
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew small γ-turn type N-primary amino terminal tripeptide organocatalyst for solvent-free asymmetric aldol reaction of various ketones with aldehydes†
Rajkumar Thiyagarajana,
Zubeda Beguma,
Chigusa Sekia,
Yuko Okuyamab,
Eunsang Kwon*c,
Koji Uwaia,
Michio Tokiwad,
Suguru Tokiwad,
Mitsuhiro Takeshitad and
Hiroto Nakano *a
*a
aDivision of Sustainable and Environmental Engineering, Graduate School of Engineering, Muroran Institute of Technology, 27-1 Mizumoto-cho, Muroran 050-8585, Japan. E-mail: catanaka@mmm.muroran-it.ac.jp
bTohoku Medical and Pharmaceutical University, 4-4-1 Komatsushima, Aoba-Ku, Sendai 981-8558, Japan
cResearch and Analytical Center for Giant Molecules, Graduate School of Sciences,Tohoku Medical and Pharmaceutical University, 4-4-1 Komatsushima, Aoba-Ku, Sendai 981-8558, Japan
dTokiwakai Group, 62 Numajiri Tsuduri-Chou Uchigo, Iwaki 973-8053, Japan
First published on 6th December 2021
Abstract
New small γ-turn type N-primary amino terminal tripeptides were synthesized and their functionality as an organocatalyst was examined in the asymmetric aldol reaction of various ketones with different aromatic aldehydes under solvent-free neat conditions to afford the desired chiral anti-aldol products in good to excellent chemical yields, diastereoselectivities and enantioselectivities (up to 99%, up to syn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :anti/13:87 dr, up to 99% ee).
:anti/13:87 dr, up to 99% ee).
1. Introduction
From the last decade, the process of developing chiral multifunctional organocatalysts and their application in asymmetric synthesis as an independent chiral source has been a new area of focus for scientists. Catalysts possessing either Lewis or Brønsted basic functionalities and a non-covalent hydrogen-bond donor group suitably positioned over a chiral scaffold have been synthesized in general for various asymmetric reactions.1 However, it is always a difficult job to design and synthesize multifunctional organocatalysts with remarkable catalytic activity in diverse asymmetric reactions. In organocatalysis, peptide catalysis has been recognized as a powerful and useful tool in the synthetic organic chemistry field, because peptides are highly modular, structurally diverse and easily accessible from nature's chiral pool. Due to their broad synthetic applicability, the studies on the development of new peptide organocatalysts have been continuously explored and attained great progress over the years.2 In the development of peptide organocatalysts, inspired by the efficient and stereospecific metal-free enzymatic processes, synthetic chemists have devoted much effort in recent years towards the development of a broad range of short peptide-based asymmetric catalysts, which mimic various qualities of enzymes. The structural diversity available with short peptide sequences and the fact that peptides offer a more strictly defined asymmetric environment to the reactants, compared to single amino acids, makes this class of molecules particularly promising towards the development of a wide range of organocatalysts with fine-tuneable structural and electronic properties. As for the short peptide organocatalyst, Inoue and co-workers reported the first cyclic dipeptide as an organocatalyst for the asymmetric cyanation reaction of hydrogen cyanide with benzaldehyde.3 Furthermore, Miller and co-workers introduced first linear peptide organocatalyst, wherein fixing by the hydrogen bonding, based on β-turn concept for asymmetric acylation reaction was reported.4 Using these pioneering studies, a great number of short peptide organocatalysts based on β-turn concept were reported over various asymmetric reactions such as aldol reactions5 and so on.6 However, to the best of our knowledge, no research group has been reported the design and use of small peptide organocatalysts in an asymmetric reaction, fixing by the hydrogen bonding based on γ-turn concept. A γ-turn type peptide organocatalyst has different conformation with β-turn type and it is expected to show interesting catalytic activity in asymmetric reactions.We designed a new small γ-turn type N-primary amino terminal tripeptide X containing the pyrrolidine ring as a backbone (Scheme 1). This peptide X has N-terminal side at nitrogen atom in pyrrolidine backbone and C-terminal exists at 2-position on the pyrrolidine ring. Amide carbonyl group at N-terminal side and amide amino group at C-terminal side are connected by γ-turn intramolecular hydrogen bonding. In addition, X has both primary amino group for the formation of enamine with substrates and secondary amide group for the activation and fixing substrates on N-terminal side, and also the amide part at C-terminal side containing polycyclic aromatic substituent as steric influence site. Furthermore, alkyl or aryl substituents on the molecule act as steric and electric influence sites. The designed catalyst X was applied in the asymmetric aldol reaction, which is known as one of the practical reaction for creating β-hydroxyl carbonyl structural motif which is observed in numerous biologically active natural products and drug molecules.7 There are several scientific reports that have proved the tremendous versatility of the chiral crossed aldol reaction using various organocatalysts, especially asymmetric crossed aldol reaction of ketones with aromatic aldehydes.8 Therefore, we anticipated that our designed tripeptide X may act as an efficient organocatalyst for an enantioselective crossed aldol reaction over a wide range of aromatic aldehydes with ketones.
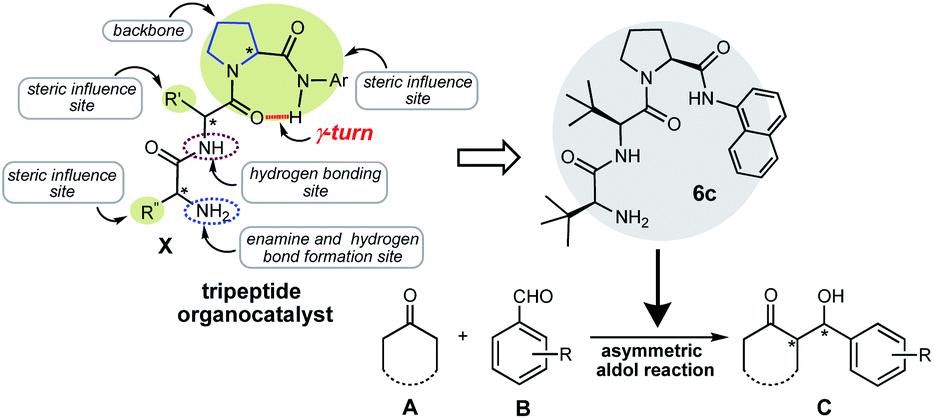 | ||
| Scheme 1 Concept of tripeptide organocatalyst design. | ||
Herein, we describe the highly efficient catalytic activity displayed by our newly prepared tripeptide organocatalyst 6c as X in the crossed aldol reaction of various ketones A with aromatic aldehydes B to afford the corresponding anti-aldol products C in good to excellent chemical yields and stereoselectivities (up to 99%, up to syn:anti = 13:87 dr, up to 99% ee) without the addition of any co-catalysts under eco-friendly solvent-free neat condition, which is still a challenging task in the asymmetric organocatalysis.
2. Results and discussion
2.1. Preparation and screening of catalysts
The tripeptide organocatalysts 6a–j were prepared easily by the condensation reaction, starting from the reaction of L-N-Boc-proline 1 with amines 2a–d as shown in Scheme 2. The first condensation of 1 with 2a–d in the presence of DCC followed by the deprotection of Boc group of the crude N-protected proline amides afforded the corresponding proline amides 3a–d at 85–95% yields. Next, the condensation of the obtained 3a–d with N-Boc amino acids 4a–e in the presence of DCC followed by the deprotection of the crude N-protected dipeptides gave the corresponding dipeptides 5a–h at 60–92% yields. As amino acids, γ-substituted amino acids that are effective towards a conformation favorable to form γ-turn intramolecular hydrogen bonding were used. Furthermore, final condensation of the dipeptides 5a–h with N-protected 4′a–g, respectively, in the presence of DCC followed by the deprotection afforded the corresponding tripeptides 6a–j at 70–93% yields. Moreover, the diastereomer 10 of 6c, tripeptides 14a and 14b with azetidine and piperidine backbones, respectively, were also prepared using the same procedure used to prepare 6a–j. Thus, the first condensation of D-N-Boc-7, L-N-Boc-11a,b with aromatic amine 2b followed by the deprotection afforded the corresponding amides 8, 12a, 12b at 79–91% yields. Next the condensations of 8, 12a, 12b with N-Boc amino acid 4′c followed by the deprotection gave the dipeptides 9, 13a, 13b at 70–89% yields. Then, the final condensation of 9, 13a, 13b with 4′c, respectively, followed by the deprotection afforded the corresponding tripeptides 10, 14a, 14b at 79–85% yields. | ||
| Scheme 2 Preparation of di- and tripeptide organocatalysts. | ||
After the synthesis of the designed peptide organocatalysts, initially, we examined the crossed aldol reaction of cyclohexanone 15a as an aldol donor and 4-nitrobenzaldehyde 16a as an aldol acceptor using the precursor dipeptides 5a–h of tripeptides 6a–h as catalysts in Et2O at 25 °C (entries 1–8, Table 1). These dipeptide organocatalysts 5a–h showed catalytic activity in this reaction, and the desired chiral aldol product 17 was obtained. However, only low to moderate chemical yields, moderate diastereoselectivities (17a: anti or 17′a: syn forms), and low to moderate enantioselectivities were obtained in this reaction condition. The absolute configurations of 17a and 17′a were identified based on comparison with literature data.8 We next tried this reaction using tripeptides 6a–j as catalysts (entries 9–18) under same reaction condition wherein catalysts 5a–h were used. Encouraged by the result of dipeptide 5c, possessing t-butyl group at N-terminal side which showed better catalytic activity, the substituent on the first amide part was fixed only to t-butyl group. Similar to 5a–h, 6a–j also showed catalytic activity in this reaction to afford the chiral aldol product anti-17a, but with low to moderate chemical yields. Interestingly, with the use of catalysts 6a–j diastereoselectivity increased in the case of almost all catalysts and the main aldol product 17 was obtained in only anti form unlike the catalysts 5a–h. Furthermore, enantioselectivity also quite increased with all the catalysts 6a–j. Particularly, catalyst 6c with L-t-Leu-L-t-Leu moiety at N-terminal side and 1-naphthyl group on amide amino moiety at C-terminal side afforded 17a in fairly good enantioselectivity (94% ee) and with good diastereoselectivity (syn:anti/22:78), although chemical yield was low (25%) (entry 11). Also, the catalyst 6i afforded 17a in fairly good enantioselectivity (93% ee) and diastereoselectivity (syn:anti/20:80) (entry 17) similar to the catalyst 6c, but owing to the toxicity and cost effectiveness of pyrenes, 6c with 1-naphthyl group may be preferred. We have also examined the catalytic activity of catalysts 6j with D-t-Leu-L-t-Leu chains at N-terminal side, 10 with D-pyrrolidine ring, 14a with four membered azetidine ring, and 14b with six membered piperidine ring, respectively in this reaction. As a result, the catalysts 6j, 10, 14a and 14b did not show better catalytic activity than 6c having L-t-Leu-L-t-Leu moiety at N-terminal side and five membered pyrrolidine ring. These results indicated that the chain length of N-terminal side, the size of backbone ring for forming γ-turn, the configuration and types of substituents at N-terminal side and the aromatic ring on amide amino group at C-terminal side are highly important for achieving satisfactory stereoselectivities, by shielding one enantiotopic face to attack from the aldol acceptor 16a in this reaction condition using ether solvent.
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yielda (%) | dr (syn:anti)b |
eec (%) | |
| syn 17′a | anti 17a | ||||
| a Isolated yields.b Diastereoselectivity was determined by 1H NMR using crude reaction mixture.c The ee value was determined by HPLC with a Daicel Chiralpak AD-H column. | |||||
| 1 | 5a | 18 | 33:67 |
18 | 34 |
| 2 | 5b | 40 | 40:60 |
22 | 7 |
| 3 | 5c | 52 | 67:33 |
63 | 10 |
| 4 | 5d | 45 | 40:60 |
35 | 6 |
| 5 | 5e | 33 | 47:53 |
35 | 8 |
| 6 | 5f | 13 | 70:30 |
54 | 11 |
| 7 | 5g | 20 | 72:28 |
54 | 13 |
| 8 | 5h | 47 | 59:41 |
37 | rac |
| 9 | 6a | 30 | 35:65 |
28 | 71 |
| 10 | 6b | 19 | 40:60 |
28 | 74 |
| 11 | 6c | 25 | 22:78 |
70 | 94 |
| 12 | 6d | 16 | 23:77 |
25 | 50 |
| 13 | 6e | 25 | 25:75 |
26 | 39 |
| 14 | 6f | 58 | 25:75 |
1 | 30 |
| 15 | 6g | 10 | 28:72 |
22 | 50 |
| 16 | 6h | 26 | 25:75 |
30 | 80 |
| 17 | 6i | 35 | 20:80 |
26 | 93 |
| 18 | 6j | 20 | 35:65 |
7 | −87 |
| 19 | 10 | 30 | 22:78 |
17 | 77 |
| 20 | 14a | 20 | 30:70 |
44 | 86 |
| 21 | 14b | 27 | 24:76 |
63 | 90 |

With these results in hand, we tried to optimize the reaction conditions using tripeptide catalyst 6c, that afforded the best enantioselectivity, to further improve the chemical yield and stereoselectivities (Table 2). An extensive screening of the reaction was further carried out by varying different parameters such as solvent, catalyst loading, and reaction temperature. First, the solvent screening was performed with different ethereal (entries 2–4), non-polar aliphatic (entries 5 and 6), aromatic (entry 7), chlorinated (entries 8 and 9), protic and aprotic polar (entries 10–15) solvents and solvent free neat condition (entry 16). The reaction was carried out in the presence of 10 mol% of 6c at 25 °C for 24 h. However, catalyst 6c did not show enough catalytic activity to afford the aldol product 17a in all solvents. On the other hand, both diastereoselectivity and the enantioselectivity were quite different according to characteristic solvents (entries 1–16). Interestingly, the chemical yield and stereoselectivities were greatly improved under neat condition to yield up to 52%, (syn:anti/22:78 dr) and 87% ee (anti) (entry 16). Although aldol reaction has been generally carried out under organic and water solvents system so far for obtaining satisfactory result, the successful examples of under neat condition have been reported by only a little group.9 Next, we examined the effect of catalyst loading by varying from 30, 20, 5% and 1 mol% under superior neat reaction condition (entries 17–20). Good result (88%, syn:anti/22:78 dr, 96% ee) was obtained in the presence of 30 mol% of catalyst (entry 17). The use of 20 mol% also afforded good chemical yield and stereoselectivities. However, chemical yield and enantioselectivity were slightly decreased (79%, 22:78, 90% ee) (entry 18). When the reaction was carried out in the presence of 5 mol% of catalyst, diastereoselectivity and enantioselectivity was maintained to 22:78 (syn:anti) and 87% ee, but chemical yield was decreased to 20% (entries 19). Furthermore, the use of 1 mol% of 6c brought about the large decrease of chemical yield and stereoselectivities (4%, syn:anti/22:78 dr, 65% ee) (entry 20). In addition, reaction temperature (0 and −25 °C) were also examined in the presence of superior 30 mol% of catalyst 6c under neat condition (entries 21, 22). Best result was obtained at 0 °C for chemical yield and stereoselectivities (96%, syn:anti/20:80 dr, 98% ee) (entry 21). However, the reaction at −25 °C brought about the decrease of chemical yield (40%), although enough stereoselectivities were maintained (syn:anti/20:80 dr, 93% ee) (entry 22). Based on these results, it was revealed that the reaction in the presence of 30 mol% of catalyst at 0 °C for 24 h under solvent-free was the best reaction condition to obtain the chiral aldol adduct 17a in satisfactory chemical yield and stereoselectivities (entry 21). Furthermore, in order to demonstrate the practical utility of the catalyst 6c, the reaction of 15a with 16a was conducted on gram scale (15a: 1 g) using the above optimized reaction conditions and the corresponding product 17a was obtained in good chemical yield (98%), diastereoselectivity (syn:anti/24:76 dr), and enantioselectivity (93% ee). This result indicated that the catalyst 6c can also be effective on practical scale (entry 23).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Cat 6c (mol%) | Temp. (°C) | Solvent | Yielda (%) | dr (syn:anti)b |
eec (%) | |
| syn 17′a | anti 17a | ||||||
| a Isolated yields.b Diastereoselectivity was determined by 1H NMR using crude reaction mixture.c The ee value was determined by HPLC with a Daicel Chiralpak AD-H column.d 1 g of substrate 15a was used. | |||||||
| 1 | 10 | 25 | Et2O | 25 | 22:78 |
70 | 94 |
| 2 | 10 | 25 | i-Pr2O | 25 | 47:53 |
30 | 81 |
| 3 | 10 | 25 | tBuOMe | 18 | 45:55 |
47 | 83 |
| 4 | 10 | 25 | THF | 11 | 45:55 |
37 | 70 |
| 5 | 10 | 25 | Pentane | 20 | 40:60 |
53 | 79 |
| 6 | 10 | 25 | Hexane | 15 | 38:62 |
14 | 78 |
| 7 | 10 | 25 | Toluene | 15 | 38:62 |
70 | 80 |
| 8 | 10 | 25 | CH2Cl2 | 13 | 28:72 |
40 | 67 |
| 9 | 10 | 25 | CHCl3 | 10 | 38:62 |
45 | 93 |
| 10 | 10 | 25 | CH3CN | 18 | 35:65 |
23 | 51 |
| 11 | 10 | 25 | DMSO | 10 | 30:70 |
02 | 30 |
| 12 | 10 | 25 | DMF | 10 | 31:69 |
7 | 51 |
| 13 | 10 | 25 | i-PrOH | 10 | 41:59 |
8 | 55 |
| 14 | 10 | 25 | MeOH | 13 | 44:56 |
69 | 24 |
| 15 | 10 | 25 | H2O | 16 | 28:76 |
10 | 65 |
| 16 | 10 | 25 | Neat | 52 | 22:78 |
64 | 87 |
| 17 | 30 | 25 | Neat | 88 | 23:77 |
60 | 96 |
| 18 | 20 | 25 | Neat | 79 | 22:78 |
64 | 90 |
| 19 | 5 | 25 | Neat | 20 | 24:76 |
60 | 87 |
| 20 | 1 | 25 | Neat | 4 | 23:77 |
48 | 65 |
| 21 | 30 | 0 | Neat | 94 | 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :80 :80 |
64 | 98 |
| 22 | 30 | −25 | Neat | 40 | 21:79 |
43 | 93 |
| 23d | 30 | 0 | Neat | 98 | 24:76 |
55 | 93 |

With best reaction conditions in hand, we tried to re-examine the catalytic activity of the prepared catalysts 10 (diastereomer of 6c), and 14a, 14b having 4, 6 membered rings as back bone under the best solvent-free reaction condition (30 mol%, 0 °C, 24 h, neat), respectively (entries 2–4, Table 3). As a result, these catalysts showed satisfactory catalytic activities to afford 17a in good chemical yields and stereoselectivities (72–90%, syn:anti/23:77–22:78 dr, 92–98% ee) in this reaction condition. Especially, the catalyst 14b having 6 membered piperidine ring (entry 4) gave 17a with excellent enantioselectivity in results similar to that of catalyst 6c having 5 membered pyrrolidine ring (entry 1), although the decrease of chemical yield was observed. From the fact that all catalysts showed satisfactory chemical yield and stereoselectivity, it is indicated that there is no restriction of ring size and the conformation to obtain the product with good chemical yield and stereoselectivity. Hence it might be possible to choose a suitable substrate in consideration of the distances between N-terminal side and C-terminal side of 6c, 14a, 14b, respectively, for obtaining the corresponding product with satisfactory chemical yield and stereoselectivity (Scheme 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Yielda (%) | drb (syn:anti) |
eec (%) | |
| syn 17′a | anti 17a | ||||
| a Isolated yields.b Diastereoselectivity was determined by 1H NMR using crude reaction mixture.c The ee value was determined by HPLC with a Daicel Chiralpak AD-H column. | |||||
| 1 | 6c | 96 | 20:80 |
60 | 98 |
| 2 | 10 | 90 | 22:78 |
44 | 96 |
| 3 | 14a | 89 | 23:77 |
33 | 92 |
| 4 | 14b | 72 | 20:80 |
38 | 98 |

 | ||
| Scheme 3 Structures of tripeptide organocatalysts. | ||
2.2. Substrate scope
After the optimization of reaction conditions, we investigated the generality of the superior catalyst 6c in the reactions of various ketones 15a–h with aldehydes 16a–j (Scheme 4). The reactions were carried out in the presence of catalyst 6c (30 mol%) under best reaction conditions (0 °C, neat, 24 h). First, the reaction of cyclohexanone 15a with nitro-substituted aromatic aldehydes 16b, 16c afforded the corresponding anti-aldol products 17b,c in good to excellent chemical yields, diastereoselectivities and enantioselectivities (17b: 78%, syn:anti/22:78 dr, 99% ee, 17c: 99%, up to syn:anti/20:80 dr, up to 99% ee). Furthermore, the reactions using the halogenated 16d–g and p-cyano 16h benzaldehydes, respectively, also afforded the corresponding anti-aldol adducts 17d–h with satisfactory results (30–63%, syn:anti/25:75–14:86 dr, 93–98% ee). However, the use of simple benzaldehyde 16i brought about the decrease of chemical yield and enantioselectivity (17i: 30%, syn:anti/25:75 dr, 60% ee). These results justified our finding that catalyst 6c enhances the generality of application towards the aldol reaction using different substituted aromatic aldehydes. The aldol reactions of other various carbocyclic 15b–d, heterocyclic 15e, 15f, and acyclic 15g,h ketones with 16a were also examined in the presence of 6c (30 mol%) under best reaction condition (0 °C, neat, 24 h). The reaction of cyclopentanone 15b with 16a proceeded and afforded the corresponding anti-aldol product 17m in fairly good chemical yield and stereoselectivity (81%, syn:anti/15:85 dr, 92% ee). The bulkier cycloheptanone 15c also gave the anti-aldol product 17n with good chemical yield, diastereoselectivity and excellent enantioselectivity (90%, syn:anti/15:85 dr, 95% ee). Moreover, the reaction using p-methyl cyclohexanone 15d also afforded anti-aldol product 17o in good chemical yield and stereoselectivity (70%, syn:anti/21:79, 90% ee). Although the use of heterocyclic 2,2-dimethyl-1,3-dioxan-5-one 15e did not afford 17p with satisfactory results (66%, syn:anti/30:70 dr, 68% ee), tetrahydropyran-4-one 15f afforded the corresponding anti-aldol product 17q in good chemical yield and diastereoselectivity with fairly good enantioselectivity (72%, syn:anti/13:87 dr, 94% ee). Finally, the reactions of acyclic acetone 15g and acetophenone 15h with 16a also examined under same reaction condition. The use of 15g gave the corresponding anti-aldol adduct 17r in moderate results (60%, 59% ee), but 15h afforded 17s in moderate chemical yield and good enantioselectivity (67%, 90% ee). The anti-aldol products 17b–j,m–s were characterized in accordance with full spectroscopic data originated from the previous report.8 And also, absolute stereochemistry of 17b–j,m–s were found to be equivalent to specific optical rotation values in literature.8 These results justified our finding that catalyst 6c enhances the generality of application towards the aldol reaction using different substituted aromatic aldehydes. However, catalyst 6c did not show catalytic activity with aliphatic aldehyde in the best reaction condition. Therefore, the reaction under different reaction conditions might need to be tried.
 | ||
| Scheme 4 Substrate scope for aldol reaction. | ||
2.3. Reaction mechanism
Based on the excellent enantiopurity (98% ee) of the obtained optically active aldol product (2S,3R)-17a from the asymmetric aldol reaction of cyclohexanone 15a with 4-nitrobenzaldehyde 16a using catalyst 6c, a model of the asymmetric reaction course is proposed as shown in Scheme 5.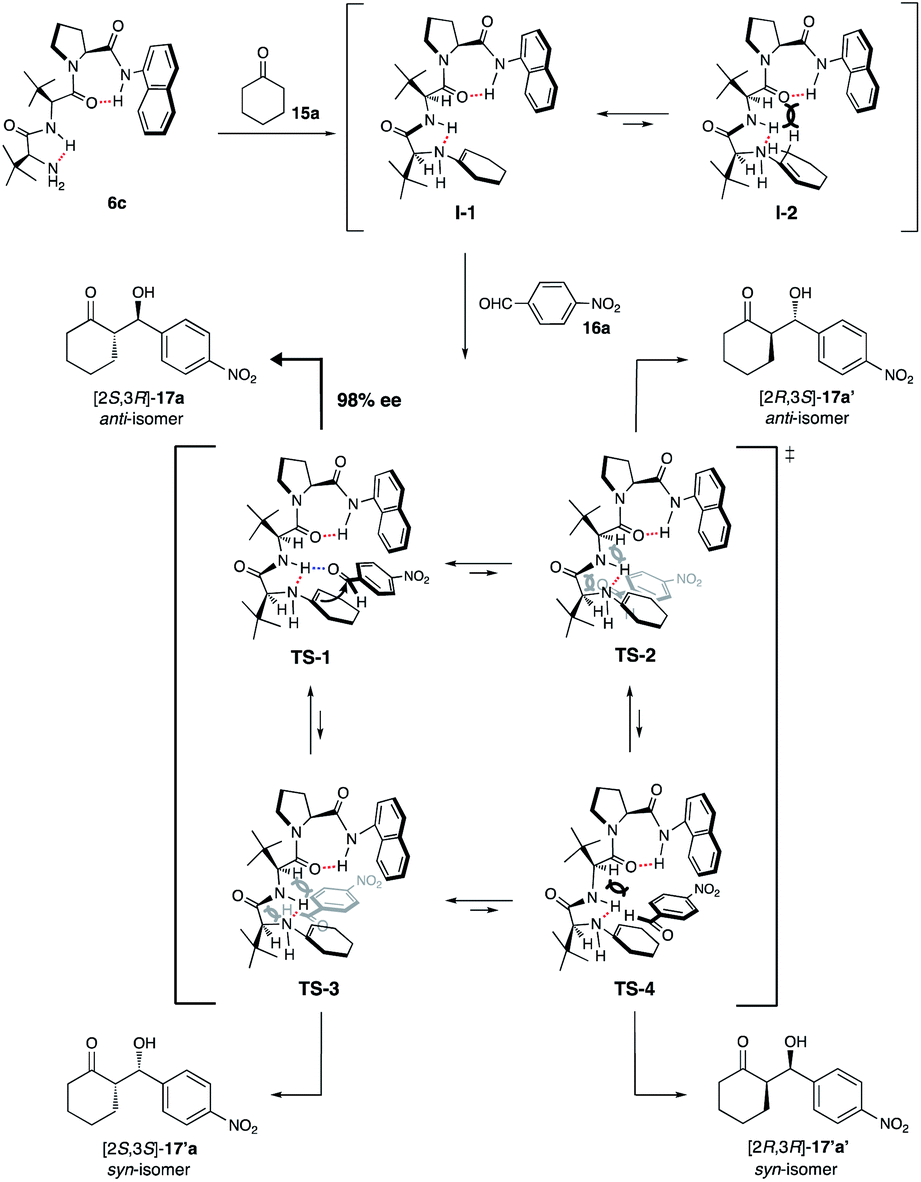 | ||
| Scheme 5 Plausible reaction course using catalyst 6c. | ||
First, the condensation of N-terminal primary amine peptide organocatalyst 6c with aldol donor 15a forms the enamine as intermediate I. The conformation of intermediate I is fixed by γ-turned intramolecular hydrogen bonding interactions between the amide carbonyl group at N-terminal side and the amide amino group at C-terminal side on pyrrolidine ring and also the amino hydrogen atom and the next amide amino group of organocatalyst species. Intermediate I might exist as I-1 which is having less steric interaction between dipeptide chain on N-terminal side and the enamine part than that of intermediate I-2. This expectation was supported by the conformational analyses using scan of total energies and DFT calculations for the enamine intermediates I-1 and I-2 and the result indicated that the conformation of I-1 is most stable (see the ESI† for details) (Fig. 1). Subsequently, more stable enamine intermediate I-1 might attack 4-nitrobenzaldehyde 16a through possible transition states TS-1 to TS-4 as shown in afford [2S,3R]-17a as a major product than those of TS-2, TS-3, Scheme 5. In the TS, the reaction might proceed through TS-1 to and TS-4, wherein the substrate 16a has steric interactions from N-terminal side and C-terminal side fixed by γ-turn intramolecular hydrogen bonding. On the other hand, aldol acceptor 16a might coordinate with amide hydrogen atom through hydrogen bonding at N-terminal side on I-1 species in TS-1. Subsequently, the enamine moiety attacks the enantiotopic face of 16a resulting 17a as the major isomer.
 | ||
| Fig. 1 Energy profile of the interconversion of 6c. | ||
3. Conclusion
We have developed new small γ-turn type N-primary amino terminal tripeptides organocatalysts 6a–j and their functionality as organocatalyst were examined in the asymmetric aldol reaction of various ketones 15a–h with different aromatic aldehydes 16a–j to afford the desired chiral aldol products 17a–s. All the catalysts showed catalytic activity in this reaction. Particularly, catalyst 6c showed good catalytic activity to afford the various chiral aldol products in satisfactory chemical yields and stereoselectivities (up to 99%, up to syn:anti/13:87 dr, up to 99% ee) under eco-friendly solvent-free condition.
4. Experimental
4.1 General information
Reagents and dry solvents were of the commercially available maximum grade and used without further purification. Reactions were performed under an inert atmosphere in flame dried and cooled glassware. The reaction progress was monitored by thin layer chromatography (TLC) using Merk silica plate gel 60 F254 aluminum sheet. The purification of products were carried out using column chromatography techniques in silica gel 60 N (40–50 μm) purchased from Kanto Chemical Company. Visualization of the products was confirmed by ultraviolet light, iodine vapor and ninhydrin stain. 1H and 13C NMR spectra were recorded on a JEOL JNM-ECA500 (1H for 500 MHz and 13C for 125 MHz). All the spectra were recorded at 21 °C. Chemical shifts (δ) are reported in parts per million (ppm) relative to the signals of tetramethylsilane (TMS) using the residual solvents signals. Report data for 1H NMR spectroscopy was reported as: chemical shift (δ ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quadruplet, dd = doublet of doublets, td = triplet of doublets, m = multiplet and br = broad), coupling constants (J) and assimilation were measured in hertz (Hz). Optical rotation was measured by JASCO DIP-360 polarimeter. The melting point was measured using a Yanaco micro melting point apparatus. High resolution mass spectra (HRMS) data was collected by electron impact (EI) using Hitachi RMG-GMG and JEOL JNK-DX303 sector instruments. The enantiomeric excess (ee) was determined using high pressure liquid chromatography (HPLC) principle by DAICEL CHIRALPAK AD-H, OD-H column.4.2 General procedure for the synthesis of peptide organocatalysts 6a–j, 10 and 14a–b
To a solution of N,N′-dicyclohexylcarbodiimide (DCC) (1.29 mmol) in dry CH2Cl2 (10 mL) were added N-Boc-L-amino acids 4′a–f (0.86 mmol) at 0 °C and the reaction mixture was stirred for 1 hour. After 1 h, corresponding dipeptides 5a–h, 9 and 13a–b (0.95 mmol) were added, and the reaction was stirred for another 24 h at rt. The insoluble by-product DCU was filtered off and the filtrate evaporated to dryness. The obtained oily residue was dissolved in CH2Cl2 and extracted sequentially with saturated aqueous NaHCO3 solution, aqueous HCl (1.0 M), and brine. The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under a reduced pressure to afford the crude products that were used for next step without further purification. To the solution of the crude products in dry CH2Cl2, TFA was added in dropwise over a period of times (0.4 mL) at 0 °C and successively stirred at room temperature (r.t.) for 4 h. After the reaction completion, DCM and TFA were removed under a reduced pressure, the residue was basified by drop-wise addition of saturated NaHCO3 solution at 0 °C and stirred for 1 h at r.t. The crude products were extracted with CHCl3, dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by flash column chromatography on SiO2 (CHCl3:MeOH = 99:1 to 95:5) to obtain the corresponding tripeptide catalysts 6a–j, 10 and 14a, 14b.
4.3 General procedure for the asymmetric aldol reaction of various ketones with aromatic aldehydes
The peptide catalyst 6c (30 mol%) was added to a solution of ketones 15a–h (0.4 mmol) and the aldehydes 16a–j (0.1 mmol) under the neat reaction condition. The reaction mixture was stirred at 0 °C for appropriate time until the reaction completion, monitored by thin layer chromatography (TLC). The reaction mixture was directly purified by flash column chromatography on SiO2 (n-hexane/CH3CO2Et) to afford the corresponding aldol products 17a–s.Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021, 14, 2785 CrossRef CAS PubMed; (b) V. V. Kouznetsov and P. Galvis, Tetrahedron, 2018, 22, 773 CrossRef; (c) Y. Nishikawa, Tetrahedron Lett., 2018, 59, 216 CrossRef CAS; (d) C. Yalamanchili, G. Amar, A. G. Chittiboyina, S. C. Kumar Rotte, J. A. Katzenellenbogen, W. G. Helferich, A. Ikhlas and I. A. Khan, Tetrahedron, 2018, 74, 2020 CrossRef CAS; (e) G. M. Ziarani, R. Moradi and N. N. Lashgari, Tetrahedron, 2018, 74, 1323 CrossRef; (f) C. E. Sleet, K. Uttam, U. K. Tambar and P. P. Maity, Tetrahedron, 2017, 73, 4023 CrossRef CAS; (g) M. M. Heravi, V. Zadsirjan, M. Dehghani and N. Hosseintash, Tetrahedron: Asymmetry, 2017, 28, 587 CrossRef CAS; (h) S. Ahmad, L. Shukla, J. Szawkalo, P. Roszkowski, K. Jan, J. K. Maurin and Z. Czarnocki, Catal. Commun., 2017, 89, 44 CrossRef CAS; (i) F. E. Held and S. B. Tsogoeva, Catal. Sci. Technol., 2016, 6, 64 RSC.
- (a) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef PubMed; (b) S. J. Miller, Acc. Chem. Res., 2004, 37, 601 CrossRef CAS PubMed; (c) J. D. Revell and H. Wennemers, Curr. Opin. Chem. Biol., 2007, 11, 269 CrossRef CAS PubMed; (d) D. R. Kelly and S. M. Roberts, Biopolymers, 2006, 84, 74 CrossRef CAS PubMed; (e) A. Berkessel, Curr. Opin. Chem. Biol., 2003, 7, 409 CrossRef CAS PubMed; (f) Y. Zhao, J. Rodrigo, A. H. Hoveyda and M. L. Snapper, Nature, 2006, 443, 67 CrossRef CAS PubMed; (g) C. A. Lewis and S. J. Miller, Angew. Chem., Int. Ed., 2006, 45, 5616 CrossRef CAS PubMed; (h) C. A. Lewis, A. Chiu, M. Kubryk, J. Balsells, D. Pollard, C. K. Esser, J. Murry, R. A. Reamer, K. B. Hansen and S. J. Miller, J. Am. Chem. Soc., 2006, 128, 16454 CrossRef CAS PubMed; (i) E. Delort, T. Darbre and J. L. Reymond, J. Am. Chem. Soc., 2004, 126, 15642 CrossRef CAS PubMed; (j) A. Clouet, T. Darbre and J. L. Reymond, Angew. Chem., Int. Ed., 2004, 43, 4612 CrossRef CAS PubMed; (k) F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2002, 124, 3510 CrossRef CAS PubMed; (l) P. Krattiger, C. McCarthy, A. Pfaltz and H. Wennemers, Angew. Chem., Int. Ed., 2003, 42, 1722 CrossRef CAS PubMed; (m) M. Wiesner, J. D. Ravell, S. Tonazzi and H. Wennemers, J. Am. Chem. Soc., 2008, 130, 5610 CrossRef CAS PubMed; (n) H. Wennemers, Chem. Commun., 2011, 47, 12036 RSC.
- (a) J. Oku and S. Inoue, Makromol. Chem., 1979, 180, 1089 CrossRef CAS; (b) J. Oku and S. Inoue, J. Chem. Soc., Chem. Commun., 1981, 229 RSC; (c) J. Oku, N. Ito and S. Inoue, Makromol. Chem., 1982, 183, 579 CrossRef CAS.
- (a) S. J. Miller, G. T. Copeland, N. Papaioannou, T. E. Horstmann and E. M. Ruel, J. Am. Chem. Soc., 1998, 120, 1629 CrossRef CAS; (b) E. R. Jarvomelissa, M. Vasbinder and S. J. Miller, Tetrahedron, 2000, 56, 9773 CrossRef; (c) S. M. Mennen, J. T. Blank, M. B. Tran-Dube, J. E. Imbriglio and S. J. Miller, Chem. Commun., 2005, 195 RSC.
- (a) O. Illa, O. Porcar-Tost, C. Robledillo, C. Elvira, P. Nolis, O. Reiser, V. Branchadell and R. M. Ortuno, J. Org. Chem., 2018, 83, 350 CrossRef CAS PubMed; (b) S. B. Tsogoeva and S. B. Jagtap, Synlett, 2004, 14, 2624 CrossRef; (c) S. B. Tsogoeva, S. B. Jagtap, Z. A. Ardemasova and V. N. Kalikhevich, Eur. J. Org. Chem., 2004, 19, 4014 CrossRef; (d) K. Yoshida, T. Shigeta, T. furuta and T. Kawabata, Chem. Commun., 2012, 48, 6981 RSC.
- (a) Z. H. Du, B. X. Tao, M. Yuan, W. J. Qin, Y. L. XU, P. Wang and C. S. Da, Org. Lett., 2020, 11, 4444 CrossRef PubMed; (b) G. Coombs, M. H. Sak and S. J. Miller, Angew. Chem., Int. Ed., 2019, 131, 2 CrossRef; (c) C. E. Grunerfelder, J. K. Kisunzu and H. Wennemers, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef; (d) K. Akagawa, T. Hirata and K. Kudo, Synlett, 2016, 27, 1217 CAS; (e) K. Akagawa, N. Sakai and K. Kudo, Angew. Chem., Int. Ed., 2015, 127, 1842 CrossRef; (f) X. Hu, D. Zhang, S. Zhang and P. Wang, RSC Adv., 2015, 5, 39557 RSC; (g) A. Bisticha, I. Triandafillidi and C. G. Kokotos, Tetrahedron: Asymmetry, 2015, 26, 102 CrossRef CAS; (h) Z. H. Du, W. Qin, B. Tao, M. Yuan and C. S. Da, Org. Biomol. Chem., 2013, 11, 1 RSC; (i) F. C. Wu, C. S. Da, Z. X. Du, Q. P. Guo, W. P. Li, L. Yi, Y. N. Jia and X. Ma, J. Org. Chem., 2009, 74, 4812 CrossRef CAS PubMed; (j) V. Delia, H. Zwicknagl and O. Reiser, J. Org. Chem., 2008, 73, 3262 CrossRef CAS PubMed; (k) J. D. Revell and H. Wennemers, Curr. Opin. Chem. Biol., 2007, 11, 269 CrossRef CAS PubMed; (l) A. Cordova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes and Y. Xu, Chem.–Eur. J., 2006, 12, 5383 CrossRef CAS PubMed; (m) P. Dziedzic, W. Zou, J. Hafren and A. Cordova, Org. Biomol. Chem., 2006, 4, 38 RSC; (n) J. Kofoed, J. Nielsen and J. L. Reymond, Bioorg. Med. Chem. Lett., 2003, 13, 2445 CrossRef CAS PubMed; (o) F. Tanaka and C. F. Barbas III, Chem. Commun., 2001, 769 RSC.
- (a) T. Ohashi and S. Hosokawa, Org. Lett., 2018, 10, 3021 CrossRef PubMed; (b) D. L. J. Clive and D. Liu, J. Org. Chem., 2009, 74, 4812 CrossRef PubMed; (c) B. Schetter and R. Mahrwald, Angew. Chem., Int. Ed., 2006, 45, 7506 CrossRef CAS PubMed; (d) Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, Germany, 2004, vol. 1 and 2 Search PubMed; (e) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassu, Chem. Rev., 2000, 100, 1929 CrossRef CAS PubMed.
- (a) M. G. Emma, A. Tamburrini, A. Martinelli, M. Lombardo, A. Quintavalla and C. Trombini, Catalysts, 2020, 10, 649 CrossRef CAS; (b) I. A. Owolabi, U. V. Subba Reddy, M. Chennapuram, C. Seki, Y. Okuyama, E. Kwon, K. Uwai, M. Tokiwa, M. Takeshita and H. Nakano, Tetrahedron, 2018, 76, 4705 CrossRef; (c) J. Dutta, N. Wakdikar and S. Tiwari, Org. Biomol. Chem., 2017, 15, 6746 RSC; (d) Z. Li, Y. Chen, C. Zheng, Y. Yin, L. Wang and X. Sun, Tetrahedron, 2017, 73, 78 CrossRef CAS; (e) V. Ashokkumar, C. Chithiraikumar and A. Siva, Org. Biomol. Chem., 2016, 14, 9021 RSC; (f) V. Ashokkumar, C. Chithiraikumar and A. Siva, Org. Biomol. Chem., 2016, 14, 9021 RSC; (g) C. L. Wu, X. K. Fu and S. Li, Eur. J. Org. Chem., 2011, 7, 1291 CrossRef; (h) C. L. Wu, X. K. Fu and S. Li, Tetrahedron, 2011, 67, 4283 CrossRef CAS; (i) C. L. Wu, X. K. Fu, X. B. Ma and S. Li, Tetrahedron: Asymmetry, 2010, 21, 2465 CrossRef CAS; (j) S. Guizzetti, M. Benaglia, L. Raimondi and G. Celentano, Org. Lett., 2007, 9, 1247 CrossRef CAS PubMed; (k) K. Sakthivel, W. Notz, T. Bui and C. F. Barbas III, J. Am. Chem. Soc., 2001, 123, 5260 CrossRef CAS PubMed.
- (a) V. D. Juarez, C. E. Escobedo, M. P. Venegas, E. Juaristi, S. P. Estrada, S. R. Lima and H. L. Ruiz, Tetrahedron, 2021, 79, 153 Search PubMed; (b) C. G. A. Ortiz, M. P. Venegas, J. V. Caporali and E. Juaristi, Tetrahedron, 2019, 60, 1749 CrossRef; (c) P. V. Milla, J. M. Sansano, C. Naajera, B. Fiser and E. G. Bengoa, Eur. J. Org. Chem., 2015, 12, 2514 Search PubMed; (d) J. G. Hernandez, V. García-Lopez and E. Juaristi, Tetrahedron, 2012, 68, 92 CrossRef CAS; (e) J. G. Hernandez and E. Juaristi, J. Org. Chem., 2011, 76, 1464 CrossRef CAS PubMed; (f) G. Ma, A. Bartoszewicz, I. Ibrahem and A. Cordova, Adv. Synth. Catal., 2011, 353, 3114 CrossRef CAS; (g) J. G. Hernandez and E. Juaristi, Tetrahedron, 2011, 67, 6953 CrossRef CAS; (h) N. Mase, Y. Nakai, N. Ohara, H. Yoda, K. Takabe, F. Tanaka and C. F. Barbas III, J. Am. Chem. Soc., 2006, 128, 734 CrossRef CAS PubMed; (i) P. Dziedzic, W. Zou, J. Hafren and A. Cordova, Org. Biomol. Chem., 2006, 4, 38 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08635a |
| This journal is © The Royal Society of Chemistry 2021 |