
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN–C bond formation between two anilines coordinated to a ruthenium center in cis-form affording a 3,5-cyclohexadiene-1,2-diimine moiety†
Nozomi Tomioka,
Shinkoh Nanbu ,
Tomoyo Misawa-Suzuki and
Hirotaka Nagao*
,
Tomoyo Misawa-Suzuki and
Hirotaka Nagao*
Department of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo, 102-8554, Japan. E-mail: h-nagao@sophia.ac.jp
First published on 15th November 2021
Abstract
Ruthenium complexes containing two anilines or its derivatives, cis-[RuII(NH2C6H5)2(bpy)2]2+ ([1]2+) and cis-[RuII(NH2C6H4(4-CH3))2(bpy)2]2+ ([2]2+), were oxidized by four molar equivalents of (NH4)4[CeIV(SO4)4]·2H2O to give N1-phenylcyclohexa-3,5-diene-1,2-diimineruthenium(II) complexes, cis-[RuII(NHC6H4NC6H5)(bpy)2]2+ ([4]2+) and cis-[RuII(NHC6H3(4-CH3)NC6H4(4-CH3))(bpy)2]2+ ([5]2+), respectively, through an N–C bond formation between two aniline ligands cis-coordinated to the ruthenium center.
Introduction
Nitrogen is one of the essential elements for all living things and nitrogen-containing compounds are synthesized and converted by various ways.1 Active nitrogen species such as amines and imines are useful for synthesis of nitrogen-containing compounds, which are present in a variety of chemical forms with the formal oxidation number of the nitrogen atom(s) ranging from –III to + V.2–4 Transition metals and transition metal complexes play important roles as reaction centers for material conversion accompanied with redox and proton transfer reactions.5,6Amines react with organic substances and are oxidized in the presence of catalysts to produce valuable organic nitrogen-containing compounds.5,7–12 Aniline and derivatives are oxidized to form an N–N bond, an N–C bond, or a C–C bond between two molecules, owing to an electron-rich phenyl group. Selective conversion reactions of aniline mediated by transition metal complexes have been reported.12 Transition metal centers function as oxidants and regulate the oxidation potential and the number of electrons involved in the reaction. One-electron oxidation of aniline occurs by metal species with one-proton dissociation affording an anilide radical. Hydrazines are formed by radical coupling and are further oxidized by the metal species to give azo compounds.13–17 For the syntheses of polyaniline via N–C bond coupling of aniline, a radical cation is formed by a one-electron removal and has stable canonical structures.18–21 The radical cation is resonance-stabilized by the π-electrons of the phenyl group, and a radical coupling reaction occurs between the carbon atom at the para-position and the aniline nitrogen, resulting in polyaniline by successive oxidation and deprotonation. The position and the electronic properties of the substituents on the phenyl group of the aniline determine whether the N–N bond coupling or the N–C bond coupling is preferred. However, the N–C bond formation between two anilines at the ortho-position is a rare reaction and it is possible to synthesize new nitrogen-containing compounds having diimine moieties.22–28
In this study, a conversion reaction of anilines on the ruthenium(II) center bearing two 2,2′-bipyridine (bpy) ligands was investigated. The coordinated anilines showed proton dissociation reactions accompanied with an oxidation reaction of the ruthenium complexes. Herein, we describe oxidation reactions of cis-[RuII(NH2C6H5)2(bpy)2]2+ ([1]2+) and cis-[RuII(NH2C6H4(4-CH3))2(bpy)2]2+ ([2]2+), affording corresponding N1-phenylcyclohexa-3,5-diene-1,2-diimine ligands on the ruthenium(II) center with N–C bond formation between two aniline ligands.
Results and discussion
Syntheses and characterization of dianiline complexes of ruthenium(II)
Ruthenium complexes having two aniline or aniline-derived ligands with two 2,2′-bipyridine (bpy) as supporting ligands, cis-[RuII(NH2C6H4R)2(bpy)2](CF3SO3)2 (R = H; [1](CF3SO3)2, R = 4-CH3; [2](CF3SO3)2) and trans-[RuII(NH2C6H3(2,6-F2))2(bpy)2](CF3SO3)2 ([3](CF3SO3)2), were synthesized in 94, 92 and 83% yield, respectively, by reactions of the carbonatobis(2,2′-bipyridine)ruthenium(II) complex, [Ru(η2-O2CO)(bpy)2]·2H2O, with aniline or its derivatives in dichloromethane containing CF3SO3H. The carbonato ligand is converted to an aqua ligand with evolution of carbon dioxide and two anilines coordinated to the ruthenium(II) center. 1H and 13C NMR spectra were measured for dianiline complexes, [1]2+ and [2]2+, in dimethylsulfoxide-d6 and [3]2+ in methanol-d4 (Fig. S1 and S2†). Signals of the aniline and the bpy ligands were observed in the normal aromatic region (9.51–6.46 ppm), indicating these aniline complexes are diamagnetic. The characteristic signals of the amine protons of the aniline ligands were also observed at 6.15 ppm for [1]2+, 6.02 ppm for [2]2+ and 5.54 ppm for [3]2+. IR spectra of the dianiline complexes show two characteristic stretching vibrational modes within the range of 3137–3270 cm−1, assigned to ν(N–H) (Fig. S3†). The intense bands in electronic spectra show characteristic absorption bands at 479 and 340 nm for [1]2+, 481 and 340 nm for [2]2+, and 487 and 341 nm for [3]2+ (Fig. S4 and Table S1†). These bands could be assigned to transitions from the metal center to the bpy ligand based on the results of time-dependent density-functional theory (TD DFT) calculations (Fig. S5†). These attributions are consistent with those of other diamine complexes bearing two bpy ligands.29Electrochemical behavior of dianiline complexes of ruthenium(II)
Dianiline complexes [1]2+ and [2]2+ showed an irreversible one-electron oxidation wave at the same potential, 0.83 V (Epa vs. Ag|0.01 M AgNO3) in nitromethane (Fig. 1), attributed to the redox couple from Ru(II) to Ru(III). On a reverse scan to the negative potential direction after the oxidation wave was observed, a reduction wave appeared at 0.57 V for [1]2+ or 0.48 V for [2]2+, respectively. In addition, no oxidation wave corresponding the cathodic current was observed during multiple scans between 0.5 and 0.7 V. The reduction waves observed around 0.5 V are attributed to the one-electron reduction of a ruthenium(II) species, which was formed via intramolecular electron transfer from the aniline moiety to the ruthenium center immediately after one-electron oxidation, rather than reduction of a species formed by deprotonation from the aniline nitrogen. The methyl group on the phenyl group of the aniline ligand won't affect the oxidation potential of ruthenium(II) species, but the reduction process of the one-electron oxidized complex, which was formed after intramolecular electron transfer, is influenced. For [3]2+, a quasi-reversible oxidation wave was observed at 0.88 V (Epa) in a short time, because of instability in nitromethane. On the other hand, the behaviors after one-electron oxidation of [1](CF3SO3)2 were different in aqueous solution because aniline protons are more easily dissociated from aniline nitrogen than in nitromethane.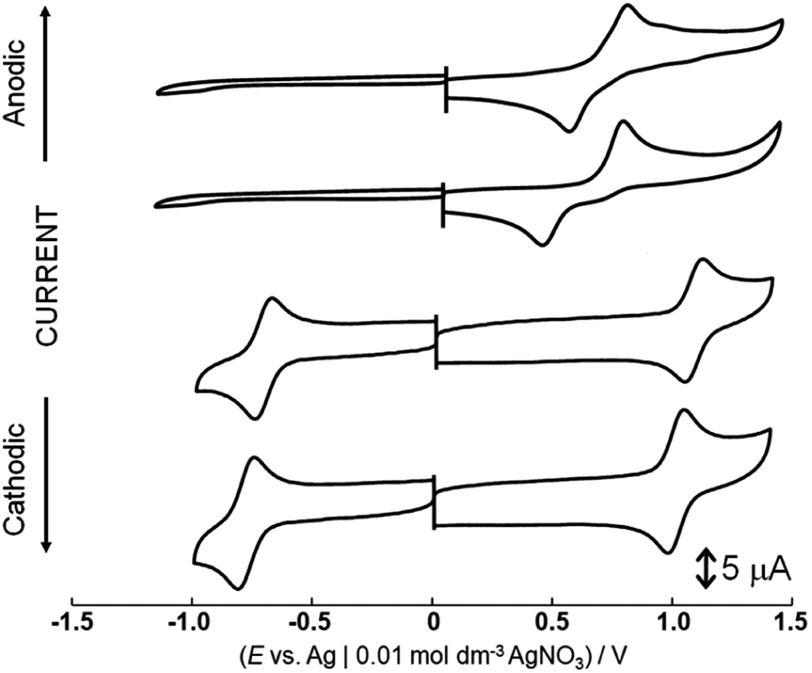 | ||
| Fig. 1 Cyclic voltammograms of [1](CF3SO3)2, [2](CF3SO3)2, [4](PF6)2 and [5](PF6)2 in nitromethane containing 0.1 mol dm−3 tetra-n-butylammonium perchlorate. | ||
Cyclic voltammograms of [1](CF3SO3)2 in water-dimethyl sulfoxide mixed solutions (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v), whose pH values were adjusted by HClO4 and NaOH are shown in Fig. 2 and S7.† The oxidation waves are irreversible even at the high scan rate (5 V s−1). Pourbaix diagrams are plotted on pH vs. potentials of each irreversible oxidation wave (Fig. S8†). The potentials of two irreversible oxidation waves at 1.00 V and 1.32 V (Epa vs. Ag|AgCl 3.0 M NaCl aq.) are almost the same regardless of the pH of the solutions. The pH independent region indicates only a change of the oxidation states of the complex. The first oxidation wave is attributed to the one-electron oxidation of the ruthenium center. Based on the results of DFT calculations, the highest occupied molecular orbital (HOMO) of the one-electron oxidized form [1]3+ is distributed over the aniline ligands (Fig. S9†). On the reverse scan from 1.22 V before the second oxidation wave was observed, the first oxidation wave was still irreversible. Therefore, it is possible that one-proton dissociates from the aniline ligand as a consequence of one-electron oxidation and a new oxidizable complex is generated under this condition. However, oxidation and deprotonation reactions do not occur concertedly because of no dependence of the oxidation potentials on pH. After that, the second oxidation reaction occurs. It could be assigned to the one-electron oxidation of the moiety consisting of the ruthenium center and the aniline radical, because the singly occupied molecular orbital (SOMO) of one-electron oxidized form ([1]3+) or one-proton dissociated form ([1]3+-H+) is contributed by the ruthenium center and the two aniline ligands or the aniline radical (Fig. S9†). The metric parameters of the 6-membered ring in the [1]3+-H+ complex consist with the quinoid-type (Table S2†).30
:3, v/v), whose pH values were adjusted by HClO4 and NaOH are shown in Fig. 2 and S7.† The oxidation waves are irreversible even at the high scan rate (5 V s−1). Pourbaix diagrams are plotted on pH vs. potentials of each irreversible oxidation wave (Fig. S8†). The potentials of two irreversible oxidation waves at 1.00 V and 1.32 V (Epa vs. Ag|AgCl 3.0 M NaCl aq.) are almost the same regardless of the pH of the solutions. The pH independent region indicates only a change of the oxidation states of the complex. The first oxidation wave is attributed to the one-electron oxidation of the ruthenium center. Based on the results of DFT calculations, the highest occupied molecular orbital (HOMO) of the one-electron oxidized form [1]3+ is distributed over the aniline ligands (Fig. S9†). On the reverse scan from 1.22 V before the second oxidation wave was observed, the first oxidation wave was still irreversible. Therefore, it is possible that one-proton dissociates from the aniline ligand as a consequence of one-electron oxidation and a new oxidizable complex is generated under this condition. However, oxidation and deprotonation reactions do not occur concertedly because of no dependence of the oxidation potentials on pH. After that, the second oxidation reaction occurs. It could be assigned to the one-electron oxidation of the moiety consisting of the ruthenium center and the aniline radical, because the singly occupied molecular orbital (SOMO) of one-electron oxidized form ([1]3+) or one-proton dissociated form ([1]3+-H+) is contributed by the ruthenium center and the two aniline ligands or the aniline radical (Fig. S9†). The metric parameters of the 6-membered ring in the [1]3+-H+ complex consist with the quinoid-type (Table S2†).30
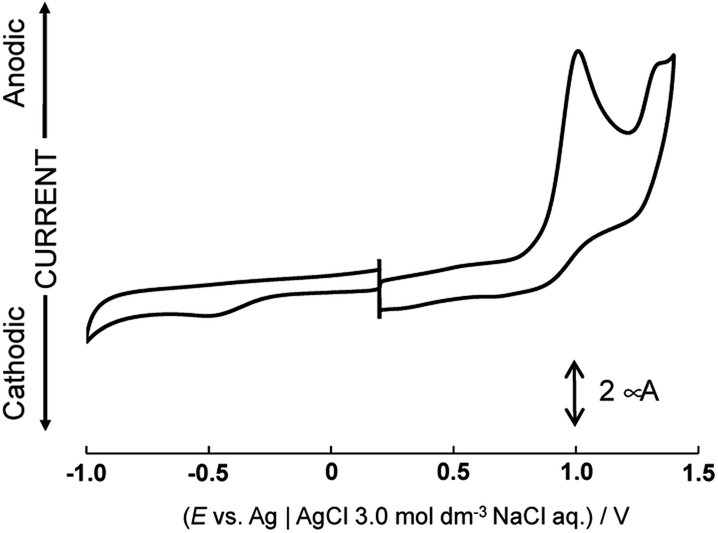 | ||
| Fig. 2 Cyclic voltammogram of [1](CF3SO3)2 in water (pH 2.77)-dimethyl sulfoxide. | ||
Oxidation of dianiline complexes of ruthenium(II)
Aqueous red solutions of [1](CF3SO3)2 or [2](CF3SO3)2 containing four molar equivalents of (NH4)4[CeIV(SO4)4]·2H2O were refluxed, affording purple solutions. By addition of NH4PF6, N1-phenyl-cyclohexa-3,5-diene-1,2-diimine complexes of ruthenium(II) (diimine complex), cis-[RuII(NHC6H3RNC6H4R)(bpy)2](PF6)2 (R = H; [4](PF6)2, R = 4-CH3; [5](PF6)2), were obtained in 93 and 98% yield, respectively, through C–H activation of the aniline ligands and N–C bond formation on the ruthenium center (Scheme 1). On the other hand, oxidation of [3](CF3SO3)2, whose geometrical configuration is a trans-form, afforded a mixture of ruthenium complexes that are different from the diimine complex. 1H NMR spectra of [4](PF6)2 and [5](PF6)2 in nitromethane-d3 revealed the diamagnetic property of the ruthenium(II) state (Fig. S1†). The signal patterns of diimine complexes are more complex than those of dianiline complexes, because of the signals of bpy, the cyclohexadiene ring, and an uncoordinated phenyl group are overlapped in the typical aromatic regions. The signals of the imine proton were observed as a singlet at 12.39 ppm for [4](PF6)2, and 12.28 ppm for [5](PF6)2, respectively. These are similar to that of the same diimine ligand (15.2 ppm for [RuCl2(bpy)(NHC6H4NPh)]24 and 13.08 ppm for [RuCl(tpy)(NHC6H4NPh)]+, tpy = 2,2′; 6′,2′′-terpyridine26). In a D2O solution, the singlet signal of the imine proton disappeared, indicating the proton was exchanged with the D atom of D2O. An IR spectra of [4](PF6)2 and [5](PF6)2 showed characteristic bands of the diimine group at 3297 and 3298 cm−1, which are assignable to ν(NH) (Fig. S3†). These are similar to the characteristic N–H stretch bands of diimine ligands.31,32 | ||
| Scheme 1 Oxidation reactions of dianiline complexes to give diimine complexes in water. | ||
Electronic spectra of [4](PF6)2 and [5](PF6)2 are shown in Fig. S4 and Table S1.† The intense band at 520 and 421 nm ([4](PF6)2) and 524 and 425 nm ([5](PF6)2) could be assigned to transitions between orbitals that are both delocalized over the ruthenium center and the diimine moiety as similar to those of the reported diimineruthenium(II) complexes.22–28 These attributions are based on TD DFT calculations (Fig. S6†). Spectral changes of dianiline complexes [1](CF3SO3)2 and [2](CF3SO3)2 in the presence of four molar equivalents of (NH4)4[CeIV(SO4)4]·2H2O in aqueous solutions are shown in Fig. 3 and S11.† Formation of [4]2+ occurred within 30 minutes without any byproduct as evidenced by the spectral monitoring for the oxidation reaction of [1](CF3SO3)2, while that of [5]2+ within 2 hours. Reaction intermediates was not confirmed by ESR spectrum of the reaction mixture.
 | ||
| Fig. 3 Electronic spectra of [1]2+ (0.05 mM, black) and after 30 minutes (blue). | ||
For the oxidation of [1](CF3SO3)2 in the presence of a radical scavenger, 2,2,6,6-tetramethylpiperidine 1-oxyl, the spectrum after the reaction showed absorption bands at 575 and 486 nm, indicating that [4]2+ is not formed (Fig. S12†). The results suggest that a radical species is formed during the oxidation reaction of [1]2+. However, the spectrum changed from [1]2+ to [4]2+ with isosbestic points, so the reaction intermediates are hardly observed and isolated in the reaction conditions. Similar spectral change was observed in a four-electron oxidation reaction of [2]2+ to afford [5]2+. Changes of absorbance at 520 nm for [1]2+ (black) and at 524 nm for [2]2+ (blue) during oxidation are shown in Fig. S13.† The reaction rate of the formation of [5]2+ is slower than that of [4]2+. For 1H NMR spectrum of [1](CF3SO3)2, the signals of the aniline ligands were observed in the higher magnetic field than those of [2](CF3SO3)2. The results indicate that the electron density of aniline nitrogen of [2](CF3SO3)2 increases due to the effect of the electron-donating methyl group, and the protons are not easily dissociated from the aniline nitrogen and the formation of diimine ligand of [5](PF6)2 occurs at a slower rate than that of [4](PF6)2.
Cyclic voltammograms of [4](PF6)2 and [5](PF6)2 showed a one-electron oxidation wave at 1.10 and 1.03 V (E1/2) and a one-electron reduction wave at −0.69 and −0.76 V (E1/2) in nitromethane (Fig. 1). Notably, the potential difference between [4](PF6)2 and [5](PF6)2 is 70 mV because of the contribution cyclohexadiene-1,2-diimine ligand to the redox center, although the oxidation waves of dianiline complexes [1](CF3SO3)2 and [2](CF3SO3)2 with or without the methyl groups of the aniline ligand were observed at the same potential. These waves of [4](PF6)2 and [5](PF6)2 are assigned to oxidation and reduction processes based on the molecular orbitals contributed by both the diimine ligand and the ruthenium center, which is supported by the results of DFT calculations (Fig. S10 and Table S3†). In other words, the diimine ligand is a non-innocent ligand. On the other hand, the HOMOs of [1](CF3SO3)2 and [2](CF3SO3)2 are mainly contributed by the ruthenium center, so the redox potential does not change even when the methyl group is introduced at the phenyl group of the aniline ligand.
X-ray structures of dianiline and diimine complexes
Single crystals were obtained by diffusion of diethyl ether into acetone solutions of [1](CF3SO3)2 and [2](CF3SO3)2, a methanol solution of [3](CF3SO3)2, a nitromethane solution of [4](PF6)2 and a methanol solution of [5](PF6)2 at room temperature. Crystallographic data and selected structural parameters are summarized in Tables 1 and S4.† Structures are shown in Fig. 4–6, S14 and S15.† These complexes have a hexa-coordinate octahedral geometry. For [1](CF3SO3)2, [2](CF3SO3)2, [4](PF6)2 and [5](PF6)2, two bpy ligands coordinate at the cis-position to each other. The structural parameters of two dianiline complexes, [1](CF3SO3)2 and [2](CF3SO3)2, are similar to one another. For [3](CF3SO3)2, due to the bulkiness of 2,6-difluoroaniline, these ligands coordinated trans to each other to avoid steric hindrance. The N–C bond distances of the aniline ligands are within the range of the single bond distances. The Ru–Naniline bond distances are longer than the Ru–Nbpy, due to the differences in nitrogen-donor properties and the radius of an N atom between amine and imine. The Ru–Naniline bond distances and the Ru–N–C bond angles are similar to those of the previously reported anilineruthenium complexes.24 For [3](CF3SO3)2, the bond angle of N3–Ru–N4 in bpy is smaller than that of [1](CF3SO3)2 and [2](CF3SO3)2 indicating repulsion between the protons at 6-position of two bpy ligands. [3](CF3SO3)2 has distorted hexa-coordinate octahedral geometry and dihedral angle of pyridine (containing N3)-pyridine (containing N4) (18.469°) is larger than those of cis-type dianiline complexes [1](CF3SO3)2 and [2](CF3SO3)2 (2.999–11.678°).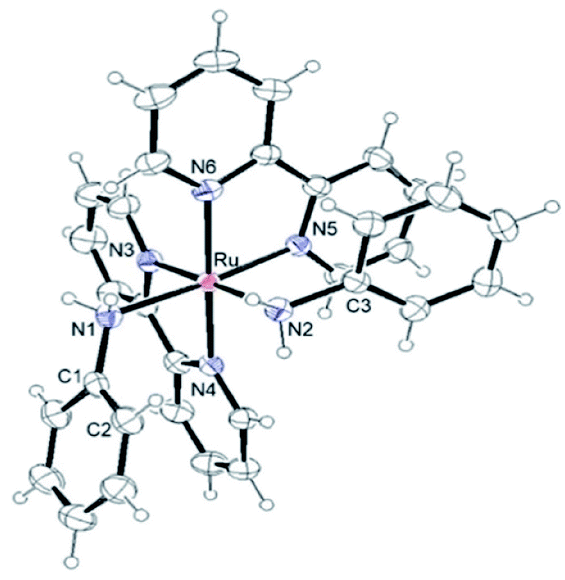 | ||
| Fig. 4 Structure of [1]2+ showing 50% thermal ellipsoid probability. | ||
 | ||
| Fig. 5 Structure of [3]2+ showing 50% thermal ellipsoid probability. | ||
 | ||
| Fig. 6 Structure of [4]2+ showing 50% thermal ellipsoid probability. | ||
For diimine complexes [4](PF6)2 and [5](PF6)2, the ruthenium center and the bidentate ligand form a five-membered ring, which consists of Ru, two nitrogen (N1 and N2) and two carbon (C1 and C2) atoms. The distance between the ruthenium center and the nitrogen atom of the imine group (Ru–N1) is shorter than those of bpy (Ru–N3, N4, N5 and N6) and the aniline ligands of [1](CF3SO3)2 and [2](CF3SO3)2 (Ru–N1 and N2) because of π back donation from the dπ orbital of ruthenium center to the imine moiety. The bond distances of N1–C1 and N2–C2 of diimine ligands are within the general N–C double bond range. The bond distance of Ru–N2 is longer than that of Ru–N1 and is similar to those of bpy nitrogen (N3, N4, N5, N6). The bond distances of Ru–N3 and Ru–N5 are longer than those of Ru–N4 and Ru–N6, because N3 and N5 contribute to the same orbital of the 5-membered ring of the diimine ligand (Table 1).
| [1]2+ | [2]2+ | [3]2+ | [4]2+ | [5]2+ | |
|---|---|---|---|---|---|
| Bond distances (Å) | |||||
| Ru–N1 | 2.195(2) | 2.192(4) | 2.145(3) | 1.978(2) | 1.984(4) |
| Ru–N2 | 2.2009(19) | 2.174(3) | — | 2.051(2) | 2.036(4) |
| Ru–N3 | 2.044(2) | 2.063(3) | 2.083(3) | 2.086(2) | 2.079(4) |
| Ru–N4 | 2.0794(19) | 2.077(4) | 2.092(3) | 2.068(2) | 2.073(3) |
| Ru–N5 | 2.0398(19) | 2.037(4) | — | 2.102(2) | 2.083(4) |
| Ru–N6 | 2.067(2) | 2.074(4) | — | 2.069(2) | 2.065(3) |
| N1–C1 | 1.448(3) | 1.446(6) | 1.420(5) | 1.303(3) | 1.304(6) |
| N2–C3 | 1.453(3) | 1.435(6) | — | 1.436(3) | 1.456(5) |
| C1–C2 | 1.376(4) | 1.380(7) | 1.390(6) | 1.453(3) | 1.458(7) |
| N2–C2 | — | — | — | 1.322(3) | 1.311(6) |
|
|||||
| Bond angles (°) | |||||
| N1–Ru–N2 | 86.69(8) | 89.03(14) | 180.0 | 77.53(9) | 77.02(16) |
| N3–Ru–N4 | 78.83(8) | 78.44(14) | 77.16(11) | 78.22(9) | 77.77(14) |
| N5–Ru–N6 | 79.10(8) | 78.48(15) | — | 77.77(8) | 78.12(15) |
| Ru–N1–C1 | 118.33(15) | 115.3(3) | 117.7(2) | 118.83(18) | 118.8(3) |
| Ru–N2–C2 | — | — | — | 115.34(16) | 117.0(3) |
| Ru–N2–C3 | — | — | — | 124.87(16) | 121.8(3) |
| N1–C1–C2 | 120.0(2) | 121.0(4) | 121.1(4) | 114.0(2) | 113.8(4) |
| N2–C2–C1 | — | — | — | 114.1(2) | 113.3(4) |
|
|||||
| Dihedral angles (°) | |||||
| pyridine(N3)–pyridine(N4) | 2.999 | 11.678 | 18.469 | 7.780 | 9.122 |
| pyridine(N5)–pyridine(N6) | 6.047 | 5.398 | 7.158 | 2.648 | |
Due to the steric hindrance of the o-position hydrogen atoms, the uncoordinated phenyl ring bounds to the nitrogen N2 of the diimine complexes makes no contribution to the cyclohexadienediimine moiety and twists from the plane of the five-membered ring (Ru, N1, N2, C1 and C2), in which the dihedral angle is 60.194°. The structural parameters of two bpy ligands are similar to those of the previously reported typical ruthenium complexes.33–36
Conclusions
Dianilineruthenium(II) complexes [1]2+, [2]2+ and [3]2+ were synthesized by reactions of the carbonato complex of Ru(II) with aniline or its derivatives. The two aniline ligands are situated on the cis position in [1]2+ and [2]2+, and on the trans position in [3]2+. The point of our research is to induce the proton dissociation reaction of coordinated aniline by oxidation of the central metal. Four-electron oxidation reaction of the cis-form dianiline complexes afforded diimineruthenium(II) complexes [4]2+ and [5]2+ with the formation of an N–C bond. At the first step in the formation of [4]2+ and [5]2+, a one-electron oxidation of the ruthenium center occurs. Due to one-electron oxidation, the dπ orbitals of the ruthenium center are more stabilized than those of the aniline ligand. As a result, the electrons of the aniline ligand are removed, and the protons will be easily dissociated. The formation of the diamine complexes involves successive oxidation, intramolecular electron transfer, and deprotonation steps, and the intermediate will be easily oxidized at a closer potential to 0 V than the starting complexes. These C–H activation and N–C formation processes accompanied with redox and proton transfer reactions will be useful in synthesis of a nitrogen-containing ligand on a metal complex. Studies on properties of the present ruthenium(II) complexes and syntheses of their derivatives containing the nitrogen-containing ligands are in progress to elucidate the details of the reaction mechanisms.Experimental
Materials
[Ru(η2-O2CO)(bpy)2]·2H2O and cis-[RuII(NH2C6H5)2(bpy)2](CF3SO3)2 ([1](CF3SO3)2) were synthesized by the procedures reported in the literature.37,38Measurements
Elemental analyses were carried out with a PerkinElmer 2400-II or were performed by the Institute of Chemical and Physical Research Analytical Facility, Wako Saitama, Japan. FAB MS spectra were measured with a JEOL JMS-700 spectrometer with m-nitrobenzyl alcohol using as matrix. 1H and 13C NMR spectra were obtained with a JEOL JML-AL300 and AL500 spectrometers. UV-vis spectra were measured on a Shimadzu MultiSpec-1500 or UV-3600 Plus. IR spectra were recorded on a Shimadzu IR Affinity-1 spectrophotometer using samples prepared as KBr disks. Electrochemical measurements were made in CH3NO2 solutions containing 0.1 mol dm−3 tetra-n-buthylammonium perchlorate (TBAP, Nakarai Tesque Ltd.) as a supporting electrolyte and an Ag|0.01 mol dm−3 AgNO3 reference or in H2O–DMSO (2:3 (v/v)) solutions containing 0.2 M HClO4–NaOH aqueous solution with a glassy carbon working electrode (ϕ = 1.6 mm), an Ag|AgCl 3.0 M NaCl reference electrode, and a platinum wire counter electrode using an ALS 630E electrochemical analyzer. At the end of each measurement, ferrocene (Fc) was added as an internal standard to correct redox potentials (E(Fc+/Fc) = 0.074 V vs. Ag|0.01 mol dm−3 AgNO3). Caution! Perchloric acid is potentially explosive and should be handled with care.
Syntheses of complexes
X-ray crystallography
Single crystals were obtained by ether diffusion into acetone solutions of [1](CF3SO3)2 and [2](CF3SO3)2, a methanol solution of [3](CF3SO3)2, a nitromethane solution of [4](PF6)2 and a methanol solution of [5](PF6)2 at room temperature. Intensity data of X-ray crystallography were measured at 298 K on a Rigaku Synergy-S diffractometer using multi-layer mirror monochromated CuKα radiation (1.54184 Å). All calculations were carried out using Olex2 1.2. Structures were solved by direct methods, expected using Fourier techniques and refined using full-matrix least-squares techniques on F2 using SHELXL97 or SHELXT Version 2014/5. Structural Figures of thermal ellipsoid plots were made by Ortep-3 for Windows (L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854). ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Associate Prof. Dr Morgan L. Thomas at Sophia University for English language editing.Notes and references
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- T. Irrgang and R. Kempe, Chem. Rev., 2019, 119, 2524–2549 CrossRef CAS PubMed.
- N. Hofmann and K. C. Hultzsch, Eur. J. Org. Chem., 2019, 3105–3111 CrossRef CAS.
- C. Ghosh, K. Kim, M. R. Mena, J.-H. Kim, R. Pal, C. L. Rock, T. L. Groy, M.-H. Baik and R. J. Trovitch, J. Am. Chem. Soc., 2019, 141, 15327–15337 CrossRef CAS PubMed.
- P. Dongare, S. Maji and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 2194–2199 CrossRef CAS PubMed.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 57, 2224–2228 CrossRef CAS PubMed.
- Y. Nishibayashi, Dalton Trans., 2018, 47, 11290–11297 RSC.
- N. Stucke, B. M. Flöser, T. Weyrich and F. Tuczek, Eur. J. Inorg. Chem., 2018, 1337–1355 CrossRef CAS.
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- P. Nagaraaj and V. Vijayakumar, Org. Chem. Front., 2019, 6, 2570–2599 RSC.
- S. E. Allen, R. Walvoord, R. Padilla-Salinas and M. C. Kozlowski, Chem. Rev., 2013, 113, 6234–6458 CrossRef CAS PubMed.
- F. Hamon, F. D-Pilard, F. Barbot and C. Len, Tetrahedron, 2009, 65, 10105–10123 CrossRef CAS.
- R. Goyal, D. Dumbre, L. N. S. Konathala, M. Pandeya and A. Bordoloi, Catal. Sci. Technol., 2015, 5, 3632–3638 RSC.
- A. Grirrane, A. Corma and H. García, Science, 2008, 322, 1661–1664 CrossRef CAS PubMed.
- C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2010, 49, 6174–6177 CrossRef CAS PubMed.
- W. Lu and C. Xi, Tetrahedron Lett., 2008, 49, 4011–4015 CrossRef CAS.
- X.-G. Li, M.-R. Huang and W. Duan, Chem. Rev., 2002, 102, 2925–3030 CrossRef CAS PubMed.
- G. Ćirić-Marjanović, E. N. Konyushenko, M. Trchová and J. Stejskal, Synth. Met., 2008, 158, 200–211 CrossRef.
- N. Toshima, H. Yan and M. Ishiwatari, Bull. Chem. Soc. Jpn., 1994, 67, 1947–1953 CrossRef.
- Q. Zhang, A. Khajo, T. Sai, I. de Albuquerque, R. S. Magliozzo and K. Levon, J. Phys. Chem. A, 2012, 116, 7629–7635 CrossRef CAS PubMed.
- K. N. Mitra, P. Majumdar, S. M. Peng, A. Castiñeiras and S. Goswami, Chem. Commun., 1997, 1267–1268 RSC.
- K. N. Mitra, S.-M. Peng and S. Goswami, Chem. Commun., 1998, 1685–1687 RSC.
- S. K. Roy, D. Sengupta, S. P. Rath, T. Saha, S. Samanta and S. Goswami, Inorg. Chem., 2017, 56, 4966–4977 CrossRef CAS PubMed.
- S. Mandal, S. Samanta, T. K. Mondal and S. Goswami, Organometallics, 2012, 31, 5282–5293 CrossRef CAS.
- C. Das, K. K. Kamar, A. K. Ghosh, P. Majumdar, C.-H. Hung and S. Goswami, New J. Chem., 2002, 26, 1409–1414 RSC.
- P. Majumdar, L. R. Falvello, M. Tomás and S. Goswami, Chem.–Eur. J., 2001, 7, 5222–5228 CrossRef CAS.
- K. N. Mitra, S. Choudhury, A. Castiñeiras and S. Goswami, J. Chem. Soc., Dalton Trans., 1998, 2901–2906 RSC.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7272 CrossRef CAS PubMed.
- X. Chen, X. Wang, Y. Sui, Y. Li, J. Ma, J. Zuo and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11878–11881 CrossRef CAS PubMed.
- A. A. Danopoulos, A. C. C. Wong, G. Wilkinson, M. Hursthouse and B. Hussain, J. Chem. Soc., Dalton Trans., 1990, 315–331 RSC.
- T. Jüstel, J. Bendix, N. Metzler-Nolte, T. Weyhermüller, B. Nuber and K. Wieghardt, Inorg. Chem., 1998, 37, 35–43 CrossRef PubMed.
- A. Enriquez-Cabrera, P. G. Lacroix, I. Sasaki, S. Mallet-Ladeira, N. Farfán, R. M. Barba-Barba, G. Ramos-Ortiz and I. Malfant, Eur. J. Inorg. Chem., 2018, 531–543 CrossRef CAS.
- K. Singha, P. Laha, F. Chandra, N. Dehury, A. L. Koner and S. Patra, Inorg. Chem., 2017, 56, 6489–6498 CrossRef CAS PubMed.
- A. Wołoszyn, C. Pettinari, R. Pettinari, G. V. B. Patzmay, A. Kwiecień, G. Lupidi, M. Nabissi, G. Santonib and P. Smoleński, Dalton Trans., 2017, 46, 10073–10081 RSC.
- S.-H. Wu, J.-Y. Shao, X. Dai, X. Cui, H. Su and Y.-W. Zhong, Eur. J. Inorg. Chem., 2017, 3064–3071 CrossRef CAS.
- E. C. Johnson, B. P. Sullivan, D. J. Salmon, S. A. Adeyemi and T. J. Meyer, Inorg. Chem., 1978, 17, 2211–2215 CrossRef CAS.
- N. Tomioka, T. Misawa-Suzuki and H. Nagao, Polyhedron, 2021, 202, 115193 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, IR spectra, UV-vis spectra, electrochemical measurements, DFT calculations and X-ray structural analysis. CCDC 2101320, 2101321, 2101323, 1972140 and 2101328. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07736h |
| This journal is © The Royal Society of Chemistry 2021 |