
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDFT study on the adsorption of 5-fluorouracil on B40, B39M, and M@B40 (M = Mg, Al, Si, Mn, Cu, Zn)†
Li Zhanga,
Zi-Dan Qia,
Ya-Ling Yea,
Xiang-Hui Libc,
Jing-Hua Chen a and
Wei-Ming Sun*a
a and
Wei-Ming Sun*a
aFujian Key Laboratory of Drug Target Discovery and Structural and Functional Research, The School of Pharmacy, Fujian Medical University, Fuzhou 350108, People's Republic of China. E-mail: sunwm@fjmu.edu.cn
bThe School of Medical Technology and Engineering, Fujian Medical University, Fuzhou 350004, Fujian, People's Republic of China
cKey Laboratory of OptoElectronic Science and Technology for Medicine of Ministry of Education, Fujian Provincial Key Laboratory of Photonics Technology, Fujian Normal University, Fuzhou 350007, Fujian, People's Republic of China
First published on 13th December 2021
Abstract
Based on density functional theory, the adsorption behavior of 5-fluorouracil (5-Fu) on B40 and its derivatives has been explored. It was observed that 5-Fu prefers to combine with the corner boron atom of the B40 cage via one of its oxygen atoms, forming a strong polar covalent B–O bond. The adsorption energy of 5-Fu on B40 was calculated to be −11.15 kcal mol−1, and thus, it can be duly released from B40 by protonation in the slightly acidic environment of tumor tissue, which makes for reducing the toxic and side effects of this drug. Additionally, the substituent and embedding effect of Mg, Al, Si, Mn, Cu, and Zn atoms on the drug delivery performance of B40 have been also considered. We hope this work could offer some implications for the potential application of boron-based nanomaterials, such as B40 in drug delivery.
1. Introduction
It is estimated that there will be more than 1.9 million new cases of colorectal cancer (CRC) and it will lead to 0.93 million deaths in 2020, representing about one in 10 cancer cases and deaths.1 Since 5-fluorouracil (5-Fu) was discovered in 1957,2 it has been widely used as a first-line therapy in colorectal cancer.3 As an analogue of uracil, 5-Fu interferes with nucleoside metabolism during DNA and RNA metabolism to produce cytotoxicity, leading to the death of tumor cells. Thus, this drug has been extensively used in chemotherapy for treating colorectal, liver, breast, pancreatic, gastric cancer, and squamous cell carcinomas arising in the head and neck.4–6 Despite the wide application of 5-Fu, its therapeutic value is also accompanied by increasing drug resistance and serious side effects, such as myelotoxicity, neurotoxicity, and cardiotoxicity, etc.7 Therefore, several efforts have been devoted to enhance the bioavailability of 5-Fu and reduce its side effects in the past decades.8–10 Among them, the introduction of delivery carriers has attracted great attention because it can effectively control the release of drug into diseased tissues, which can not only reduce the drug toxicity but also improve the therapeutic efficiency.11 For example, the adsorption of 5-Fu on the B36 nanosheet and metalloborospherenes has been detailedly investigated by Shakerzadeh12,13 in recent years. Accordingly, it is of great importance to explore more proper drug delivery systems for the 5-Fu drug.Among miscellaneous nanostructures, fullerene and its derivatives are known as suitable candidates for drug delivery owing to their high loading capability, less side effects, and high efficiency to pass the cell membrane.14–17 However, the biological application of nanocarbon fullerene family is limited by their inherent hydrophobic characteristics18 and possible toxicity.19 To overcome these disadvantages, tremendous endeavors have been devoted to searching for novel water-soluble fullerene-like carriers composed of non-carbon elements, such as MgnOn,20,21 BxNy,22–25 etc. More recently, an intriguing all-boron fullerene B40 was successfully synthesized by Zhai et al.,26 which has been extensively used in many fields, including gas sensing, hydrogen storage and separation applications and drug delivery.27–30 Besides its unique physical and chemical features, this intriguing B40 cage also possesses high stability due to its double aromatic character.31 Therefore, it is expected that the B40 and its derivatives could serve as a new kind of drug delivery materials.
Inspired by the above information, the interaction between 5-Fu and B40 nanocage was systematically investigated by density functional theory (DFT) calculations to study the potential of B40 as a new drug delivery vehicle for 5-Fu in this work. Our results show that 5-Fu is adsorbed on B40 by chemisorption with the adsorption energy of −11.15 kcal mol−1. Meanwhile, the drug can be easily released from the B40 surface by protonation in the slightly acidic microenvironment of tumor tissue, which denotes that B40 cage can be used as a nice carrier to deliver the 5-Fu drug. Moreover, the adsorption behavior of 5-Fu drug on M@B40 and B39M (M = Mg, Al, Si, Mn, Cu, and Zn) have been also examined to reveal the influence of foreign atoms on the adsorption performance of B40 for this drug. These six foreign atoms were selected because they are either harmless trace elements in human body or the commonly used dopants in improving the performance of delivery systems.11,32–36 Due to the changes of electronic properties of encapsulated M@B40 and substituted B39M cages, it is believed that such embedding and substituent strategies could be used to regulate the drug adsorption on B40. Hence, we hope this work could not only provide a thorough understanding on the drug delivery performance of B40 and its derivatives, but also offer some clues for seeking more effective boron-based nanomaterials to deliver antitumor drugs.
2. Computational details
The neutral geometric structures of 5-Fu, B40, M@B40, and B39M as well as their complexes 5-Fu@B40, 5-Fu@[M@B40], and 5-Fu@B39M (M = Mg, Al, Si, Cu, Mn, and Zn) with all real frequencies were optimized by using the B3LYP method in conjunction with the SDD effective core potential and corresponding basis set for Cu, Mn, Zn atoms and 6-31G(d) basis set for the other atoms. The functional B3LYP has been widely used for studying boron clusters and was extensively used in many reported works.37,38 By considering different spin states in structural optimization, the quartet and quintet structures are found to be the most stable configurations of Mn@B40 and MnB39 cages as well as their complexes with 5-Fu, respectively (see Table S1†), whereas the rest nanocages and their complexes are all stabilized at the lowest spin state on their respective potential energy surface (i.e., singlet states for closed shell systems and doublet states for open shell systems). In this work, the self-consistent reaction field (SCRF) with the solvation model based on density (SMD)39 was used to consider the solvent effect of water and dispersion corrected DFT-D3 was employed to consider the long-range interactions.40The enthalpy change (ΔH) and Gibbs free energy change (ΔG) for the adsorption of 5-Fu on these nanocages were computed under 298 K and 1 atm at B3LYP-D3/6-31G(d)&SDD level. The adsorption behaviors, electronic properties, and quantum theory of atoms in molecule (QTAIM), localized molecular orbitals (LMOs), and electron localization function (ELF) of these studied complexes were systemically investigated at the SMD-B3LYP-D3/6-311+G(d, p)&SDD level. The adsorption energy (Ead) of 5-Fu drug on the surface of each nanocage was calculated by the following equation,
| Ead = E5-Fu@cage − Enanocage − E5-Fu + EBSSE | (1) |
| Eint = E5-Fu@cage − Ecage in complex − E5-Fu in complex | (2) |
| Edef = Ecagedef + Edrugdef = (Ecage in complex − Ecage) + (E5-Fu in complex − E5-Fu) | (3) |
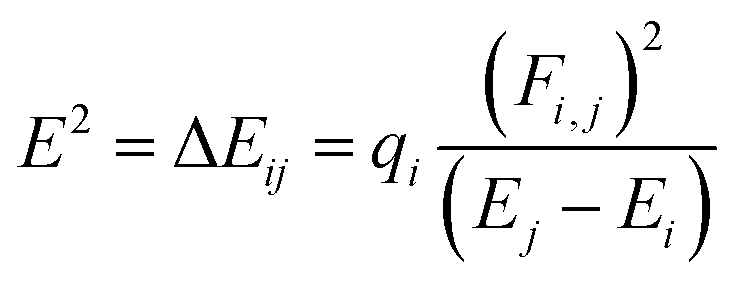 | (4) |
All the calculations were performed by using Gaussian 16 software.44 Dimensional plots of molecular configurations, electrostatic potential (ESP), and frontier molecular orbital diagrams were generated with the GaussView program.45 To reveal the bonding nature of the linkage bonds between 5-Fu and nanocages in the resulting 5-Fu@cage complexes, WBI, QTAIM, LMOs, and ELF analyses were carried out by using Multiwfn program package46 based on the output files from Gaussian software.
3. Results and discussion
3.1 Adsorption of 5-Fu on B40
Firstly, the optimized geometric structures and electrostatic potential (ESP) plots of 5-Fu and B40 are shown in Fig. 1. It is observed from Fig. 1a that the lengths of N3–C6, C6–O2, and C6–N4 bonds are 1.392 Å, 1.216 Å, and 1.390 Å, respectively, which are in good agreement with the corresponding experimental values of 1.408 Å, 1.233 Å, and 1.385 Å.47 The good consistency between the calculated parameters and corresponding experimental values demonstrates that the method and basis set used in this work are reliable.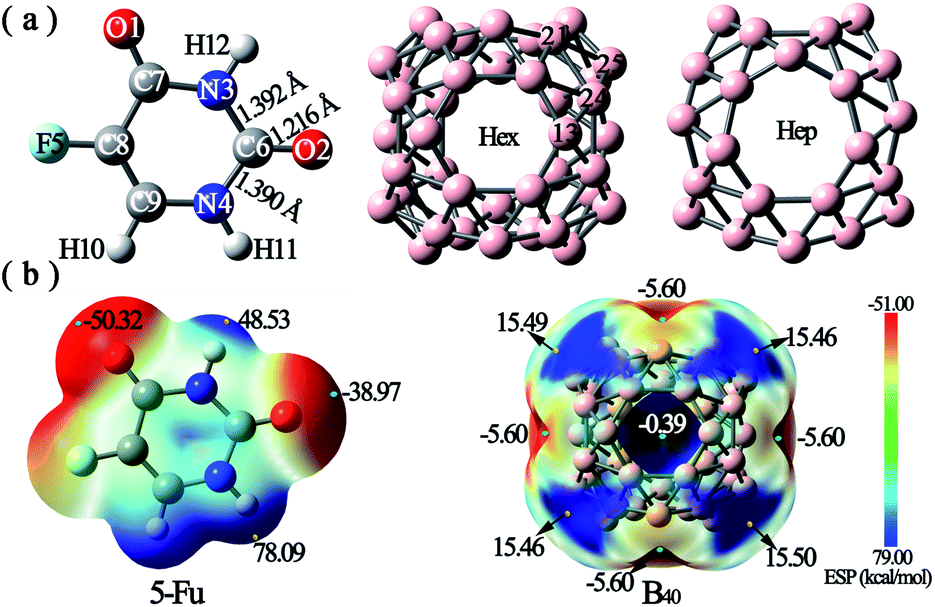 | ||
| Fig. 1 (a) Optimized structures and (b) electrostatic potential (ESP) plots of 5-Fu and B40. The ESP values of several critical points are also given in the ESP figures. | ||
As shown in Fig. 1b, the ESP plot of 5-Fu shows that the H atom connected with N4 possesses the largest positive potential of 78.09 kcal mol−1, while the negative electrostatic potentials are mainly distributed around O1 and O2 atoms. This indicates that the oxygen atoms of 5-Fu drug are more suitable for nucleophilic attack, while its hydrogen atoms tend to attack the electron-rich region. As for B40 cluster, the heptagonal holes are the reactive sites for electrophilic attack with ESP of −5.60 kcal mol−1, whereas the corner sites with positive electrostatic potentials of ca. 15.5 kcal mol−1 tend to be attacked by the oxygen atoms of 5-Fu drug. Hence, different possible initial configurations are considered by approaching the oxygen atoms of 5-Fu drug to the surface region with positive ESP of B40 cage to obtain the most stable structure of 5-Fu@B40. After full optimization, six low-lying isomers of 5-Fu@B40 complexes, which are named A, B, C, … according to their increasing energies are presented in Fig. 2.
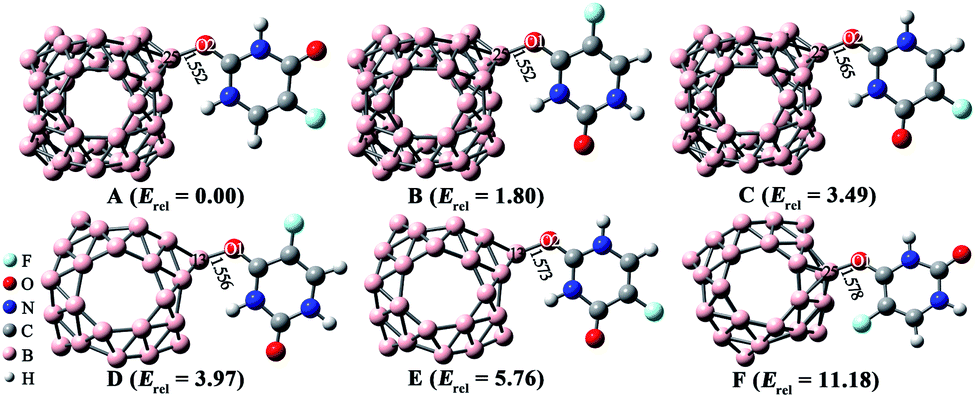 | ||
| Fig. 2 Optimized geometries of low-lying 5-Fu@B40 complexes. The lengths of newly formed bonds (in Å) and zero-point energy (ZPE)-corrected relative energies (Erel, in kcal mol−1) are also given. | ||
As implied in Fig. 2, among these six low-lying isomers, 5-Fu always combines with B40 via its oxygen atoms. To be specific, in complexes A and C, the O2 atom of 5-Fu interact with the B25 atom of B40 cluster. Herein, the length of newly formed B–O bond in isomer A is 1.552 Å, which is a bit shorter than the corresponding bond of 1.565 Å for isomer C. This is because that the H atom connected with N4 atom has the most positive ESP value, and thereby, it is more likely to attack the hexagonal holes of B40, forming the most stable isomer A. As for isomers B and F, 5-Fu is connected to B25 atom of B40 via O1 atom. Though the length of B–O bond (1.552 Å) in isomer B is equal to that in A, the isomer A is more stable by 1.80 kcal mol−1 than isomer B, indicating that B40 cage prefers to interact with the O2 site rather than the O1 atom of 5-Fu. Similar phenomenon is also observed for 5-Fu adsorbed on the B24N24 cage reported in previous works.48 Differently, isomer D is obtained by linking O1 atom of 5-Fu drug to the B13 atom of B40, while the isomer E is formed by attaching O2 atom of drug to the B13 atom of B40. The newly formed B–O bonds in isomers D and E are 1.556 and 1.573 Å, respectively.
In order to evaluate whether B40 cage could be used as a good carrier for drug delivery of 5-Fu, the adsorption behavior of 5-Fu on B40 has been analyzed on the basis of most stable isomer A of 5-Fu@B40. Hereafter, 5-Fu@B40 always refers to its isomer A. The adsorption energy (Ead), interaction energy (Eint), deformation energy (Edef), enthalpy change (ΔH), and Gibbs free energy change (ΔG) of 5-Fu@B40 are tabulated in Table 1. As shown in Table 1, the adsorption energy of 5-Fu onto B40 is computed to be −11.15 kcal mol−1, indicating that the adsorption process is exothermic. The interaction energy between 5-Fu and B40 in the resulting 5-Fu@B40 is −26.66 kcal mol−1, which suggests that these two parts are tightly combined together in this complex. Additionally, the ΔH and ΔG values for the adsorption of 5-Fu on B40 are both negative, which denotes that the adsorption of 5-Fu on the surface of B40 cage is exothermic, and thus, it is thermodynamically possible to form this complex.49
| Complexes | Ead | Eint | Edef | ΔH | ΔG | Q5-Fu | qB/M | qO |
|---|---|---|---|---|---|---|---|---|
| 5-Fu@B40 | −11.15 | −26.66 | 13.86 | −20.06 | −7.24 | 0.391 | 0.189 | −0.611 |
| 5-Fu@[Mg@B40] | −15.65 | −34.60 | 17.10 | −25.46 | −11.87 | 0.419 | 0.279 | −0.599 |
| 5-Fu@[Al@B40] | −15.79 | −33.22 | 15.61 | −25.85 | −13.25 | 0.427 | 0.307 | −0.615 |
| 5-Fu@[Si@B40] | −11.97 | −27.55 | 13.87 | −20.81 | −7.02 | 0.390 | 0.201 | −0.613 |
| 5-Fu@[Mn@B40] | −13.60 | −27.71 | 12.44 | −18.63 | −5.09 | 0.369 | 0.244 | −0.647 |
| 5-Fu@[Cu@B40] | −15.01 | −31.37 | 14.52 | −23.83 | −11.45 | 0.436 | 0.312 | −0.620 |
| 5-Fu@[Zn@B40] | −12.38 | −24.42 | 10.30 | −15.77 | −2.55 | 0.383 | 0.209 | −0.616 |
| 5-Fu@B39Mg | −22.49 | −21.66 | −2.23 | −40.51 | −29.68 | 0.150 | 1.522 | −0.745 |
| 5-Fu@B39Al | −29.75 | −35.44 | 4.34 | −35.66 | −23.14 | 0.212 | 1.718 | −0.774 |
| 5-Fu@B39Si | −8.99 | −24.27 | 13.83 | −14.81 | −2.25 | 0.390 | 0.261 | −0.620 |
| 5-Fu@B39Mn | −11.86 | −8.63 | −4.96 | −23.20 | −12.57 | 0.162 | 0.937 | −0.684 |
| 5-Fu@B39Cu | −13.12 | −17.67 | 3.16 | −13.54 | −3.32 | 0.116 | 0.887 | −0.735 |
| 5-Fu@B39Zn | −12.29 | −13.65 | −0.05 | −20.60 | −9.05 | 0.147 | 1.558 | −0.728 |
To gain insights into the nature of interactions between 5-Fu and B40 cage, the natural bonding analysis (NPA) charges on 5-Fu as well as B/M and O atoms involved in the linkage bonds of the most stable 5-Fu@B40 have been calculated by the NBO calculations and shown in Table 1. As evidenced in Table 1, the NPA charge on the drug (Q5-Fu) is as large as 0.391|e|, which implies that there is obvious charge transfer from 5-Fu to B40 during the adsorption process. It is reported that Wiberg bond index (WBI) can be utilized to demonstrate the strength of the covalent character of bond.50,51 Hence, based on NBO calculations, the WBI values have been obtained and listed in Table 2. It is found that the WBI value of the newly formed B–O bond in 5-Fu@B40 is as large as 0.574, which is even larger than that of B21–B24 bond (0.482) of B40 in this complex, implying that the interaction between 5-Fu and B40 cage is quite strong. This can be verified by the results of second order perturbation stabilization energy (E2) related to the interaction between 5-Fu and B40 as shown in Table S2.† From Table S2,† it can be observed that the E2 values for interaction between the lone pair (LP) of O2 atom and LP* of B25 atom is as large as 359.57 kcal mol−1, which demonstrates that 5-Fu combines with B40 via donating the lone pair of O2 atom to the empty p orbital of B25 atom, resulting in a B–O coordinate bond.
| Complexes | Bonds | WBI | ρr | ∇2ρr | Gr | Vr | Hr |
|---|---|---|---|---|---|---|---|
| 5-Fu@B40 | B25–O2 | 0.574 | 0.119 | 0.468 | 0.189 | −0.262 | −0.072 |
| 5-Fu@[Mg@B40] | B36–O1 | 0.613 | 0.134 | 0.592 | 0.232 | −0.315 | −0.084 |
| 5-Fu@[Al@B40] | B26–O1 | 0.635 | 0.148 | 0.685 | 0.267 | −0.363 | −0.096 |
| 5-Fu@[Si@B40] | B35–O2 | 0.576 | 0.120 | 0.471 | 0.191 | −0.265 | −0.073 |
| 5-Fu@[Mn@B40] | B7–O2 | 0.585 | 0.122 | 0.494 | 0.198 | −0.272 | −0.074 |
| 5-Fu@[Cu@B40] | B25–O1 | 0.648 | 0.157 | 0.714 | 0.284 | −0.390 | −0.106 |
| 5-Fu@[Zn@B40] | B17–O2 | 0.575 | 0.120 | 0.486 | 0.194 | −0.267 | −0.073 |
| 5-Fu@B39Mg | Mg52–O2 | 0.237 | 0.044 | 0.349 | 0.074 | −0.061 | 0.013 |
| 5-Fu@B39Al | Al40–O2 | 0.344 | 0.065 | 0.437 | 0.106 | −0.103 | 0.003 |
| 5-Fu@B39Si | B17–O2 | 0.592 | 0.119 | 0.506 | 0.197 | −0.267 | −0.070 |
| 5-Fu@B39Mn | Mn52–O2 | 0.354 | 0.055 | 0.334 | 0.081 | −0.078 | 0.002 |
| 5-Fu@B39Cu | Cu52–O2 | 0.189 | 0.081 | 0.562 | 0.140 | −0.140 | 0.000 |
| 5-Fu@B39Zn | Zn52–O2 | 0.223 | 0.070 | 0.418 | 0.104 | −0.102 | 0.001 |
In addition, QTAIM52,53 is usually employed to characterize the bonding nature by calculating the topological parameters, including electron density (ρr), Laplacian of electron density (∇2ρr), the density of potential energy (Vr), the density of kinetic energy (Gr) and the density of total energy (Hr) at the bond critical point (BCPs). Herein, the values of topological parameters at the BCP of the newly formed B–O bond are tabulated in Table 2. It is known that the strength of a chemical bond is strongly correlated to the electron density (ρr) at the BCP.54 Generally, the BCP of a bond with ρr > 0.20 a.u. is defined as covalent bonding, and if ρr < 0.10 a.u., can be regarded as the close-shell type, including ionic and van der Waals interactions.54 As illustrated in Table 2, the ρr value of B–O bond is 0.119 a.u., which is slightly larger than 0.10 a.u. but is much smaller than 0.20 a.u., implying that the B–O bond of 5-Fu@B40 should be classified as a strong polar covalent bond with obvious ionic characteristic. This can be proved by the fact that B25 atom carries opposite NPA charge (0.189|e|) to that of −0.611|e| on the O2 atom involved in the linkage B–O bond (see Table 1).
It is reported that the bond should be covalent in nature when the ∇2ρr < 0 and Hr < 0, and should be partially covalent if ∇2ρr > 0 and Hr < 0.55,56 As a result, the formed B–O bond in 5-Fu@B40 can also be confirmed to be partially covalent character considering its BCP with ∇2ρr > 0 and Hr < 0. The polar covalent bond characteristic of this linkage bond can be further verified by its corresponding LMO, where the localized electrons mainly located between the O2 and B25 atoms (see Fig. 3a). In addition, as shown in Fig. 3a, the shared electron pair is mostly derived from the 2p electrons of O2 in 5-Fu, which again rationalizes the high polarity of this linkage bond between B40 and drug.57 This can be also verified by the ELF figure of 5-Fu@B40, where the localized electrons between O2 and B25 atoms are closer to the electronegative O atom (see Fig. 3b). Based on the above information, it can be concluded that the formed B–O bond in the most stable 5-Fu@B40 can be regarded as strongly polar covalent bond.
 | ||
| Fig. 3 (a) LMO related to the B–O bond in 5-Fu@B40 complex, in which the percentage contribution of linkage atoms to LMO are also listed; (b) ELF plot of 5-Fu@B40 complex. | ||
To further investigate the ability of B40 cluster as a carrier to deliver 5-Fu, the electronic properties, including the energies (EHOMO and ELUMO) of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), HOMO–LUMO energy gap (Eg), dipole moment (μ), solvation energies (Esol), global hardness (η), and electrophilicity index (ω) of 5-Fu, B40, and 5-Fu@B40 are summarized in Table 3. It can be observed that the Eg of 2.90 eV for B40 is slightly decreased to 2.66 eV for 5-Fu@B40 complex because the adsorption of 5-Fu on B40 significantly raises the HOMO level of B40 and slightly raises its LUMO level. The change of HOMO and LUMO energy levels can be clearly seen from the DOS plots for 5-Fu and B40 before and after interaction in Fig. 4. Moreover, the disappearance of the peak corresponding to the HOMO of 5-Fu at −6.97 eV in the DOS of the resulting 5-Fu@B40 clearly demonstrates that this drug indeed donates its electron densities to B40 in this complex, as mentioned above.
| Complexes | EHOMO | ELUMO | Eg | η | ω | μ | Esol |
|---|---|---|---|---|---|---|---|
| 5-Fu | −6.97 | −1.62 | 5.35 | 2.673 | 3.451 | 6.26 | −14.75 |
| B40 | −5.72 | −2.82 | 2.90 | 1.449 | 6.284 | 0.00 | −5.59 |
| 5-Fu@B40 | −5.39 | −2.73 | 2.66 | 1.332 | 6.193 | 8.81 | −18.82 |
| 5-Fu@[Mg@B40] | −3.96 | −2.66 | 1.31 | 0.653 | 8.396 | 20.96 | −38.49 |
| 5-Fu@[Al@B40] | −3.87 | −2.85 | 1.02 | 0.508 | 11.121 | 20.59 | −17.75 |
| 5-Fu@[Si@B40] | −4.54 | −2.83 | 1.71 | 0.854 | 7.945 | 7.88 | −18.40 |
| 5-Fu@[Mn@B40] | −4.32 | −2.72 | 1.60 | 0.799 | 14.889 | 8.08 | −42.57 |
| 5-Fu@[Cu@B40] | −3.60 | −2.92 | 0.68 | 0.342 | 15.509 | 9.64 | −31.73 |
| 5-Fu@[Zn@B40] | −3.79 | −2.66 | 1.13 | 0.567 | 9.173 | 7.77 | −33.51 |
| 5-Fu@B39Mg | −4.99 | −3.39 | 1.60 | 0.800 | 10.975 | 12.98 | −29.57 |
| 5-Fu@B39Al | −5.41 | −2.70 | 2.71 | 1.357 | 6.063 | 11.38 | −21.11 |
| 5-Fu@B39Si | −5.39 | −3.62 | 1.77 | 0.886 | 11.456 | 11.29 | −21.96 |
| 5-Fu@B39Mn | −4.88 | −2.82 | 2.06 | 1.028 | 7.208 | 18.26 | −32.84 |
| 5-Fu@B39Cu | −5.26 | −2.76 | 2.50 | 1.249 | 6.443 | 8.46 | −30.49 |
| 5-Fu@B39Zn | −5.08 | −3.43 | 1.65 | 0.826 | 10.967 | 11.89 | −41.37 |
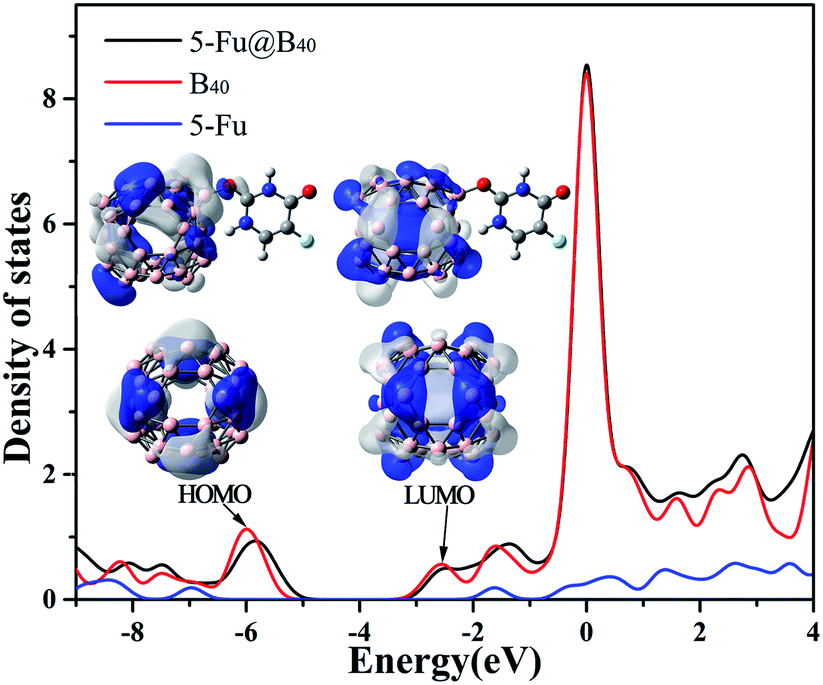 | ||
| Fig. 4 DOS figure for 5-Fu, B40, and 5-Fu@B40 with HOMO and LUMO orbitals (isovalue = 0.02 a.u.) of B40 and 5-Fu@B40 shown as inset. | ||
How to understand the variation of HOMO and LUMO levels before and after the adsorption of 5-Fu on B40? As shown in the inset of Fig. 4, the uniformly distributed HOMO of B40 has been greatly redistributed to the side close to the adsorption position of 5-Fu, which leads to the shift of the HOMO level of 5-Fu@B40 to a higher energy level. As for the LUMO of 5-Fu@B40, the electron cloud is symmetrically located on the B40 cage and the introduction of 5-Fu hardly affect the distribution of electron cloud, and thus the LUMO level of 5-Fu@B40 has been only slightly changed. As a result, the Eg value of 5-Fu@B40 is a bit smaller than that of B40. Even so, the Eg of 5-Fu@B40 is still remarkably larger than that (1.57 eV) of experimentally synthesized C60,58 indicating that this drug delivery system is stable enough to be transported in the biological environment.
To further study the stability of these studied complexes, the chemical reactivity parameters, including hardness (η) and electrophilicity index (ω) were also calculated based on the Koopman's theorem, which are defined as follows,59–61
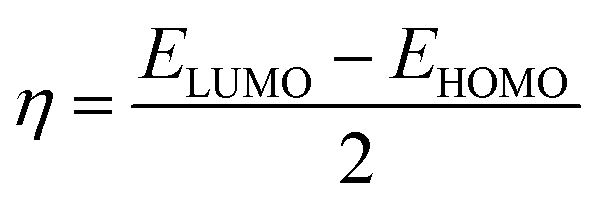 | (5) |
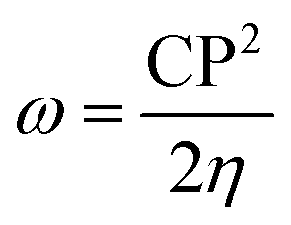 | (6) |
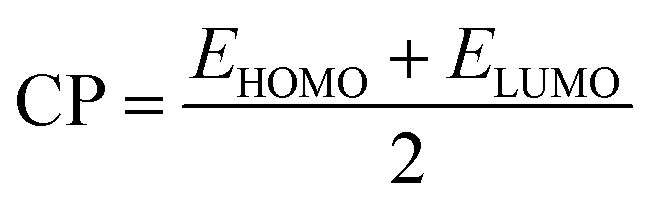 . Commonly, the global hardness (η) indicates resistance to electron cloud change of the chemical system, the high values of hardness suggests that the molecule is more stable or less reactive.62 As displayed in Table 3, the η value of 5-Fu@B40 is 1.332 eV, which is remarkably higher than those of 0.486–0.503 eV for multiple sclerosis drug adsorbed on Ni-doped graphene nanosheet,63 and 0.210 eV for 5-Fu adsorbed on graphyne nanosheet,64 suggesting that 5-Fu@B40 still exhibit much higher chemical stability than those reported drug delivery systems.63,64 In addition, the ω value of 5-Fu@B40 is as large as 6.193 eV, which is even larger than that of 5.361 eV for the reported ciclopirox@Se–B12N12,65 implying that 5-Fu@B40 possesses a high stability to avoid the accumulation in the biological systems.
. Commonly, the global hardness (η) indicates resistance to electron cloud change of the chemical system, the high values of hardness suggests that the molecule is more stable or less reactive.62 As displayed in Table 3, the η value of 5-Fu@B40 is 1.332 eV, which is remarkably higher than those of 0.486–0.503 eV for multiple sclerosis drug adsorbed on Ni-doped graphene nanosheet,63 and 0.210 eV for 5-Fu adsorbed on graphyne nanosheet,64 suggesting that 5-Fu@B40 still exhibit much higher chemical stability than those reported drug delivery systems.63,64 In addition, the ω value of 5-Fu@B40 is as large as 6.193 eV, which is even larger than that of 5.361 eV for the reported ciclopirox@Se–B12N12,65 implying that 5-Fu@B40 possesses a high stability to avoid the accumulation in the biological systems.
From Table 3, it can be seen that the 5-Fu@B40 complex exhibits much larger dipole moment of 8.81 debye than those of 6.26 and 0.00 debye for 5-Fu and B40, respectively, which suggests that the adsorption of 5-Fu molecule on B40 cage increases the polarity of the whole system, and thus, enhances the solubility of the resulting 5-Fu@B40 in an aqueous medium.62 This can be proved by the larger solvation energy of −18.82 kcal mol−1 for 5-Fu@B40 than those of −14.75 and −5.59 kcal mol−1 for 5-Fu and B40, respectively, suggesting that this complex is quite stable in aqueous environment.66
Moreover, the release of 5-Fu from the B40 carrier in target cells is another most vital step during the drug delivery process. The pH-depended drug release mechanism has been proposed by Hazrati and his colleagues in their theoretical work.48 Considering that the intracellular environment of a malignant cell has a lower pH (<6) than the normal cells (pH = 7.35–7.45),67 Shakerzadeh has applied this mechanism to prove Na@B40 and Ca@B40 to be promising carriers for the delivery of MP drug.68 Thereby, the influence of pH on the 5-Fu@B40 complex is further examined by approaching a proton to the O2 atom of 5-Fu in 5-Fu@B40. During the structural optimization, as plotted in Fig. 5, the distance between the O2 and B25 atom greatly increases from 1.552 Å to 4.052 Å. As a result, the adsorption energy of 5-Fu@B40 sharply decreases from −11.15 kcal mol−1 to −5.41 kcal mol−1 in the slightly acidic environment, reflecting that the interaction between 5-Fu and B40 cage is obviously weakened under the attack of a single proton. Therefore, it is expected that 5-Fu drug can be easily released from the B40 carrier in the tumor micro-environment.
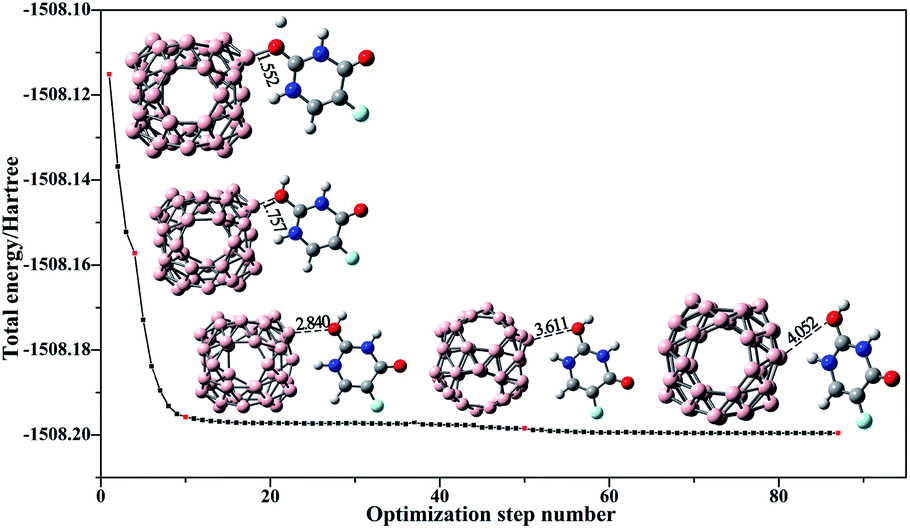 | ||
| Fig. 5 The optimization process for the protonation of 5-Fu drug adsorbed on B40 cage. The illustrated structures correspond to the red points and the distances (in Å) between B25 and O2 atoms are also given. | ||
3.2 Adsorption of 5-Fu on M@B40 (M = Mg, Al, Si, Mn, Cu, and Zn)
As mentioned above, the B40 cage is indeed capable of serving as potential carrier of 5-Fu. Then, one interesting question emerges: can the adsorption behavior of 5-Fu on this cage be tuned by introducing various foreign atoms? To answer this question, the influence of encapsulation of Mg, Al, Si, Mn, Cu, and Zn atoms into B40 cages on its adsorption behavior to 5-Fu has been studied in this section. Firstly, the geometric structures of M@B40 (M = Mg, Al, Si, Mn, Cu, and Zn) were optimized and the corresponding ESP plots have been given in Fig. S1.† It can be observed that the maximum positive potential values of 15.31–22.92 kcal mol−1 for the encapsulated M@B40 cages are mostly larger than that of 15.50 kcal mol−1 for B40 cage. From this point of view, it is speculated that the embedding effect of Mg, Al, Si, Mn, Cu, and Zn atoms could make the B40 cage easier to be attacked by the 5-Fu molecule.By placing 5-Fu near to the site with the largest ESP of each encapsulated cage, different initial configurations were constructed. After optimization, the most stable structures of 5-Fu@[M@B40] (M = Mg, Al, Si, Mn, Cu, and Zn) complexes are displayed in Fig. 6. Herein, the 5-Fu@[Mg@B40], 5-Fu@[Al@B40], and 5-Fu@[Cu@B40] are obtained by linking the O1 atom of 5-Fu to the corner B atom of Mg@B40, Al@B40, and Cu@B40, while 5-Fu@[Si@B40], 5-Fu@[Mn@B40], and 5-Fu@[Zn@B40] are formed by the combining this drug with the Si@B40, Mn@B40, and Zn@B40 cages via its O2 atom. In addition, the lengths of newly formed B–O bonds in 5-Fu@[M@B40] complexes are in the range of 1.445–1.550 Å, which are a bit shorter than the corresponding linkage bond of 1.552 Å for 5-Fu@B40. Thus, it can be inferred that 5-Fu could strongly interact with these resulting M@B40 cages. This conclusion can also be further supported by the fact that the interaction energies of −24.42 to −34.60 kcal mol−1 for 5-Fu@[M@B40] complexes are comparable to or a bit larger than that of −26.66 kcal mol−1 for 5-Fu@B40.
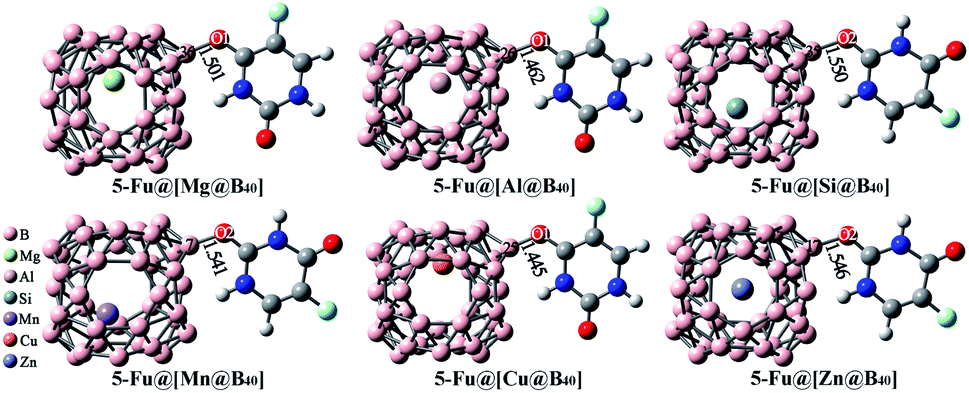 | ||
| Fig. 6 Optimized geometries of the most stable 5-Fu@[M@B40] (M = Mg, Al, Si, Mn, Cu, and Zn) complexes. The lengths of newly formed bonds (in Å) are also given. | ||
As shown in Table 1, the Ead values of these 5-Fu@[M@B40] complexes decrease in the order −15.79 kcal mol−1 (5-Fu@[Al@B40]) > −15.65 kcal mol−1 (5-Fu@[Mg@B40]) > −15.01 kcal mol−1 (5-Fu@[Cu@B40]) > −13.60 kcal mol−1 (5-Fu@[Mn@B40]) > −12.38 kcal mol−1 (5-Fu@[Zn@B40]) > −11.97 kcal mol−1 (5-Fu@[Si@B40]) > −11.15 kcal mol−1 (5-Fu@B40), which means that the adsorbency of 5-Fu drug on such M@B40 cages is more favorable than that on the pristine B40. Besides, it is noted that the descending order of Eint is slightly different from the trend of adsorption energy. This can be understood by the structural deformation of cages and drug in these complexes, as reflected by the Edef in Table 1. Additionally, it is observed that the ΔH and ΔG values of these encapsulated 5-Fu@[M@B40] complexes are all negative, indicating that the adsorption of 5-Fu on M@B40 is spontaneous and exothermic. It is worth noting that all of the energetic properties of 5-Fu@[Si@B40] are very similar to those of 5-Fu@B40, demonstrating that the encapsulation of Si atom has little effect on 5-Fu@B40.
Then, the NBO, WBI, QTAIM, LMOs, and ELF analyses have been carried out to gain insight into the nature and strength of the newly formed B–O bonds. Table 1 shows that the NPA charges on the drug are all positive and the corresponding values fall in the range of 0.369–0.436|e|, exhibiting the strong electron interaction between 5-Fu and M@B40. This can also be proved by the calculated E2 values for the critical donor–acceptor NBO interactions as listed in Table S2.† It is found that E2 values corresponding to the interactions between LP of O atom in drug and LP* of B atom in cage are as large as 188.53–368.35 kcal mol−1, suggesting that the strong adsorption of 5-Fu on M@B40 are originated from the newly formed B–O linkage bonds. In addition, it is observed that the LP* of boron of M@B40 always acts as the acceptor, which can rationalize the above-mentioned positive charges (Q5-Fu) of 5-Fu in these complexes.
As shown in Table 2, the WBI values (0.575–0.648) of B–O bonds in 5-Fu@[M@B40] are larger than that of 0.574 for the corresponding B–O bond in 5-Fu@B40, which implies that these B–O linkage bonds has been enhanced to different degrees upon introducing the foreign atoms. Similarly, these B–O bonds formed in 5-Fu@[M@B40] complexes also show relatively larger ρ values (0.120–0.157 a.u.) than that of 0.119 a.u. for the B–O bond in 5-Fu@B40. This indicates that the former has more obvious covalent characteristics than the latter. Additionally, all the ∇2ρr value at the BCPs of B–O bonds in 5-Fu@[M@B40] are much larger than zero, suggesting that these linkage bonds also possess ionic characteristics. This can be supported by the fact that the boron atoms involved in the linkage B–O bonds always carry positive NPA charges (0.201–0.312|e|) while the O atoms have the negative charges of −0.599|e| to −0.647|e|. More interestingly, the qB and qO of 5-Fu@[M@B40] are respectively larger than those of 5-Fu@B40, indicating that there is stronger electrostatic interaction between the B and O atoms involved in the newly formed B–O bonds of 5-Fu@[M@B40]. Even so, the B–O bonds also possess partially covalent character considering their negative Hr values at the BCPs. The covalent characteristics of these linkage bonds can be ensured by the LMOs of 5-Fu@[M@B40] as depicted in Fig. S2.† However, the percentage contribution analysis reveals that the shared electron pair is mostly derived from the oxygen atoms (69.2–71.0%) of 5-Fu, reflecting the large polarity of these bonds. This can be further verified by the ELF plots, where the localized electrons between B and O atoms are closer to the O atom (see Fig. S3†).
As compared with 5-Fu@B40, the HOMO levels of 5-Fu@[M@B40] have been obviously raised by the encapsulation of foreign atoms, resulting in their much smaller Eg values (0.68–1.71 eV) that that of 2.66 eV for 5-Fu@B40. This can be attributed to the fact that the electron clouds in these HOMO orbitals of 5-Fu@[Cu@B40] are mainly located around the introduced M atoms (see Fig. S4†), which makes such HOMO levels unstable. The decrease of HOMO–LUMO gaps demonstrates that the encapsulation of M atoms reduces the chemical stability of 5-Fu@[M@B40] complexes, which can be further proved by the reactivity parameters. As shown in Table 3, the 5-Fu@[M@B40] complexes are more reactive and softer than 5-Fu@B40 because the η value of 5-Fu@B40 is decreased from 1.332 eV to 0.342–0.854 eV of 5-Fu@[M@B40]. Even though, it should be mentioned that the smallest Eg value of 0.68 eV for 5-Fu@[Cu@B40] is still larger than that of 0.42 eV for the reported 5-Fu/GpNS,64 implying that these 5-Fu@[M@B40] complexes are still stable enough to be transported in a biological environment.
Moreover, as displayed in Table 3, the μ values of 5-Fu@[M@B40] are 20.96, 20.59, 7.88, 8.08, 9.64, and 7.77 debye for M = Mg, Al, Si, Mn, Cu, and Zn, respectively. Compared with 5-Fu@B40, the μ values of 5-Fu@[Mg@B40], 5-Fu@[Al@B40], and 5-Fu@[Cu@B40] are increased, indicating that the encapsulation of Mg, Al, and Cu atoms will make their corresponding complexes more soluble in polar solvents. Meanwhile, the negative Esol values show that these 5-Fu@[M@B40] are stable in the presence of water. Besides, the Esol values (−31.73 to −42.57 kcal mol−1) of 5-Fu@[M@B40] (M = Mg, Mn, Cu, and Zn) are much larger than that (−18.82 kcal mol−1) of 5-Fu@B40, suggesting that the solubility of this drug delivery system has been improved by the encapsulation of Mg, Mn, Cu, and Zn atoms into the B40 cage. Thus, it is believed that the encapsulation of selected atoms can effectively modulate the interaction between 5-Fu and B40.
3.3 Adsorption of 5-Fu on B39M (M = Mg, Al, Si, Mn, Cu, and Zn)
In addition to the embedding effect, we also detect the substituent effect of an exotic atom on the adsorption performance of 5-Fu on B40. Herein, the most positive B25 atom of B40 was replaced with Mg, Al, Si, Mn, Cu, and Zn atoms, respectively. As shown in Fig. S5,† the substitution of Mg, Al, Mn, Cu, or Zn atom for one boron atom poses a great effect on the ESP of B40, whereas the introduction of Si atom hardly affects the ESP distribution of B40. Based on the ESP plots, a lot of initial configurations of 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn) have been constructed via approaching the nucleophilic sites of 5-Fu to the mostly positive sites, and the obtained global minima have been depicted in Fig. 7.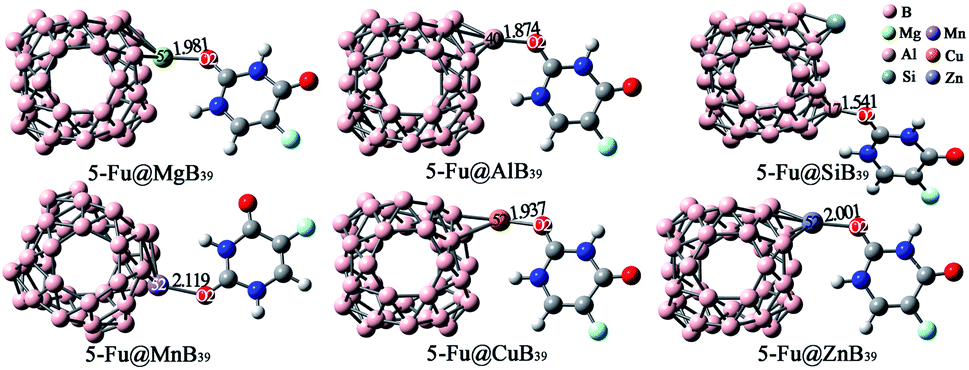 | ||
| Fig. 7 Optimized geometries of the most stable 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn) complexes. The lengths of newly formed bonds (in Å) are also given. | ||
As shown in Fig. 7, B39M cages prefer to bind with the O2 atom of 5-Fu via M sites in the 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) because that these Mg, Al, Mn, Cu, and Zn sites have large ESP values of 259.33, 45.21, 128.10, 81.72, and 149.72 kcal mol−1, respectively. Differently, the 5-Fu@B39Si complex is formed by attaching the O2 atom of 5-Fu to the B17 atom of B39Si cage because the Si site with negative ESP is not conducive to be attacked by 5-Fu. Upon adsorption, the lengths of M–O bonds in 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) complexes are 1.874–2.119 Å, which are much longer than that of 1.541 Å for the B–O bond in 5-Fu@B39Si. That can be attributed to the fact that these M atoms have much larger atomic radii of 2.17–2.42 Å than that of 2.05 Å for boron atom.69
As presented in Table 1, the Ead values of these 5-Fu@B39M complexes decrease in the order −29.75 kcal mol−1 (5-Fu@B39Al) > −22.49 kcal mol−1 (5-Fu@B39Mg) > −13.12 kcal mol−1 (5-Fu@B39Cu) > −12.29 kcal mol−1 (5-Fu@B39Zn) > −11.86 kcal mol−1 (5-Fu@B39Mn) > −11.15 kcal mol−1 (5-Fu@B40) > −8.99 kcal mol−1 (5-Fu@B39Si), indicating that the adsorbency of 5-Fu on B39M cages are more favorable than pristine B40 except for B39Si. However, it is observed that these Eint values reduce in the trend −35.44 kcal mol−1 (5-Fu@B39Al) > −26.66 kcal mol−1 (5-Fu@B40) > −24.27 kcal mol−1 (5-Fu@B39Si) > −21.66 kcal mol−1 (5-Fu@B39Mg) > −17.67 kcal mol−1 (5-Fu@B39Cu) > −13.65 kcal mol−1 (5-Fu@B39Zn) > −8.63 kcal mol−1 (5-Fu@B39Mn), which is a bit different from the order of adsorption energy. This can be attributed to complexes 5-Fu@B40 and 5-Fu@B39Si show much larger deformation energies than the other compounds. It is worth mentioning that all the Ead, Eint, and Edef values of 5-Fu@B39Si are comparable to those of 5-Fu@B40. This is because that, similar to 5-Fu@B40, 5-Fu is also adsorbed on the B site rather than the Si site on B39Si in 5-Fu@B39Si. Moreover, the adsorption of 5-Fu on the B39M surface is also spontaneous in view of the negative ΔH and ΔG values of 5-Fu@B39M.
From Table 1, it can be seen clearly that the total NPA charges on the drugs in these 5-Fu@B39M complexes are in the range of 0.116–0.390|e|. Amongst, only 5-Fu@B39Si possesses a comparable Q5-Fu value of 0.390|e| to that of 0.391|e| for 5-Fu@B40, whereas the rest 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) show much smaller Q5-Fu values of 0.116–0.212|e| than those of 0.369–0.436|e| for 5-Fu@B40 and 5-Fu@[M@B40] (M = Mg, Al, Si, Mn, Cu, and Zn). This clear demonstrates that the electron transfer from 5-Fu to the substituted B39M cages is weakened in these 5-Fu@B39M complexes except for 5-Fu@B39Si. Similar phenomenon has been also observed for the E2 results related to the interaction between 5-Fu and B39M cages. As shown in Table S2,† the E2 values corresponding to the interaction between the LP of O2 atom and LP* of M atoms for 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) are 8.89–79.96 kcal mol−1, which are much smaller than those of 188.53–368.35 kcal mol−1 corresponding to the interaction between LP of O atom in drug and LP* of B atom in the cages for 5-Fu@B39Si, 5-Fu@B40, and 5-Fu@[M@B40]. Even so, the LP of oxygen atoms in 5-Fu always plays a role as electron donor, which further verifies the positive Q5-Fu values.
From Table 2, it can be seen clearly that the WBI values of 0.237, 0.344, 0.354, 0.189, and 0.223 for Mg–O, Al–O, Mn–O, Cu–O, and Zn–O bonds in 5-Fu@B39M complexes are all lower than that of 0.574 for B–O in 5-Fu@B40. These results show that, as compared with 5-Fu@B40, the covalency of these M–O linkage bonds is reduced to different degrees upon the introduction of M atom. In contrast, the calculated WBI value of 0.592 for B–O bond in 5-Fu@B39Si is slightly larger than that of B–O in 5-Fu@B40. Resultantly, only the ρr value of 5-Fu@B39Si is larger than 0.10 a.u., whereas the formed M–O bonds in rest 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) complexes possess quite small ρr values of 0.044–0.081 a.u. at the BCPs. This phenomenon indicates that the M–O bonds in 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) possess more obvious ionic characteristics than the B–O bond in 5-Fu@B39Si, which is reasonable considering the metallic identities of M atoms. This can be confirmed by the fact that the B–O bond in 5-Fu@B39Si possesses negative Hr of −0.070 a.u., whereas the Hr values for the M–O bonds in 5-Fu@B39M are larger than zero. However, all these linkage bonds exhibit certain ionic characteristics because their ∇2ρr are always larger than zero. This can be confirmed by the truth that the M/B atoms at the adsorbed sites carry positive NPA charge of 0.261–1.718|e| while the O2 atoms have negative charges of −0.620 to −0.774|e|.
In addition, the covalent characteristics of M/B–O bonds can be further verified by the LMOs of 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn) as shown in Fig. S6.† The contribution analysis reveals that the shared electron pair is mostly derived from the oxygen atoms (71.2–81.7%) of 5-Fu drug, resulting in the large polarity of these bonds. The differences in contribution (62.5–72.4%) from M and O atoms to the shared localized electrons are much larger than that from B and O atoms (51.0%), indicating that the M–O bonds of 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) possess relatively larger polarity than the B–O bond of 5-Fu@B39Si. Similarly, the dark blue gaps between M and O atoms in 5-Fu@B39M (M = Mg, Al, Mn, Cu, and Zn) are more obvious than that between B and O atom in 5-Fu@B39Si, as shown in the ELF plots in Fig. S7.†
Table 3 also lists the electronic properties of 5-Fu@B39M. It is observed that the HOMO–LUMO gaps of these 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn) complexes are as large as 1.60–2.71 eV, indicating that these systems are quite stable. From Fig. S8,† it is found that the electron clouds in HOMO and LUMO orbitals of 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn) complexes are mainly located on the B39M cages, which is similar to the orbital distributions of 5-Fu@B40. Herein, the Eg value of 2.71 eV for 5-Fu@B39Al is even larger than that of 2.66 eV for 5-Fu@B40, implying that 5-Fu@B39Al complex have slightly higher chemical stability than 5-Fu@B40. Moreover, from Table 3, it can be seen that the Eg and η values of 5-Fu@B39M are generally larger than those of 5-Fu@[M@B40], which suggests that 5-Fu@B39M may serve as the better drug delivery systems than 5-Fu@[M@B40].
From Table 3, it is found that 5-Fu@B39M also have considerable μ values of 8.46–18.26 debye, most of which are even higher than that of 8.81 debye for 5-Fu@B40, indicating that these 5-Fu@B39M complexes also exhibit certain polarity. Furthermore, Esol values of −21.11 to −41.37 kcal mol−1 for 5-Fu@B39M are larger than that of −18.82 kcal mol−1 for 5-Fu@B40, demonstrating that the introduction of foreign atoms has substantially enlarged the solubility of 5-Fu@B40. Therefore, it can be concluded that substituting an exogenous atom for a boron atom of B40 can modulate the interaction between B40 and 5-Fu, as well as the stability and solubility of the resulting complexes, which could provide an effective strategy to tune the drug delivery performance of such nanocages.
4. Conclusions
In this work, DFT calculations have been performed to investigate the interaction between B40 with 5-Fu drug to examine the ability of B40 to serve as a novel delivery vehicle for antitumor drug. The results show that 5-Fu is adsorbed on B40 cage by chemisorption with adsorption energy of −11.15 kcal mol−1. In particular, under the proton attack, 5-Fu drug can be easily separated from the B40 surface and delivered to the target cells, and thus the pristine B40 can act as a suitable carrier for 5-Fu drug. Subsequently, the embedding and substituent effects of Mg, Al, Si, Mn, Cu, and Zn atoms on the drug delivery performance of B40 are considered. The calculated results demonstrate that 5-Fu tends to combine with B39M or M@B40 (M = Mg, Al, Si, Mn, Cu, and Zn) cages via its oxygen atom with the adsorption energies of −8.99 to −29.75 kcal mol−1. Meanwhile, the NBO, AIM, LMO, and ELF analyses reveal that these newly formed B–O and M–O (M = Mg, Al, Mn, Cu, and Zn) linkage bonds can be regarded as strongly polar covalent bonds, and the M–O bonds exhibit more ionic components than B–O bonds. Therefore, the encapsulation and substitution of foreign atoms can be regarded as two effective strategies to control the interaction between 5-Fu and B40. We hope that this study could not only provide a useful reference on the interaction between antitumor drug and boron nanocages, but also encourage more experimental attempts to select appropriate boron-based nanomaterials for practical trials of drug delivery.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Natural Science Foundation of Fujian Province (Grant number: 2021J01682), National Natural Science Foundation of China [Grant number: 21603032], and Joint Funds for the Innovation of Science and Technology, Fujian Province [Grant number: 2017Y9119, 2019Y9008]. The authors also acknowledge the National Supercomputing Center in Shenzhen for providing computational resources.Notes and references
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- C. Heidelberger, N. K. Chaudhuri, P. Danneberg, D. Mooren, L. Griesbach, R. Duschinsky, R. J. Schnitzer, E. Pleven and J. Scheiner, Nature, 1957, 179, 663–666 CrossRef CAS PubMed.
- M. Ducreux, O. Bouche, J. P. Pignon, M. Mousseau, J. L. Raoul, P. Cassan, B. Leduc, C. Berger, A. Dunant, J. Fournet and L. Bedenne, Oncology, 2006, 70, 222–230 CrossRef CAS PubMed.
- P. Papanastasopoulos and J. Stebbing, Anticancer Res., 2014, 34, 1531–1535 CAS.
- A. Khajeh and H. Modarress, Biophys. Chem., 2014, 187–188, 43–50 CrossRef CAS PubMed.
- C.-H. Wu, C.-C. Wu and Y.-S. Ho, J. Cancer Mol., 2007, 3, 15–22 CAS.
- Z. Dou, Y. Xu, H. Sun and Y. Liu, Nanoscale, 2012, 4, 4624–4630 RSC.
- B. Blicharska and T. Kupka, J. Mol. Struct., 2002, 613, 153–166 CrossRef CAS.
- J. B. MacNaughton, R. G. Wilks, J. S. Lee and A. Moewes, J. Phys. Chem. B, 2006, 110, 18180–18190 CrossRef CAS PubMed.
- F. Kossoski, M. H. F. Bettega and M. T. d. N. Varella, J. Chem. Phys., 2014, 140, 024317 CrossRef CAS PubMed.
- M. K. Hazrati and N. L. Hadipour, Phys. Lett. A, 2016, 380, 937–941 CrossRef CAS.
- E. Shakerzadeh, J. Mol. Liq., 2017, 240, 682–693 CrossRef CAS.
- E. Shakerzadeh, J. Mol. Liq., 2021, 343, 116970 CrossRef CAS.
- J. Fan, G. Fang, F. Zeng, X. Wang and S. Wu, Small, 2013, 9, 613–621 CrossRef CAS PubMed.
- R. Bakry, R. M. Vallant, M. Najam-Ul-Haq, M. Rainer and G. K. Bonn, Int. J. Nanomed., 2007, 2, 639–649 CAS.
- M. Raoof, Y. Mackeyev, M. A. Cheney, L. J. Wilson and S. A. Curley, Biomaterials, 2012, 33, 2952–2960 CrossRef CAS PubMed.
- J. Shi, H. Zhang, L. Wang, L. Li, H. Wang, Z. Wang, Z. Li, C. Chen, L. Hou, C. Zhang and Z. Zhang, Biomaterials, 2013, 34, 251–261 CrossRef CAS PubMed.
- J. Tong, M. C. Zimmerman, S. Li, X. Yi, R. Luxenhofer, R. Jordan and A. V. Kabanov, Biomaterials, 2011, 32, 3654–3665 CrossRef CAS PubMed.
- M. D. Ganji, H. Yazdani and A. Mirnejad, Phys. E, 2010, 42, 2184–2189 CrossRef CAS.
- N. M. El-Sawy, A. I. Raafat, N. A. Badawy and A. M. Mohamed, Int. J. Biol. Macromol., 2020, 142, 254–264 CrossRef CAS PubMed.
- I. Ravaei, M. Haghighat and S. M. Azami, Appl. Surf. Sci., 2019, 469, 103–112 CrossRef CAS.
- A. R. Juárez, M. S. Villanueva, D. Cortés-Arriagada and E. C. Anota, J. Mol. Model., 2019, 25, 21 CrossRef PubMed.
- A. R. Juárez, F. Ortíz-Chi, R. Pino-Ríos, G. Cárdenas-Jirón, M. S. Villanueva and E. C. Anota, Chem. Phys. Lett., 2020, 741, 137097 CrossRef.
- A. R. Juárez, F. Ortiz-Chi, M. Borges-Martinez, G. Cardenas-Jiron, M. S. Villanueva and E. C. Anota, Phys. E, 2019, 111, 118–126 CrossRef.
- A. D. O. Muñoz, A. Escobedo-Morales, E. Skakerzadeh and E. C. Anota, J. Mol. Liq., 2021, 322, 114951 CrossRef.
- H.-J. Zhai, Y.-F. Zhao, W.-L. Li, Q. Chen, H. Bai, H.-S. Hu, Z.-A. Piazza, W.-J. Tian, H.-G. Lu, Y.-B. Wu, Y.-W. Mu, G.-F. Wei, Z.-P. Liu, J. Li, S.-D. Li and L.-S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- Y. Zhang and X. Cheng, Int. J. Hydrogen Energy, 2018, 43, 15338–15347 CrossRef CAS.
- R. Chandiramouli and V. Nagarajan, Vacuum, 2017, 142, 13–20 CrossRef CAS.
- C. Li, W. Tang and V. Vahabi, Phys. E, 2020, 120, 114038 CrossRef CAS.
- E. Shakerzadeh, Comput. Theor. Chem., 2021, 1202, 113339 CrossRef CAS.
- N. D. Charistos and A. Muñoz-Castro, Phys. Chem. Chem. Phys., 2019, 21, 20232–20238 RSC.
- R. Gherbi, M. Benamira and Y. Bessekhouad, J. Alloys Compd., 2021, 851, 156797 CrossRef CAS.
- Ö. Alver, M. Bilge, N. Atar, C. Parlak and M. Şenyel, J. Mol. Liq., 2017, 231, 202–205 CrossRef.
- M. Lin, D. Wang, S. Li, Q. Tang, S. Liu, R. Ge, Y. Liu, D. Zhang, H. Sun, H. Zhang and B. Yang, Biomaterials, 2016, 104, 213–222 CrossRef CAS PubMed.
- X. Li, X. Zhang, Y. Zhao and L. Sun, J. Inorg. Biochem., 2020, 202, 110887 CrossRef CAS PubMed.
- J. Zhu and Z. Nan, J. Phys. Chem. C, 2017, 121, 9612–9620 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- V. S. Bernales, A. V. Marenich, R. Contreras, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2012, 116, 9122–9129 CrossRef CAS PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- E. Nemati-Kande, R. Karimian, V. Goodarzi and E. Ghazizadeh, Appl. Surf. Sci., 2020, 510, 145490 CrossRef CAS.
- J. Chocholoušová, V. Špirko and P. Hobza, Phys. Chem. Chem. Phys., 2004, 6, 37–41 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, N. Rega, J. Gao, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford, 2016 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 6, Semichem Inc, Shawnee Mission, KS, 2016 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- L. Fallon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 2549–2556 CrossRef CAS.
- M. K. Hazrati, Z. Javanshir and Z. Bagheri, J. Mol. Graphics Modell., 2017, 77, 17–24 CrossRef CAS PubMed.
- S. Bagheri Novir and M. R. Aram, Chem. Phys. Lett., 2020, 757, 137869 CrossRef CAS PubMed.
- K. K. Pandey and G. Frenking, Eur. J. Inorg. Chem., 2004, 2004, 4388–4395 CrossRef.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- M. Shahabi and H. Raissi, J. Inclusion Phenom. Macrocyclic Chem., 2015, 84, 99–114 CrossRef.
- C. F. Matta, Hydrogen–hydrogen bonding: The non-electrostatic limit of closed-shell interaction between two hydro, hydrogen bonding-new insights, Springer, 2006, pp. 337–375 Search PubMed.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed. Engl., 1984, 23, 627–628 CrossRef.
- X.-L. Zhang, L. Zhang, J.-H. Chen, C.-Y. Li and W.-M. Sun, ACS Omega, 2020, 5, 15325–15334 CrossRef CAS PubMed.
- X.-B. Wang, C.-F. Wang and L.-S. Wang, J. Chem. Phys., 1999, 110, 8217–8220 CrossRef CAS.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512–7516 CrossRef CAS.
- R. G. Parr, L. Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922–1924 CrossRef CAS.
- R. G. Parr, R. A. Donnelly, M. Levy and W. E. Palke, J. Chem. Phys., 1978, 68, 3801–3807 CrossRef CAS.
- M. Vatanparast and Z. Shariatinia, J. Mol. Graphics Modell., 2019, 89, 50–59 CrossRef CAS PubMed.
- N. Dastani, A. Arab and H. Raissi, Comput. Theor. Chem., 2021, 1196, 113114 CrossRef CAS.
- V. Nagarajan and R. Chandiramouli, J. Mol. Liq., 2019, 275, 713–722 CrossRef CAS.
- S. Kaviani, S. Shahab, M. Sheikhi, V. Potkin and H. Zhou, Comput. Theor. Chem., 2021, 1201, 113246 CrossRef CAS.
- F. Safdari, H. Raissi, M. Shahabi and M. Zaboli, J. Inorg. Organomet. Polym., 2017, 27, 805–817 CrossRef CAS.
- P. Swietach, R. D. Vaughan-Jones, A. L. Harris and A. Hulikova, Philos. Trans. R. Soc., B, 2014, 369, 20130099 CrossRef PubMed.
- E. Shakerzadeh, Appl. Organomet. Chem., 2021, e6411 CAS.
- M. Rahm, R. Hoffmann and N. W. Ashcroft, Chem.–Eur. J., 2016, 22, 14625–14632 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Relatively energies of Mn@B40 and B39Mn with various spin multiplicities; E2 and Cartesian coordinates of 5-Fu@B40; ESP plots for M@B40 and B39M cages; E2, LMOs, ELF, HOMO, LUMO, and Cartesian coordinates of 5-Fu@[M@B40] and 5-Fu@B39M (M = Mg, Al, Si, Mn, Cu, and Zn). See DOI: 10.1039/d1ra08308b |
| This journal is © The Royal Society of Chemistry 2021 |