag-3 site (occupancy 0.09(1)), which has longer bond distances from the generally strong anchored BASs. It is found coordinated with three framework oxygen atoms but at longer Ag–O distances (O2, O3 and O20). Notice that the average CN and bond distances of these metal ions derived from the SXRD and EXAFS measurements are consistent with each other. The associated XANES (Tables S4–S6†) matched with the form charges of Ag+, Fe3+, Zn2+, respectively, replacing the corresponding number(s) of H+ from BAS sites.
Thus, from the above structural studies, there are at least one or more deprotonated O2− of T6 sites (labelled O with underscore) in close proximity to these metal ions presumably due to proton exchange to offer stabilization of these metal ions with balanced charge. Also, importantly, their locations are within molecular distances with the remaining O2− sites adjacent to Al(T6) site in metal-doped H-ZSM-5 structure (as shown in Fig. S14†). We envisage that these metal ions can act as frustrated like Lewis acid and one of these O2− as frustrated like Lewis base, which can activate small substrate molecules cooperatively to give products.
Gates et al. pioneered the identification of single metal atom as a doper in zeolites, followed by others.21,22 A selected high-angle annular dark-field (HAADF) STEM image of Ag-ZSM-5 is shown in Fig. 3. We can rule out the formation of nanoparticle or aggregate especially in the case of Ag and confirm the homogeneous dispersion of the metal ions from our synthesis as that of Gates and co-workers in the zeolites. The image indeed shows the highly crystalline microporous structure with isolated heavy Ag-containing species near the sinusoidal channels. Both STEM imaging and electron diffraction patterns are also consistent with the structural model derived from SXRD refinement (see further enlarged images in Fig. S10 and S11 in ESI†), suggesting the isolated Ag species.
 | ||
Fig. 3 Aberration-corrected HAADF-STEM image of Ag-ZSM-5 along the [0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] axis. (a) Experimental image area (b) image is denoised using a nonlinear filtering algorithm (scale bar: 10 nm). (c) A structural model based on SXRD refinement, blue: Si or Al atoms as the TO4 tetrahedra, and grey is silver ions. (d) Higher magnification of denoised HAADF-STEM image of Ag-ZSM-5; yellow dotted line label as a cyclic annular (scale bar: 8.15 Å). 0] axis. (a) Experimental image area (b) image is denoised using a nonlinear filtering algorithm (scale bar: 10 nm). (c) A structural model based on SXRD refinement, blue: Si or Al atoms as the TO4 tetrahedra, and grey is silver ions. (d) Higher magnification of denoised HAADF-STEM image of Ag-ZSM-5; yellow dotted line label as a cyclic annular (scale bar: 8.15 Å). | ||
To further provide evidence the metal modified ZSM-5 containing isolated Lewis acid M ion and Lewis base O2− as FLP-like sites, a small quantity of isotope 13CH3OH was pre-adsorbed on the Zn-ZSM-5 catalyst and cross polarization-magic angle spinning (CP-MAS) nuclear magnetic resonance (NMR) spectroscopy was measured. The result in Fig. 4 shown below presents a very broad peak in 13C NMR at about 54 ppm. This chemical shift value does not correspond to the typical sharp peak of protonated methanol on BASs in zeolite without Zn at 50.4 ppm,16 but it gives a significant quantity of methanol species in the form of methoxy moiety on Zn17 (54.4 ppm) when Zn-ZSM-5 is used. It clearly suggests that the presence of Zn species (Lewis acid) can interact strongly with the 13CH3O moiety (Lewis base) where the residue BAS is likely to interact with the protonic H of the methanol.
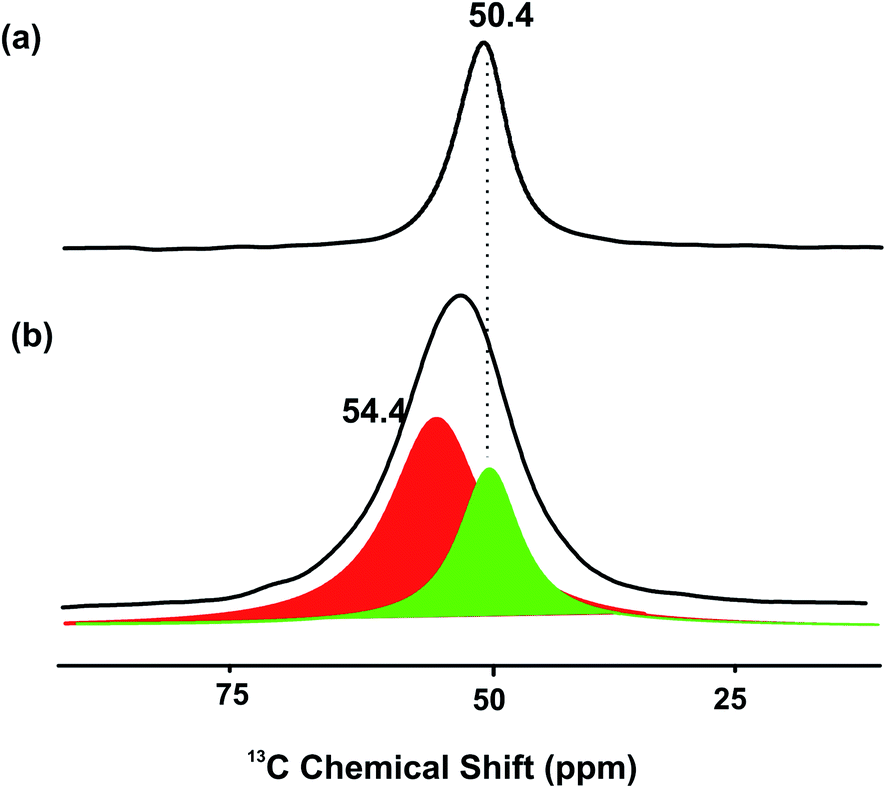 | ||
| Fig. 4 CP-MAS NMR 13C-methanol adsorbed on (a) H-ZSM-5 to give a sharp peak and (b) Zn-ZSM-5 to give a broad peak at 25 °C ((comprising sub-peaks of (a) (green) and peak (red) of CH3OZn)). | ||
As a result, we have carried out high-resolution neutron powder diffraction (NPD) studies on the deuterated-methanol (CD3OD) pre-adsorbed on both Ag-ZSM-5 and Zn-ZSM-5 to specifically ‘locate’ the positions of methoxy and proton moieties from the adsorbed methanol, since the neutron-based technique is more sensitive to light elements (these experimental details in ESI†) than the SXRD that we performed. It is worth noting that high-resolution NPD is of paramount importance to study our catalyst that contains both heavy (Ag) and light (C, H, Si, Al, O) elements. Unlike X-ray, neutron signal does not depend on the atomic number (Z) but the coherent neutron scattering cross-sections (σc). As the σc values of O, C, H, Si, and Al are significantly different from each other (4.2, 5.6, 1.8, 2.2, and 1.5 barns, respectively),23 signals from light elements such as C and H will not be disguised by those from heavy elements, as in the case of X-ray and electron scattering. Nevertheless, based on high-resolution NPD data, we can unambiguously determine the nature, location, and quantity of deuterated methanol molecule on these zeolites following the indication of the Fourier difference map.
As seen from the Rietveld refinement data shown in Fig. 5, the labelled methanol of type 2 species is in the form of methoxy moiety that indeed interacts directly to the metal site (M–OCD3). The resolved M–Omethanol bond lengths for Ag and Zn on T6 atom are 1.78 Å and 1.76 Å, respectively whereas the deuterium (of CD3O–D) interacts with the nearest Lewis base O2−, the O18 of T6 (the O–D bond is 1.83 Å and 1.73 Å for Ag and Zn, respectively) as anticipated by the heterolytic activation over the ion pair. Thus, the neutron results are consistent to those from the synchrotron results about the locations of the metal and the nearest O2− sites (T6). We understand that the precise bonding, coordination environment, and geometry of ex situ characterization techniques could be different from that when the zeolite-bound transition-metal complexes are exposed to reaction conditions and in situ or operando characterization should be attempted.24 However, the present data show that activated methanol binds on the ion-pair as synergetic active sites to offer insights for cooperative effect for the initial metal–acid site heterolytic activation of the small molecule in the metal-doped zeolites.
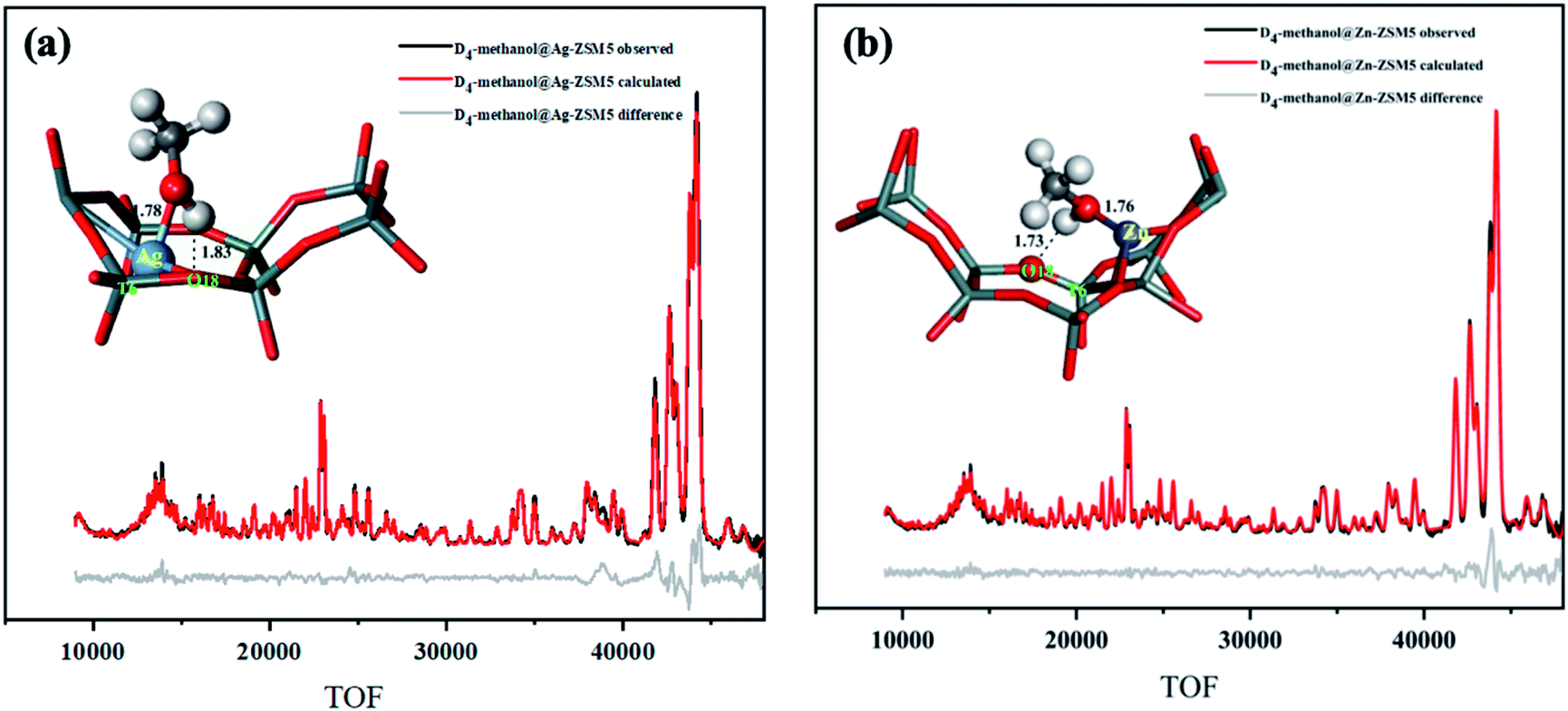 | ||
| Fig. 5 NPD data and Rietveld refinement collected at 25 °C of (a) Ag-ZSM-5, (Rwp = 3.98%, χ2 = 2.54), and (b) Zn-ZSM-5, (Rwp = 3.03%, χ2 = 2.02). The corresponding Rietveld derived crystal structure of site 2 is shown, respectively. Some symmetry and other metal site(s) were disregarded for clarity. See main text for the detailed bond distance descriptions. The atomic and crystallographic parameters are summarised in Tables S11–S13.† Atoms are represented in ball/sticks: white = D, cyan = Si, red = O, blue = Ag, and grey = Zn. | ||
As stated, although metal-doped acidic zeolites are long known to be active for a wide range of catalytic reactions, it is not clear how the transition metal ions and BASs work in synergy to account the catalysis. For example, DME is thought to be the key intermediate in methanol conversion over metal-doped zeolites before higher hydrocarbons/aromatics are formed, but the fundamental pathway for the methanol activation is not yet known.8,9 As seen in Fig. S13,† these transition metal ions doped ZSM-5 are also active for the hydrolytic cleavage of gamma-valerolactone to aromatics (HCA), suggesting their ability to activate water molecule to initiate the hydrolysis reaction. Without engaging the detail mechanisms for the upgrading of the organic substrates to products, these types of catalysts are thus able to initiate activate small molecules including methanol, water or other polar organic molecules. On the other hand, the ability to activate small polar molecules have recently been revealed in homogeneous systems25,26 involving the concept of spatially separated ion pair in molecular distances (see Fig. S15†). In the classical frontier molecular orbital theory, the Lewis base with a low-energy HOMO can accept H+ while the metal as Lewis acid possessing a high-energy LUMO accept CH3O− moiety from methanol. Such heterolytic cleavage of small polar molecules by the unquenched FLP-like ion pair within molecular distance could substantially reduce the bond cleavage energy, justifying the low-temperature activations over the zeolite-based catalysts.27,28
Indeed, according to the structural and atomic information derived from SXRD, NPD and STEM images, the isolated metal M ions (type 2) can substitute protons and are held in position in compromise with electrostatic/covalent interactions between proton-depleted O2− on BASs as well as the chelating interactions with other surface oxygen atoms. There are remaining protonated O2− and deprotonated of T6 sites in this structure in close proximity to the metal ions at variable molecular distances (as shown in Fig. S14†). It is anticipated that the late transition metal ions with higher effective nuclear charges (Zeff) will give higher Lewis acidity, which explains our observation. For H-ZSM-5 with higher Al/Si more ion pairs at variable molecular distances could be created, which may activate small molecules of different sizes. These deprotonated oxygens, according to conjugated acid–base theory, will act as the Lewis bases while the metal ions with vacant higher orbitals serve as the Lewis acids in a broad sense. As a result, the steric clash does not allow the formation of a classical adduct between the anchored metal ion as Lewis acid and rigid surface Lewis base O2− with depleted proton resulting in unquenched reactivity to a small molecule. Such structural rigidity within small molecular distances fulfils the rigid FPL-like ion pair requirements for the heterolytic activation.
Further, we have also performed the computational calculations on this catalytic reaction over the metal modifiers in synergy with the BAS. The clusters, their Cartesian coordinates and total energies calculations are shown in Table S14.† The reaction mechanism of BAS activated MeOH (methyl) with incoming methanol → dimethyl ether (DME) on the ion-pair was thus considered by density functional theory calculations, as shown in Fig. 6(a). The doped metal ions in type 2 site (M = Ag and Zn) on ZSM-5 (M-ZSM-5) plays an important role in the formation of DME. The C–O bond for incoming MeOH is significantly weakened by coordinating with the M site as Lewis acid, which lowers the energy barrier to a greater extent than that in H-ZSM-5, as shown in Fig. 6(b). Fig. 6(c) shows the typically Bader charge analysis, which depicts the remarkable charge transfer from methoxy moiety to Ag (electron transfer from Lewis base to acid), agreeing with the heterolytic activation of methanol. Using 2-D electron localization function contours,29 the focussed region of CH3O–Ag in Fig. 6(d) also indicates the charge transfer (asymmetrical red region at the O–Ag) to Ag which acts as Lewis acid while the H moiety interacts with the BAS O2− as Lewis base in the ion pair.
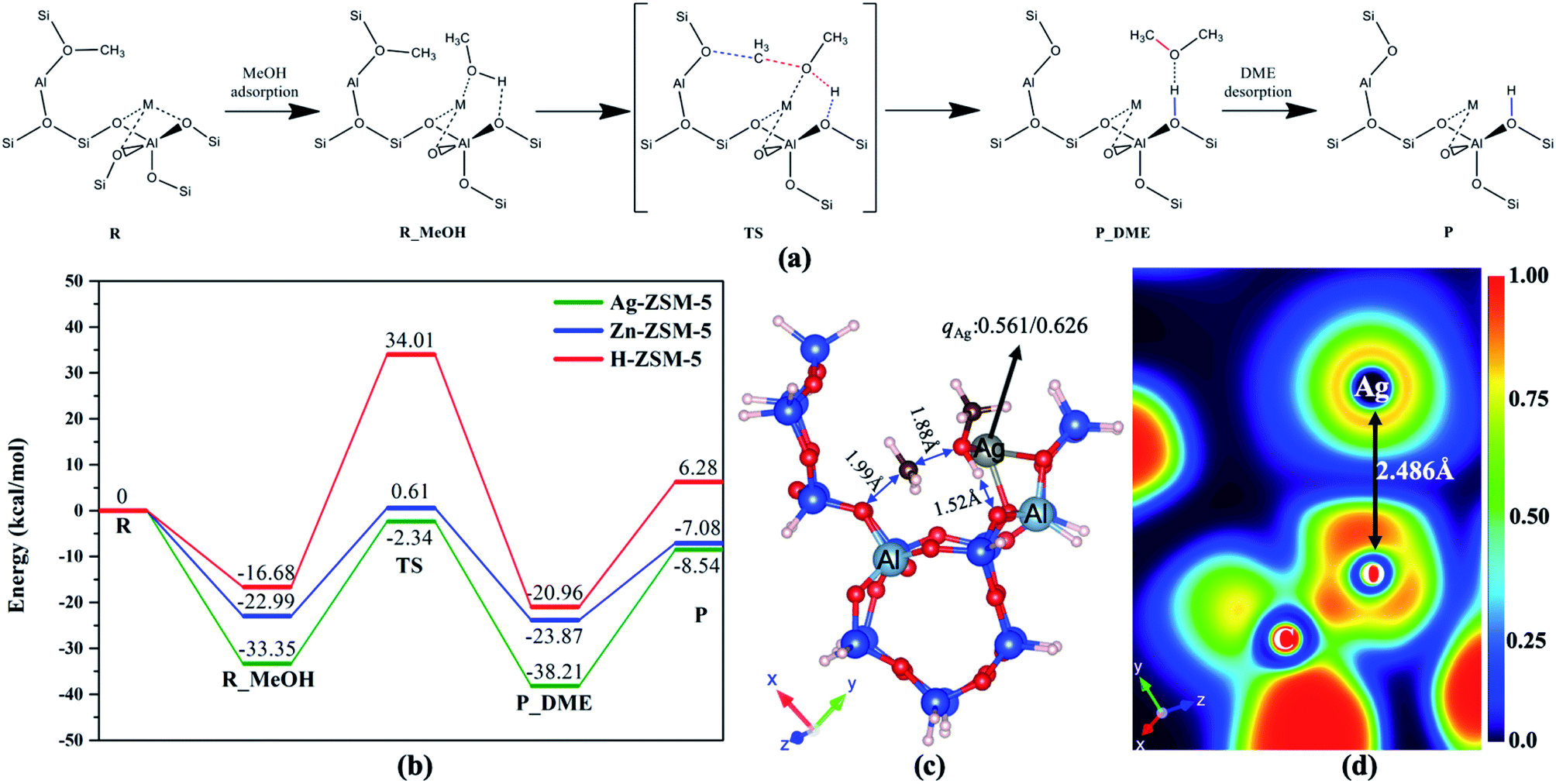 | ||
| Fig. 6 (a) Reaction scheme and (b) potential energy surface of M-ZSM-5–MeOH + MeOH (–OMe+via a rapid dehydration of MeOH2+ over BAS) → M-ZSM-5 + DME (M = Ag, Zn and H), (c) transition state (TS) structure and charge value of Ag (qM) for R_MeOH and for TS, (d) electron location function (ELF) contour of (c). | ||
As presented in Scheme 1, we therefore propose from the present structural study that an anchored transition metal ion in type 2 site works in close proximity to O from BAS with depleted proton can activate methanol heterolytically by the corresponding ion pair. This incorporation of transition metal ion will offer additional electronic effect to pull the methoxy from methanol while the protonic H of the methanol will be activated by the typical BAS. As a result, the negatively charged methoxy species can favourably be combined with methyl cations from dehydration of protonated methanol to form the initial DME (as previously identified30–36) before further C–O and C–H activations.
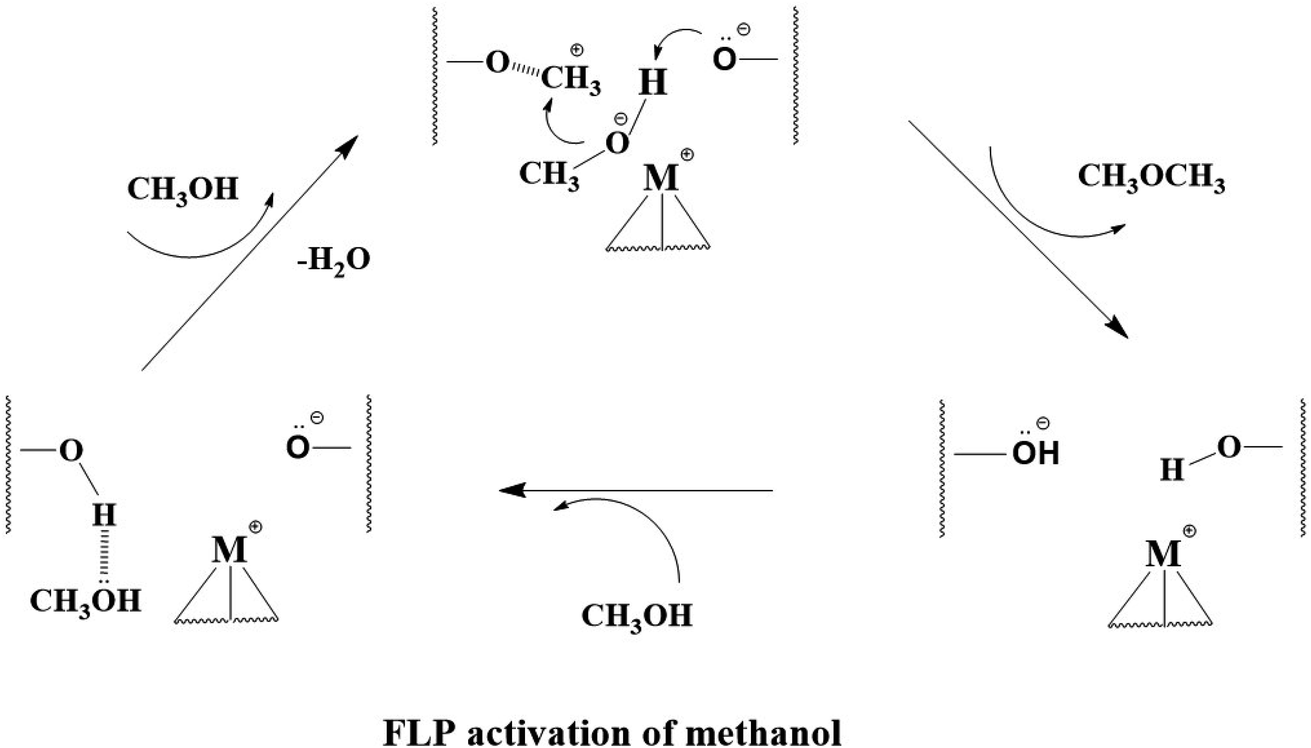 | ||
| Scheme 1 The proposal ion pair mechanism for the formation DME from methanol over M-ZSM-5. The wavy lines represent the zeolite framework. | ||
The mechanism for methanol to aromatics (MTA)/hydrocarbons (MTH) is a very complex system with a lot of key intermediates in forms of rather complicated network.37 As stated in our introduction and discussion of the original manuscript, it is emphasized that we aim in this paper to provide rationale and insights for the initial chemical bond activation of methanol by the metal–BAS ion pair in the structural elucidation using synchrotron and neutron diffraction and DFT calculations without providing the comprehensive account for subsequent reactions to products in the zeolite cage. But, we would like to contribute some discussion which could be useful to link our results in the context of the rich literature spanned over the past two decades. Particularly, we have referred to the recent review article from U. Olsbye et al. who have presented a comprehensive summary on the MTA/MTH mechanisms.37
As presented in Fig. 1, S2, S3 and S13† of our manuscript, the conversion of methanol over H-ZSM-5 is notably increased upon the incorporation of Fe, Zn and Ag metal sites. By combining a series of our investigations, the FLPs can promote the heterolytic cleavage of methanol (CH3O(δ−)–H(δ+)); in this case, methanol is activated and heterolytically cleaved to form a metal–methoxy species (M–OCH3) (or called surface methoxy species) and framework (O–H) before the formation of MTA (and DME). Similarly, from our previous study over SAPO-34,38 we have also found the formation of surface methoxy species by either dominant associative process on BAS sites at low temperature or dissociative process at high temperature to form MTH (olefinic hydrocarbons with DME). It is therefore evident from our studies that the formation of –OCH3 is the primary reason causing a more efficient conversion of the methanol reaction substrate. As discussed in this manuscript based on computational evidence, this pairs with subsequent methanol reactions to form hydrocarbons or aromatic products at high selectivity as that observed experimentally.
We have further compared the product selectivity with different framework-Al and doped-metal contents to investigate the structure–activity relationship of the FLPs. Briefly, H-ZSM-5 of Al2O3-to-SiO2 ratios of 25 and 38 were used (denoted as ‘H-ZSM-5(25) and H-ZSM-5(38)’). Different contents of doped-metal were achieved by performing an additional ion-exchange procedure with the corresponding metal nitrate precursor, where these samples are denoted as ‘2Fe/Zn-ZSM-5(25/38)’. The product distribution is presented in Fig. S3.† Upon increasing Fe content, it can be seen that the selectivity toward aliphatic products increases. The selectivity toward aromatic products, in contrast, increases with the Zn content. This clearly suggests that the role of the metal sites does not merely contribute toward the formation of M–OCH3 (as shown by structural analysis) but gives an additional impact towards either aliphatic or aromatic products.
It'd be good to see the direct correlation between our formed methoxy on metal by FLP with the methanol activity. However, it is a still huge challenge because it involves many elementary reactions occurring in both competing and consecutive ways throughout the whole process. The theoretical calculations presented in literature in fact show that all the proposed direct mechanisms of surface methoxy, e.g. carbene, oxonium ylide and methane-formaldehyde mechanisms, cannot generate a C–C bond from methanol and DME via methoxy because of very high activation energies and unstable intermediates.37 Thus, the hydrocarbon pool (HCP) mechanism has been widely accepted to dominate the steady process, and significant progress has been achieved recently. Also, the dual cycles, namely aromatics-based cycle and olefin-based cycle are thought to take place over ZSM-5 based materials within inter-related mechanism that have been extensively discussed.39 Nevertheless, the debate on the direct mechanism in methanol conversion to aromatics/hydrocarbons is still going because the HCP mechanism cannot account for the origin of initial methoxy species or the formation of the first C–C bond, although it gives a reasonable interpretation of the induction period.
From the literature that the methoxy may not be the only key C1 immediate for the methanol to aromatics/hydrocarbons processes. However, recently, a combination of experimental and DFT calculation results led to a possible direct route for the formation of initial C–C bonds. This direct route involves the formation of methoxymethyl cation (CH3OCH2+) intermediates from SMS and DME.40 The methoxymethyl cation further couples with another DME or methanol molecule to form C–C bond-containing 1,2-dimethoxy ethane or 2-methoxyethanol, which gives propene as an initial alkene product through the formation of a series of oxonium cations via methylation, deprotonation, dealkylation, H-shift and elimination. It was particularly found that the propene, not ethene, induced the occurrence of the HCP mechanism which also links with the aromatics via the dual cycles. Notice the high stability of CH3OCH2+ species has recently enabled its detection by in situ IR spectroscopy over carefully controlling the conditions of the reaction of DME and methoxy.40
As seen from Fig. 6 of the main manuscript that the reaction mechanism of MeOH → DME on FLP was considered by density functional theory, the loaded metal (M = Ag and Zn) on ZSM-5 (M/ZSM-5) clearly plays an important role in the formation of methoxy on M and H on Si–OH followed by DME formation. The C–O bond in MeOH is weakened by coordinating with M, which results to the lowering of energy barrier than that of H-ZSM-5. Fig. 6(c) shows the typically Bader charge analysis, which depicts the remarkable charge transfer from methoxy to Ag, agreeing with the FLP activation of methanol. Using 2-D electron localization function contours, the focus region of OCH3–Ag in Fig. 6(d) also indicates the charge transfer (asymmetrical red region at the O–Ag) to Ag which acts as the Lewis acid in FLP. As a result, the negatively charged methoxy species can favourably be combined with methyl cations from dehydration of protonated methanol to form the initial DME (as previously identified30–36) before further C–O and C–H activations. Thus, our direct proof for the existence of methoxy and DME could support the recent methoxymethyl cation (CH3OCH2+) mechanism linking to the dual cycles, as discussed.
In addition, we have also identified the quenched Zn-ZSM-5 catalyst from hydrolytic cleavage of gamma-valerolactone the Zn–OH extra-framework terminal group from the NMR and Raman spectra (see Fig. S16†). This matches with heterolytic water molecule activation by the Zn2+ and O2− ion pair to form Zn–OH and BAS, as similarly shown in Scheme 1. It should be noted that the initial “activation” of methanol or water in GVL in reaction cascades for their upgrading to aromatics requires subsequent C–H bond activation and C–C bond formation steps that proceed with slower rates than initial rates which involve relatively facile activation of a polar O–H bond by zeolites with ion pairs over these polar molecules. We are not yet able to address the fully complex reaction schemes. Also, we want to stress that it is still debated in the literature on the intermediary role of methoxymethyl cation for the formation of initial C–C bonds and the propene may be not necessary to be the unique initial alkene in MTH reaction. This present study in this manuscript can only offer insights from our diffraction and DFT studies on the initial FLP step for methanol activation to form methoxy/DME. Whether the FLP affect fundamental dehydrogenation or aromatization route is not yet known. We believe that some careful planned time- and space-resolved reactor studies,41 operando kinetic analysis and kinetic models in the understanding of the time-on-stream behaviour catalysts should be carried out for a more vigorous elucidation of the reaction mechanism in the future.
Conclusions
In this paper, we show for-the-first-time that the incorporation of transition metal ions into acidic H-ZSM-5 by ion-exchange can create catalytically active ion pair within molecular distances in the internal cavity of the H-ZSM-5 structure with BAS, as shown by labelled probe-NMR, SXRD, NPD and computational simulations. The internal surface anchored metal ion as unquenched Lewis acid site and O2− from nearby BAS in proximity as an anchored base is established. As a result, active and selective catalytic routes over this simple metal ion-doped ZSM-5 for small molecule activations such as methanol, water and small polar organic molecules are hereby accounted.Data availability
The data that support the findings of this study are available from the corresponding author on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Diamond Light Source, Ltd. UK (I11 beamline/EE16358-1) for SXRD beamline and China Spallation Neutron Source for neutron beamtime (beamline GPPD). The STEM facilities were provided by the David Cockayne Centre for Electron Microscopy of University of Oxford with discussion from Dr Chen Huang in the Diamond Light Source (UK) and Dr Hongchu Du in the Juelich Research Centre (Germany). BL wishes to acknowledge Research Grants Council (25300918) for financial support.References
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS.
- J. Lin, A. Q. Wang, B. T. Qiao, X. Y. Liu, X. F. Yang, X. D. Wang, J. X. Liang, J. X. Li, J. Y. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS.
- M. Cargnello, N. L. Wieder, T. Montini, R. J. Gorte and P. Fornasiero, J. Am. Chem. Soc., 2010, 132, 1402–1409 CrossRef CAS.
- S. H. Sun, G. X. Zhang, N. Gauquelin, N. Chen, J. G. Zhou, S. L. Yang, W. F. Chen, X. B. Meng, D. S. Geng, M. N. Banis, R. Y. Li, S. Y. Ye, S. Knights, G. A. Botton, T. K. Sham and X. L. Sun, Sci. Rep., 2013, 3, 9 Search PubMed.
- S. Abate, K. Barbera, G. Centi, P. Lanzafame and S. Perathoner, Catal. Sci. Technol., 2016, 6, 2485–2501 RSC.
- (a) S. M. T. Almutairi, B. Mezari, P. Magusin, E. A. Pidko and E. J. M. Hensen, ACS Catal., 2012, 2, 71–83 CrossRef CAS; (b) P. Gao, Q. Wang, J. Xu, G. D. Qi, C. Wang, X. Zhou, X. L. Zhou, N. D. Feng, X. L. Liu and F. Deng, ACS Catal., 2018, 8, 69–74 CrossRef CAS.
- I. Wender, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- P. Tian, Y. X. Wei, M. Ye and Z. M. Liu, ACS Catal., 2015, 5, 1922–1938 CrossRef CAS.
- M. Bjorgen, F. Joensen, M. S. Holm, U. Olsbye, K. P. Lillerud and S. Svelle, Appl. Catal., A, 2008, 345, 43–50 CrossRef.
- Y. Ono and T. Mori, J. Chem. Soc., Faraday Trans. 1, 1981, 77(9), 2209–2221 RSC.
- V. Van Speybroeck, K. Hemelsoet, K. De Wispelaere, Q. Qian, J. Van der Mynsbrugge, B. De Sterck, B. M. Weckhuysen and M. Waroquier, ChemCatChem, 2013, 5(1), 173–184 CrossRef CAS.
- J. Van der Mynsbrugge, S. L. C. Moors, K. De Wispelaere and V. Van Speybroeck, ChemCatChem, 2014, 6(7), 1906–1918 CrossRef CAS.
- P. G. Moses and J. K. Nørskov, ACS Catal., 2013, 3(4), 735–745 CrossRef CAS.
- B. T. W. Lo, L. Ye, C. A. Murray, C. C. Tang, D. Mei and S. C. E. Tsang, J. Catal., 2018, 365, 145–152 CrossRef CAS.
- S. Zhang, Z.-Q. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C.-R. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266 CrossRef CAS.
- W. C. Lin, L. Ye, S. Wu, B. Lo, Y. K. Peng, P. Zhao, I. McPherson and S. C. E. Tsang, ChemSusChem, 2018, 11, 4214–4218 CrossRef CAS.
- L. Ye, Q. Song, B. T. W. Lo, J. L. Zheng, D. J. Kong, C. A. Murray, C. C. Tang and S. C. E. Tsang, Angew. Chem., Int. Ed., 2017, 56, 10711–10716 CrossRef CAS.
- P. Zhao, L. Ye, Z. Y. Sun, B. T. W. Lo, H. Woodcock, C. Huang, C. Tang, A. I. Kirkland, D. H. Mei and S. C. E. Tsang, J. Am. Chem. Soc., 2018, 140, 6661–6667 CrossRef CAS.
- F. L. Liao, X. P. Wu, J. W. Zheng, M. M. J. Li, A. Kroner, Z. Y. Zeng, X. L. Hong, Y. Z. Yuan, X. Q. Gong and S. C. E. Tsang, Green Chem., 2017, 19, 270–280 RSC.
- B. T. W. Lo, L. Ye and S. C. E. Tsang, Chem, 2018, 4, 1778–1808 CAS.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and A. Katz, Catal. Sci. Technol., 2017, 7(19), 4259–4275 RSC.
- N. Kosinov, C. Liu, E. J. M. Hensen and E. A. Pidko, Chem. Mater., 2018, 30, 3177–3198 CrossRef CAS.
- V. F. Sears, Neutron News, 1992, 3(3), 26–37 CrossRef.
- B. E. R. Snyder, M. L. Bols, R. A. B. F. Sels and E. I. Solomon, Chem. Rev., 2018, 118, 2718–2768 CrossRef CAS.
- D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS.
- M. W. Anderson and J. Klinowski, J. Am. Chem. Soc., 1990, 112, 10–16 CrossRef CAS.
- J. Xu, A. Zheng, X. Wang, G. Qi, J. Su, J. Du, Z. Gan, J. Wu, W. Wang and F. Deng, Chem. Sci., 2012, 3, 2932–2940 RSC.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S.-B. Liu and A. Zheng, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS.
- P. G. Moses and J. K. Nørskov, ACS Catal., 2013, 3(4), 735–745 CrossRef CAS.
- A. Ghorbanpour, J. D. Rimer and L. C. Grabow, ACS Catal., 2016, 6(4), 2287–2298 CrossRef CAS.
- A. J. Jones and E. Iglesia, Angew. Chem., Int. Ed., 2014, 53(45), 12177–12181 CrossRef CAS.
- Y. Jiang, M. Hunger and W. Wang, J. Am. Chem. Soc., 2006, 128(35), 11679–11692 CrossRef CAS.
- M. Hunger and T. Horvath, J. Am. Chem. Soc., 1996, 118(49), 12302–12308 CrossRef CAS.
- W. Wang, A. Buchholz, M. Seiler and M. Hunger, J. Am. Chem. Soc., 2003, 125(49), 15260–15267 CrossRef CAS.
- T. R. Forester and R. F. Howe, J. Am. Chem. Soc., 1987, 109(17), 5076–5082 CrossRef CAS.
- U. Olsbye, S. Svelle, K. P. Lillerud, Z. H. Wei, Y. Y. Chen, J. F. Li, J. G. Wang and W. B. Fan, Chem. Soc. Rev., 2015, 44, 7155–7176 RSC.
- B. T. W. Lo, L. Ye, G. G. Z. Chang, K. Purchase, S. Day, C. C. Tang, D. Mei and S. C. E. Tsang, Appl. Catal., B, 2018, 237, 245–250 CrossRef CAS.
- X. Sun, S. Mueller, H. Shi, G. L. Haller, M. Sanchez-Sanchez, A. C. van Veen and J. A. Lercher, J. Catal., 2014, 314, 21–31 CrossRef CAS.
- J. Li, Z. Wei, Y. Chen, B. Jing, Y. He, M. Dong, H. Jiao, X. Li, Z. Qin, J. Wang and W. Fan, J. Catal., 2014, 317, 277–283 CrossRef CAS.
- D. Wragg, M. G. O'Brien, F. L. Bleken, M. Di Michiel, U. Olsbye and H. Fjellvag, Angew. Chem., Int. Ed., 2012, 51, 7956–7959 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc04058d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |