
Improved batch and flow syntheses of the nonsteroidal anti-inflammatory COX-2 inhibitor celecoxib†
Chantal
Scholtz
 and
Darren L.
Riley‡
*
and
Darren L.
Riley‡
*
Department of Chemistry, University of Pretoria, Pretoria, 0002, South Africa. E-mail: darren.riley@up.ac.za
First published on 20th October 2020
Abstract
The comparison of an improved conventional batch mode synthesis of the nonsteroidal anti-inflammatory COX-2 inhibitor celecoxib with its flow chemistry alternative is reported. The stepwise and continuous flow synthesis of celecoxib was achieved by means of a Claisen condensation to access 4,4,4-trifluoro-1-(4-methyl-phenyl)-butane-1,3-dione followed by a cyclo-condensation reaction with 4-sulfamidophenylhydrazine hydrochloride to obtain the pyrazole moiety. A batch process was developed with improved work-up and purification (90% yield). This was successfully translated to flow in yields of 90–96% with greatly shortened reaction times (20 h vs. 1 h) and reduced chemical exposure.
Introduction
Celecoxib [1] is a nonsteroidal anti-inflammatory drug (NSAID) that is used in the treatment of arthritis and is often prescribed for acute pain.1 It has also been shown to possess anti-cancer properties.2–4 Typically NSAIDs work by inhibition of the cyclooxygenase (COX) enzymes,5 which are involved in arachidonic acid metabolism leading to the synthesis of thromboxanes6 and prostaglandins (PGs) – the major mediators of pain.2,6 Conventional NSAIDs inhibit both COX-1 and COX-2, with COX-1 being inhibited to a greater extent, leading to undesirable side effects such as gastrointestinal bleeding and ulcers with long term use.2,4,5 The selective inhibition of COX-2, however, results in anti-inflammatory effects without the undesirable gastrotoxic side effects.3 Two major classes of compounds have been developed as selective COX-2 inhibitors; the “coxibs” of which celecoxib [1] is a classic example and the methanesulfonamide type compounds such as nimesulide [2] (Fig. 1).1,2,7 | ||
| Fig. 1 Structures of celecoxib [1] and nimesulide [2]. | ||
Celecoxib (1) was first prepared by Penning and co-workers8 by means of a Claisen condensation between 4-methylacetophenone (4-MAP, 3) and ethyl trifluoroacetate (ETFA, 4) in methanol to obtain dione [5] followed by a cyclo-condensation reaction with (4-sulfamoylphenyl)hydrazine hydrochloride [6] to obtain celecoxib [1] in a yield of 46% (Scheme 1). The same group determined that using the hydrochloride salt of the hydrazine in the cyclo-condensation reaction resulted in the regioselective formation of the 1,5-diarylpyrazole. This approach is still used industrially,9 however, several optimizations primarily focusing on the use of more appropriate solvents for both the Claisen, the cyclo-condensation as well as improved work-up and product isolation have resulted in more acceptable yields in the range of 73–89%.10–14 In addition, several alternative approaches using different synthetic routes have been reported with yields in the range of 35–80%.9,15–18
 | ||
| Scheme 1 Claisen condensation and cyclo-condensation synthesis route for celecoxib [1]. | ||
Currently, our research group has interests in the utilization of modern manufacturing technologies such as flow chemistry to improved process routes to pharmaceuticals. Regionally, in South Africa, where we are based, celecoxib [1] represents an attractive target for manufacturing as high costs associated with the drug mean that it is currently only available through the private sector.19a Furthermore, due to an imbalanced two-tier market structure in South Africa this translates into only 16% of the total population having access to the drug,19a despite it currently being one of the safest options available for the treatment of inflammatory diseases and pain.19b
We believed that the original process reported by Penning and co-workers,8 was well suited for flow translation and as such we embarked on developing an improved flow-based synthesis of celecoxib [1]. The synthesis of structurally related pyrazoles have previously been reported under flow conditions,20–27 with the Ley group recently having reported a four-step flow synthesis of several pyrazoles including the first flow synthesis of celecoxib [1] in 48% yield via a metal-free amine-redox process.27
Herein, we describe an improved batch and continuous flow processes for the synthesis of celecoxib [1], which could potentially be modified to access a range of pyrazoles. We demonstrate an efficient Claisen condensation to obtain the required 1,3-dicarbonyl adduct followed by cyclo-condensation with the appropriate hydrazine across two steps and we evaluate the advantages of the flow synthesis when compared to the conventional batch processes.
Results and discussion
The preferred synthetic route to access celecoxib [1] (Scheme 1) involved a Claisen condensation between 4-methylacetophenone [3] and ethyl trifluoroacetate [4] to form dione [5] (stage 1). This was followed by a cyclo-condensation with 4-sulfamidophenylhydrazine hydrochloride [6] to obtain celecoxib [1] (stage 2). Batch optimizations were performed to both assess the suitability of the reactions for flow translation as well as investigate if there were any improvements to be made to the reported protocols.Batch optimization of stage 1
The majority of published procedures report heating 4-MAP [3] and ETFA [4] in the presence of a base at about 80 °C for a period of 10–20 hours resulting in yields ranging from 40% pure to 95% crude.8,28 It was decided that a more consistent first stage would be beneficial to the overall synthesis, thus the reaction was optimized in terms of solvent, temperature, duration and concentration. The work-up procedures were also critically evaluated as this appeared to be a phase where product loss occurred. Initially a basic solvent screen using low molecular weight protic solvents was performed, with the base source derived from the in situ generation of the corresponding sodium salts (Table 1). The selection of low molecular weight protic solvents was driven by their routine use in Claisen condensations and their ease of use and storage on industrial scale. In addition, their compatibility with the second stage of the process was also a deciding factor as it would negate the need for a solvent swop between stages when translating the process to flow. Furthermore, the inherent greenness of these solvents was also seen as being attractive.| Solvent | Temperature (°C) | Yield (%) | |
|---|---|---|---|
| Standard conditions: 4-MAP [3] (1.28 M), ETFA [4] (1.1 equiv.), Na (1.6 equiv.), 16 h. a Reaction abandoned due to extensive time taken to form the sodium salt of iso-propanol. b Purification by trituration. Isolated yields reported. | |||
| 1 | MeOH | 80 | 15 |
| 2 | EtOH | 80 | 5 |
| 3 | i-PrOHa | 80 | — |
| 4 | MeOHb | 80 | 72 |
The Claisen condensation proceeded satisfactorily as monitored by TLC, however, the isolated yields (entries 1 and 2, Table 1) were disappointingly low. It was speculated that the poor yields obtained were a result of the work-up and purification protocols adapted from literature.8,28 An improved and simplified protocol was developed which involved the simple removal of the protic solvent in vacuo followed by trituration of the resulting solid residue in hexane. This afforded dione [5] as a fine, light brown solid which could be isolated by vacuum filtration. When performed in methanol, the modified work-up and purification allowed the isolation of [5] in 72% yield (entry 4, Table 1).
A temperature screen was then performed using methanol as solvent. When utilizing the new work-up and purification protocol, dione [5] was isolated in quantitative yield in the range of 25–50 °C, above which temperature, the yield began to steadily decrease (Fig. 2). The process was rescreened in absolute ethanol at room temperature affording a lower yield of 83%, although with a notable reduction in reaction time (17 h vs. 5.5 h). The improved rate of reaction and lower toxicity profile of ethanol when compared to methanol prompted us to proceed with the batch optimizations using absolute ethanol despite the slightly lower yield.
 | ||
| Fig. 2 Study of the effect of reaction temperature on formation of [5]. Standard conditions: 4-MAP [3] (1.28 M), ETFA [4] (1.1 equiv.), Na (1.6 equiv.), MeOH, 16 h. Isolated yields reported. | ||
Thereafter, an assessment of the reaction time in ethanol (Fig. 3) showed an almost quantitative yield (97%) for [5] after 3 hours reaction time. Notably, the isolated yields then steadily decreased as the reaction time increased ultimately affording only a 5% isolated yield when left to react for 24 h.
 | ||
| Fig. 3 Study of the effect of reaction duration on formation of [5]. Standard conditions: 4-MAP [3] (1.28 M), ETFA [4] (1.1 equiv.), Na (1.6 equiv.), EtOH, RT. Isolated yields reported. | ||
Finally, in a lead up to translating the reaction to flow an assessment was performed to determine the lowest stoichiometric amount of base that could be used (entries 1–4, Table 2) and the highest concentration that the reaction could be performed at relative to 4-MAP (3) (entries 5–7, Table 2).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Equiv. 4-MAP [3] | Equiv. ETFA [4] | Equiv. NaOEt | [4-MAP] (M) | Yield (%) |
| Standard conditions: 1.0 equiv. 4-MAP [3] [0.71–0.94 M], 1.1 equiv. [4], 1–1–1.6 equiv. NaOEt, EtOH, RT, 3 h. Isolated yields reported. | |||||
| 1 | 1.0 | 1.1 | 1.6 | 0.71 | 46 |
| 2 | 1.0 | 1.1 | 1.4 | 0.71 | 100 |
| 3 | 1.0 | 1.1 | 1.3 | 0.71 | 80 |
| 4 | 1.0 | 1.1 | 1.1 | 0.71 | 78 |
| 5 | 1.0 | 1.1 | 1.4 | 0.76 | 100 |
| 6 | 1.0 | 1.1 | 1.4 | 0.85 | 98 |
| 7 | 1.0 | 1.1 | 1.4 | 0.94 | 97 |
The lowest stoichiometric amount of base that could be used without compromising the yield was found to be 1.4 equivalents relative to 4-MAP [3] (entry 2, Table 2). This information was then used to determine the concentration of 4-MAP [3] at which the reaction could be performed without reduced yields and loss of homogeneity.
The final optimized batch conditions for the Claisen condensation involved a reaction time of 3 hours at 25 °C using 1.0 equiv. 4-MAP [3], 1.1 equiv. ETFA [4] and 1.4 equiv. NaOEt in absolute ethanol at a concentration between 0.71–0.94 M relative to 4-MAP [3]. This was followed by a work-up/purification involving concentration, trituration in hexane and vacuum filtration to obtain a quantitative yield of [5] which was of adequate purity to be carried forward into stage 2.
Batch optimization of stage 2
The current literature procedures available generally reflux [5] and [6] together for extended periods of time (>20 h) in various solvents followed by an extractive work-up and crystallization resulting in reported yields ranging from 46–84%.8,10–14 We again assessed the reaction in terms of solvent, temperature, duration and concentration. The work-up procedure was also evaluated as this again appeared to be an area which could benefit from improvement. The solvents chosen were based on compatibility with the stage 1 Claisen condensation.The most promising procedures reported, made use of either ethanol or 50% ethyl acetate/water as a solvent system.8,13 Unfortunately, in our hands we were only able to obtain low yields of 8–12% when following the reported protocols. The yields obtained were a concern, especially since TLC analysis indicated good reaction progression and it was again speculated that the extractive work-up and subsequent recrystallization was the problem. The solubility of celecoxib was examined in various solvents and then compared to the solubility of [5] and [6] in the same solvents. Celecoxib was found to be soluble in ethyl acetate while [5] and [6] were insoluble. As such the work-up was adjusted by simply removing the reaction solvent in vacuo, re-dissolving the resulting residue in ethyl acetate and removing the unreacted [5] and [6] by vacuum filtration. The filtrate was then concentrated in vacuo affording celecoxib [1] as a pure solid. This method resulted in substantial improvement in the isolated yields (50–60%) of celecoxib [1]. As absolute ethanol matched the solvent utilised for stage 1 it was decided to continue with batch optimizations using the same solvent.
A temperature screen was performed in the range of 25–100 °C (Fig. 4) identifying an optimal temperature for conversion between 70–80 °C affording yields of 81–82%. As in the case of stage 1 a sharp decrease in the yields was observed at higher temperatures together with noticeable darkening of the reaction mixture and precipitation of an unidentified brown solid. A subsequent optimization of the reaction time afforded a yield of 90% after 17 h (Fig. 5). The darkening and brown precipitates were also observed with longer reaction times, again with significantly lower isolated yields, suggesting possible loss of product due to decomposition with extended heating.
 | ||
| Fig. 4 Study of the effect of reaction temperature on the formation of [1]. Standard conditions: 1.1 equiv. [3], 1.0 equiv. [4], EtOH, 17 h. Isolated yields reported. | ||
 | ||
| Fig. 5 Study of the effect of the reaction duration on formation of [1]. Standard conditions: 1.1 equiv. 3, 1.0 equiv. 4, EtOH, 80 °C. Isolated yields reported. | ||
Unfortunately, the low solubility of hydrazine [6] in absolute ethanol necessitated that the reaction be performed under dilute conditions (0.08 M) to both avoid the formation of unwanted precipitates and a drop-off in the yield. (Fig. 6).
 | ||
| Fig. 6 Study of the effect of concentration of [5] on formation of [1]. Isolated yields reported. | ||
The optimised batch conditions for the cyclo-condensation reaction were found to be 17 hours at 80 °C using 1.1 equiv. of [5] and 1.0 equiv. of [6], at a concentration of 0.08 M in absolute ethanol relative to [5]. This was followed by a work-up involving concentration, suspension in ethyl acetate, vacuum filtration and concentration of the filtrate to obtain [1] in a yield of 90%.
Flow translation and optimization of stage 1
All flow experiments were performed using a Uniqsis FlowSyn stainless steel platform.29 The reactor set-up for stage 1 had reservoirs of 4-MAP (3, 2.68 M) in absolute ethanol and ETFA (4, 2.99 M) and sodium ethoxide (3.82 M) in absolute ethanol connected to two HPLC pumps. The HPLC pumps fed the reagent streams through a 2 mL mixing chip with a 100 psi back pressure regulator (BPR) fitted at the output of the flow stream.An initial assessment of residence time was performed at 50 °C (Table 3) which afforded a best isolated yield of 43% with a residence time of 4 minutes (entry 2, Table 3). A subsequent temperature screen in the range of 30 to 150 °C with the same residence time showed no further improvement in the yield (Fig. 7). It was observed that reaction temperatures below 50 °C were inefficient with yields <5%, as with the batch process route, the isolated yields also decreased steadily when increasing the temperature above 50 °C.
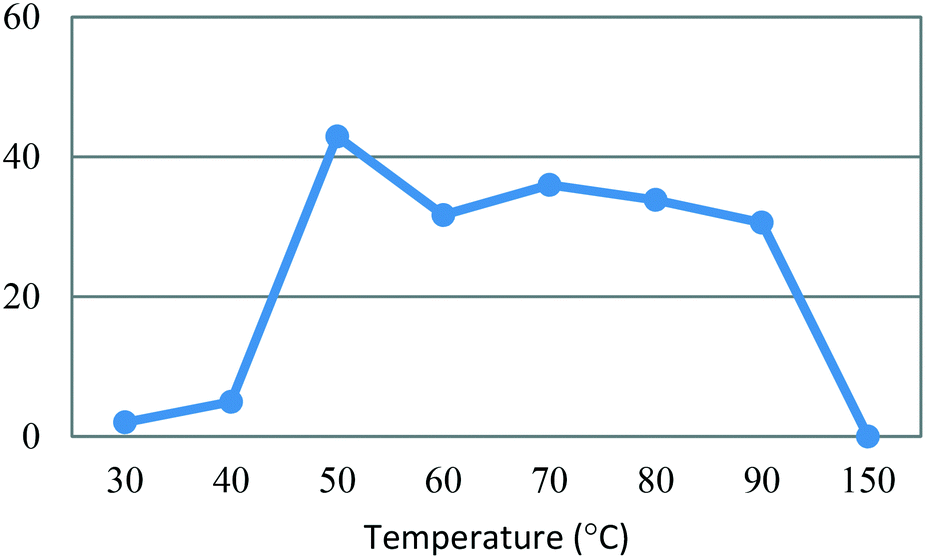 | ||
Fig. 7 Study of the effect of reaction temperature on formation of dione [5] standard conditions: [3] (2.68 M), [4] (2.99 M), NaOEt (3.82 M), 0.50 mL min−1, pump ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Isolated yields reported. :1. Isolated yields reported. | ||
In an attempt to increase the reaction yield the stoichiometric ratio of ETFA [4]/NaOEt relative to 4-MAP [3] was increased (Table 4) with the flow rate maintained at 0.50 mL min−1 at 50 °C. A stoichiometric ratio of 2.5:1 ETFA [4]/NaOEt:4-MAP [3] corresponding to 2.8 and 3.5 equivalents of ETFA [4] and NaOEt respectively, and a concentration of 0.75 M with respect to 4-MAP [3] showed complete conversion by TLC and afforded a 93% isolated yield of dione [5] (Scheme 2).
| Ratio A:B |
Solution A (M) | Solution B (M) | Yield (%) | |
|---|---|---|---|---|
| 4-MAP [3] | ETFA [4] | NaOEt | ||
| Standard conditions: 2 mL mixing chip, stock solutions of [3] (2.68 M), [4] (2.99 M), NaOEt (3.82 M), 50 °C, 0.50 mL min−1, new work-up. Isolated yields reported. | ||||
| 1:1 |
2.68 | 2.99 | 3.82 | 43 |
| 1:1.5 |
2.68 | 4.49 | 5.73 | 83 |
| 1:2.5 |
2.68 | 7.48 | 9.55 | 93 |
| 1:3 |
2.68 | 8.97 | 11.46 | 89 |
 | ||
| Scheme 2 Stage 1 flow schematic with optimised conditions. | ||
Flow translation and optimization of stage 2
The second stage reactor setup made use of two HPLC pumps, a 2 mL mixing chip, a 14 mL HT PTFE coil reactor and a 100 psi back-pressure regulator fitted at the output flow stream. It was found that a 2 mL mixing chip at RT was beneficial to improve mixing of the reagents before reaction in the heated coil reactor. Under flow conditions it was necessary to solubilise the hydrazine [6] stock solution by dissolution in a 75% EtOH/H2O solvent mix. This allowed the preparation of a homogeneous 0.15 M stock solution of hydrazine [6] which upon mixing with the stock solution of dione [5] allowed the reaction to be performed at 0.075 M.As the batch optimizations showed that temperature plays a crucial role in the formation of celecoxib [1] an initial temperature screen was performed in the range of 50–140 °C with a residence time of 32 min (Fig. 8). An isolated yield of 52–53% was obtained between 90 and 100 °C, once again, as observed in the batch optimizations there is a rapid decrease in isolated yields at higher temperatures. Finally, optimization of residence time was performed at 90 °C (Table 5), affording a quantitative conversion of dione [5] to celecoxib [1] with a residence time of 64 min (entry 2, Table 5) (Scheme 3).
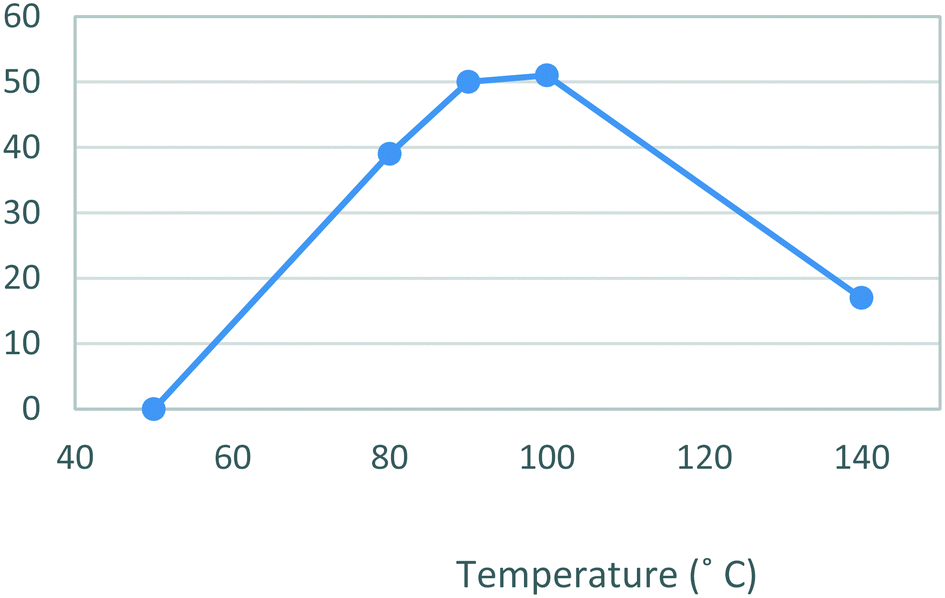 | ||
| Fig. 8 Study of the effect of reaction temperature on formation of [1] standard conditions: 0.17 M [5] in EtOH (0.14 mL min−1), 0.15 M [6] in EtOH (0.36 mL min−1). Isolated yields reported. | ||
| Entry | Flow rate (mL min−1) | Chip residence time (min) | Coil residence time (min) | Yield (%) |
|---|---|---|---|---|
| Standard conditions: 2 mL mixing chip, 14 mL HT PTFE coil, standard solutions of [5] (0.17 M) in EtOH, [6] (0.15 M) in EtOH, 90 °C. Isolated yields reported. | ||||
| 1 | 0.20 | 10 | 70 | 88 |
| 2 | 0.25 | 8 | 56 | 100 |
| 3 | 0.50 | 4 | 28 | 58 |
 | ||
| Scheme 3 Stage 2 flow schematic showing optimised conditions. | ||
Telescoped process
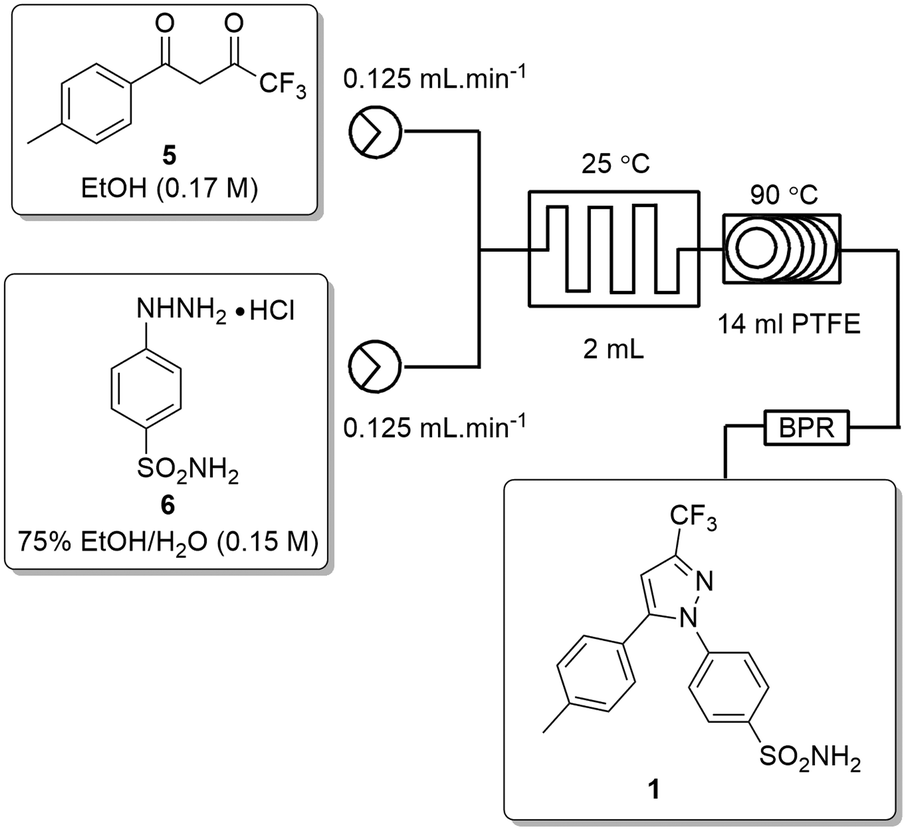
Ideally, we desired a continuous synthesis of [1], that did not require manual work-up, purification or physical isolation of [5]. To realise this, we needed to neutralize the excess base from the Claisen condensation prior to passage into stage 2. In addition, we also needed to remove any unreacted ETFA [4] and adjust the concentration of the product stream of [5] to ∼0.16–0.17 M.The base was neutralized by passage through an OmniFit® column housing Amberlyst-15 (hydrogen-form). Conveniently, passage across the Amberlyst-15 column resulted in dispersion of the reagent stream resulting in a solution of [5] which was only slightly lower in concentration than the 0.17 M required for stage 2. As a result, we elected to employ a simple in-line concentration step which involved passing the product stream into a pre-calibrated beaker heated at 80 °C with a stream of air blowing across the surface of the solution. In doing so, unreacted ETFA [4] (bp 60–62 °C) was effectively removed and a concentrated solution of [5] could then be pumped directly into stage 2. The reactor set-up for the second stage was modified slightly by replacing the 2 mL mixing chip with a T-piece mixer prior to passage through the 14 mL PTFE coil and 100 psi BPR (Scheme 4).
 | ||
| Scheme 4 Flow schematic for the telescoped flow synthesis of [1]. | ||
The telescoped process was successfully validated on a 1 g scale affording celecoxib [1] in an isolated yield of 90% after employing the work-up/purification developed during the batch optimization of stage 2.
We next wished to scale-up the process to produce 10 g of [1], unfortunately, we did not have access to a suitably sized column reactor to house the Amberlyst-15 (hydrogen-form). As such, we elected to split the process into two distinct stages with a manual concentration adjustment. In the case of stage 1 we collected the reaction stream in a beaker containing the Amberlyst 15. The resin was then removed by filtration and the concentration of the filtrate was manually adjusted to 0.17 M before being fed into stage 2 and processed as described for the telescoped process above. Interestingly, when performed in this manner we were able to produce celecoxib [1] in a 96% isolated yield.
The improvement in the yield suggests that the in-line concentration step utilized in the 1 g telescoped process may not be as efficient as required to afford [5] at the desired concentration of 0.17 M. It is conceivable that on large scale the concentration of the stream of [5] in the telescoped process could be more accurately controlled and automated through the integration of an inline FlowIR or a Liqui-Sonic sensor.31 Furthermore, it is also conceivable that the final off-line work-up and purification could be integrated through the use of an in-line triturator and the direct trituration of the reaction stream.32
Conclusion
We have successfully demonstrated an improved batch process and a high yielding continuous flow alternative for the synthesis of celecoxib [1]. In the case of the former, we were able to isolate [1] in 90% yield with a total reaction time of 20 h. Notably, we simplified the previously reported downstream work-up and purification of both stages using simple triturating techniques.In the case of the flow translation, we were able to demonstrate a telescoped process affording [1] in 90% yield, which did not require the handling of intermediates, was conducted in a green solvent system and has a greatly reduced residence time relative to existing batch processes (64 min residence time vs. 20 h reaction time). When run as two standalone stages, the yield was improved to 96%, the increase arguably being due to more accurate manual concentration adjustment between stages 1 and 2.
Finally, the flow approach developed could also conceivably be used for the rapid synthesis of libraries of pyrazole analogues if required.
Experimental section
All reagents were purchased from Sigma-Aldrich. 1H and 13C NMR data were recorded on a Bruker AVANCE-III 400 MHz spectrometer with the residual solvent peak as an internal reference (d6-DMSO = 2.54 and 40.45 ppm for 1H and 13C NMR respectively). Automated flow reactions were performed using a Uniqsis FlowSyn stainless steel platform. All yields quoted represent isolated yields.Batch syntheses
![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif)
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif) CO); 2.31 (s, 3H, C
CO); 2.31 (s, 3H, C![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif) ). 13C NMR (100 MHz d6-DMSO) 185.62, 169.27*, 168.99*, 168.71*, 168.44*, 139.89, 138.96, 128.70, 126.66, 123.78†, 120.88†, 117.98†, 115.07†, 86.84, 20.91. 19F NMR (377 MHz, d6-DMSO) −74.42. *†Signals exhibit splitting due to coupling with fluorine.
); 8.96 (s, 1H, NNH2); 7.74 (d, 2H, J 12.00, Ar–H); 7.24 (s, 2H, NHN); 7.11 (d, 2H, J 8.00, Ar–H). 13C NMR (100 MHz d6-DMSO) 149.41, 137.05, 127.89, 114.31.
); 7.21–7.19 (m, 4H, Ar–H); 7.18 (s, 1H, C); 2.31 (s, 3H, C). 13C NMR (100 MHz d6-DMSO) 154.77, 153.49, 152.23†, 151.85†, 151.49†, 151.10†, 150.61, 148.61, 138.92, 138.28, 136.30, 135.50, 134.86, 135.06*, 132.13*, 129.46*, 126.79*, 115.65, 30.29. 19F NMR (377 MHz, d6-DMSO) −60.86. HRMS m/z (ESI) C17H14F3N3O2S 382.094 ([M + H]+ requires 382.0837) †*signals exhibit splitting due to coupling with fluorine.
). 13C NMR (100 MHz d6-DMSO) 185.62, 169.27*, 168.99*, 168.71*, 168.44*, 139.89, 138.96, 128.70, 126.66, 123.78†, 120.88†, 117.98†, 115.07†, 86.84, 20.91. 19F NMR (377 MHz, d6-DMSO) −74.42. *†Signals exhibit splitting due to coupling with fluorine.
); 8.96 (s, 1H, NNH2); 7.74 (d, 2H, J 12.00, Ar–H); 7.24 (s, 2H, NHN); 7.11 (d, 2H, J 8.00, Ar–H). 13C NMR (100 MHz d6-DMSO) 149.41, 137.05, 127.89, 114.31.
); 7.21–7.19 (m, 4H, Ar–H); 7.18 (s, 1H, C); 2.31 (s, 3H, C). 13C NMR (100 MHz d6-DMSO) 154.77, 153.49, 152.23†, 151.85†, 151.49†, 151.10†, 150.61, 148.61, 138.92, 138.28, 136.30, 135.50, 134.86, 135.06*, 132.13*, 129.46*, 126.79*, 115.65, 30.29. 19F NMR (377 MHz, d6-DMSO) −60.86. HRMS m/z (ESI) C17H14F3N3O2S 382.094 ([M + H]+ requires 382.0837) †*signals exhibit splitting due to coupling with fluorine.
Flow syntheses
4-Methyl acetophenone in ethanol. A stock solution of 4-methyl acetophenone [3] was prepared by dissolving [3] (14.3 mL, 107 mmol) in absolute ethanol (40 mL).
Ethyl trifluoroacetate 4/sodium ethoxide in ethanol. A stock solution of ethyl trifluoroacetate [4] and sodium ethoxide was prepared by dissolving sodium metal (4.39 g, 191 mmol) in absolute ethanol (50 mL) followed by the addition of [4] (17.8 mL, 150 mmol).
4-Sulfamidophenylhydrazine hydrochloride in ethanol. A stock solution of 4-sulfamidophenylhydrazine hydrochloride [6] was prepared by dissolving [6] (1.32 g, 5.9 mmol) in 75% ethanol/water (40 mL).
Flow synthesis of 4,4,4-trifluoro-1-(4-methyl-phenyl)-butane-1,3-dione [5]. The 4-methyl acetophenone [3] stock solution (1.48 mL) and the ethyl trifluoroacetate [4]/sodium ethoxide stock solution (3.70 mL) were pumped at flow rates of 0.14 mL min−1 and 0.36 mL min−1 respectively through a 2 mL glass mixing chip heated to 50 °C (Scheme 2). The output of the reactor was collected until no further product was eluted and the solvent removed in vacuo. The solid obtained was suspended in hexane (15 mL) with vigorous stirring followed by vacuum filtration and washing with hexane (2 × 2 mL) to obtain [5] as a pure cream coloured solid (0.84 g, 3.7 mmol, 93%).
Flow synthesis of celecoxib [1] from [5]. A 4,4,4-trifluoro-1-(4-methyl-phenyl)-butane-1,3-dione [5] stock solution (7 mL) and 4-sulfamidophenylhydrazine hydrochloride [6] stock solution (7 mL) were each pumped at a flow rate of 0.125 mL min−1 through a 2 mL glass mixing chip at room temperature followed by a 14 mL PTFE coil heated to 90 °C (Scheme 3). The output of the reactor was collected until no further product was eluted and the solvent removed in vacuo. The solid obtained was suspended in ethyl acetate (30 mL) followed by vacuum filtration. The filtrate was concentrated to obtain a pale yellow solid (0.393 g, 1.0 mmol, 99%). Rf = 0.33 (20% methanol/dichloromethane).
Telescoped flow synthesis of celecoxib [1]. A 4-methyl acetophenone [3] (1.48 mL) stock solution and the ethyl trifluoroacetate [4]/sodium ethoxide (3.70 mL) stock solution were pumped at flow rates of 0.14 mL min−1 and 0.36 mL min−1 respectively through a 2 mL glass mixing chip heated to 50 °C followed by an Omnifit ® column packed with Amberlyst-15 resin hydrogen form (1.25 g, 5.9 mmol). The output (4,4,4-trifluoro-1-(4-methyl-phenyl)-butane-1,3-dione 5) of the reaction was collected in a pre-calibrated beaker held at 80 °C with an air stream blowing over the surface to concentrate the solution down to 16.4 mL (0.17 M relative to 5). The concentrated solution of 5 (15.0 mL) and a 4-sulfamidophenylhydrazine hydrochloride 6 stock solution (15.0 mL) were both pumped at flow rates of 0.125 mL min−1 and mixed via a stainless-steel T-piece followed by reaction in a 14 mL HT PTFE coil reactor heated to 90 °C (Scheme 4). The output of the reactor was collected until no further product was eluted and the solvent removed under vacuum. The solid obtained was suspended in ethyl acetate (30 mL) followed by vacuum filtration. The filtrate was concentrated in vacuo to obtain a pale yellow solid (0.76 g, 2.0 mmol, 90%).
Flow synthesis of celecoxib [1] with manual concentration adjustment between steps one and two. A 4-methyl acetophenone [3] (12 mL) stock solution and the ethyl trifluoroacetate [4]/sodium ethoxide (30 mL) stock solution were pumped at flow rates of 0.14 mL min−1 and 0.36 mL min−1 respectively through a 2 mL glass mixing chip heated to 50 °C. The output stream was then passed through a 100 psi back pressure regulator into a beaker containing Amberlyst-15 resin hydrogen form (14 g, 66.1 mmol) until no further product was eluted. The Amberlyst-15 was then removed by filtration and the volume of the filtrate adjusted to 200 mL affording a 0.17 M stock solution of [5]. The stock solution of [5] (176 mL) and a 4-sulfamidophenylhydrazine hydrochloride [6] stock solution (176 mL) were then both pumped at flow rates of 0.125 mL min−1 and mixed via a stainless steel T-piece followed by reaction in a 14 mL HT PTFE coil reactor heated to 90 °C. The output of the reactor was collected until no further product was eluted and the solvent removed under vacuum. The solid obtained was suspended in ethyl acetate (300 mL) followed by vacuum filtration. The filtrate was concentrated in vacuo to obtain a pale yellow solid (9.60 g, 25.3 mmol, 96%).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Research Foundation of South Africa (grant number 87893), the University of Pretoria (University, Science Faculty Research Councils and Research and Development Program), South Africa and Pelchem Pty Ltd. Opinions expressed in this publication and the conclusions arrived at, are those of the authors, and are not necessarily attributed to the NRF. The author(s) would like to acknowledge Eric Palmer and Mamoalosi Selepe for NMR spectroscopy services, Ms Madelien Wooding (University of Pretoria) for LC-MS services and Uniqsis Ltd for Flow Equipment.References
- S. M. Gaulier, R. McKay and N. A. Swain, Tetrahedron Lett., 2011, 52, 6000 CrossRef CAS.
- S. K. Singh, P. G. Reddy, K. S. Rao, B. B. Lohray, P. Misra, S. A. Rajjak, Y. K. Rao and A. Venkateswarlu, Bioorg. Med. Chem. Lett., 2004, 14, 499 CrossRef CAS.
- D. Boschi, L. Lazzarato, B. Rolando, A. Filieri, C. Cena, A. Di Stilo, R. Fruttero and A. Gasco, Chem. Biodiversity, 2009, 6, 369 CrossRef CAS.
- D. Desai, N. Kaushal, U. H. Gandhi, R. J. Arner, C. D'Souza, G. Chen, H. Vunta, K. El-Bayoumy, S. Amin and K. S. Prabhu, Chem.-Biol. Interact., 2010, 188, 446 CrossRef CAS.
- G. Menozzi, L. Merello, P. Fossa, L. Mosti, A. Piana and F. Mattioli, Farmaco, 2003, 58, 795 CrossRef CAS.
- Y. Kuge, Y. Katada, S. Shimonaka, T. Temma, H. Kimura, Y. Kiyono, C. Yokota, K. Minematsu, K. Seki, N. Tamaki, K. Ohkura and H. Saji, Nucl. Med. Biol., 2006, 33, 21 CrossRef CAS.
- M. Gao, M. Wang, K. D. Miller, G. D. Hutchins and Q.-H. Zheng, Appl. Radiat. Isot., 2009, 67, 2019 CrossRef CAS.
- T. D. Penning, J. J. Talley, S. R. Bertenshaw, J. S. Carter, P. W. Collins, S. Docter, M. J. Graneto, L. F. Lee, J. W. Malecha, J. M. Miyashiro, R. S. Rogers, D. J. Rogier, S. S. Yu, G. D. Anderson, E. G. Burton, J. N. Cogburn, S. A. Gregory, C. M. Koboldt, W. E. Perkins, K. Seibert, A. W. Veenhuizen, Y. Y. Zhang and P. C. Isakson, J. Med. Chem., 1997, 40, 1347 CrossRef CAS.
- L. M. Oh, Tetrahedron Lett., 2006, 47, 7943 CrossRef CAS.
- (a) B. Zhi and M. Newaz, Process for preparing 3-haloalkyl-1H-pyrazoles, US Pat., Patent no: 5892053, 1999 Search PubMed; (b) B. Zhi and M. Newaz Process for preparing 3-haloalkyl-1H-pyrazoles. US Pat., Patent no: 5910597, 1999 Search PubMed.
- P. O'Shea, R. D. Tillyer, X. Wang, S.-D. Clas and C. Dalton, Synthesis of 4-[5-substituted or unsubstituted phenyl)-3-substituted-1H-pyrazol-1-yl]benzenesulfonamides, US Pat., Patent no: 6150534, 2000 Search PubMed.
- L. J. Letendre, W. D. McGhee, C. Snoddy, G. Klemm and H. T. Gaud, Synthesis of diaryl pyrazoles, US Pat., Patent no: 7141678B2, 2006 Search PubMed.
- A. R. Reddy, A. Sampath, G. Goverdhan, B. Yakambaram, K. Mukkanti and P. P. Reddy, Org. Process Res. Dev., 2009, 13, 98 CrossRef CAS.
- V. R. R. Ambati, S. Garaga, S. P. S. Mallela and S. Meenakshisunderam, An improved process for the preparation of celecoxib. Patent no: WO2010/095024A2, Aug 2010.
- R. E. Beveridge, D. Fernando and B. S. Gerstenberger, Tetrahedron Lett., 2010, 51, 5005 CrossRef CAS.
- R. Reddy, V. Ramana and S. C. Bell, Chem. Abstr., 2003, 138, 271677 Search PubMed.
- J. Hu, S. Chen, Y. Sun, J. Yang and Y. Rao, Org. Lett., 2012, 14, 5030 CrossRef CAS.
- (a) Y. Shang, J. Du and H. Cui, Org. Prep. Proced. Int., 2014, 46, 132 CrossRef CAS; (b) K. Sano and S. Hara, Heterocycles, 2010, 80, 349 CrossRef CAS.
- (a) D. L. Riley, I. Strydom, R. Chikwamba and J.-L. Panayides, React. Chem. Eng., 2019, 4, 457–489 RSC; (b) https://www.npr.org/sections/health-shots/2018/04/25/605226604/fda-panel-affirms-safety-of-painkiller-celebrex (Accessed 20/01/2020).
- C. J. Smith, F. J. Iglesias-Sigüenza, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2007, 5, 2758 RSC.
- (a) I. R. Baxendale, S. C. Schou, J. Sedelmeier and S. V. Ley, Chem. – Eur. J., 2010, 16, 89 CrossRef CAS; (b) H. Lange, C. F. Carter, M. D. Hopkin, A. Burke, J. G. Goode, I. R. Baxendale and S. V. Ley, Chem. Sci., 2011, 2, 765 RSC.
- J. R. Breen, G. Sandford, D. S. Yufit, J. A. K. Howard, J. Fray and B. Patel, Beilstein J. Org. Chem., 2011, 7, 1048 CrossRef CAS.
- D. Obermayer, T. N. Glasnow and C. O. Kapper, J. Org. Chem., 2011, 76, 6657 CrossRef CAS.
- B. Li, D. Widlicka, S. Boucher, C. Hayward, J. Lucas, J. C. Murray, B. T. O'Neil, D. Pfisterer, L. Samp, J. VanAlsten, Y. Xiang and J. Young, Org. Process Res. Dev., 2012, 16, 2031 CrossRef CAS.
- A. DeAngelis, D.-H. Wang and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 3434 CrossRef CAS.
- C. Battilocchio, B. J. Deadman, N. Nikbin, M. O. Kitching, I. R. Baxendale and S. V. Ley, Chem. – Eur. J., 2013, 19, 7917 CrossRef CAS.
- J.-S. Poh, D. L. Browne and S. V. Ley, React. Chem. Eng., 2016, 1, 101 RSC.
- (a) M. M. Ahlström, M. Ridderström, I. Zamora and K. Luthman, J. Med. Chem., 2007, 50, 4444 CrossRef; (b) S. Bűttner, A. Riahi, I. Hussain, M. A. Yawer, M. Lubbe, A. Villinger, H. Reinke, C. Fischer and P. Langer, Tetrahedron, 2009, 65, 2124 CrossRef; (c) G. Szabó, J. Fischer, A. Kis-Varga and K. Gyires, J. Med. Chem., 2008, 51, 142 CrossRef.
- www.uniqsis.com (Accessed 19/02/2020).
- R. Soliman, J. Med. Chem., 1979, 22, 321 CrossRef CAS.
- https://www.sensotech.com/en/products/systems/liquisonic (Accessed 19/02/2020).
- N. C. Neyt and D. L. Riley, React. Chem. Eng., 2018, 3, 17–24 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0re00346h |
| ‡ Present Address: University of Pretoria, Department of Chemistry, Natural and Agricultural Sciences, 2 Lynnwood Road, Hatfield, 0002, Gauteng, South Africa. |
| This journal is © The Royal Society of Chemistry 2021 |