
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alternating polarity for enhanced electrochemical synthesis†
Christiane
Schotten
a,
Connor J.
Taylor
a,
Richard A.
Bourne
b,
Thomas W.
Chamberlain
 a,
Bao N.
Nguyen
a,
Nik
Kapur
*c and
Charlotte E.
Willans
*a
a,
Bao N.
Nguyen
a,
Nik
Kapur
*c and
Charlotte E.
Willans
*a
aSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK. E-mail: C.E.Willans@leeds.ac.uk
bSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
cSchool of Mechanical Engineering, University of Leeds, Leeds LS2 9JT, UK. E-mail: N.Kapur@leeds.ac.uk
First published on 29th October 2020
Abstract
Synthetic electrochemistry has recently become an exciting technology for chemical synthesis. The majority of reported syntheses use either constant current or constant potential, however a few use non-linear profiles – mostly alternating polarity – to maintain efficiency throughout the process, such as controlling deposits on electrodes or ensuring even use of electrodes. However, even though parameters that are associated with such profiles, such as the frequency, can have a major impact on the reaction outcome, they are often not investigated. Herein, we report the crucial impact that the applied frequency of the alternating polarity has on the observed reaction rate of Cu(I)–NHC complex formation and demonstrate that this can be manipulated to give enhanced yield that is stable over extended reaction times.
Introduction
Electrochemistry has recently attracted renewed interest as a sustainable technology for synthetic chemistry due to its mild, selective and atom efficient nature.1 The increasing number of continuous electrochemical reactors developed has further stimulated the application of electrochemistry as a synthetic method.2The use of non-constant current and potential methods, such as sinusoidal and square wave profiles, are of interest (see ESI† for explanation of each type) and are extensively employed in other electrochemistry areas such as electroplating and electroanalytics,3 though they remain an under-investigated parameter in electrochemically-driven synthetic chemical reactions. In electrosynthesis, non-constant methods are mostly used to maintain efficiency throughout the process, enabling the even use of both electrodes, removal of electrodeposited contaminants, e.g. via gas evolution, and regular renewal of the Nernst layer. In this manuscript we refer to alternating current as a sinusoidal curve and alternating polarity as a square wave profile (see ESI† for further details).
Alternating current was first used for the electrolysis of water by De la Rive in 1837, who found that it was most effective at low frequencies and current densities.4 Since then it has also been employed for organic syntheses such as the Kolbe reaction.5 Drechsel was the first to postulate that alternating current could be used to determine reaction rates, as the frequency determines whether the reverse reaction or a productive reaction occurs; however, only the scale of the reaction rate was determined.6 Others have investigated this approach but, in common with earlier work, these studies do not derive reaction rates or rate determining steps.7 There are only a small number of recent studies that use non-constant currents or potential, with the majority using it to maintain electrode efficiency by removing deposits on the electrode surface.8 However, accurate control of the waveform is important. For example, Reid and co-workers recently reported a significant change in selectivity towards a different product when applying alternating polarity but ultimately attributed this to the switching circuitry causing an asymmetry in the waveform, rather than an underlying mechanistic effect.8c Even fewer examples investigate the use of non-constant currents or potential further in terms of efficiency or selectivity and in relation to varying the frequency.9 Hibino and co-workers report high dependency of the applied frequency on the production of phenol from benzene under alternating current, which affects efficiency and selectivity.9a Modestino and co-workers applied a pulsed current in the synthesis of adiponitrile, addressing over-reduction by giving the product time to diffuse away from the electrode during pulses.9d Wessling and co-workers have investigated the use of alternating polarity in the synthesis of Metal Organic Frameworks (MOFs) from a process technology point of view, primarily to avoid passivation of the electrodes.9c Luo and co-workers demonstrated the advantage of alternating polarity in the electrochemical trifluoromethylation of arenes to avoid degradation of unstable intermediates, which increased yield from 13 to 84%.9e
We have recently demonstrated the electrochemical synthesis of metal-N-heterocyclic carbene (NHC) complexes under both batch and continuous conditions (Fig. 1), where the electrode material acts as the metal source and hydrogen gas is the only by-product.10 This avoids the use of strong bases and metal precursors traditionally required for their synthesis. The substrate, an imidazolium salt, also acts as the electrolyte, allowing for a very direct and clean synthesis, where reaction mixtures can be used directly for catalysis. The electrochemical method therefore results in milder reaction conditions and improved substrate tolerance, higher atom efficiency, and simplified downstream processing. The continuous electrochemical setup allows for higher throughput and higher efficiency due to the small interelectrode gap.
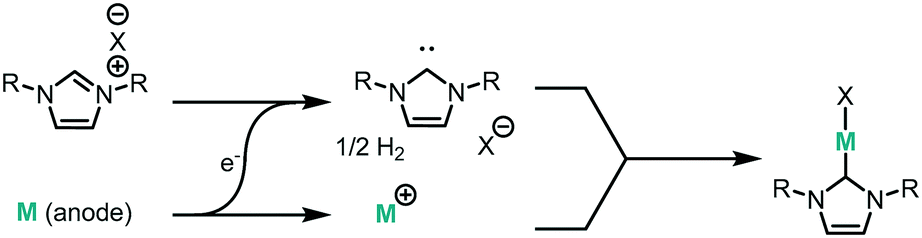 | ||
| Fig. 1 Electrochemical synthesis of metal–NHC complexes. | ||
We envisaged the use of electrogenerated metal–NHC complexes for direct screening in catalysis and developed a scaled down version of our reactor (Fig. 2A). We evaluated its properties using copper electrodes and IMes·HCl as a model reaction to form IMes–Cu–Cl 1 and obtained full conversion at 1.8 V and a residence time of 27.3 min (Fig. 2B). However, when running the reaction for longer (>2 h), reproducibility proved to be difficult due to inconsistent behaviour and current spiking. In some cases, we observed particles being flushed out of the reactor. We attributed this to excess metal cations, i.e. metal that was oxidised from the anode and not used up in a metal-complex, being redeposited on the cathode, which can form dendrites over time. These dendrites can cause short circuiting and therefore decrease efficiency. We envisaged the use of alternating polarity to use both electrodes evenly. This is similar to reports by Wessling and co-workers, who observe that a conductive precipitate causes short circuiting in the synthesis of MOFs.9c In addition, we expected an improvement in reaction performance through alternating polarity due to altered mass transfer (Fig. 3). In addition to constantly renewing the Nernst layers and reducing concentration gradients, we also produce both intermediates, copper(I) and free NHC, on both electrodes, resulting in shorter diffusion pathways for them to meet and react. Normally, the NHC and the copper(I) are produced on opposite sides of the reactor channel on each electrode (red 1 and black 1, Fig. 3A) and must move towards each other to form the complex. When alternating polarity is employed, they are still formed on opposite electrodes, but after the polarity is switched the opposite product is formed on each electrode (red 2 and black 2, Fig. 3B), avoiding the need for diffusion across the channel from one electrode to the other. This is particularly important in laminar flow regimes, such the continuous flow conditions employed here, where substrate movement is diffusion dominated.11 However, if the frequency of the polarity switch is not optimised, the reverse reaction, in this case most likely the reduction of copper(I) to copper(0), will compete (black 2, Fig. 3C) reducing the efficiency of the reaction.
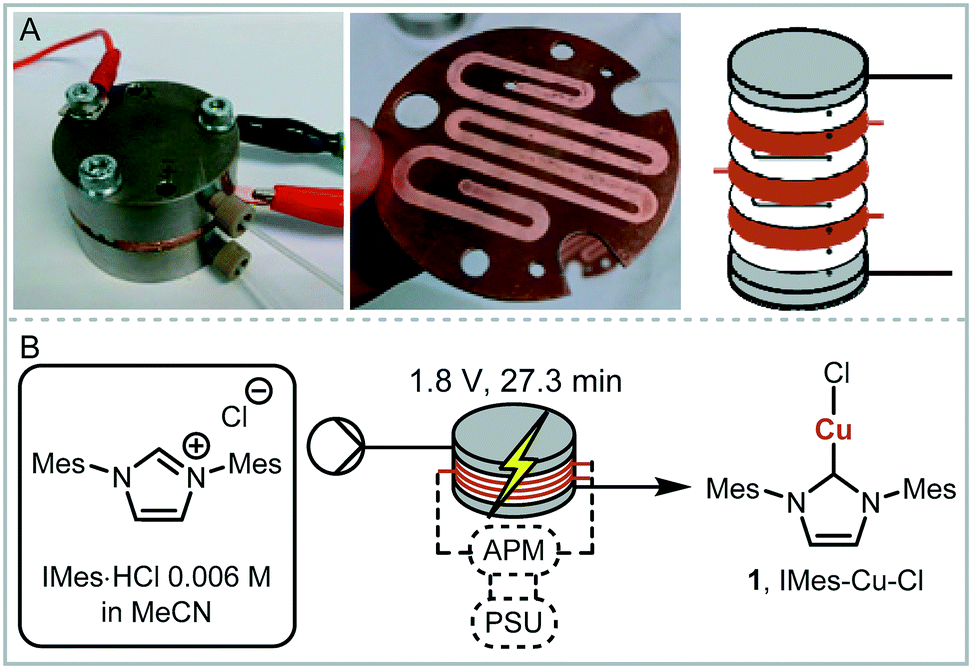 | ||
| Fig. 2 A: Scaled down electrochemical reactor; B: optimised model reaction with full conversion, APM: alternating polarity microcontroller, PSU: power supply unit. | ||
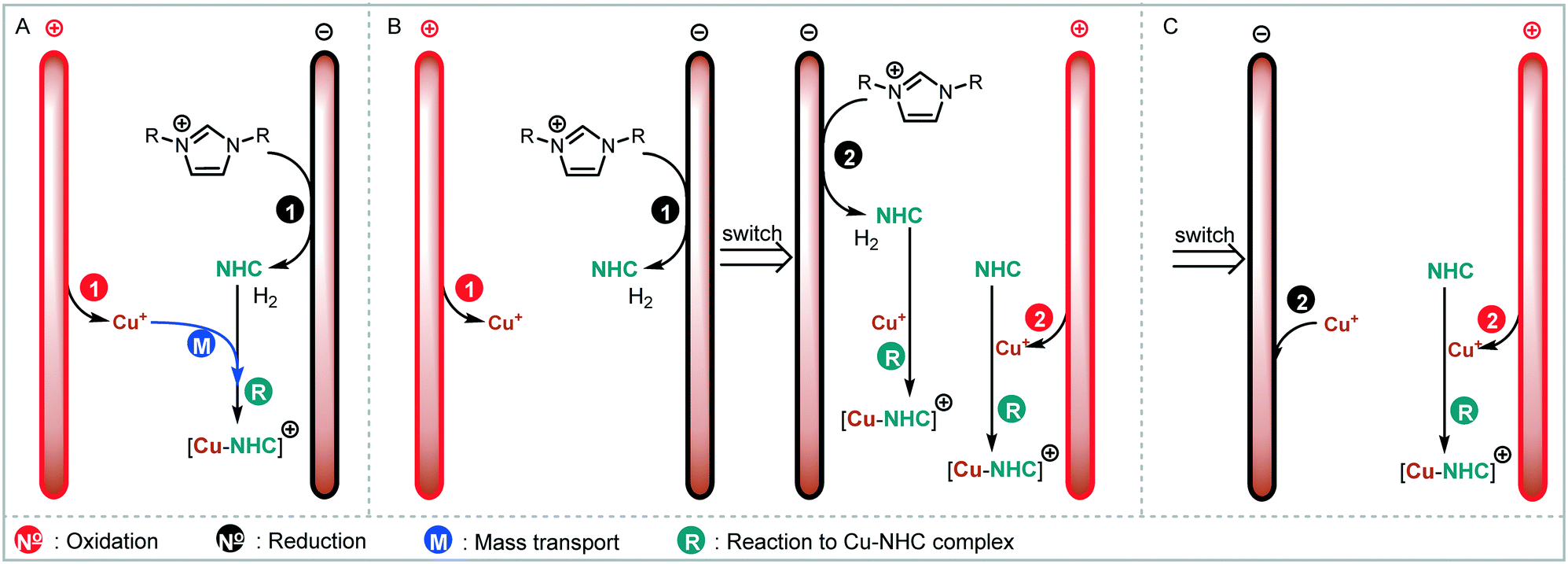 | ||
| Fig. 3 Considerations for altered mass transfer, A: normal reaction dependent on mass transport (blue M); B: switch avoids mass transport by forming intermediates on both electrodes; C: reverse reaction of copper cation possible. | ||
Herein, we demonstrate the impact of alternating polarity, applying a square wave profile of the potential over time, and explore the effect of the frequency of switching on the success of a model synthetic reaction. In addition, reaction rates for a range of frequencies have been calculated.
Results and discussion
As a model reaction the formation of IMes–Cu–Cl (1) from IMes·HCl and copper electrodes was investigated (Fig. 2B). A solution of the imidazolium salt in acetonitrile was pumped through the reactor using a syringe pump, and a range of voltages were screened at a 27.3 minute residence time. Under initial optimised conditions (Fig. 4A, orange, 1.8 V), the reaction was not reproducible, showing spikes in the current at varying time points (typically after 30 min to 3 h) and a significant decrease in conversions (one example shown in Fig. 4B, orange). In addition, copper particles were sometimes flushed out. In line with Wessling and co-workers, we envisaged that the use of alternating polarity would use the electrodes more evenly and avoid the build-up of dendrites.9c To alternate the polarity an Arduino MKRZero board with a mechanical relay, which is inexpensive and open source, was connected between the power supply and the reactor (see ESI† for further information). The time for each polarity and a pause can be programmed onto the APM. A square wave profile with equal times on each polarity, opposed to a sinusoidal profile, was selected for this study to ensure an accurate control over the potential (Fig. 4C). Frequencies given correspond to a full cycle as indicated by the red arrow (e.g. positive 2.5 s, negative 2.5 s gives a 5 s period and a 1/5 s−1 = 0.2 Hz frequency). The potential screen was repeated with a range of frequencies from 5 Hz to 1/60 Hz (Fig. 4A). At a high frequency (5 Hz, yellow, Fig. 4B) the overall conversion is significantly lower when compared to the case without the use of alternating polarity. This can be rationalised by assuming that the charged species in the reaction mixture, such as the imidazolium starting material, do not have time to migrate to either electrode prior to switching, and are essentially stagnant between the electrodes. In addition, copper(I) formed at the anode is still very close to the electrode and can be reduced back after the polarity switch, instead of forming the desired product (black 2 in Fig. 3C). At medium frequencies (1 Hz, grey, Fig. 4A) the reaction performs at similar levels to the reaction without alternating polarity. However, at low frequencies (1/60 Hz, 1 min period, blue, Fig. 4A) the reaction outperforms that without alternating polarity at low voltages and reaches full conversion at 1.8 V. Most importantly, the reaction is now stable over a long period of time without any loss in conversion over 7 h (Fig. 4B, blue). The experiment was repeated for 24 h using HPLC monitoring with no significant decrease in conversion or yield. Interestingly, after each change in polarity the current increased significantly up to ∼50 mA and then reduced exponentially over ∼30 s to an equilibrium value similar to that found without the use of alternating polarity of ∼0.9 mA. This is attributed to the renewal of the Nernst layers at the electrodes resulting in substrate being available in high concentrations, and therefore a peak in current, after a polarity switch. and therefore results in a peak in current. Discharging and then charging of the electrochemical double layer is typically observed to occur on much shorter timescales (ms) so can be neglected on the time scale used.3a,5a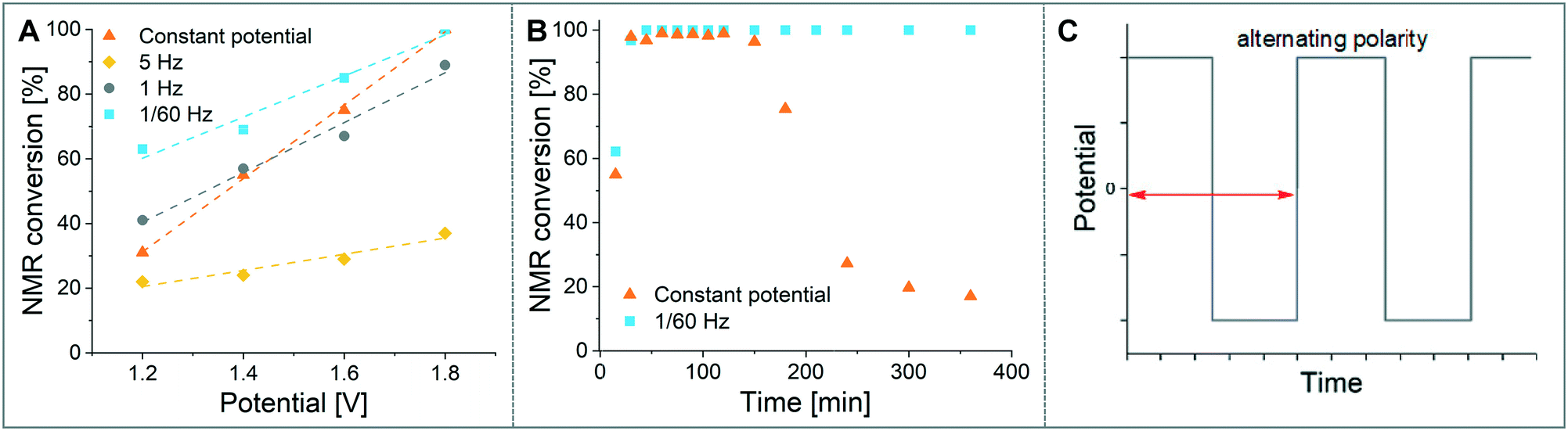 | ||
| Fig. 4 A: Single sweep voltammetry, 1H NMR conversions of IMes·HCl to IMes–Cu–Cl determined at different frequencies; B: long-term reaction stability, 1H NMR conversions of IMes·HCl to IMes–Cu–Cl; C: principle of alternating polarity, red arrow indicates one period. | ||
The influence of the frequency on the reaction performance was further investigated. Different frequencies were screened at both 1.2 V and 1.8 V from 5 Hz to 1/120 Hz (Fig. 5A). The reaction volume was halved for the frequency screen at 1.8 V to decrease the conversion so that changes in performance can be readily observed. Without alternating polarity at a constant potential (orange line) a conversion of 31% at 1.2 V was observed. The use of alternating polarity benefitted the reaction when the frequency was higher than 2.5 Hz. At very low frequencies (0.01 Hz, period 120 s) the reaction performed as if it were under a constant potential. The highest conversion (57%) was achieved at 5 Hz. At 1.8 V, introduction of alternating polarity shows a similar trend. For frequencies higher than 0.2 Hz the reaction performs worse compared to an 87% conversion at constant potential, whereas for lower frequencies the conversion is similar to those observed using constant potential. Generally, at high frequencies the reaction underperforms, at mid frequencies it reaches an optimum and at low frequencies the switching is too slow to affect the conversion. As the frequency influences the overall reaction rate, we set out to investigate the kinetics at different frequencies at 1.8 V, and determine the observed reaction rates with and without alternating polarity using the kinetic fitting software Compunetics.12 The precursor solution was pumped through the reactor at different flow rates and the conversion determined (Fig. 5B). After each run the reactor was disassembled and cleaned prior to screening the next frequency. With constant potential (orange), the reaction reaches full conversion at 15.5 minutes; however, as observed previously, the long-term stability of the reactor prevented further study. The reaction conversions at 1/60 Hz (light blue) are similar to those observed using a constant potential, but now shows improved stability. The kinetic profile was repeated with half the reactor volume and double the flow rates to determine the influence of mixing (dark blue with asterisk). As both curves overlap, it appears that mixing due to changes in flow rate at this scale do not affect the outcome of the reaction significantly. When increasing the frequency to 1 Hz and 5 Hz (grey and yellow), the reaction performs much worse, plateauing at about 60% and 50%, respectively. These trends are also visible in the observed reaction rates (values and errors in Table S7 in ESI†). The observed reaction rates with constant potential and with switching at 1/60 Hz are comparable within error, though the rates are slightly lower with the APM. These results indicate that either the switch in polarity does not have a large effect on the mass transfer of the reaction or that the reaction is not mass transfer limited. This is probably due to using a charged starting material that migrates to the electrode and is not only dependent on diffusion and convection alone. Instead, the use of alternating polarity gives slightly lower observed reaction rates, possibly due to a small amount of copper(I) being reduced back to copper(0). The reactions with higher frequencies show significantly reduced observed reaction rates suggesting that the reaction progress is limited by charged species being stagnant in solution and intermediates not having the time to move away from the electrodes before doing the reverse reaction. In addition, reactions at higher frequencies suffer from reproducibility issues, while the yield at 1/60 Hz typically shows an error under 5%. Without further optimisation the reaction conditions were applied to a range of other imidazolium salts and produced the copper–NHC complexes in quantitative yields, including Cu4 which we found to be inaccessible via traditional chemical routes (Fig. 5C).
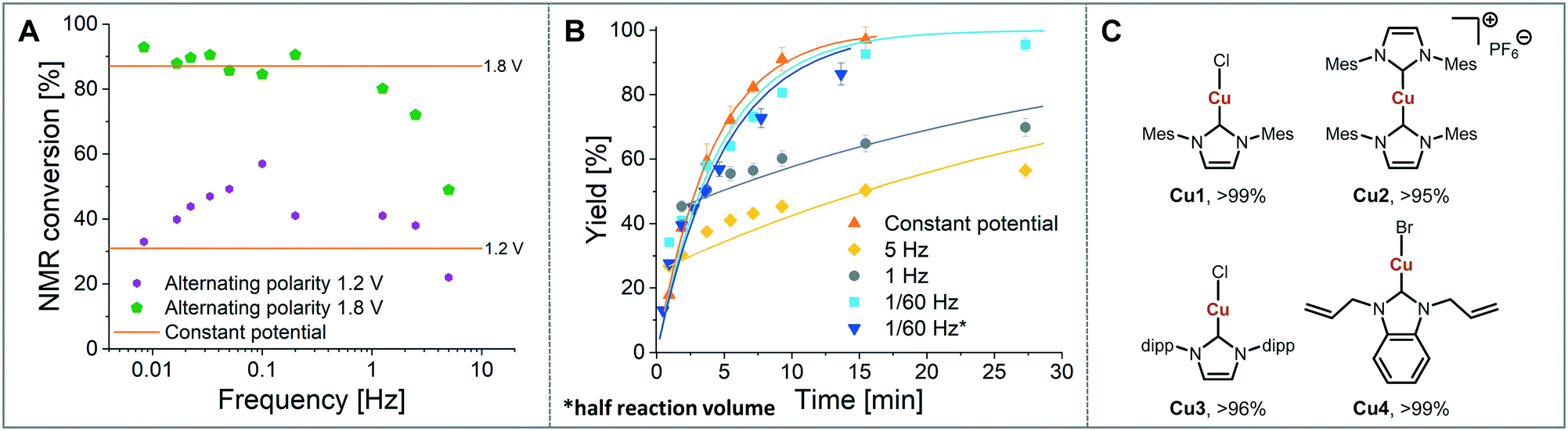 | ||
| Fig. 5 A: Frequency screen at 1.2 V and 1.8 V with orange line for constant potential, 1H NMR conversions of IMes·HCl to IMes–Cu–Cl for each frequency; B: kinetic profile with points for experimental data and lines for fitting, HPLC yield at each residence time, HPLC yield was calculated from comparing peaks against an internal standard, 1,3,5-trimethoxybenzene; C: substrate scope with NMR conversions. 1H NMR conversions were determined by comparing product and starting material peaks. | ||
Conclusions
These results highlight that the use of alternating polarity parameters can significantly enhance the performance of the reaction in terms of yield and long-term stability. Operated under an optimum frequency, alternating polarity increases the stability of the continuous synthesis of Cu(I)–NHC complexes studied here, through the even use of both surfaces and avoiding the build-up of metal dendrites that cause short-circuiting. However, the choice of frequency is crucial and high frequencies appear to promote the reverse reaction (for the exemplar reactions studied here Cu(I) to Cu(0)), resulting in lower yields and observed reaction rates. A frequency of 1/60 Hz proved to be optimal in our system, resulting in an observed reaction rate of 3.6 (±0.3) × 10−3 s−1 and proving capable of operating as a flow process with long-term stability that was not seen when run without alternating potential.Since the underlying electrochemical mechanisms are common amongst many synthetic reactions including those studied here, these findings are potentially widely applicable. Consequently, it is important to consider alternating polarity and its frequency as a reaction parameter that requires careful consideration in electrochemical synthesis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from EPSRC (EP/R009406/1).References
- (a) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020 10.1039/d0sc01848a; (b) C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC; (c) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS; (d) P. Gandeepan, L. H. Finger, T. H. Meyer and L. Ackermann, Chem. Soc. Rev., 2020, 49, 4254–4272 RSC.
- (a) J.-I. Yoshida, Electrochem. Soc. Interface, 2009, 18, 40–45 CAS; (b) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS; (c) M. Atobe, H. Tateno and Y. Matsumura, Chem. Rev., 2018, 118, 4541–4572 CrossRef CAS; (d) K. Mitsudo, Y. Kurimoto, K. Yoshioka and S. Suga, Chem. Rev., 2018, 118, 5985–5999 CrossRef CAS; (e) S. Maljuric, W. Jud, C. O. Kappe and D. Cantillo, J. Flow Chem., 2020, 10, 181–190 CrossRef CAS; (f) A. A. Folgueiras-Amador and T. Wirth, in Flow Chemistry: Integrated Approaches for Practical Applications, The Royal Society of Chemistry, 2020, pp. 153–198, 10.1039/9781788016094-00153.
- (a) M. S. Chandrasekar and M. Pushpavanam, Electrochim. Acta, 2008, 53, 3313–3322 CrossRef CAS; (b) C. Larson and J. P. G. Farr, Trans. Inst. Met. Finish., 2012, 90, 20–29 CrossRef CAS; (c) H. Adamson, A. M. Bond and A. Parkin, Chem. Commun., 2017, 53, 9519–9533 RSC; (d) L.-L. Henry, R. Martin, H. Alice, R. F. Jack, Z. Jie, B. Alan, P. Alison, G. David, Y. Nick and E. Darell, ChemRxiv. Preprint, 2020, DOI:10.26434/chemrxiv.12919367.v1; (e) T. D. Hall, M. E. Inman and E. J. Taylor, Electrochem. Soc. Interface, 2020, 29, 49–54 CrossRef CAS.
- (a) A. De La Rive, Compt. Rend., 1837, 4, 835 Search PubMed; (b) A. De La Rive, Compt. Rend., 1837, 4, 909 Search PubMed.
- (a) C. L. Wilson and W. T. Lippincott, J. Electrochem. Soc., 1956, 103, 672–675 CrossRef CAS; (b) M. Fleischmann, J. R. Mansfield, H. R. Thirsk, H. G. E. Wilson and L. Wynne-Jones, Electrochim. Acta, 1967, 12, 967–982 CrossRef CAS.
- (a) E. Drechsel, J. Prakt. Chem., 1880, 22, 476–488 CrossRef; (b) E. Drechsel, J. Prakt. Chem., 1884, 29, 229–252 CrossRef; (c) E. Drechsel, J. Prakt. Chem., 1888, 38, 75–77 CrossRef.
- (a) P. S. Fedkiw and W. D. Scott, J. Electrochem. Soc., 1984, 131, 1304–1315 CrossRef CAS; (b) M. Le Blanc and K. Schick, Z. Physiol. Chem., 1903, 46U, 213 Search PubMed; (c) A. Brochet and J. Petit, Z. Elektrochem. Angew. Phys. Chem., 1904, 10, 909–922 CrossRef CAS; (d) A. Brochet and J. Petit, Z. Elektrochem. Angew. Phys. Chem., 1905, 11, 441–453 CrossRef.
- (a) Y. Takahira, M. Chen, Y. Kawamata, P. Mykhailiuk, H. Nakamura, B. K. Peters, S. H. Reisberg, C. Li, L. Chen, T. Hoshikawa, T. Shibuguchi and P. S. Baran, Synlett, 2019, 30, 1178–1182 CrossRef CAS; (b) P. L. Norcott, C. L. Hammill, B. B. Noble, J. C. Robertson, A. Olding, A. C. Bissember and M. L. Coote, J. Am. Chem. Soc., 2019, 141, 15450–15455 CrossRef CAS; (c) A. G. Wills, D. L. Poole, C. M. Alder and M. Reid, ChemElectroChem, 2020, 7, 2771–2776 CrossRef CAS.
- (a) B. Lee, H. Naito, M. Nagao and T. Hibino, Angew. Chem., Int. Ed., 2012, 51, 6961–6965 CrossRef CAS; (b) C. W. Lee, N. H. Cho, K. T. Nam, Y. J. Hwang and B. K. Min, Nat. Commun., 2019, 10, 1–8 CrossRef; (c) J. Vehrenberg, M. Vepsäläinen, D. S. Macedo, M. Rubio-Martinez, N. A. S. Webster and M. Wessling, Microporous Mesoporous Mater., 2020, 303, 110218 CrossRef CAS; (d) D. E. Blanco, B. Lee and M. A. Modestino, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17683–17689 CrossRef CAS; (e) S. Rodrigo, C. Um, J. C. Mixdorf, D. Gunasekera, H. M. Nguyen and L. Luo, Org. Lett., 2020, 22(17), 6719–6723 CrossRef CAS.
- (a) B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little and C. E. Willans, Chem. Commun., 2012, 48, 4887–4889 RSC; (b) M. R. Chapman, Y. M. Shafi, N. Kapur, B. N. Nguyen and C. E. Willans, Chem. Commun., 2015, 51, 1282–1284 RSC; (c) M. R. Chapman, S. E. Henkelis, N. Kapur, B. N. Nguyen and C. E. Willans, ChemistryOpen, 2016, 5, 351–356 CrossRef CAS; (d) H. R. Stephen, C. Schotten, T. P. Nicholls, M. Woodward, R. A. Bourne, N. Kapur and C. E. Willans, Org. Process Res. Dev., 2020, 24, 1084–1089 CrossRef CAS; (e) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS; (f) T. P. Nicholls, C. Schotten and C. E. Willans, Curr. Opin. Green Sustain. Chem., 2020, 26, 100355 CrossRef.
- F. Amemiya, D. Horii, T. Fuchigami and M. Atobe, J. Electrochem. Soc., 2008, 155, E162–E165 CrossRef CAS.
- Compunetics, http://www.compunetics.net/, (accessed 22/06/2020).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00399a |
| This journal is © The Royal Society of Chemistry 2021 |