DOI:
10.1039/D0SC05452F
(Edge Article)
Chem. Sci., 2021,
12, 220-226
Preparation of α-amino acids via Ni-catalyzed reductive vinylation and arylation of α-pivaloyloxy glycine†
Received
2nd October 2020
, Accepted 26th October 2020
First published on 27th October 2020
Abstract
This work emphasizes easy access to α-vinyl and aryl amino acids via Ni-catalyzed cross-electrophile coupling of bench-stable N-carbonyl-protected α-pivaloyloxy glycine with vinyl/aryl halides and triflates. The protocol permits the synthesis of α-amino acids bearing hindered branched vinyl groups, which remains a challenge using the current methods. On the basis of experimental and DFT studies, simultaneous addition of glycine α-carbon (Gly) radicals to Ni(0) and Ar–Ni(II) may occur, with the former being more favored where oxidative addition of a C(sp2) electrophile to the resultant Gly–Ni(I) intermediate gives a key Gly–Ni(III)–Ar intermediate. The auxiliary chelation of the N-carbonyl oxygen to the Ni center appears to be crucial to stabilize the Gly–Ni(I) intermediate.
Introduction
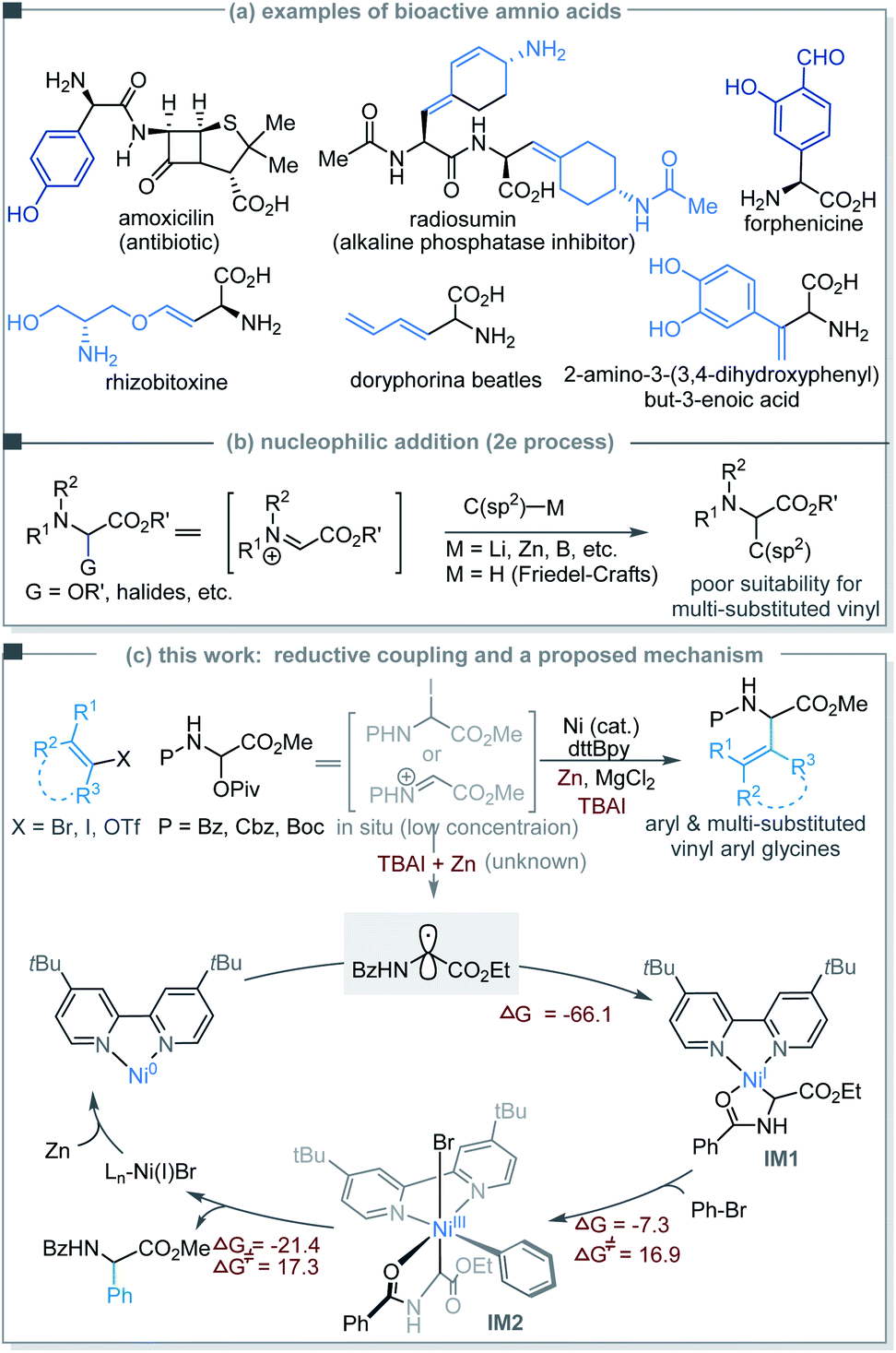
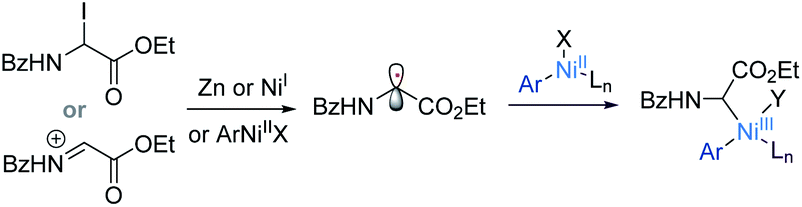
Ni-catalyzed cross-electrophile coupling has emerged as a powerful tool for expeditious creation of C(sp3)–C bonds,1 in which alkyl radical intermediates are generally involved. However, to the best of our knowledge, application of this strategy to the reductive coupling of α-glycinyl electrophiles, which enables direct decoration of the α-carbon of glycine derivatives, remains unexplored.2 A prominent obstacle may be the rapid conversion of α-glycinyl electrophiles to imino or iminium esters that are well-established for addition reactions.3,4 Reduction of an α-glycinyl electrophile to generate a glycinyl α-C(sp3)-radical was hitherto unknown. Realization of such a radical forming process may invoke a reductive coupling protocol for facile structural enrichment of α-amino acids.2
Unusual and unnatural α-vinyl and -aryl amino acids have seen a broad range of applications in drug discovery, biomaterials, and protein engineering.5–7 Selected examples include amoxicillin,8 forphenicine,9 the trypsin inhibitor radiosumin,10 the phytotoxin rhizobitoxine,11 butadienylglycine (found in the defensive secretion of beetles),12 and 2-amino-3-(3,4-dihydroxyphenyl)but-3-enoic acid as an antidepressant (Scheme 1a).13 Numerous synthetic methods have been developed to access α-aryl and -vinyl amino acids based on two-electron addition (e.g., R-Li, -Zn and -B and electron-rich arenes) to glycine cation equivalents such as iminoesters (Scheme 1b).14–16 Nevertheless, decoration of glycine α-carbons with branched vinyl groups bearing multiple substituents remains a challenge.17
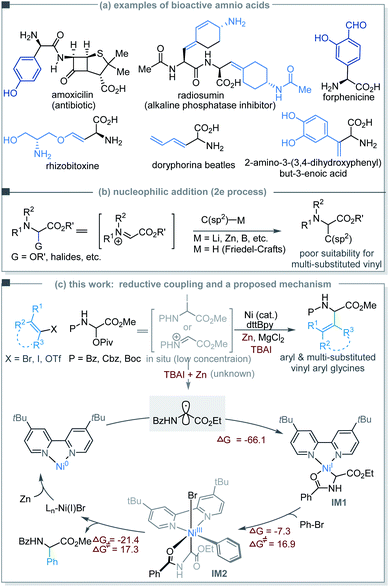 |
| | Scheme 1 (a) Examples of bioactive compounds consisting of unusual amino acids. (b) C(sp2)-functionalization of glycine cation equivalents using 2e addition strategies. (c) Ni-catalyzed reductive coupling strategy for the preparation of unusual α-amino acids. | |
Herein, we describe the Ni-catalyzed reductive coupling of N-carbonyl-protected α-pivaloyloxy glycine derivatives with aryl and vinyl halides/triflates to afford unusual α-aryl/vinyl amino acids. The use of α-pivaloyloxy glycine proved crucial for the coupling reaction probably because it converts in situ into the active α-iodoglycine or iminium ester at low concentrations. Upon SET reduction or halide abstraction, a glycinyl (Gly) radical intermediate was produced. DFT studies suggest that the Gly radical may simultaneously add to Ni(0) and Ar–Ni(II), but with the former being more favored to give a highly stable Gly–Ni(I) chelate wherein the auxiliary chelation of the N-carbonyl oxygen to the Ni center appears to be pivotal. Oxidative addition of an aryl halide to the Ni(I) species forms a key Gly–Ni(III)–Ar intermediate (Scheme 1c). The necessity of the N-carbonyl protecting groups may provide useful information about coupling reactions of functionalized alkyl precursors which are currently essential for Ni-catalyzed stereoselective synthesis of C–C bonds.18
Results and discussion
Optimization of the reaction conditions
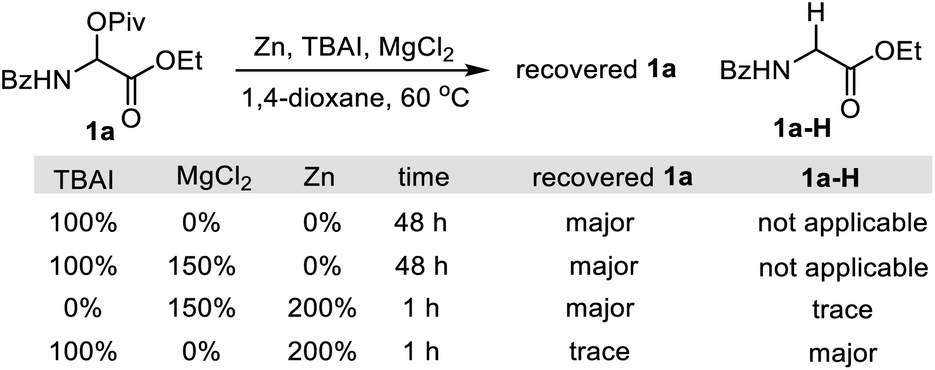
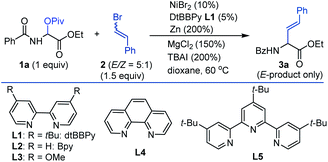
We began our studies by investigating the coupling of benzoyl protected α-pivaloyloxy glycine ethyl ester 1a and (2-bromovinyl)benzene 2 (E/Z = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). An extensive survey of experimental parameters led us to identify the optimal reaction conditions at 60 °C comprising NiBr2/dtBBpy in combination with the reductant Zn, the additives MgCl2 and TBAI, and the solvent 1,4-dioxane.19 The vinyl glycine product 3a was isolated in 75% yield consisting only of the E-isomer (Table 1, entry 1).20 Control experiments indicated that NiBr2, Zn, MgCl2, and TBAI all were essential reagents (entries 2–5). Other nickel sources, ligands or temperatures gave inferior results (entries 6–10 in Table 1). The reaction could be performed on a gram scale with slightly diminished efficiency (entry 11). The need for 5 mol% ligand (L1) was tentatively explained by the inhibited formation of hydro-reduction product 1a-H. The remaining non-ligand chelated 5 mol% Ni may be stabilized by weak coordination with 1a, which could ensure the formation of a decent amount of Ni–L1 complex for efficient catalysis, provided formation of inactive Ni species is possible over the course of the reaction.
:1). An extensive survey of experimental parameters led us to identify the optimal reaction conditions at 60 °C comprising NiBr2/dtBBpy in combination with the reductant Zn, the additives MgCl2 and TBAI, and the solvent 1,4-dioxane.19 The vinyl glycine product 3a was isolated in 75% yield consisting only of the E-isomer (Table 1, entry 1).20 Control experiments indicated that NiBr2, Zn, MgCl2, and TBAI all were essential reagents (entries 2–5). Other nickel sources, ligands or temperatures gave inferior results (entries 6–10 in Table 1). The reaction could be performed on a gram scale with slightly diminished efficiency (entry 11). The need for 5 mol% ligand (L1) was tentatively explained by the inhibited formation of hydro-reduction product 1a-H. The remaining non-ligand chelated 5 mol% Ni may be stabilized by weak coordination with 1a, which could ensure the formation of a decent amount of Ni–L1 complex for efficient catalysis, provided formation of inactive Ni species is possible over the course of the reaction.
Table 1 Optimization for the reaction of 1a with 2
|
NMR yield using 2,5-dimethylfuran as the internal standard.
Isolated yield.
The reaction was run on a gram scale.
|
|
| Entry |
Variation from the standard conditions |
Yield%a |
| 1 |
No changes |
65 (75)b |
| 2 |
w/o Ni |
Trace |
| 3 |
w/o Zn |
Not detected |
| 4 |
w/o TBAI |
Trace |
| 5 |
w/o MgCl2 |
Trace |
| 6 |
NiCl2 |
10 |
| 7 |
L2 instead of L1 |
Trace |
| 8 |
L3 instead of L1 |
62 |
| 9 |
L4 instead of L1 |
18 |
| 10 |
L5 instead of L1 |
Not detected |
| 11 |
1a (8 mmol) |
63c |
Substrate scope
The established vinylation conditions (Table 1, entry 1) proved to be general for various vinyl halides (Fig. 1). The substituents on the phenyl rings of styrenyl bromides include both electron-donating and -withdrawing groups as evidenced by 3b–3f. For 3d, 20% of the enamine tautomer was also detected.19 The naphthyl, anthranyl, furyl, and thiyl-conjugated vinyl bromides all delivered the corresponding vinyl glycines (4–7) in good to excellent yields. The dienyl glycine 8 was obtained in 75% yield, which can be used for further transformations (e.g., Diels–Alder reactions). The alkyl-substituted 1-vinyl bromides (E and Z mixtures) en route to 9 and 10 were also viable, wherein conversion of the Z-vinyl bromides to the E-products was much less effective than that observed for 2 (Table 1). The 2,2-disubstituted vinyl bromides appeared to be slightly more effective than the mono-substituted ones (e.g., 11–15). An outstanding feature of this vinylation protocol was its competency in the cases of α-vinyl bromide substrates, which produced the phenyl, alkyl, and silyl-substituted vinyl glycines 16–20 in moderate to good yields. The low yield for 18 was due to homocoupling of the α-vinyl bromide. The 1,2-disubstituted Z-alkenyl bromides were compatible with the coupling conditions, affording 21–24 in moderate to good yields. To our delight, the trimethyl substituted vinyl bromide delivered 25 in 44% yield, which is difficult to access with concurrent methods.
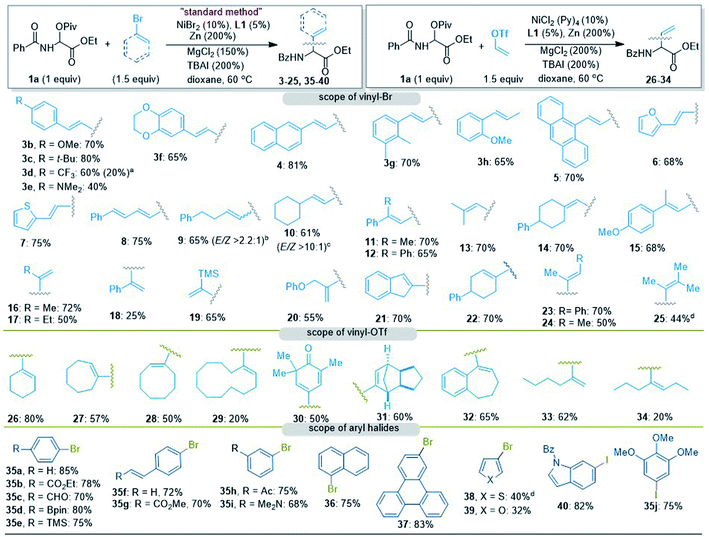 |
| | Fig. 1 Coupling of 1a with vinyl bromides and triflates, and aryl halides using the conditions in Table 1, entry 1 with isolated yields. aYield of isomerization to α-enamine determined by 1H NMR analysis after purification. bA mixture of E/Z vinyl–Br in a ratio of 1.8:1 was used. cA mixture of E/Z vinyl–Br in a ratio of 10:1 was used. dNiCl2(DME). | |
Vinyl triflates that could be readily obtained from ketones or alkynes were viable (Fig. 1). By replacing NiBr2 with NiCl2(Py)4, coupling of 1a with cyclohexenyl triflate produced 26 in 80% yield. Products 27 and 28 bearing cycloheptenyl and cyclooctenyl groups were obtained in moderate yields, while the formation of cyclododecenyl amino acid 29 was of lower yield. The conjugated 3,3,5-trimethyl-4-oxocyclohexa-1,5-dien-1-yl, sterically hindered 2,3,3a,4,7,7a-hexahydro-1H-4,7-methanoindenyl and phenyl-conjugated vinyl triflates within a cycloheptene ring resulted in 30–32 in moderate yields. While 2-hexenyl glycine 33 was obtained in 62% yield, the more hindered heptenyl glycine 34 was produced in 20% yield.
The vinylation method was also extended to the generation of arylated glycines 35–40 (Fig. 1). The reactions exhibited excellent functional group compatibility, retaining such functionalities as aldehyde in 35c and vinyl in 35f and 35g. The naphthyl and anthrenyl bromides afforded 36 and 37 in high yields. Electron-rich heteroaromatics such as 3-bromofuran and thiophene gave 38 and 39 in low yields. This problem could be solved using the iodo analogs, as exemplified by 40 and 35j.
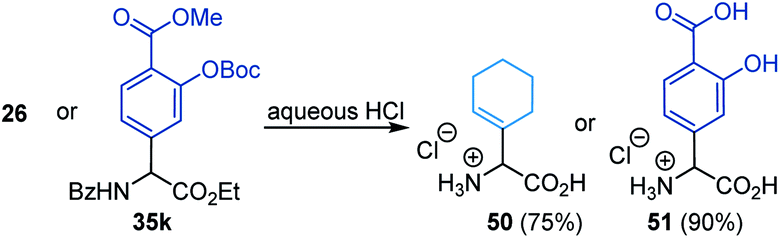
The N-carbonyl protecting groups ranging from benzoyl groups decorated with electron-donating and -withdrawing substituents, to thiophenyl, Cbz, and Boc were suitable as evidenced in 41–45 (Fig. 2). In the case of arylation, Fmoc was more effective than Boc and Cbz groups (see 46–48). In contrast, N-tert-butylsulfinyl was ineffective. Moreover, hydrolysis of 26 and 35k with hydrochloric acid afforded the amino acid salts 50 and 51 in good yields (eqn (1)).
| | 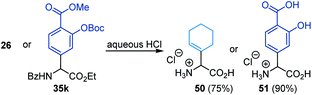 | (1) |
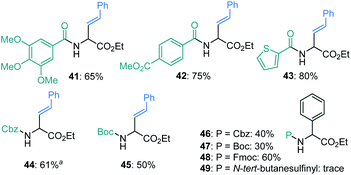 |
| | Fig. 2 Variation of amine-protecting groups. aNiCl2 was used in place of NiBr2. | |
Mechanistic and computational studies
(2) Formation of an α-glycinyl carbon radical intermediate.
More importantly, α-cylcopropyl imine 54 resulted in the radical ring-opening product 55 under Zn/MgCl2/TBAI conditions (eqn (5)). The catalytic reaction as in Table 1, entry 1 was completely inhibited upon introduction of 2 equiv. of TEMPO.19 These results suggest that the catalytic reaction involves generation of a glycine α-carbon radical via single-electron reduction/halide abstraction of the in situ-formed iminium intermediate or α-iodoglycine by MgCl2-activated Zn, or Ln–NiI (eqn (6)).22–24 The Ln–NiI species can be generated from reduction of Ln–NiII by Zn or from the reductive elimination of a Gly–(Ln)NiIII–Ar intermediate (Scheme 1c).| |  | (5) |
(3) Addition of the α-glycinyl carbon radical to ArNi(0) vs. ArNi(II).
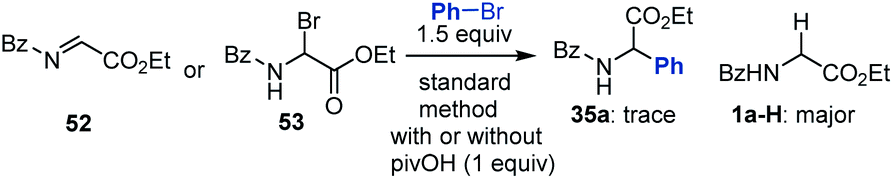
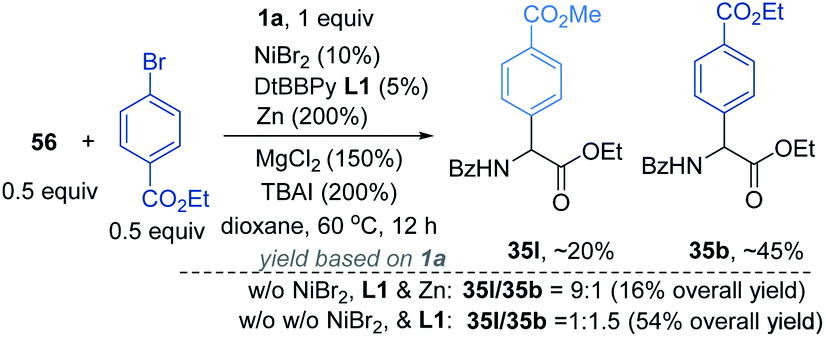
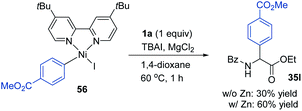
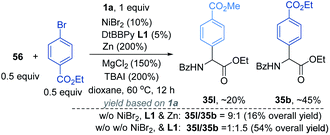
According to a well-established radical-chain mechanism, the putative Gly radical could be trapped by an Ar–Ni(II) species (eqn (6) and Scheme S4†).24 Thus, we performed stoichiometric reactions of the Ar1–NiII complex 56 with 1a with and without Zn (eqn (7)).19,25 The reactions resulted in 35l in 30% and 60% yields, respectively, suggesting that C(sp2)–NiII complexes might engage in the coupling event.24 However, in a deliberately designed competition reaction of 1a with an equimolar mixture of 55 and ethyl bromobenzoate (Ar2Br), 35b was formed in preference to 35l (2.3:1) (eqn (8)). Without Ni/L1, 35b remained the dominant product when Zn was used. The preferential formation of 35b should not be directed by addition of a Gly radical to the in situ formed Ar2–Ni(II), which would instead give 35I as the major product. Thus, a competing process that bypasses the aryl–NiII species may occur and be more favored. This idea was further confirmed by the reaction profiles shown in Fig. 3. The formation of 35b becomes much faster than that of 35l after an induction period, during which Ni(0) species is presumably generated.26 Likewise, the reaction cannot be dictated by Ar–Ni(I) that is most likely generated from single electron reduction of Ar–Ni(II).27,28 This would again warrant 35I as the major product, which opposes the observation in eqn (8).| | 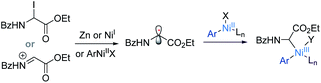 | (6) |
| |  | (7) |
| |  | (8) |
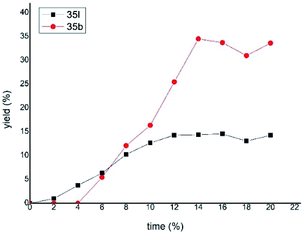 |
| | Fig. 3 The reaction profile for the formation of 35l and 35b in eqn (8) in the presence of Zn. Yield based on 1 equiv. of 1a. | |
The seemingly unusual observation in eqn (8) led us to consider the viability of addition of a Gly radical to Ni(0), which would generate a Gly–Ni(I) species.29 Subsequent oxidative addition of an aryl halide to Gly–Ni(I) would give the key Gly–Ni(III)–Ar intermediate (Scheme 1). DFT computations provide support for this mechanistic proposal (Scheme 1 and Fig. S4†).19 There is a large decrease of free energy of 66 kcal mol−1 as the Ni(0) catalyst binds the Gly radical carbon, forming the highly stable Gly–Ni(I)–Ln intermediate IM1 wherein the chelation of benzoyl oxygen to the Ni(I) center is prominent. Oxidative addition of bromobenzene to IM1 requires overcoming an energy barrier of 16.9 kcal mol−1 to give the Ni(III) intermediate IM2.
The subsequent reductive elimination proved to be rate-determining with an activation energy of 17.3 kcal mol−1. In comparison, DFT studies indicate that the radical-chain mechanism as in Scheme S4† was disfavored, because the precursor complex I5 to the oxidative addition of bromobenzene to Ln–Ni(0) would be much less stable than complex IM1 (Fig. 4).19 The Ni(0) catalyst would proceed to IM1, which is completely irreversible.
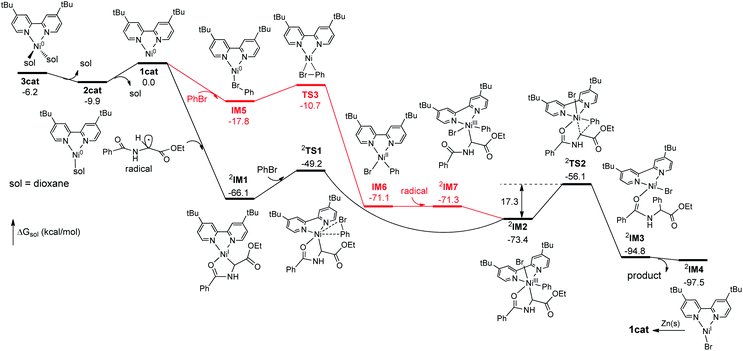 |
| | Fig. 4 Free energy profile comparing the favored Ni0 → NiI → NiIII → NiI pathway (black) and the Ni0 → NiII → NiIII → NiI pathway (red) with upper left superscripts indicating the spin states of open-shell, paramagnetic species. | |
Conclusions
In summary, we have demonstrated that α-pivaloyloxy glycine effectively coupled with vinyl and aryl halides/triflates to afford C(sp2)-functionalized α-amino acids under Ni-catalyzed reductive conditions. This method displays unique competency for incorporating hindered α- and tri-substituted vinyl moieties into the α-position of glycine, which is unattainable by previous methods. Mechanistically, a glycine α-carbon radical is thought to arise from the reduction of an in situ generated iminium or α-iodoglycine by Zn or a Ni(I) species, which then participates in the coupling process by addition to Ni(0) and Ar–Ni(II). DFT calculations showed that addition of the glycinyl carbon radical to Ln–Ni0 to give Ln–NiI–Gly followed by oxidative addition to ArX is possibly a more favored process. The auxiliary chelation of N-carbonyl to the Ni center appears to play a profound role in stabilizing the Ni intermediate, which may become crucial in developing an asymmetric version of this amino-acid forming event, which is ongoing in our laboratory and will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support was provided by the National Natural Science Foundation of China (No. 21871173). We are grateful to Dr Deli Sun for preparation of compound 54.
Notes and references
- For reviews on cross-electrophile couplings, see:
(a) C. E. I. Knappke, S. Grupe, D. Gärtner, M. Corpet, C. Gosmini and A. J. Wangelin, Chem.–Eur. J., 2014, 20, 6828–6842 CrossRef CAS;
(b) T. Moragas, A. Correa and R. Martin, Chem.–Eur. J., 2014, 20, 8242–8252 CrossRef CAS;
(c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767–1775 CrossRef CAS ; 43–72;
(d) J. Gu, X. Wang, W. Xue and H. Gong, Org. Chem. Front., 2015, 3, 1411–1421 RSC;
(e) J. Liu, Y. Ye, J. L. Sessler and H. Gong, Acc. Chem. Res., 2020, 53, 1833–1845 CrossRef CAS.
- The involvement of glycine α-carbon radicals has also been proposed in the recent photo-induced or oxidative C–H bond radical fragmentation of glycine derivatives, wherein electron-rich substituents on the amino moieties are curial, see: comment: The generation and utility of glycine α-carbon radicals have also been proposed in the recent photo-induced or oxidative dehydride of the C–H bonds of glycine derivatives, wherein electron-rich substituents on the amine are crucial, see:
(a) C. Wang, M. Guo, R. Qi, Q. Shang, Q. Liu, S. Wang, L. Zhao, R. Wang and Z. Xu, Angew. Chem., Int. Ed., 2018, 57, 15841–15846 CrossRef CAS;
(b) Y. Matsumoto, J. Sawamura, Y. Murata, T. Nishikata, R. Yazaki and T. Ohshima, J. Am. Chem. Soc., 2020, 142, 8498–8505 CrossRef CAS.
- For a Petasis borono–Mannich reaction, see:
(a) Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770–13773 CrossRef CAS ; for a Rh-catalyzed hydrogen-mediated reductive coupling of conjugated alkynes with ethyl (N-sulfinyl)iminoacetates, see:;
(b) J.-R. Kong, C.-W. Cho and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 11269–11276 CrossRef CAS.
- For an example of addition of alkyl radicals to sulfinyl iminoesters, see:
(a) S. Ni, A. F. Garrido-Castro, R. R. Merchant, J. N. de Gruyter, D. C. Schmitt, J. J. Mousseau, G. M. Gallego, S. Yang, M. R. Collins, J. X. Qiao, K.-S. Yeung, D. R. Langley, M. A. Poss, P. M. Scola, T. Qin and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 14560–14565 CrossRef CAS ; for an example of addition of alkyl radicals to imines, see:;
(b) J. Yi, S. O. Badir, R. Alam and G. A. Molander, Org. Lett., 2019, 21, 4853–4858 CrossRef CAS.
- For a selected review on protein engineering, see: K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS.
- R. Liu and G. A. Hudalla, Molecules, 2019, 24, 1450 CrossRef CAS.
-
(a) Y. Mehellou, H. S. Rattan and J. Balzarini, J. Med. Chem., 2018, 61, 2211–2226 CrossRef CAS;
(b) A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS.
- F. Mori, A. Cianferoni, S. Barni, N. Pucci, M. E. Rossi and E. Novembre, J. Allergy Clin. Immunol., 2015, 3, 375–380 CrossRef.
- H. Morishima, J. Yoshizawa, R. Ushijima, T. Takeuchi and H. Umezawa, J. Antibiot., 1982, 35, 1500–1506 CrossRef CAS.
- Radiosumin: H. Noguchi, T. Aoyama and T. Shioiri, Tetrahedron Lett., 1997, 38, 2883–2886 CrossRef CAS.
- U. Sahm, G. Knobloch and F. Wagner, J. Antibiot., 1973, 26, 389–390 CrossRef CAS.
- M. Timmermans, T. Randoux, D. Daloze, J. C. Braekman, J. M. Pasteels and L. Lesage, Biochem. Syst. Ecol., 1992, 20, 343–349 CrossRef CAS.
- For bioactive alpha-styryl aminoacids, see: S. J. Friedman, D. E. Frankhouser, R. A. MacDonald and G. D. Street, US Pat., 4346110, 1982.
-
(a) N. Sakai, H. Hori, Y. Yoshida, T. Konakahara and Y. Ogiwara, Tetrahedron, 2015, 71, 4722–4729 CrossRef CAS;
(b) N. Sakai, J. Asano, Y. Kawada and T. Konakahara, Eur. J. Org. Chem., 2009, 917–922 CrossRef CAS;
(c) R. Frauenlob, C. García, G. A. Bradshaw, H. M. Burke and E. Bergin, J. Org. Chem., 2012, 77, 4445–4449 CrossRef CAS;
(d) M. A. Beenen, D. J. Weix and J. A. Ellman, J. Am. Chem. Soc., 2006, 128, 6304–6305 CrossRef CAS;
(e) C. Haurena, E. Le Gall, S. Sengmany, T. Martens and M. Troupel, J. Org. Chem., 2010, 75, 2645–2650 CrossRef CAS.
- Friedel–Crafts:
(a) C. F. Bigge, J. T. Drummond, G. Johnson, T. Malone, A. W. Probert Jr, F. W. Marcoux, L. L. Coughenour and L. J. Brahce, J. Med. Chem., 1989, 32, 1580–1590 CrossRef CAS;
(b) J. Halli and G. Manolikakes, Eur. J. Org. Chem., 2013, 7471–7475 CrossRef CAS.
-
(a) G. Dyker, Angew. Chem., Int. Ed. Engl., 1997, 36, 1700–1702 CrossRef CAS ; Petasis borono–Mannich reactions are known as three-component reactions of amine, glyoxylic acid and organoboron reagents wherein coordination of the carboxylate group and boron is key:;
(b) N. R. Candeias, F. Montalbano, P. M. S. D. Cal and P. M. P. Gois, Chem. Rev., 2010, 110, 6169–6193 CrossRef CAS.
-
(a) Z. He and A. K. Yudin, J. Am. Chem. Soc., 2011, 133, 13770–13773 CrossRef CAS;
(b) J.-R. Kong, C.-W. Cho and M. J. Krische, J. Am. Chem. Soc., 2005, 127, 11269–11276 CrossRef CAS.
-
(a) H. Huo, B. J. Gorsline and G. C. Fu, Science, 2020, 367, 559–564 CrossRef CAS;
(b) X. Wei, W. Shu, A. García-Domínguez, E. Merino and C. Nevado, J. Am. Chem. Soc., 2020, 142, 13515–13522 CrossRef CAS.
- See the ESI for details.†.
- The coupling of pure (Z)-2a with 1a under the standard reaction conditions led to a trace amount of 3a along with dimerization and a substantial amount of unknown products that may arise from oligomerization of styrene via hydrodebromination of (Z)-2a, suggesting that (Z)-vinyl bromides are not suitable for the coupling event.
- For examples of coupling of activated benzyl C–OPiv via oxidative addition to Ni(0), see:
(a) Q. Zhou, H. D. Srinivas, S. Dasgupta and M. P. Watson, J. Am. Chem. Soc., 2013, 135, 3307–3310 CrossRef CAS;
(b) E. J. Tollefson, L. E. Hanna and E. R. Jarvo, Acc. Chem. Res., 2015, 48, 2344–2353 CrossRef CAS.
- Our previous studies indicate that the necessity of MgCl2 in the catalytic process was attributed to activating Zn and generating a more stable Ni–Cl bond by halide exchange.
- The redox potentials of the 2,2′:6′,2′′-terpyridine–Ni(I)–Me complex (−1.32 V) and 4,4′,4′′-tri-tert-butyl-2,2′:6′,2′′-terpyridine–Ni(I)–Me (−1.44 V) have been reported (both vs. Ag/Ag+ in THF solution):
(a) G. D. Jones, C. McFarland, T. J. Anderson and D. A. Vicic, Chem. Commun., 2005, 4211–4213 RSC ; for additional discussion of a discrete L1–Ni(I) complex that can react with Ph3CCl via an outer-sphere halide abstraction process to afford Ph3C radical, see:;
(b) M. M. Beromi, G. W. Brudvig, N. Hazari, H. M. C. Lant and B. Q. Mercado, Angew. Chem., Int. Ed., 2019, 58, 6094–6098 CrossRef.
- Other than by Ni(I), abstraction of halides by ArNi(II)X to generate an alkyl radical is possible. S. Biswas and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 16192–16197 CrossRef CAS.
- For details of the reaction profile including formation of Ar1I in the presence of Zn, see Schemes S1 and S2 and the discussions in the ESI.†.
- Reduction of Ni(II) to Ln–Ni(0) by Zn can be significantly accelerated by MgCl2:
(a) S. Ni, N. M. Padial, C. Kingston, J. C. Vantourout and P. S. Baran,
et al.
, J. Am. Chem. Soc., 2019, 141, 6726–6739 CrossRef CAS;
(b) C. Zhao, X. Jiao, X. Wang and H. Gong, J. Am. Chem. Soc., 2014, 136, 17645–17651 CrossRef CAS.
- In the Ni-catalyzed reductive coupling of aryl halides with imines, addition of aryl–NiI to an imine was proposed by Doyle. The radical nature of the alkyl group also led us to conclude that such a mechanism may not operate in the present amino acid forming process. C. Heinz, J. P. Lutz, E. M. Simmons, M. M. Miller, W. R. Ewing and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 2292–2300 CrossRef CAS.
- Generation of Ar–Ni(I) by single electron reduction of Ar–Ni(II) has been reported, see: Q. Lin and T. Diao, Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes, J. Am. Chem. Soc., 2019, 141, 17937–17948 CrossRef CAS.
- O. Gutierrez, C. Tellis, J. D. N. Primer, G. A. Molander and M. C. Kozlowski, J. Am. Chem. Soc., 2015, 137, 4896–4899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc05452f |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc and
Hegui
Gong
*bc and
Hegui
Gong