
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of vinyl-1,2,3-triazole derivatives under transition metal-free conditions†
Menghan Cui a,
Changhui Suc,
Rong Wanga,
Qing Yangb and
Chunxiang Kuang*a
a,
Changhui Suc,
Rong Wanga,
Qing Yangb and
Chunxiang Kuang*a
aShanghai Key Lab of Chemical Assessment and Sustainability, School of Chemical Science and Engineering, Tongji University, Shanghai 200092, China. E-mail: kuangcx@tongji.edu.cn
bState Key Laboratory of Genetic Engineering, Department of Biochemistry, School of Life Sciences, Fudan University, Shanghai 200438, China
cSchool of Chemistry and Life Science, Nanjing University, Jinling College, Nanjing 210089, China
First published on 6th December 2021
Abstract
Herein, we describe a novel and green route for the direct synthesis of vinyl triazole derivatives with alkynes and triazoles promoted by an inorganic base under transition metal-free conditions. The base shows great catalytic activity for the anti-Markovnikov stereoselective hydroamination of alkynes. Moreover, good yields with excellent functional group tolerance are successfully achieved for a range of substrates, including aryl and heteroaryl groups, terminal alkynes and internal alkynes, and various triazole derivatives. This work presents an advanced concept for the synthesis of alkenyl triazole with a versatile and cost-efficient approach.
Introduction
1,2,3-Triazole compounds are an important class of nitrogen-containing heterocyclic compounds that are often introduced into existing drugs or lead compounds as pharmacodynamic groups to improve their pharmacological activity.1,2 In addition, their derivatives have a variety of biological activities, such as antibacterial,3 anti-malaria,4 anti-HIV,5 anti-fungal,6 anticancer7 and anti-inflammatory,8 activities (Fig. 1), which are indispensable in the field of new drug research and development.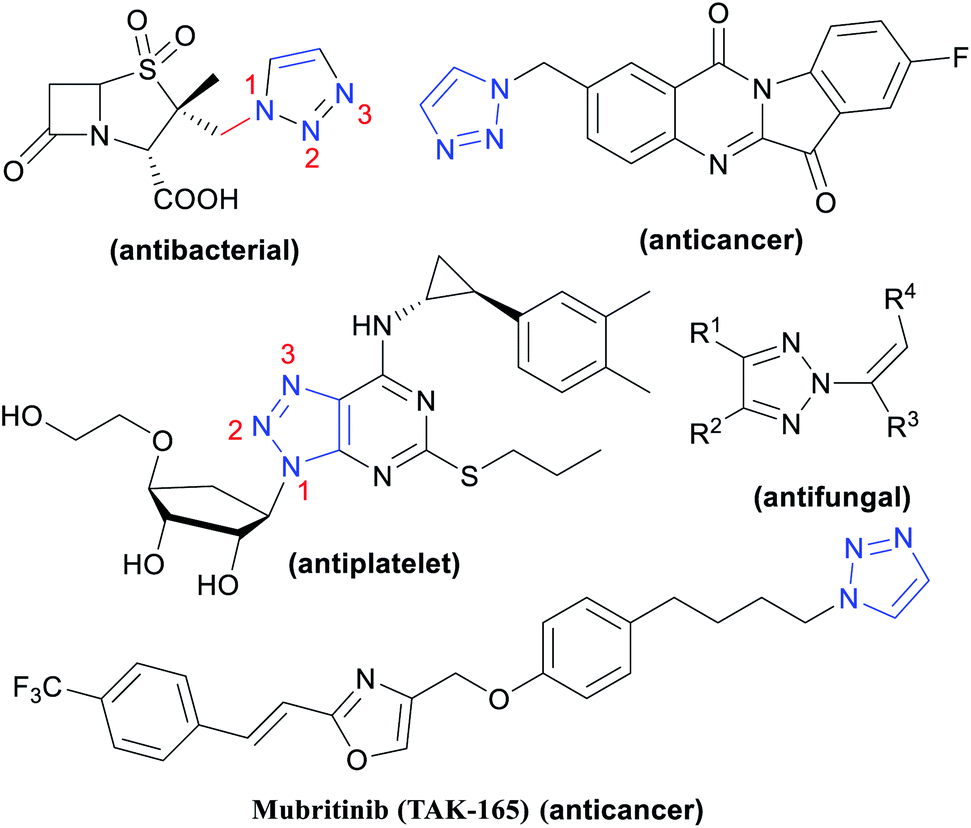 | ||
| Fig. 1 Biologically active compounds featuring 1,2,3-triazole. | ||
When vinyl is used as a substituent in triazole, its desirable features including aromaticity, acid–base stability, large dipole moment, structural diversity of substituents, vinyl functional groups and regioisomers have attracted increasing attention.9 Therefore, vinyl-1,2,3-triazole is used as an important precursor for functionalized polymers in industry. The industrial production of electron-rich polymers can be realized by using the properties of high activity and easy polymerization, which are widely used in the production of polymerizable functional materials and polymer materials.10 Therefore, vinyl-1,2,3-triazole shows great application potential in the application of cations in biomaterials, ion exchange membranes, drug delivery, and metal-containing polymers for depollution and metal recovery.11
At present, a well-known method for the synthesis of 1,2,3-triazole compounds, azide–alkyne click chemistry (CuACC reaction), is considered to be the most extensive approach and has the characteristics of simple operation, high yields and compatibility with functional groups (Scheme 1a).10d Such methodologies rely on Cu-catalyzed cycloaddition reactions as a key step and form vinyl substituents through elimination or Wittig-type reactions (Scheme 1c).12 However, this method, which uses organic azide or sodium azide as raw materials for synthesis, is potentially explosive and is not suitable for large-scale industrial production.13 Therefore, there is academic and practical significance for the development of alternative methods other than azide–alkyne click chemistry. Vinyl triazole has been prepared using diazonium as a raw material. Yan prepared a variety of N-vinyl-1,2,3-triazole monomers from 2-aminoethanol and α-diazo-β-oxoamides through modified Wolff's cyclocondensation reactions (Scheme 1d). This method enjoys not only the advantages of Wolff's 1,2,3-triazole cyclocondensation but also replacement for click chemistry due to its high yields and feasibility.9 To date, 1,2,3-triazole as a substrate is also an effective method for the direct synthesis of triazole derivatives (Scheme 1b).14 Kizhnyaev reported that 1,2,3-triazole and alkenyl compounds are directly reacted to synthesize N-vinyl-1,2,3-triazole derivatives under the catalysis of mercury acetate. Unfortunately, a toxic mercury acetate catalyst is used in the reaction, and the reaction easily generates unknown nitro byproducts.15 Duan, Zhao studied the use of precious metal complexes to add 1,2,3-triazole to alkynes (Scheme 1e and f).16 Besides, alkali metal hydroxides as have already been employed for the addition of nitrogen derivatives to alkynes, such as anilines, imidazoles and pyrroles.17 However, the synthesis of alkenyl triazoles without transition metal promoted has not yet been studied.
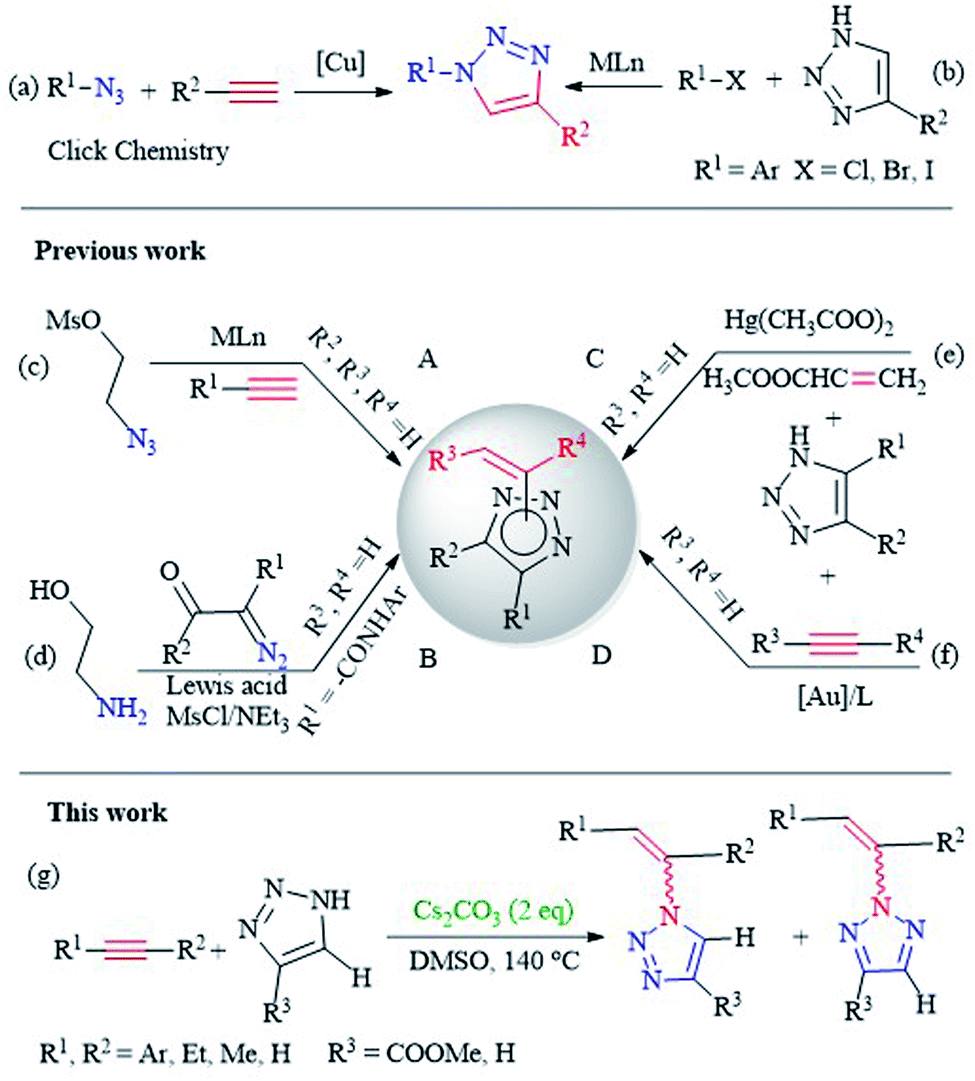 | ||
| Scheme 1 Previous and present synthesis of vinyl-1,2,3-triazole. | ||
Our research is mainly devoted to the development of environmentally friendly and waste minimization procedures without metal conversion and aims to report the transition metal-free hydrogenation amination of 1,2,3-triazole on alkynes.
Results and discussion
To commence our studies, reaction conditions were examined in the reaction of 1,2,3-triazole (2a) with aryl acetylene (1a) (Table 1). Initially, the reaction was screened in DMSO under the catalyzed conditions for the organic bases piperidine and triethylamine, but no reaction was observed at 120 °C for 24 hours (Table 1, entries 1 and 2). Then, the reaction was explored in the presence of KOH, EtONa and Cs2CO3 under the abovementioned conditions (Table 1, entries 3–5). The results show that with Cs2CO3 (2 eq.) as the base catalyst, the yield is 73%, and the 3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a ratio is equal to 88:12 (Table 1, entry 5). Unsubstituted 1,2,3-triazole is well known for rapid tautomerization. When 1H-1,2,3-triazole was used, mixtures of tautomeric addition products were obtained, mainly N-1 substituted products (Scheme 2). When the reaction equivalent of Cs2CO3 was changed, the yield was significantly reduced (Table 1, entries 6–8).
:4a ratio is equal to 88:12 (Table 1, entry 5). Unsubstituted 1,2,3-triazole is well known for rapid tautomerization. When 1H-1,2,3-triazole was used, mixtures of tautomeric addition products were obtained, mainly N-1 substituted products (Scheme 2). When the reaction equivalent of Cs2CO3 was changed, the yield was significantly reduced (Table 1, entries 6–8).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Base | T (°C) | Yieldb (%) | 3aa:4aac |
| a Reactions were performed by using 1a (0.5 mmol), alkyne 2a (1 mmol), and base (0.05–1 mmol) in solvent (2 mL) at 80–150 °C for 12 h.b Yield of mixture of isolated product.c Regioselective isomer ratio. | |||||
| 1 | Pyridine (2 eq.) | DMSO | 120 | None | — |
| 2 | Et3N (2 eq.) | DMSO | 120 | None | — |
| 3 | KOH (2 eq.) | DMSO | 120 | 21 | 4753 |
| 4 | EtONa (2 eq.) | DMSO | 120 | 42 | 8020 |
| 5 | Cs2CO3 (2 eq.) | DMSO | 120 | 73 | 8812 |
| 6 | Cs2CO3 (0.1 eq.) | DMSO | 120 | 22 | 4456 |
| 7 | Cs2CO3 (1 eq.) | DMSO | 120 | 47 | 6931 |
| 8 | Cs2CO3 (1.5 eq.) | DMSO | 120 | 54 | 8614 |
| 9 | Cs2CO3 (2 eq.) | DMSO | 140 | 92 | 8119 |
| 10 | Cs2CO3 (2eq.) | DMSO | 150 | 69 | 7327 |
| 11 | Cs2CO3 (2 eq.) | DMSO | 100 | 6 | — |
| 12 | Cs2CO3 (2 eq.) | DMSO | 80 | 14 | — |
| 13 | Cs2CO3 (2eq.) | DMF | 140 | 59 | 8515 |
| 14 | Cs2CO3 (2eq.) | NMP | 140 | 24 | — |
| 15 | Cs2CO3 (2eq.) | DMA | 140 | 65 | 8614 |
| 16 | — | DMSO | 140 | None | — |

 | ||
| Scheme 2 Tautomerization in unsubstituted 1,2,3-triazole. | ||
We examined the influence of different temperatures on the formation of 3a and 4a (Table 1, entries 9–12). To our surprise, a satisfactory result was achieved when Cs2CO3 was used as the base at 140 °C (92% yield, 3aa:4aa = 81:19; Table 1, entry 9). Then, various solvents, such as N,N-dimethylformamide (DMF), N-methyl pyrrolidone (NMP) and N,N-dimethylacetamide (DMA), were tested (Table 1, entries 13–15), and the yields were not significantly improved. In a control experiment, no product was detected in the absence of the base, as expected (Table 1, entry 16). Finally, the best reaction conditions were determined to be those shown in entry 9 (Table 1).
With the optimized conditions in hand, we then explored the substrate scope for synthesis of vinyl-1,2,3-triazole derivatives via a variety of aromatic alkynes and triazole derivatives. In all cases, anti-Markovnikov products were formed, and alkynes with electron-rich and electron-deficient substituents were well tolerated (Scheme 3). Alkynes containing electron-donating groups on the phenyl ring gave the corresponding better yield. With the enhancement of electron donation, products were obtained in moderate to good yields (Scheme 3, 3/4ba–da, 78%, 89% and 92% yield). The N-1/N-2 ratio for the addition products was approximately 80/20 in terms of location selectivity. When alkynes with electron-drawing groups were used, the reaction proceeded with moderate conversion values (Scheme 3, 3/4ea–ga, 61%, 69% and 48% yield). However, the N-1/N-2 ratio for the addition products with electron withdrawing groups trends towards the conversion of N-2-vinyl-triazole. Interestingly, the addition of alkyne with stronger electron withdrawing substituents –CF3 and –CN on the phenyl ring afforded E-isomer products completely (Scheme 3, 3/4ha–ia, 91% and 96% yield), presumably due to the decreased electron density at the distal end of the C–C bond. The cyano-substituted product was regioselectively obtained as the N-2 substitution product. We can see that the strength of the electron withdrawing group on the phenyl ring directly affects the stereoselectivity and regioselectivity of the product. Similarly, the addition product for alkyne with disubstituted trifluoromethyl groups only produces N-2-vinyl-triazole, and the yield is as high as 94% (Scheme 3, 4ma). We also investigated the effect of substituting substrates at different positions on the phenyl ring. The yield of the meta-substituent alkyne addition product is relatively low compared to the para-substituent alkyne (Scheme 3, 3/4ja–la, 85%, 82% and 59% yield). In addition to aryl acetylenes, we also studied the reaction of aliphatic alkynes with triazoles. Alkynes containing ester groups completely generate trans products with poor position selectivity (Scheme 3, 3/4pa, 82% yield).
 | ||
| Scheme 3 1,2,3-Triazolation of terminal alkynes. | ||
The addition of heteroaromatic alkynes was studied for their potential applications in bioactive molecules and synthetic drugs. When the reaction was conducted using bulkier 2-ethynyl-naphthalene, products were obtained in 68% yield with N-1-vinyl-triazole regioselectivity (Scheme 3, 3/4na). Consistent with above results, alkyne with electron-donating thiophene group gave completely Z-stereoselective and N-1-regioselective adducts (Scheme 3, 3oa).
N-Functionalised azoles are important molecular scaffolds for pharmaceuticals and natural products, show excellent biological activity. To our delight, we used internal alkynes as the source of alkynes to synthesis with triazole derivatives and got better results. At first, diphenylacetylene and bis(4-bromophenyl) acetylene were reacted with 1,2,3-triazole lead to the corresponding Z-products 5qa/5qa′ and 5ra/5ra′ in good yields (Scheme 4, 92% and 66% yield). The reaction afforded preferentially the N-1 addition product 5qa, the N-2 tautomer product 5qa′ being minor. The ethyl 2-butynoate with 1,2,3-triazole only afforded E-isomers, but the yield is 13% (Schemes 4 and 5 sa).
 | ||
| Scheme 4 Triazolation of internal alkynes and alkenylation of triazole. | ||
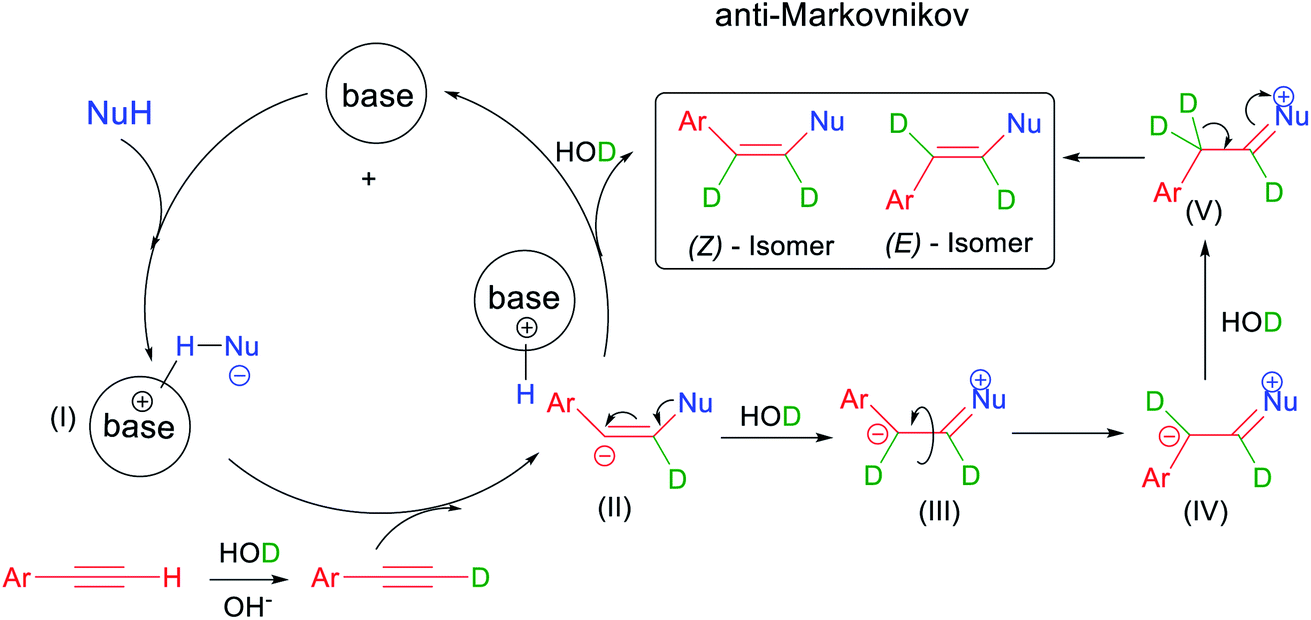 | ||
| Scheme 5 Proposed reaction mechanism. | ||
To expand the universality, a variety of triazole substrates were examined in the addition reaction under these optimized reaction conditions. We were delighted to find that the addition reaction of 1,2,4-triazole to phenylacetylene and diphenylacetylene proceeded smoothly to give the Z-isomers in good to excellent yields (Scheme 4 5ab, 5qb, 93% and 61% yield). Subsequently, methyl 1,2,3-triazole-4-carboxylate was also tested as a substrate and afforded the desired products with ethyl propiolate. This result is consistent with the previous conclusion that ester groups as alkynes afford E-isomer products in moderate yields (Scheme 4 5pb, 5pe, 40% and 66% yield). However, reaction with aromatic acetylenes did not give products.
To explore the reaction mechanism, we used DMSO-d6 as a solvent to mark the styryl position of compound 1a/h. The results show that the source of the styrene matrix proton in the addition reaction is H in the solvent. Therefore, we preliminarily deduced the possible reaction mechanism: the base can efficiently activate N-nucleophiles and DMSO-d6 under alkaline conditions. Then, kinetically stable alkenyl anion III is initiated by the attack of ion pair I on alkyne-d6, prioritizing conversion to the Z-isomer. In addition, intermediate II is converted into intermediate III after deuterium substitution due to migration of the triazole lone pair toward the adjacent carbon. Intermediate V was formed via bond transformation and deuteration. Furthermore, the migration of the triazole lone pair for intermediate V generated the compound E-isomer. The transformation of Z-isomers and E-isomers proceeds in an anti-Markovnikov fashion. The possible reason for this is that intermediate III is more stable.
Conclusions
In summary, an efficient base-mediated stereoselective and regioselective addition method for imidazoles (electron-deficient heterocycles) onto alkynes involves the synthesis of a broad range of functionalized vinyl triazole derivatives. This approach utilizes a simple and economical basic system composed of Cs2CO3/DMSO for the addition of terminal and internal alkynes, avoiding the use of expensive catalysts and ligands. The catalytic system, which is low cost, benign and easily available, makes this method of potential practical value. The present process represents a safe, green and atom-economical alternative approach for existing synthesis methods to assemble a variety of functional vinyl triazole derivatives.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Center for Instrumental Analysis, Tongji University, China.Notes and references
- (a) P. Wu and V. V. Fokin, Aldrichimica Acta, 2007, 40, 7–17 CAS; (b) K. Kacprzak, I. Skiera, M. Piasecka and Z. Paryzek, Chem. Rev., 2016, 116, 5689–5743 CrossRef CAS PubMed.
- (a) G. Schneider, Nat. Rev. Drug Discovery, 2010, 9, 273–276 CrossRef CAS PubMed; (b) H. M. Li, F. O. Cheng, A. M. Duft and A. Adronov, J. Am. Chem. Soc., 2005, 127, 14518–14524 CrossRef CAS PubMed; (c) T. Gadzikwa, O. K. Farha, C. D. Malliakas, M. G. Kanatzidis, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 13613–13615 CrossRef CAS PubMed; (d) S. E. Motika, Q. Wang, X. Ye and X. Shi, Org. Lett., 2015, 17, 290–293 CrossRef CAS PubMed.
- (a) S. Eswaran, A. V. Adhikari and N. S. Shetty, Eur. J. Med. Chem., 2009, 44, 4637–4647 CrossRef CAS PubMed; (b) Y.-L. Fan, X. Ke and M. Liu, J. Heterocycl. Chem., 2018, 55, 791–802 CrossRef CAS.
- R. Gujjar, A. Marwaha, F. El Mazouni, J. White, K. L. White, S. Creason, D. M. Shackleford, J. Baldwin, W. N. Charman, F. S. Buckner, S. Charman, P. K. Rathod and M. A. Phillips, J. Med. Chem., 2009, 52, 1864–1872 CrossRef CAS PubMed.
- B. A. Johns, J. G. Weatherhead, S. H. Allen, J. B. Thompson, E. P. Garvey, S. A. Foster, J. L. Jeffrey and W. H. Miller, Bioorg. Med. Chem. Lett., 2009, 19, 1802–1806 CrossRef CAS PubMed.
- D. Dheer, V. Singh and R. Shankar, Bioorg. Chem., 2017, 71, 30–54 CrossRef CAS PubMed.
- (a) Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848–2866 RSC; (b) Y. Y. Li, S. N. Zhang, R. Wang, M. H. Cui, W. Liu, Q. Yang and C. X. Kuang, Bioorg. Med. Chem. Lett., 2020, 30, 127–159 Search PubMed; (c) R. Wang, M. H. Cui, Q. Yang and C. X. Kuang, Synthesis, 2021, 53, 978–982 CrossRef CAS.
- E. Caselli, C. Romagnoli, R. Vahabi, M. A. Taracila, R. A. Bonomo and F. Prati, J. Med. Chem., 2015, 58, 5445–5458 CrossRef CAS PubMed.
- Z. K. Wang, Y. H. Tao, Z. Wang and J. L. Yan, Polym. Chem., 2016, 7, 3172–3178 RSC.
- (a) E. V. Khavula, V. A. Kuznetsov, V. N. Verezhnikov and G. V. Shatalov, Polym. Sci., Ser. B, 2003, 45, 26–31 Search PubMed; (b) N. A. Tsypina, V. N. Kizhnyaev, F. A. Pokatilov and A. I. Smirnov, Polym. Sci., Ser. B, 2003, 45, 41–44 Search PubMed; (c) V. N. Kizhnyaev, F. A. Pokatilov and L. I. Vereshchagin, Polym. Sci., Ser. C, 2008, 50, 1–21 CrossRef; (d) S. Beghdadi, I. A. Miladi, D. Addis, H. Ben Romdhane, J. Bernard and E. Drockenmuller, Polym. Chem., 2012, 3, 1680–1692 RSC.
- (a) C. J. Hawker and K. L. Wooley, Science, 2005, 309, 1200–1205 CrossRef CAS PubMed; (b) F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Haw ker, Nat. Chem., 2010, 2, 207–212 CrossRef CAS PubMed; (c) B. J. Adzima, S. C. Taylor, H. He, D. R. Luebke, K. Matyjaszewski and H. B. Nulwala, J. Polym. Sci., Part A: Polym. Chem., 2013, 52, 417–423 CrossRef; (d) C. Ye, G. L. Gard, R. W. Winter, R. G. Syvret, B. Twamley and J. n. M. Shreeve, Org. Lett., 2007, 9, 3841–3844 CrossRef CAS PubMed.
- K. Takizawa, H. Nulwala, R. J. Thibault, P. Lowenhielm, K. Yoshinaga, K. L. Wooley and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2897–2912 CrossRef CAS.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2010, 44, 5188–5240 CrossRef PubMed.
- S. Ueda, M. Su and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8944–8947 CrossRef CAS PubMed.
- V. N. Kizhnyaev, F. A. Pokatilov, N. A. Tsypina, G. V. Ratovskii and A. I. Smirnov, Russ. J. Org. Chem., 2002, 38, 1056–1059 CrossRef CAS.
- (a) H. Duan, W. Yan, S. Sengupta and X. Shi, Bioorg. Med. Chem. Lett., 2009, 19, 3899–3902 CrossRef CAS PubMed; (b) C. Sun, X. Yuan, Y. Li, X. Li and Z. Zhao, Org. Biomol. Chem., 2017, 15, 2721–2724 RSC.
- D. Tzalis, C. Koradin and P. Knochel, Tetrahedron Lett., 1999, 40, 6193–6195 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra08322h |
| This journal is © The Royal Society of Chemistry 2021 |