DOI:
10.1039/D1RA05670K
(Paper)
RSC Adv., 2021,
11, 36651-36662
Effects of inorganic particles on the crystallization, mechanical properties and cellular structure of foamed PP composites in the IMD/MIM process
Received
24th July 2021
, Accepted 25th October 2021
First published on 12th November 2021
Abstract
Foamed polypropylene (PP) composites containing inorganic particles were prepared using a combined in-mold decoration and microcellular injection molding (IMD/MIM) method, and supercritical fluid-nitrogen was used as the physical blowing agent. Calcium carbonate (CaCO3) and montmorillonite (MMT) with particle size that varied from nanometer to micron were selected as reinforcement particles. The effect of inorganic particle type and size on the crystallization, mechanical properties, and cellular structure of the parts were investigated. The results showed that the polymer chains were not well inserted into the MMT layer, and there was almost no increase in the interlayer spacing. As the particle size decreased, the relative amount of β-crystal and mechanical properties increased. The addition of CaCO3 was more effective than MMT at increasing the volume fraction of the β-crystal form when the particle size was reduced to the nanometer level. In the vertical and parallel section, the average cell diameter of the composites decreased and the cell density increased as the particle size decreased. As the particle size of the inorganic filler decreased, the deformation degree of cells gradually increased.
1. Introduction
Microcellular plastics have been widely used in many fields because of their good heat insulation, impact resistance, and weight reduction effects.1–3 However, their low strength and poor surface quality limit their applications to some extent.
In order to further improve the mechanical properties, many methods have been tried. Adding various types of fillers to polymers as a reinforcing phase has been considered as a recognized method, so various fillers have been tested. Among the inorganic fillers such as calcium carbonate (CaCO3), montmorillonite (MMT), talc, carbon nanotubes, etc. MMT with an exfoliation layer can effectively improve the mechanical properties of materials, which has attracted extensive attention in recent years. Zhu et al. studied the effect of layered silicate on microcellular foam polypropylene (PP). Their results showed that the layer spacing increased with the addition of layered silicate, and the effect of exfoliated silicate was better than that of aggregated layered silicate.4 Chandra investigated the effect of adding MMT on the cellular structure of the foamed polyamide (PA). The results indicated that the exfoliated MMT was distributed in the PA matrix, and the cellular structure was improved after the addition of the MMT particles.5 Hwang investigated the effect of adding MMT on the mechanical properties and cellular structure of the microcellular injection foamed polymer. Their studies suggested that the mechanical properties and cellular structure were also improved after adding MMT.6–10 CaCO3 was also an efficient inorganic filler. Ding et al. investigated the impact of CaCO3 on the foaming behavior of the batch foaming polypropylene. Their studies showed that the addition of nanoscale CaCO3 could improve the heterogeneous nucleation efficiency, thus modifying the cellular structure and improving the strength.11,12 Xi investigated the influence of filler content on the microstructure morphology of iPP/nano-CaCO3 microcellular samples and found that the bubble size of the microcellular samples could be effectively decreased while the cell density increased for iPP/nano-CaCO3 composites.13 Huang investigated the effects of different concentrations of nano-CaCO3 on the polymer blend of HDPE and PP, and the cell structures of foams were analyzed. Their studies showed that a foam PP/nano-CaCO3 composite with 5 wt% nano-CaCO3 exhibited the largest cell density.14 Mao investigated the effects of the nano-CaCO3 content on the crystallization, mechanical properties, and cell structure of PP nanocomposites in microcellular injection molding, and it was found that the mechanical properties, cell structure, and the crystallinity of the material improved after adding nano-CaCO3.15
Some other researchers focused on improving the surface quality of microcellular plastics. Kuo investigated six injection molds with different locations of cooling channels and six injection molds with different degrees of surface roughness of cooling channels were fabricated for low-pressure wax injection molding, and found that the surface roughness of the cooling channel wall is as small as possible when the flow of the coolant is completely turbulent.16 Wang investigated the influences of the cavity surface temperature just before filling on part surface appearance and texture in rapid heat cycle moulding, and it was found that as the cavity surface temperature increased, the aesthetic quality of the molded part was greatly improved by reducing the surface roughness, increasing surface gloss and reducing weld marks.17 In other words, the surface quality of plastic parts was significantly improved by increasing the melt flow performance during microinjection molding. The technology of microcellular injection molding assisted by external field heating such as electric heating was developed consequently and the expected surface quality was obtained.18–20 The combination of gas back-pressure and microcellular foaming was also used to improve the surface quality, which prevented cell nucleation during the filling stage.21–23 High-pressure gas was injected into the mold cavity prior to the injection of the melt, and the high pressure in the cavity prevented cell nucleation during the filling stage, thereby improving the surface quality. However, two methods needed additional equipment, which greatly increased the experimental cost. Co-injection combined with microcellular injection molding can also improve the surface quality.24,25 To obtain parts with a good surface appearance, the solid melts and foamed melts were successively injected into the mold cavity to form a sandwich structure with the solid as the surface layer and the foam as the core layer. In addition to the above moldings, inserting a thermal insulation film into the mold cavity may be a convenient way to improve the surface quality of microcellular injection molded parts. Lee and Chen investigated the effect of insulation film thickness on the surface quality of microcellular injection parts, and they found that the surface quality of the parts became better as the film thickness increased.26,27
However, little research has been done on the effects of inorganic fillers on the surface quality of foamed parts. In this study, a combined in-mold decoration and microcellular injection molding (IMD/MIM) method was studied and used to improve the surface quality of foamed parts. The effects of inorganic filler on the crystallization, mechanical properties, and cellular structure of foamed parts were investigated, respectively.
2. Mathematical model
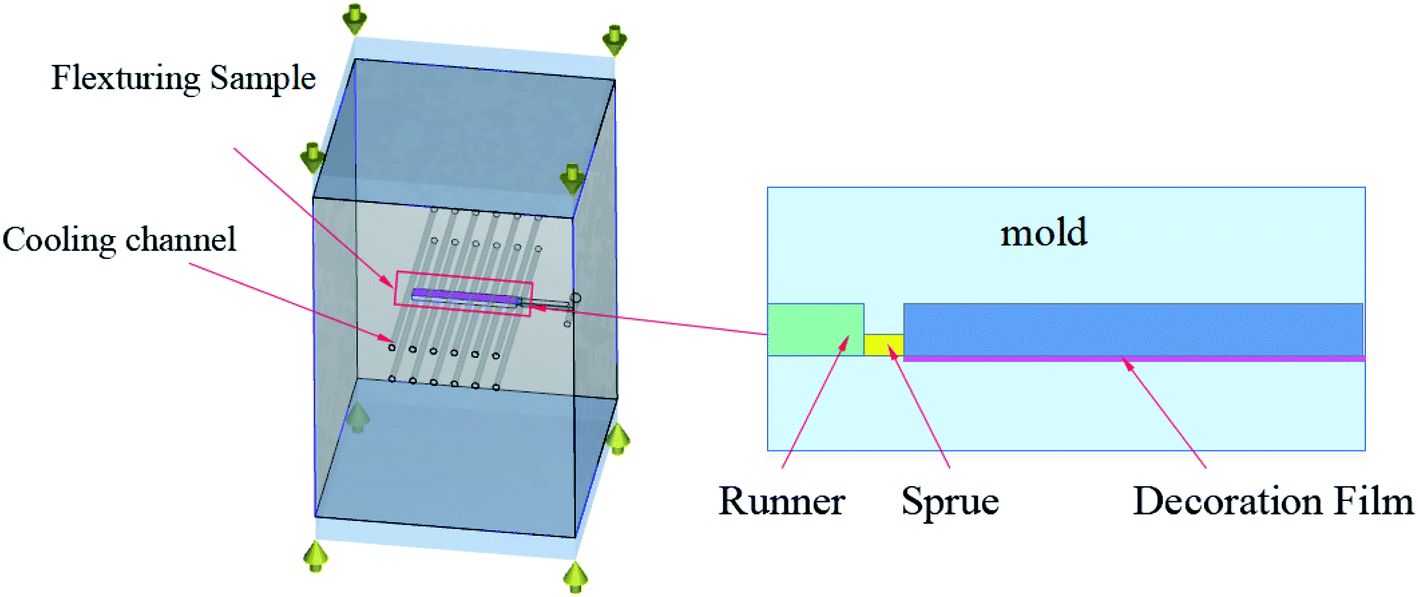
The process of IMD/MIM is shown in Fig. 1. The supercritical fluid nitrogen (N2) was injected into the barrel through the gas inlet, and the SCF-N2 and polymer melt were mixed with a screw to form a single-phase melt. The PET film with a thickness of 0.2 mm adhered to the surface of the mold cavity before the injection of single-phase melt into the mold cavity. The large pressure drop caused the cells to nucleate and the melt was cooled by the mold and film.
 |
| | Fig. 1 The process of IMD/MIM. | |
The lower thermal conductivity of the film led to a higher melt temperature on the film side, resulting in asymmetric melt flow. Higher temperature helps to improve the surface quality because of the lower melt viscosity and film smoothing function. The surface defects were caused by the formation of so-called bubble marks by the deformed bubbles reaching the surface of the part in the filling stage. The lower melt viscosity helps to reduce the friction between the mold and the part, and the higher temperature helped to smooth the bubble marks to improve the surface quality. Both cell size and degree of deformation were affected by the asymmetric temperature distribution.
As shown in Fig. 2, the cell deformation is caused by shear forces between different melt layer flows; the shearing force can be calculated by the following equation:
| |
 | (1) |
where

is the velocity gradient, and
η is the melt viscosity. The cross-WLF viscosity model was used to calculate the polymer melt viscosity:
| |
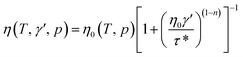 | (2) |
where
T is the temperature of the polymer melt (K),
γ′ is the shear rate of melt (s
−1),
p is the pressure of the polymer melt (Pa),
τ* is the critical shear stress (Pa), and
η0(
T,
p) is the melt viscosity when the shear rate of the melt is zero:
| |
 | (3) |
 |
| | Fig. 2 The shearing force field of melt flow and cell deformation of the IMD/MIM process. | |
The shearing force model could be modified as below:
| |
 | (6) |
Let the minimum shear stress of a single cell be τ1 until it gets deformed, so the region of the core layer could be calculated by the following equation:
| |
 | (7) |
According to this equation, we could calculate the position of the core layer and transition layer.
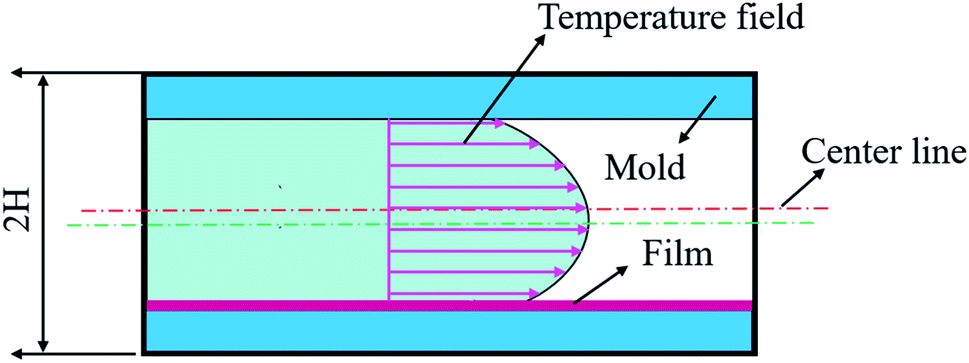
As shown in Fig. 3, the center of the core layer deviates from the center of the mold cavity, and the deviation distance affects the mechanical properties in some way. According to the cell growth model, cell growth is badly affected by the melt temperature. The highest temperature of the mold cavity leads to the lowest melt viscosity, and the center of the core layer exists in the position of the highest temperature.
 |
| | Fig. 3 The temperature field of the IMD/MIM process. | |
Assuming that heat is transferred only along the thickness of the part, the simplified heat transfer model of the IMD/MIM process is shown in Fig. 4. The temperature field of the part is a one-dimensional transient heat transfer. The Fourier heat conduction equation is as follows:
| |
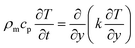 | (8) |
where
cp,
ρm,
k are the thermal capacity, density, and thermal conductivity of the polymer, respectively.
T is the temperature, and
y is the independent variable that represents the coordinate measured at the
y-axis. There is a softening range rather than a sharp melting point in this approach, so it is applied in glassy polymers.
28 The thickness of the mold H, is much thinner than other dimensions of the part, so the conduction in the thickness direction will be considered, and convection in the other axial direction is often ignored.
29 The boundary conditions for the energy equation are given as follows:
| |
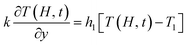 | (10) |
| |
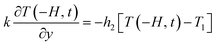 | (11) |
| |
 | (12) |
| |
 | (13) |
| |
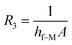 | (14) |
| |
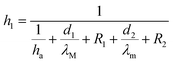 | (15) |
| |
 | (16) |
where
TM is the melt temperature,
T1 is the melt initial temperature.
h1 is the convective heat transfer coefficient on the film side,
h2 is the convective heat transfer coefficient on the non-film side.
d1 and
d2 are the thickness of the mold and decorative film, respectively.
hf–m,
hf–M, and
ha are the convective heat transfer coefficient between the melt and the film, between the melt and the mold, between the mold and the air, respectively.
λM and
λm are the thermal conductivity of the mold and the film, respectively. Let

,
T(
y,
t) =
Y(
y)
ξ(
t). The temperature field of the polymer melt obtained in the thickness direction of the cavity is given as follows:
| |
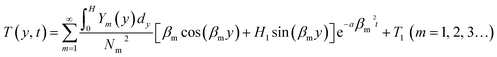 | (17) |
 |
| | Fig. 4 The simplified heat transfer model of the IMD/MIM process: 1-mold; 2-decorative film; 3-sample; 4-mold. | |
To determine the location of the highest melt temperature, it can be concluded that when y is equal to  , the melt temperature is the highest by deriving the temperature field.
, the melt temperature is the highest by deriving the temperature field.
3. Experimental
3.1. Finite element
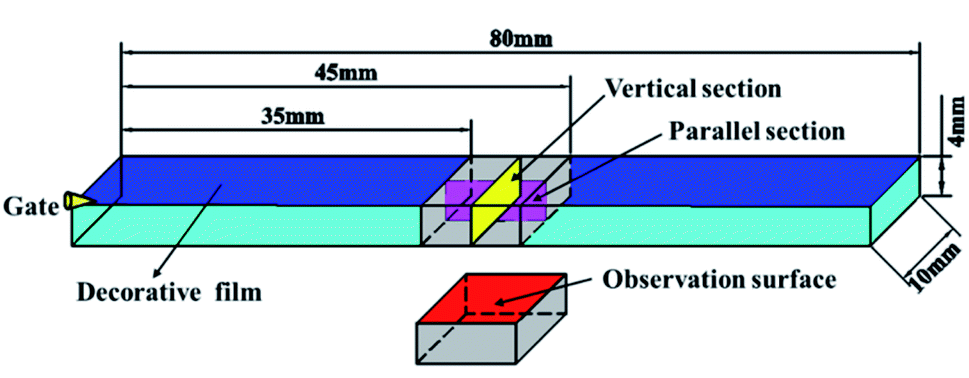
In order to better explain the experimental phenomenon, the corresponding finite element model was established for numerical simulation, and the flexural sample (80 mm × 10 mm × 4 mm) was selected as the research object, which is shown in Fig. 5. The melt temperature, mold temperature, and coolant temperature were set at 493 K, 313 K, and 298 K, respectively. The melt flow rates were 0.08, 0.12, 0.16, 0.2 and 0.24 m s−1, respectively. The content of the foaming agent (N2) was 0.5%.
 |
| | Fig. 5 Finite element model. | |
3.2. Preparation of composites
The MIM process flow chart is shown in Fig. 6. All materials were dried in a drying oven (101A-1, Guangdi Instrument Equipment Co. Ltd, Shanghai, China) when preparing composites. The materials were then put into the mixer (SHR-10, Yiyang Plastic Machinery Co. Ltd, Wuhan, China) according to the established proportions. CaCO3 and MMT were respectively selected as the inorganic filler with a content of 4 wt% and nucleation agent with particle sizes of 19 μm, 3.4 μm, 1.6 μm and 70 nm, and 5 wt% PP-g-mah was added as the compatibilizer. The composites were extruded by the twin-screw extruder (SHJ-20, Giant Machinery Co. Ltd, Nanjing, China) and then pelletized by the pelletizer (LQ-20, Giant Machinery Co. Ltd, Nanjing, China). Materials were dried for 24 hours at 80 °C. The temperature of the extruder was set into five stages of 443 K, 453 K, 453 K, 453 K, and 458 K, respectively. The inorganic fillers were supplied by Changshan Jinxiong Co. Ltd (Changshan, China). The polypropylene (K8303) was supplied by Sinopec Beijing Yanshan Co. Ltd (Beijing, China), with a 2.0 g per 10 min melt flow index. The PP-g-mah (1.2% grafting) was provided by Dongyuan Ziheng Plastics Co. Ltd (Dongguan, China). The industrial N2 (99% purity) used as the blowing agent was provided by Wuhan XiangYun Industry Co. Ltd (Wuhan, China). The film was provided by the China Gwell Machinery Co. Ltd (Shanghai, China) and its thermal conductivity was 0.248 W m−1 K−1.
 |
| | Fig. 6 The MIM process flow chart. | |
3.3. Foaming process setup
As shown in Fig. 6, during the foaming process, the industrial N2 was pressurized to over 28 MPa by a pressure pump (GBL-200/350, CHN-top Machinery Group Co. Ltd, Beijing, China) to become the supercritical fluid (SCF). The barrel temperature of the injection molding machine (HDX50, Haida Plastic Machinery Co. Ltd, Ningbo, China) was divided into four stages, 443 K, 453 K, 453 K, and 453 K, respectively. The injection pressure of SCF-N2 was controlled by a microcellular foaming console (CHN-top Machinery Group Co. Ltd, Beijing, China). The SCF-N2 (15 MPa) was injected into the barrel during the plasticizing process of the composites. The injection time of SCF-N2 was 2 s and its content was 0.5%. The composites and SCF-N2 formed a single-phase melt after being mixed by screws. The injection pressure and back-pressure of the injection machine were 80 MPa and 10 MPa, respectively. The decorative film should be stuck to the mold cavity surface before the melt is injected. Due to the large pressure drop in the cavity, the composite began to foam and the sample was obtained after cooling for 30 s.
3.4. Characterizations
The microstructure and mechanical properties of injection molded parts were analyzed. A D8 Advance (Bruker Co. Ltd Karlsruhe, Germany) X-ray diffractometer was employed to analyze the crystallization behavior and the intercalation effect of MMT. The scanning range was from 3 to 30° with a scanning rate of 5°·min−1.
The relative content of the β-crystal form was calculated by the following equation:
| |
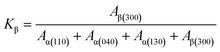 | (18) |
where
Kβ represents the relative content of the β-crystal form relative to the α-crystal form,
Aα and
Aβ refer to the areas of different diffraction peaks.
A JSM-IT300 (JEOL Co. Ltd Tokyo, Japan) scanning electron microscope (SEM) was used to characterize the structure and distribution of cell and surface quality. The sample was broken after immersing in liquid nitrogen for 3 h, and a gold layer was plated onto the fracture surface before SEM observation. The parallel and vertical sections (10 mm × 4 mm) were taken from the flexural sample as shown in Fig. 7. The middle section of the flexural sample was selected to observe the surface quality.
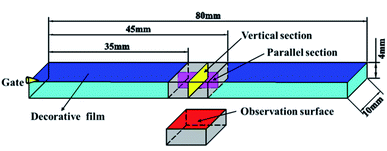 |
| | Fig. 7 Preparation of vertical and parallel sections and the observation surface from the flexural sample. | |
The average cell diameter could be calculated by the following equation:
| |
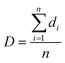 | (19) |
where
di is the diameter of the
ith cell in the given area, and
n is the number of cells, and the cell density
Nf can be calculated using the following equation:
| |
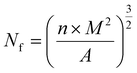 | (20) |
M is the magnification of the SEM picture,
A is the area of the SEM image.
In the parallel section, the cells deformed due to the melt flow are shown in Fig. 8. The ratio of length-diameter (d) was calculated using the following equation:
| |
 | (21) |
where
L and
B are the major axis and minor axis of cells, respectively. The angle between the major axis of the cell and the melt flow direction is defined as the tilt angle (
θ), as shown in
Fig. 8.
 |
| | Fig. 8 Schematic diagram of the ratio of length to diameter and tilt angle. | |
The electromechanical universal test machine (MTS SYSTEMS CMT6104, China) was used to measure the tensile properties and flexural properties. The tensile test method was based on ISO 527-1:1993 with a crosshead speed of 50 mm min−1. The flexural test method follows the standard of ISO 178:2001 with a speed of 2 mm min−1. The impact strength was measured using an impact tester (XJUD-5.5, Chengde Jinjian Testing Instrument Co., Ltd, Chengde, China) according to ISO 180:2000. The average of the five samples calculated was taken as the value of the mechanical properties.
4. Results and discussion
4.1 Temperature field distribution
The simulation results of the composite-forming process suggested that the core layer temperature was higher than the transition layer, as shown in Fig. 9. Besides, the existence of the thermal insulation film resulted in different heat transfer behaviors on both sides of the die, which led to a higher temperature on the side of the parts with the film. At the same time, the asymmetric temperature distribution resulted in the asymmetric flow of the melt, and the front end of the melt flow tended toward the film side. The influences of the asymmetrical distribution of temperature on the vertical section on the crystallization behavior of PP and cellular structure are illuminated below.
 |
| | Fig. 9 (a) Temperature distribution of the melt on the vertical section at the end of filling. (b) Asymmetric flow state of melt. | |
4.2. Microstructure
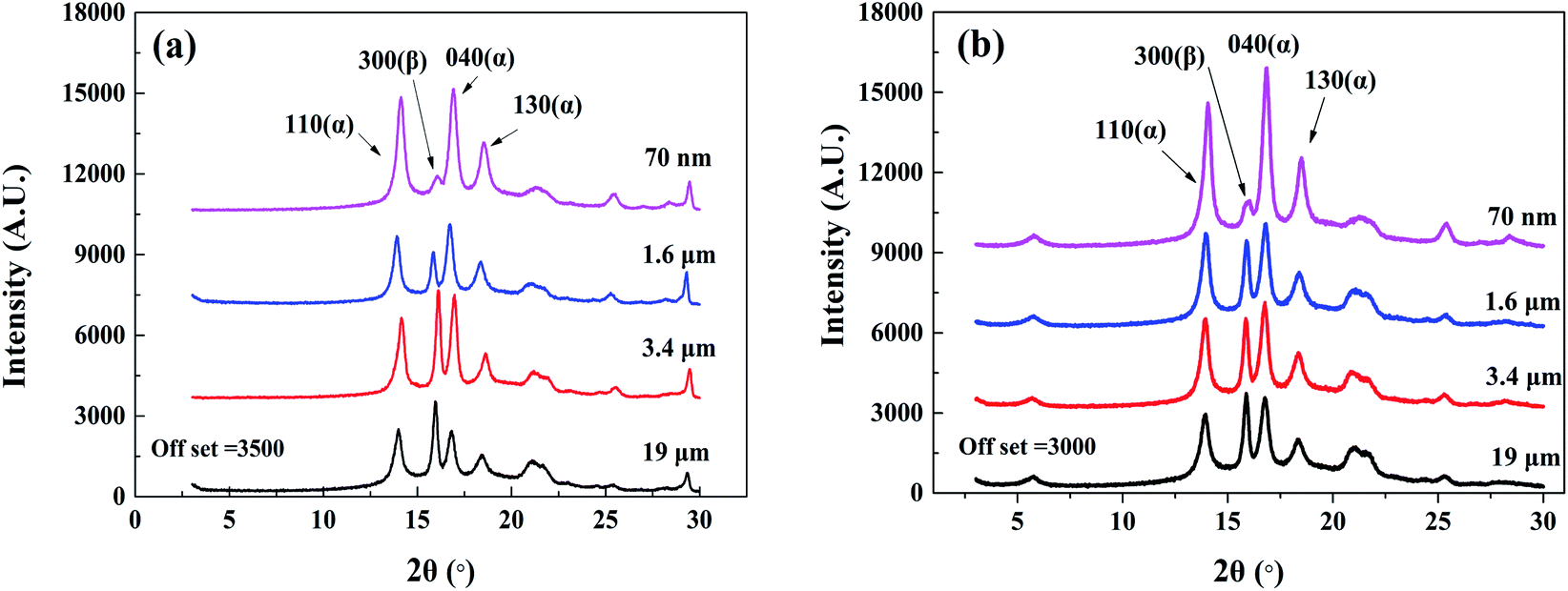
4.2.1. Crystallization. The XRD results of the plastic parts with two reinforcing phases are shown in Fig. 10. The characteristic peak of the α-crystal of the (110), (040), and (130) planes appeared at 2θ = 14.1°, 16.9°, and 18.5°, respectively, and the characteristic β-crystal peak of the (300) plane was located at 2θ = 15.9°. The appearance of β-crystal in all plastic parts indicated that both MMT and nano-CaCO3 acted as nucleating agents and promoted the formation of β-crystal.
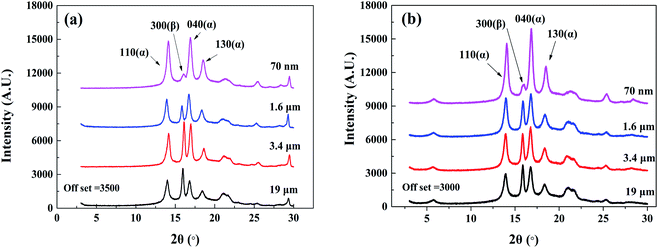 |
| | Fig. 10 The XRD spectra of composites with different particle sizes of inorganic particles added. (a) PP/CaCO3. (b) PP/MMT. | |
The relative β-crystal content was calculated according to formula (18) and listed in Table 1. It can be seen that as the particle size of the inorganic particles decreased, the relative content of the β-crystal form showed an increasing trend. The results indicate that the decrease in particle size increased the contact area between PP and inorganic particles and promoted heterogeneous nucleation, resulting in the β-crystal form. However, the addition of CaCO3 had a greater relative content of the β-crystal form as compared to the addition of MMT when the particle size was reduced to the nanometer level. Compared with layered montmorillonite, spherical calcium carbonate with the same size has a larger specific surface area, resulting in a larger contact area with polypropylene, which is more conducive to the formation of β-crystal.
Table 1 The relative content of the β-crystal form of different composites
| Particle type |
19 μm (%) |
3.4 μm (%) |
1.6 μm (%) |
70 nm (%) |
| CaCO3 |
2.13 |
12.07 |
21.77 |
26.14 |
| MMT |
3.06 |
14.64 |
16.67 |
20.58 |
A local amplification of Fig. 10(b) at a lower diffraction angle was implemented to analyse the intercalation effect of MMT. The diffraction peak of PP/nano-MMT was located at 2θ = 5.73°, which was consistent with pure nanoscale MMT. Besides, the diffraction peak was almost unchanged as the particle size increased, indicating that the variation of particle size did not affect the intercalation of MMT. It is suggested that the efficiency of preparing PP/MMT composites with unmodified montmorillonite by the melt mixing method is not enough to insert the polymer chain into the montmorillonite layer. The MMT particles were distributed in the polymer in an aggregated form rather than in an exfoliated or intercalated form.
4.2.2. Cellular structure on vertical sections. The cellular structures of vertical sections of different composites are presented in Fig. 11. In general, the distribution of cells became more dense and cell merging gradually occurred as the particle size of the inorganic particles decreased. To better analyze the cellular structure, the vertical sections of samples were artificially divided into three layers according to the cell size (one core layer and two transition layers). The thickness of the transition layer on the film side was smaller than that of the transition layer on the non-film side. The higher temperature of the transition layer on the film side caused the core layer to shift toward the film side.
 |
| | Fig. 11 The cellular structure of vertical sections of different composites: (a) 19 μm CaCO3; (b) 3.4 μm CaCO3; (c) 1.6 μm CaCO3; (d) 70 nm CaCO3; (e) 19 μm MMT; (f) 3.4 μm MMT; (g) 1.6 μm MMT; (h) 70 nm MMT. | |
The cell diameter and density of vertical sections of different composites are shown in Fig. 12. As the particle size decreased, the average cell diameter of the composites decreased and the cell density increased. Compared with the transition layer, the core layer had a larger cell size and a lower cell density. Moreover, the transition layer on the film side had a larger cell diameter and density than the transition layer on the non-film side. It is suggested that the smaller particle size helped to increase the cell nucleation efficiency, resulting in the small, dense cellular structure. Besides, a higher temperature makes the cells in the transition layer on the film side have more growth time, while higher temperature reduces the solubility of gas, enhances the thermodynamic instability of gas, and improves the nucleation rate of cells.
 |
| | Fig. 12 The cell diameter and density of vertical sections of different composites. (a) Average cell diameter of PP/CaCO3 composites. (b) Cell density of PP/CaCO3 composites. (c) Average cell diameter of PP/MMT composites. (d) Cell density of PP/MMT composites. | |
4.2.3. Cellular structure on the parallel sections. The parallel sections were also divided into three layers (one core layer and two transition layers) depending on the degree of cell deformation. As shown in Fig. 13, the cell diameters of different composites decreased gradually with the decrease in the particle size. Besides, the cells in the core layer were slightly deformed due to the low shear force, while the cells in the transition layers were severely deformed because of the high shear forces between the melt and the mold.
 |
| | Fig. 13 The cellular structure of parallel sections of different composites: (a) 19 μm CaCO3; (b) 3.4 μm CaCO3; (c) 1.6 μm CaCO3; (d) 70 nm CaCO3; (e) 19 μm MMT; (f) 3.4 μm MMT; (g) 1.6 μm MMT; (h) 70 nm MMT. | |
The statistical results of cell diameters and density of parallel sections of different composites are presented in Fig. 14. The results show that with the decrease in the particle size, the cell diameter and density of PP/CaCO3 and PP/MMT composites decrease and increase, which is consistent with the cell distribution of the vertical section.
 |
| | Fig. 14 The cell density and cell diameter of the core layers. (a) Cell diameter. (b) Cell density. | |
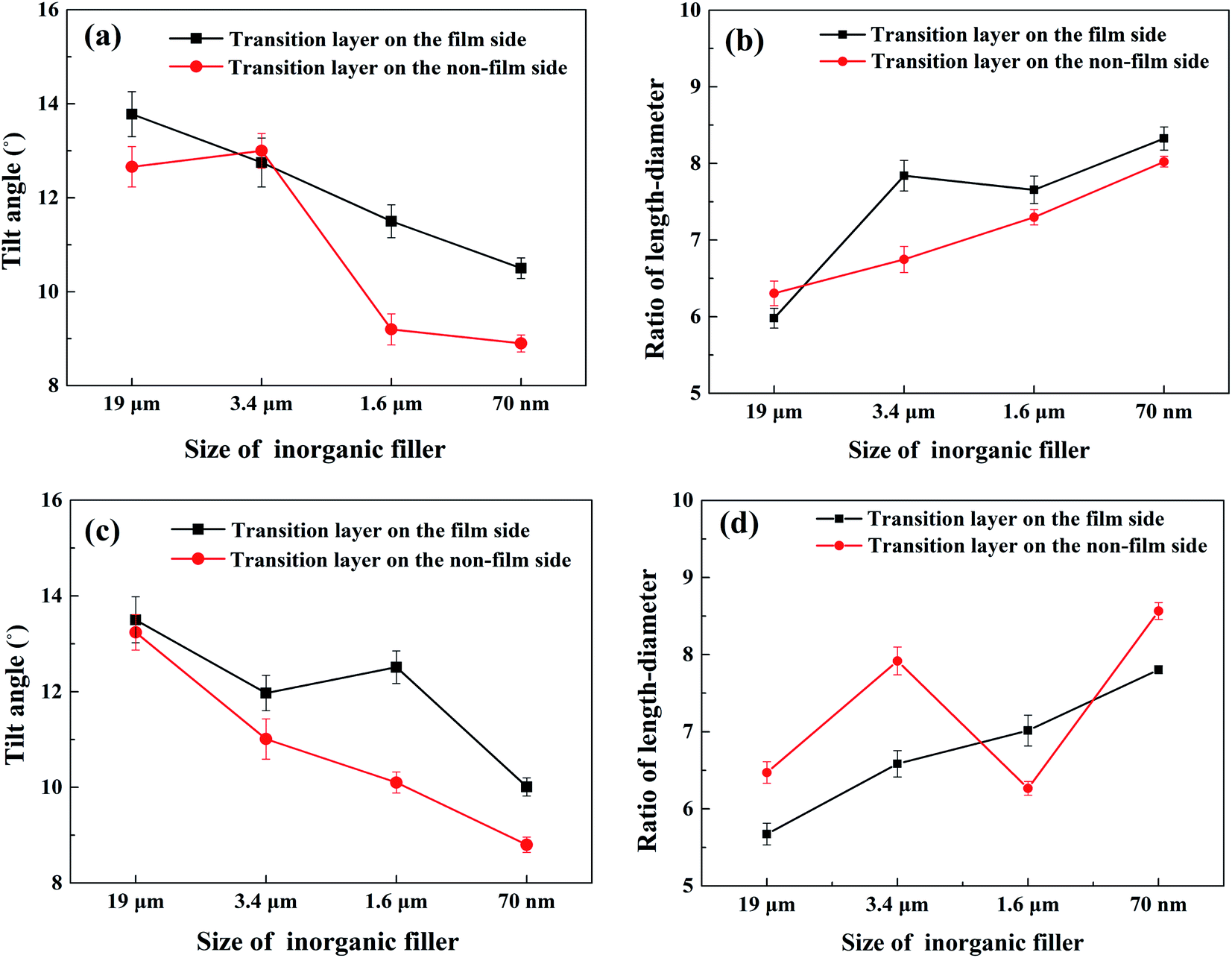
The cell tilt angle and ratio of the length-diameter of transition layers of different composites are shown in Fig. 15. A large ratio of length-diameter and small tilt angle means large cell deformation. We found that the degree of cell deformation increased with the decrease in the inorganic filler particle size. The smaller the particle size, the more uniform the distribution of inorganic particles in the polymer melt, which helps to increase the melt viscosity, resulting in a larger deformation degree of cells during the filling stage. Besides, as the cell density gradually increases, the cell walls between the cells become thinner, and the cells are more easily deformed by the shear force.
 |
| | Fig. 15 Cell tilt angles and ratios of length-diameter of transition layers. (a) Tilt angles of PP/CaCO3 composites. (b) Ratios of length-diameter of PP/CaCO3 composites. (c) Tilt angles of PP/MMT composites. (d) Ratios of length-diameter of PP/MMT composites. | |
As shown in Fig. 16, the thickness of the transition layer on the film side was slightly smaller than the thickness of the transition layer on the non-film side. The higher temperature on the film side drove the core layer to shift toward the film side. The thickness of the core layer gradually decreased and the thickness of both transition layers increased to a small extent as the particle size of the inorganic particles decreased. The increasing viscosity caused by the decreased particle size led to a higher shear force, so the core layer thickness was correspondingly reduced.
 |
| | Fig. 16 The layer thickness of parallel sections. (a) PP/CaCO3 composites. (b) PP/MMT composites. | |
4.3. Mechanical properties
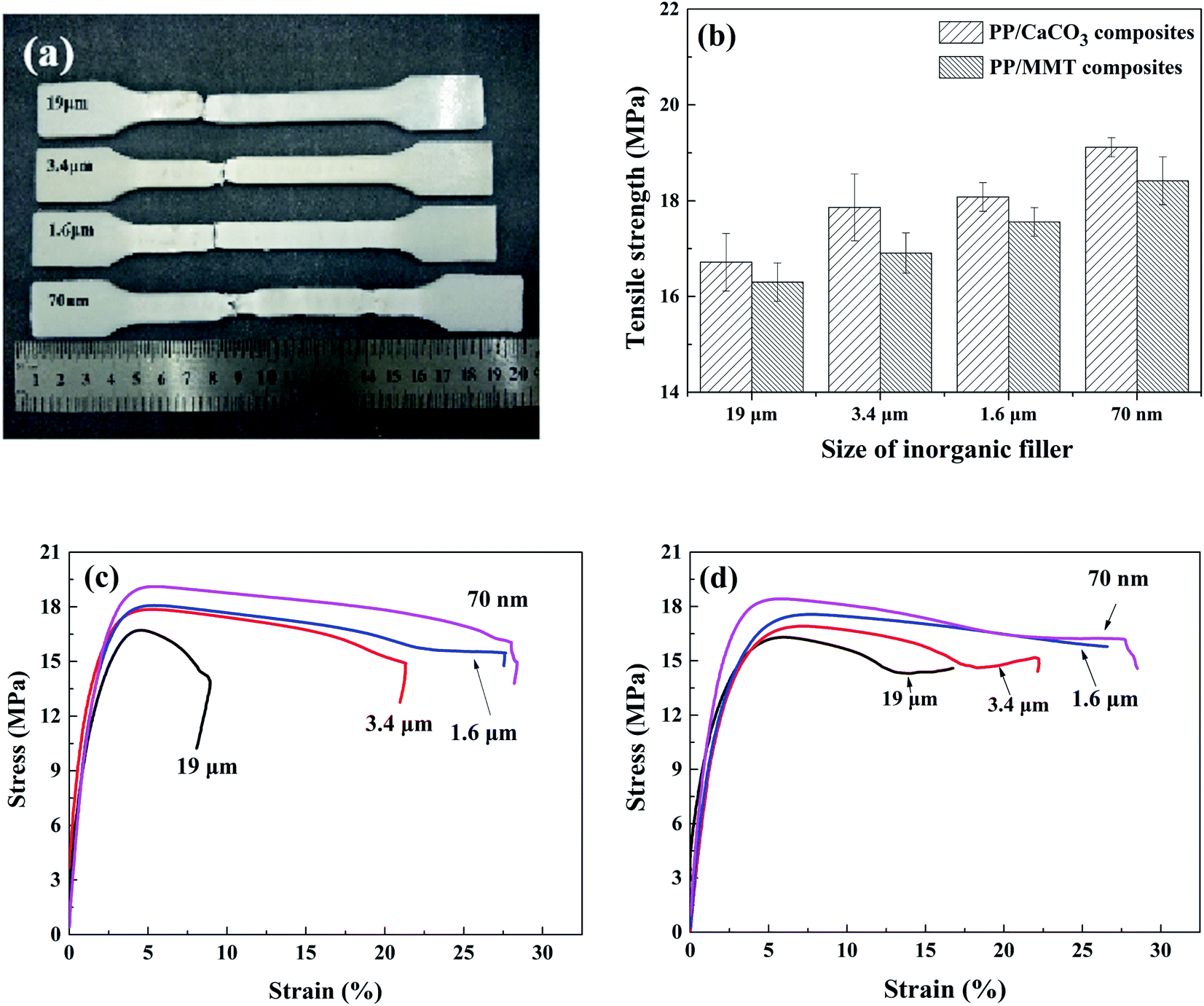
4.3.1. Tensile properties. The effect of particle size on the tensile strength of composites is shown in Fig. 17. The tensile strength of parts added with CaCO3 or MMT increased with the decrement of the particle size. Particles of larger size can cause a large stress concentration in the polymer matrix, thereby weakening the tensile strength. As the particle size decreased, the particles were more evenly distributed in the material matrix and increased the contact area with the polymer matrix. In addition, the smaller inorganic particles could help to delay crack propagation during the stretching process, thereby improving the tensile properties. At the same time, the decrease in particle size is conducive to cell nucleation, resulting in more heterogeneous nucleation. Small and dense elements provide a more uniform stress distribution on the stressed surface, which improves the tensile properties of the parts.
 |
| | Fig. 17 Tensile test results. (a) Partial samples after tensile strength. (b) Tensile strength. (c) Tensile stress–strain curve of PP/CaCO3. (d) Tensile stress–strain curve of PP/MMT. | |
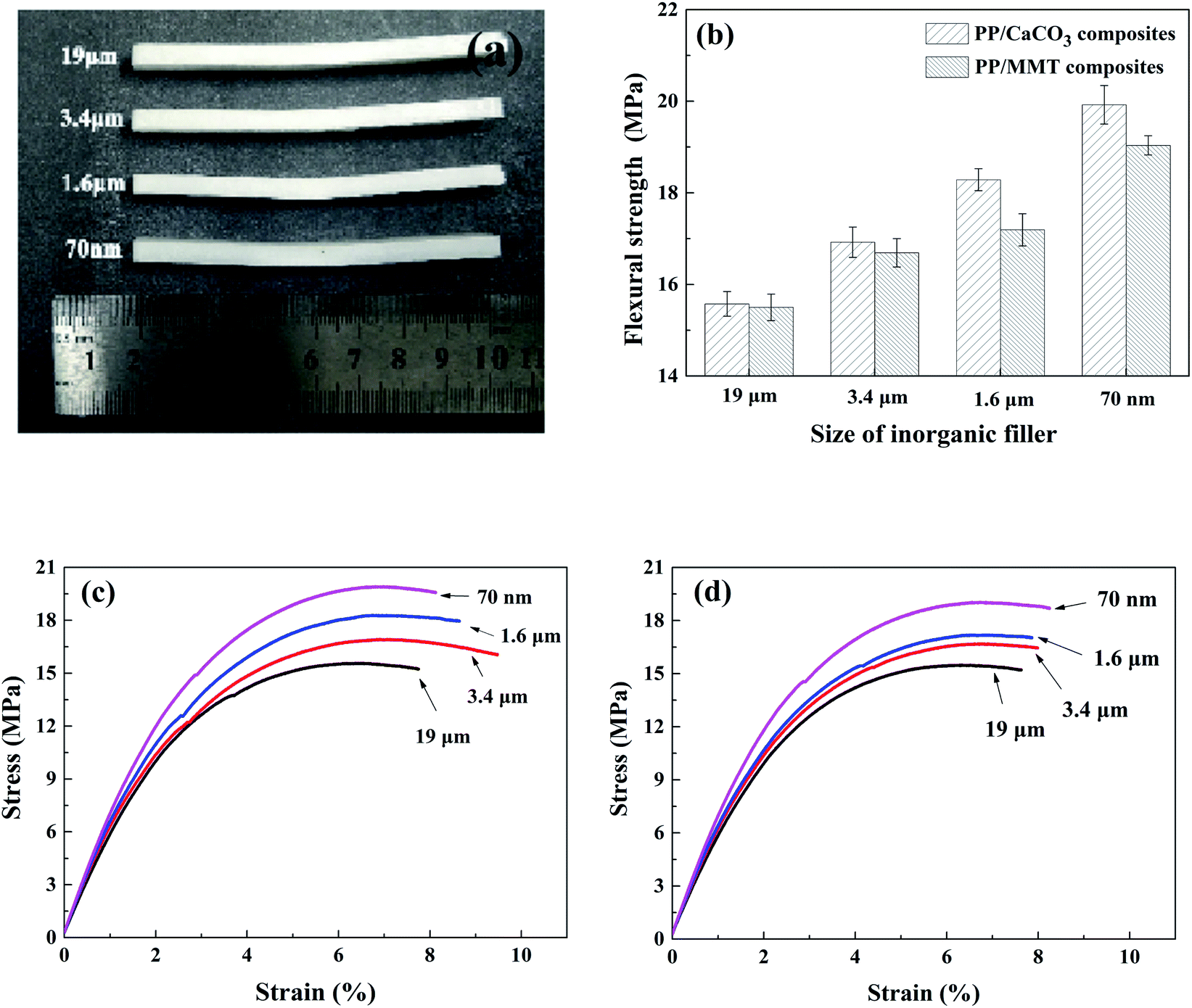
4.3.2. Flexural properties. The effect of particle size on the flexural strength of composites is shown in Fig. 18. The results showed that the deflection of inorganic particle samples with different particle sizes had little difference, which was mainly due to the large rebound after the bending test. The bending strength increased with the decrease of particle size. When inorganic particles of the same mass fraction were added, the smaller particle size led to better improvement in the cellular structure to improve the flexural strength. As shown in Fig. 18(c) and (d), the reduction of the particle size had an obvious effect on the improvement of the flexural strength of samples, and as the particle size decreased, the stress and strain slope of the sample increased continuously; that is, the flexural modulus of the samples increased continuously, which also showed that the reduction in particle size helped to increase the stiffness of the composites.
 |
| | Fig. 18 Flexural test results. (a) Partial samples after bending experiment. (b) Flexural strength. (c) Flexural stress–strain curve of PP/CaCO3. (d) Flexural stress–strain curve of PP/MMT. | |
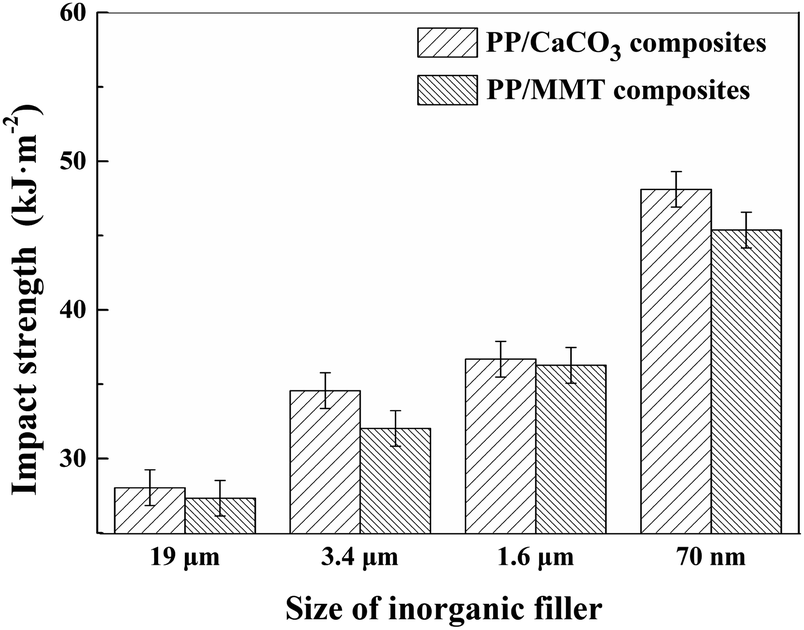
4.3.3. Impact properties. As shown in Fig. 19, as the particle size of the inorganic particles was reduced, the impact strength of the composites was remarkably improved. Adding large-sized inorganic particles to the polymer matrix is more likely to generate a larger stress concentration. As the particle size decreases, small cracks between the particles and the matrix help to absorb the impact energy while small-sized particles help to prevent crack propagation. Besides, the reduction in particle size increases the heterogeneous nucleation efficiency. During the impact load, the deformation of the cells helps to absorb a large amount of the impact energy. The cells in the polymer matrix change the crack propagation path, hinder the crack propagation, and improve the impact strength. The denser and smaller cells will further increase the impact strength.
 |
| | Fig. 19 The impact test results of different composites. | |
5. Conclusion
The effects of inorganic particles on crystallization, mechanical properties, and cellular structure of PP foamed composites were investigated in a combined in-mold decoration and microcellular injection molding (IMD/MIM) process. The results showed that as the particle size of the inorganic particles decreased, the relative content of the β-crystal form showed an increasing trend. However, the addition of CaCO3 had a greater relative content of β-crystal form as compared to the addition of MMT when the particle size was reduced to the nanometer level. The mechanical properties increased with the decrement of the particle size. In the vertical section, the cell size and density of the transition layer on the film side were larger than that on the non-film side. As the particle size decreased, the average cell diameter of the composites decreased and the cell density increased. In the parallel section, the cell ratio of length to diameter on the film side was smaller than that on the non-film side, and the cell tilt angle was larger than that on the non-film side. Changes in cell density and size in the core layer were consistent with those in the vertical section. As the particle size of the inorganic filler decreased, the deformation degree of cells gradually increased.
Author contributions
Formal analysis, W. G.; investigation, T. F.; software, W. R.; supervision, B. H., H. M.; writing original draft, Q. Z.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We would like to thank the National Natural Science Foundation of China Youth Fund (No. 51605356), 111 Project (B17034), Innovative Research Team Development Program of Ministry of Education of China (No. IRT_17R83), the Fundamental Research Funds for the Central Universities (WUT: 2019III112CG).
References
- C. Brondi, M. R. Di Caprio, E. Di Maio, T. Mosciatti, S. Cavalca, V. Parenti and S. Iannace, Int. Polym. Process., 2020, 35, 326–330 CrossRef CAS.
- M. Y. Wang, L. S. Xie, B. Qian, Y. L. Ma and N. Q. Zhou, J. Appl. Polym. Sci., 2016, 133, 41 Search PubMed.
- W. Gong, Y. He, C. Zhang, J. H. Zhu and L. He, Appl. Mech. Mater., 2011, 117–119, 256–261 Search PubMed.
- B. Zhu, W. Zha, J. Yang, C. Zhang and L. J. Lee, Polymer, 2010, 51, 2177–2184 CrossRef CAS.
- A. Chandra, S. Gong, M. Yuan, L. S. Turng and H. Cordes, Polym. Eng. Sci., 2005, 45, 52–61 CrossRef CAS.
- S. S. Hwang, S. P. Liu, P. P. Hsu, J. M. Yeh, K. C. Chang and Y. Z. Lai, Int. Commun. Heat Mass Transfer, 2010, 37, 1036–1043 CrossRef CAS.
- S. S. Hwang, P. P. Hsu, J. M. Yeh, J. P. Yang, K. C. Chang and Y. Z. Lai, Int. Commun. Heat Mass Transfer, 2009, 36, 471–479 CrossRef CAS.
- S. S. Hwang, S. P. Liu, P. P. Hsu, J. M. Yeh, J. P. Yang and K. C. Chang, Int. Commun. Heat Mass Transfer, 2011, 38, 1219–1225 CrossRef CAS.
- S. S. Hwang, P. P. Hsu, J. M. Yeh, K. C. Chang and Y. Z. Lai, Polym. Compos., 2009, 30, 1625–1630 CrossRef CAS.
- S. S. Hwang, S. P. Liu, P. P. Hsu, J. M. Yeh, J. P. Yang and C. L. Chen, Int. Commun. Heat Mass Transfer, 2012, 39, 383–389 CrossRef CAS.
- J. Ding, J. Shangguan, W. Ma and Q. Zhong, J. Appl. Polym. Sci., 2013, 128, 3639–3651 CrossRef CAS.
- J. Ding, W. Ma, F. Song and Q. Zhong, J. Mater. Sci., 2013, 48, 2504–2511 CrossRef CAS.
- Z. Xi, J. Chen, T. Liu, L. Zhao and L. S. Turng, Chin. J. Chem. Eng., 2016, 24(1), 180–189 CrossRef CAS.
- H. X. Huang and J. K. Wang, J. Appl. Polym. Sci., 2007, 106(1), 505–513 CrossRef CAS.
- H. Mao, B. He, W. Guo, L. Hua and Q. Yang, Polymers, 2018, 10(10), 1160 CrossRef PubMed.
- C. C. Kuo, Z. F. Jiang and J. H. Lee, Int. J. Adv. Manuf. Technol., 2019, 103(5–8), 2169–2182 CrossRef.
- G. L. Wang, G. Q. Zhao and X. X. Wang, Int. J. Adv. Manuf. Technol., 2013, 68(5–8), 1293–1310 CrossRef.
- C. L. Xiao, H. X. Huang and X. Yang, Appl. Therm. Eng., 2016, S1359431116301880 Search PubMed.
- L. Zhang, G. Zhao, G. Wang, G. Dong and H. Wu, Int. J. Heat Mass Transfer, 2017, 104, 1246–1258 CrossRef CAS.
- S. C. Chen, Y. W. Lin, R. D. Chien and H. M. Li, Adv. Polym. Technol., 2008, 27, 224–232 CrossRef CAS.
- S. Li, G. Zhao, G. Wang, Y. Guan and X. Wang, J. Cell. Plast., 2014, 50, 415–435 CrossRef CAS.
- G. L. Wang, G. Q. Zhao, J. C. Wan and L. Zhang, Polym. Eng. Sci., 2015, 55, 807–835 CrossRef CAS.
- S. C. Chen, P. S. Hsu and S. S. Hwang, J. Appl. Polym. Sci., 2013, 127, 4769–4776 CrossRef CAS.
- K. Y. Zhang, V. Nagarajan, N. Zarrinbakhsh, A. K. Mohanty and M. Misra, Macromol. Mater. Eng., 2014, 299, 436–446 CrossRef CAS.
- E. Suhartono, S. C. Chen, Y. H. Chang, J. A. Chang and K. H. Lee, Int. J. Plast. Technol., 2017, 21, 239–251 CrossRef CAS.
- J. Lee and L. S. Turng, Polym. Eng. Sci., 2010, 50, 1281–1289 CrossRef CAS.
- S. C. Chen, H. M. Li, S. S. Hwang and H. H. Wang, Int. Commun. Heat Mass Transfer, 2008, 35, 822–827 CrossRef CAS.
- C. Gutfinger, E. Broyer and Z. Tadmor, Polym. Eng. Sci., 1975, 15, 515–524 CrossRef CAS.
- A. Arefmanesh and S. G. Advani, Polym. Eng. Sci., 1995, 35, 252–260 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abcde,
Huajie Mao*abf,
Bo Heabf and
Wei Ruang
*abcde,
Huajie Mao*abf,
Bo Heabf and
Wei Ruang

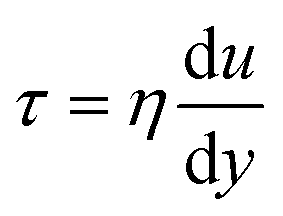
 is the velocity gradient, and η is the melt viscosity. The cross-WLF viscosity model was used to calculate the polymer melt viscosity:
is the velocity gradient, and η is the melt viscosity. The cross-WLF viscosity model was used to calculate the polymer melt viscosity: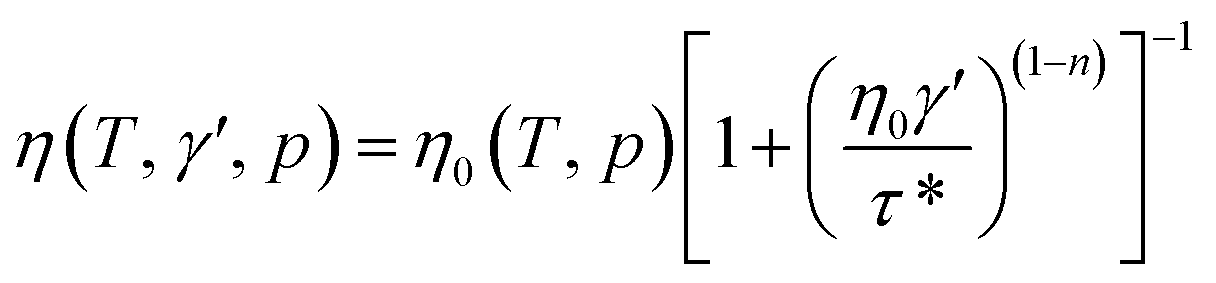
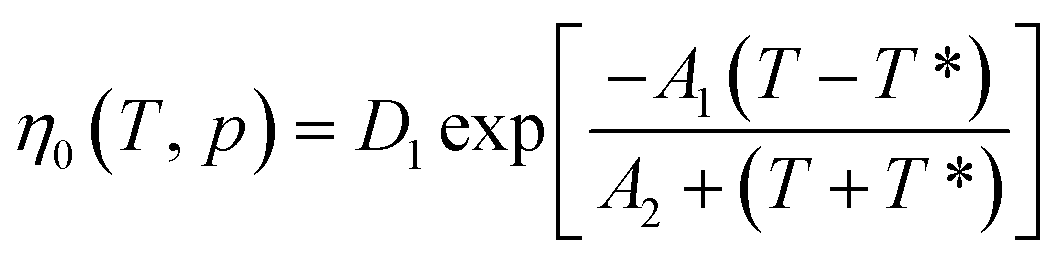

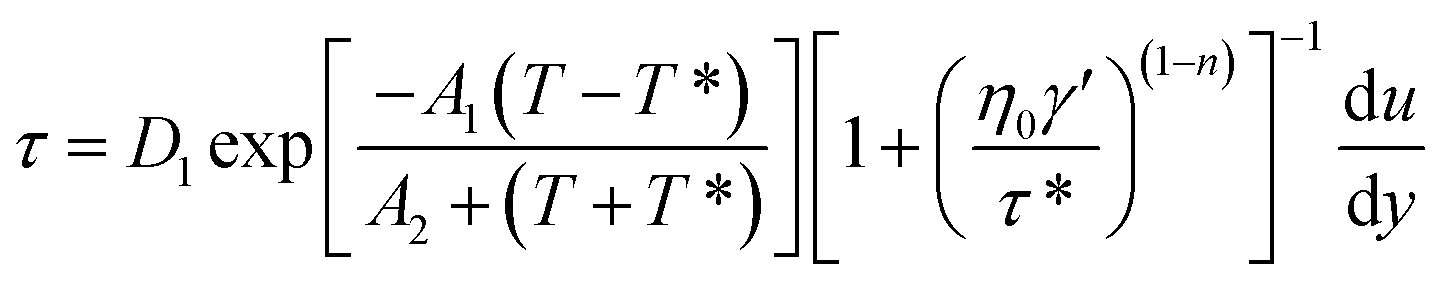


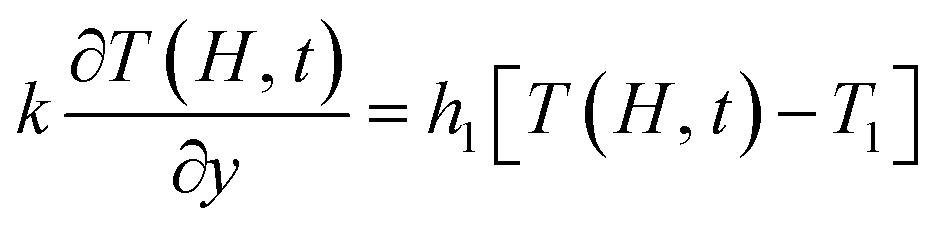






 , T(y, t) = Y(y)ξ(t). The temperature field of the polymer melt obtained in the thickness direction of the cavity is given as follows:
, T(y, t) = Y(y)ξ(t). The temperature field of the polymer melt obtained in the thickness direction of the cavity is given as follows:

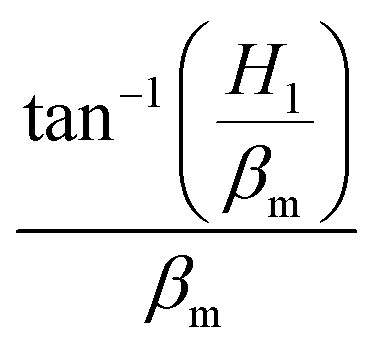 , the melt temperature is the highest by deriving the temperature field.
, the melt temperature is the highest by deriving the temperature field.