
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction of polyoxotantalate by Mg–Fe layered double hydroxides: elucidation of sorption mechanisms†
Rana Choumanea,
Victor Carpentier b and
Grégory Lefèvre*a
b and
Grégory Lefèvre*a
aPSL University, Chimie ParisTech_CNRS, Institut de Recherche de Chimie Paris, Paris, 75005, France. E-mail: gregory.lefevre@chimieparistech.psl.eu
bTND, ZAC du Val de la Deûle, rue de la filature, 59890 Quesnoy sûr Deûle, France
First published on 17th November 2021
Abstract
The extraction of Ta(V) as polyoxometallate species (HxTa6O19(8−x)−) using Mg–Fe based Layered Double Hydroxide (LDH) was evaluated using pristine material or after different pre-treatments. Thus, the uptake increased from 100 ± 5 mg g−1 to 604 ± 30 mg g−1, for respectively the carbonated LDH and after calcination at 400 °C. The uptake with calcined solid after its reconstruction with Cl− or NO3− anions has also been studied. However, the expected exchange mechanism was not found by X-ray Diffraction analysis. On the contrary, an adsorption mechanism of Ta(V) on LDH was consistent with measurements of zeta potential, characterized by very negative values for a wide pH range. Moreover, another mechanism was identified as the main contributor to the uptake by calcinated LDH, even after its reconstruction with Cl− or NO3−: the precipitation of Ta(V) with magnesium cations released from MgO formed by calcination of the LDH. This latter reaction has been confirmed by the comparison of the uptake of Ta(V) in dedicated experiments with solids characterized by a higher magnesium solubility (MgO and MgCl2). The obtained precipitate has been analyzed by X-ray diffraction (XRD) and would correspond to a magnesium (polyoxo)tantalate phase not yet referenced in the powder diffraction databases.
Introduction
Tantalum is classified as a critical raw material by the European Union due to its strong demand, mostly for the fabrication of superalloys used in chemistry, aeronautics and surgery, and as a non-substitutable element in capacitors. Despite its high supply risk, its recycling is very limited, with less than 1% of the tantalum present in Waste Electrical and Electronic Equipment (WEEE) recovered.1–3 However, its recycling from capacitors is important for the sustainability of its use.4,5 Several hydrometallurgical processes have been proposed in this purpose based on an acid leaching, for example with hydrofluoric acid and/or sulfuric acid followed by liquid–liquid extraction with methyl isobutyl ketone (MIBK).4,6 Pyrometallurgy has been developed as an alternative process based on heat treatment at 725–773 K in air atmosphere followed by remelting or extraction of Ta from transition metals in the metallic state (liquid metal extraction),7–9 or extraction of tantalum from tin slags by chlorination and carbochlorination using Cl2 + N2 and Cl2 + CO + N2 gas mixtures.10 Despite the efficiency of these techniques, their industrialization is limited by the high investment costs due to the particular chemical conditions needed: the use of highly corrosive and toxic solutions of concentrated HF,11 combined with H2SO4 or HCl,12 organic solvents such as MIBK,13 or the different purification steps needed. Therefore, these processes represent a serious health hazard for the humans and the environment and recent efforts were focused on alkaline matrices to generate polyoxotantalates (POTas), which have a high solubility in alkaline medium.14,15 Tantalum, whether in metallic or oxidized form (Ta2O5), is insoluble in solution but its alkaline fusion produces a POTa that is soluble in a basic medium. It is present as highly charged polyoxometallates (POM) such as H2Ta6O196−, HTa6O197− or Ta6O198−.16 It is well known that POMs can be formed by refractory metals (V, Mo, W) leading to species with a high overall negative charge.17,18 This specificity could be used in hydrometallurgical processes based on ion exchange. Amongst ion exchangers, layered double hydroxides (LDHs) have been the subject of many studies because they can be considered as green sorbents and their synthesis is cheap.18,19 Moreover, a strong interaction between POMs and LDHs has been shown due to their high negative charge.18,20–23 LDHs structure consists in positively charged brucite sheets whose charge is balanced by intercalation of anions in the hydrated interlayer, with general formula [M1−x2+ Mx3+(OH)2]x+ (An−)x/n·mH2O. M2+ (Mg2+, Fe2+, Co2+…), M3+ (Al3+, Cr3+, Fe3+…) and An− (CO32−, Cl−, NO3−…). They display a high surface area and a high anion exchange capacity since their flexible interlayer region is accessible to various anionic species.24,25 To develop a process with a low environmental impact and based on cheap chemicals, the extraction of dissolved tantalum by LDH deserves to be investigated. However, several mechanisms have been proposed to extract anions from solution by LDHs: surface adsorption, interlayer anion exchange and reconstruction of calcined LDH precursors by “memory effect”.26–29 This term describes the ability to recover the original layered structure when the mixed oxides MII(MIII)O, obtained by mild calcination of the LDH precursor, is immersed in a solution of the anion to be intercalated.30 It should be noticed that this term has been criticized in a recent study where the authors explained that the reconstruction is not based on the memory effect, but it would be simply a direct synthesis based on the reaction of these solid with the anions present in solution.31Amongst the POMs formed by refractory metals (V, Mo, W, Nb, Ta), no study focused on Ta has been published, for the most of our knowledge. The reason can be that the basic pH range required for POM stability constrains the choice of the LDH: the relative stability of MII–MIII–An− LDH is dependent on both the structural M(II) M(III) cations and the interlayer anions (An−) building the structure, and also the electrostatic interactions between the metal hydroxide layer and the interlayer anions.32 Several methods are reported in the literature to study the relative stabilities of LDHs, like the measurement of the equilibrium constants for anion exchange reaction,33 microcalorimetric measurement,34 pH-metric titrations of mixtures of trivalent and divalent metal salt solutions with alkali.35 Miyata33 indicated that CO32− anions is the most preferred divalent anions for structurally more stable LDHs. Boclair and Braterman35 reported that using Fe(III) as M(III) leads to the best stability for LDHs. For these reasons, Mg–Fe–CO3 type composition was used for this study due to its stability at high pH.35 Thus, the main objective of this work was to investigate the possibility of extraction of tantalum as a POM by Mg–Fe–CO3 LDH. The different parameters which can influence the sorption capacities have been investigated and the extraction mechanisms have been identified.
Experimental
Reagents and synthesized solids
All solutions were prepared using Milli-Q water with a resistivity of 18.2 MΩ cm. MgCl2·6H2O (Sigma Aldrich), FeCl3·6H2O (Alfa Aesar), Na2CO3 (Sigma Aldrich), Ta2O5 (Sigma Aldrich, 99.99% purity), NaOH(s) (VWR), H2O2 (50%, VWR), NaCl (VWR), NaNO3 (Sigma Aldrich) MgO (Sigma Aldrich), NaOH (1 M) (Merck), HNO3 (1 M) and concentrated 65% (VWR) were used without further purification.Mg–Fe–CO3 LDH with Mg/Fe molar ratio of 3, general formula Mg0.168Fe0.056(CO3)0.049(OH)0.407·0.199H2O, was prepared by a co-precipitation method according to a procedure described elsewhere.36,37 Briefly, a metal salt solution (150 mL) of MgCl2·6H2O (0.15 mol) and FeCl3·6H2O (0.05 mol) was added to a vigorously stirred alkaline solution containing 150 mL NaOH (0.45 mol) and Na2CO3 (0.15 mol). The pH value of the mixture was adjusted at a value slightly higher than 9 by NaOH (0.1 mmol L−1). After heating at 80 °C for 24 h the solid phase was filtrated and washed several time by Milli-Q water until the pH value of the solution was 8. Then the obtained solid was dried at 80 °C overnight. The material was identified by XRD powder and TGA analyses to confirm its structure and composition. XRD shows that all LDH characteristic peaks are present (according to the ICDD PDF 00-024-1110 file); see Fig. S1A in ESI.† The TGA plot shows, as expected, three steps of mass loss: (1) dehydration (14%), (2) dehydroxylation and removing of carbonate ions from the interlayer (26%), (3) continuous dehydroxylation, decarbonatation, and formation of oxide metals (4%) (Fig. S2†).
Mg–Fe CLDH was obtained by calcining Mg–Fe–CO3 LDH in an oven at the temperature of 400 °C for 4 h.
Na8Ta6O19·24.5H2O(s) was synthesized by alkaline fusion using Ta2O5 as started materials according to.38 Briefly, Ta2O5 (5.7 mmol) was melted with NaOH (0.11 mol) in a Ni crucible at 400 °C for 5 h. The resulting solid was mixed with cold water (30 mL) and then washed 3 times with cold water (40 mL each time). The filtrated solid was dried in vacuum. The resulting white powder has been dissolved in 80 mL of water and then heated for 2 h at 85 °C. After filtration, the filtrate was placed in a refrigerator for a day. A white precipitation has been formed, which was dried in vacuum. The product was identified by XRD powder to confirm its structure (ICDD PDF 00-024-0948 file) (Fig. S1B†).
Batch experiments
Sorption experiments were carried out with tantalum solution prepared from polyoxotantalate (POTa) salt, Na8Ta6O19·24.5H2O(s). All experiments were performed at room temperature under vigorous stirring. 25 mg of sorbent (LDH or CLDH) was suspended into a cell with a 50 mL POTa solution. The pH was adjusted by adding NaOH (1 M) or HNO3 (1 M) if necessary. pH measurements were performed with 827 pH-Lab (Metrohm). Preliminary studies have shown that 24 h was enough to reach the sorption equilibrium.The effect of initial pH on sorption capacity was studied in a solution with 350 mg L−1 of Ta. The range of the studied pH varies between 10 and 12, to stay in the solubility range of Ta.16 The sorption was not performed below these values to avoid the precipitation of Ta, and not above to avoid too concentrated alkaline solutions.
The sorption isotherms were performed in a range of concentration from 20 to 420 mg L−1. The study was performed at pH 12 to be sure that POTa is totally soluble in solution without any precipitation.
In the last part of the study, a test was performed consisting of mixing Mg(II) in the form of solid MgO or MgCl2 in a cell containing 50 mL of POTa with [Ta] = 300 mg L−1 at pH close to 10.4.
After each experiment, the suspensions were centrifuged. The recovered materials were washed by Milli-Q water. The aqueous phases were filtered with 0.45 μm syringe filter followed by the addition of H2O2 3% and HNO3 5% to avoid precipitation14,15 and then the concentrations of Ta were determined by ICP-OES using an iCAP 6000 series spectrometer (Thermo Scientific).
The quantity of Ta sorbed after 24 h (Qe) and percentage of Ta removed (R%) was calculated by the following equations (1) and (2):
 | (1) |
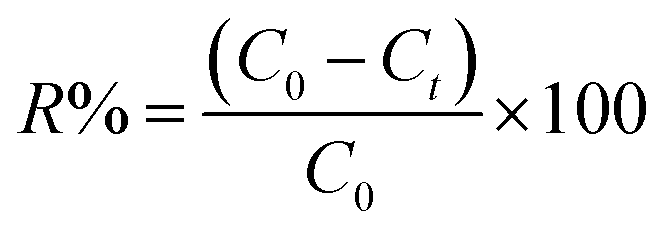 | (2) |
The materials were analyzed before and after sorption by XRD or zeta potential and observed by optical microscopy. Powder X-ray diffraction patterns (PXRD) have been recorded with a Bruker AXS model D8 Advance diffractometer at 40 kV and 35 mA using Co Kα radiation (λ = 1.7902 Å) with a Bragg angle ranging from 5° to 80° and step size 0.029. Zeta potential measurements were carried out with Malvern Nano ZS Zetasizer. 0.3 g of a suspension, either dried Mg–Fe LDH after synthesis or after sorption of Ta, was introduced in 100 mL of KCl (10−2 mol L−1) as electrolyte in a cell. The suspension was dispersed using ultrasound for 10 min. Then, the zeta potential was measured at a pH varied from 12 to 3 by adding NaOH (1 M) and HNO3 (1 M) as required. The observation by optical microscopy was done with magnification of ×25.
Thermogravimetric analyses (ATG) were performed with a Netzsch ATD/TG STA 449 under N2 atmosphere with a heating rate of 5 °C min−1 from 27 °C to 900 °C.
The Brunauer–Emmett–Teller (BET) surface area (SBET) of the powders was measured by nitrogen adsorption–desorption in Belsorp max (BEL, Japan) apparatus. The adsorption branch of the nitrogen adsorption–desorption isotherm was used to determine the pore size distribution and surface area using the Barrett–Joyner–Halenda (BJH) method.
Other classical characterization methods have been performed for the samples before and after sorption to go further in the understanding of the sorption mechanism such as FITR, Raman and SEM but no additional conclusions have been drawn from these results, so it will be not mentioned in this work.
Results and discussion
Sorption studies
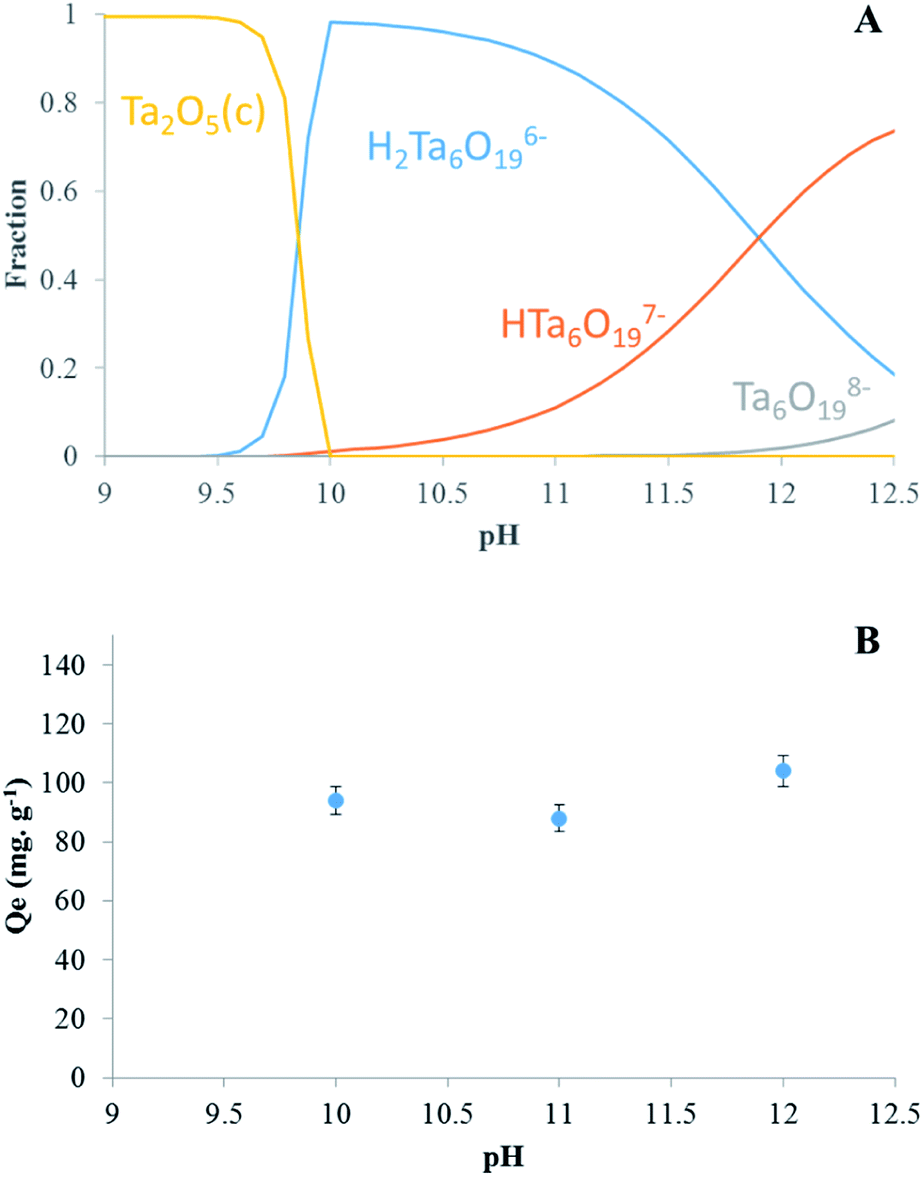 | ||
| Fig. 1 (A) Distribution diagram of Ta in solution as function of pH and (B) effect of initial pH on Ta sorption by LDH Mg–Fe–CO3. | ||
Moreover, the charge of the surface of the LDH can play a role, since a positive surface would be favorable to the adsorption of POTa. A point of zero charge (pzc) of Mg–Fe–CO3 has been found at 8.9 in a previous work,40 which seems low considering that ferric (hydr)oxides are characterized by pzc around 9.1,41 while those MgO is higher than 12.42 Our measurements of zeta potentials (see below) are in agreement with those latter values, with a positive potential up to pH 11. According to Fig. 1B, the results show that a sorption takes place, with a low dependence on pH between 10 and 12. It can be consistent either with an ion exchange or with an adsorption phenomenon.
Effect of initial Ta concentration
In order to determine the maximal sorption capacity of Ta by LDH Mg–Fe–CO3, a study was done by increasing the initial concentration of Ta in solution from 20 to 420 mg L−1. The maximal sorption capacity is equal to 100 ± 5 mg g−1, Fig. 2A. A plateau is not reached, but the sorbed amount can be considered to level off. If we assumed that the most charged POTa species is exchanged as observed with heptamolybdate and heptatungstate,21–23 the sorption quantity can be compared to the anion exchange capacity (AEC/%) of LDH calculated from its general formula, Mg0.168Fe0.056(CO3)0.049(OH)0.407·0.199H2O.36 An AEC exchange equal to 100% would correspond to the total exchange of 4 CO32− anions by one Ta6O198−. Thus, our results indicate that contacting LDH Mg–Fe–CO3 with a POTa solution results in the sorption of tantalum up to 14 ± 5% of its AEC, Fig. 2A.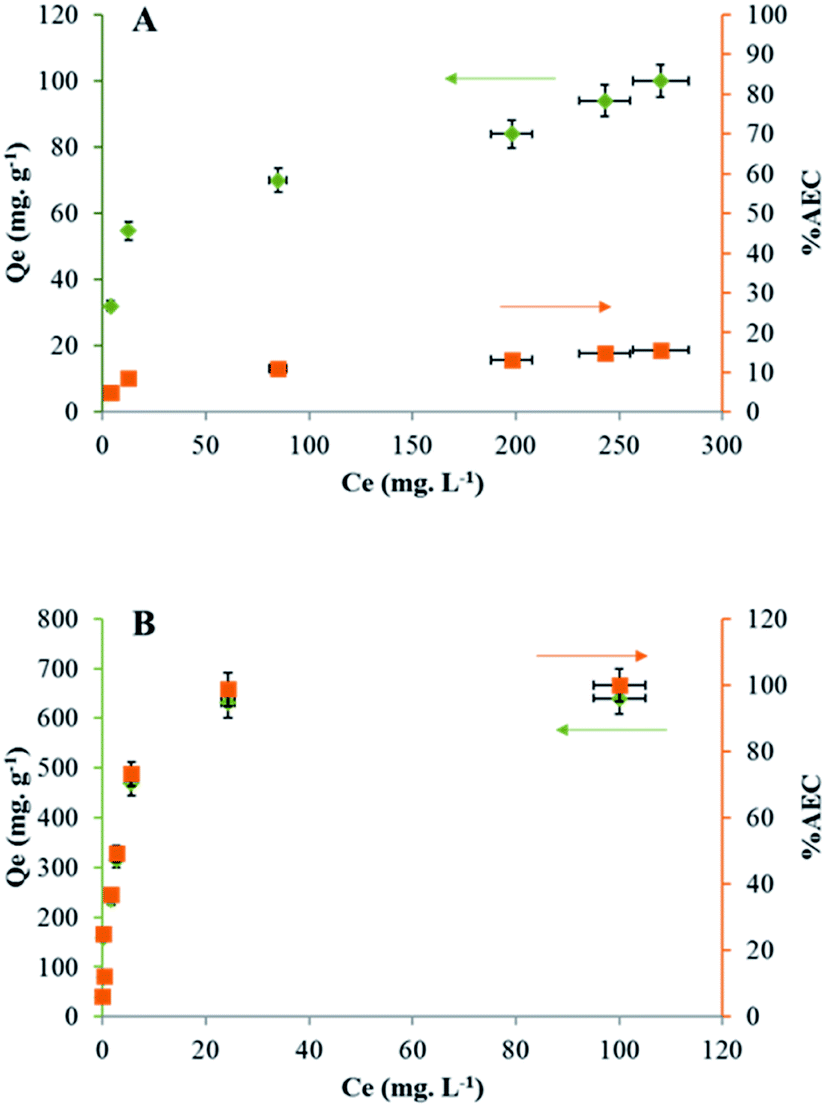 | ||
| Fig. 2 Sorption capacity of Ta (Qe/mg g−1, ●) reported to the anion exchange capacity (AEC/%, ■) by (A) LDH Mg–Fe–CO3 and (B) CLDH Mg–Fe. | ||
Based on literature studies, it is known that a strong affinity exists between LDHs and carbonate18,43 and this weak capacity could be related to the difficulty for POTa to expel the carbonate ions from the LDH structure. To avoid this problem, the LDH Mg–Fe–CO3 has been calcinated at 400 °C (called CLDH Mg–Fe), in the purpose to release carbonate ions as gaseous CO2. In this case, the sorption of POTa is expected to take place by the reconstruction of the CLDH incorporating POTa anions. The measured sorbed amount is shown in Fig. 2B. A plateau is obtained at 604 ± 30 mg g−1, what is equivalent to 100 ± 5% of the AEC. Despite the value consistent with an exchange mechanism, structural characterizations have been performed to verify the sorption mechanism (detailed below). Another approach has been followed to avoid the competition with carbonate ions. After calcination, CLDH has been reconstructed in a solution of sodium chloride or nitrate overnight with a nitrogen blanket. The sorption amount has been measured following the same protocol than above. The maximal sorption capacity determined from the plateau is 342 ± 17 mg g−1 and 140 ± 7 mg g−1 for LDH Mg–Fe–Cl and LDH Mg–Fe–NO3 respectively (Fig. S3†) (Table 1). Thus, the behavior of these solids is between those of LDH and CLDH.
| Sorbent | Qe (mg g−1) | % AEC (±5%) |
|---|---|---|
| LDH Mg–Fe–CO3 | 100 ± 5 | 14 |
| CLDH Mg–Fe | 604 ± 30 | 100 |
| CLDH Mg–Fe reconstructed with Cl− | 342 ± 17 | 56 |
| CLDH Mg–Fe reconstructed with NO3− | 140 ± 7 | 23 |
Characterization of materials: identification of the mechanism(s)
To interpret the measured values of sorption on the four solids, the structure and the surface of the solids have been characterized. XRD characterization of LDH Mg–Fe–CO3 before and after Ta extraction shows no change in its diffractogram (Fig. 3A).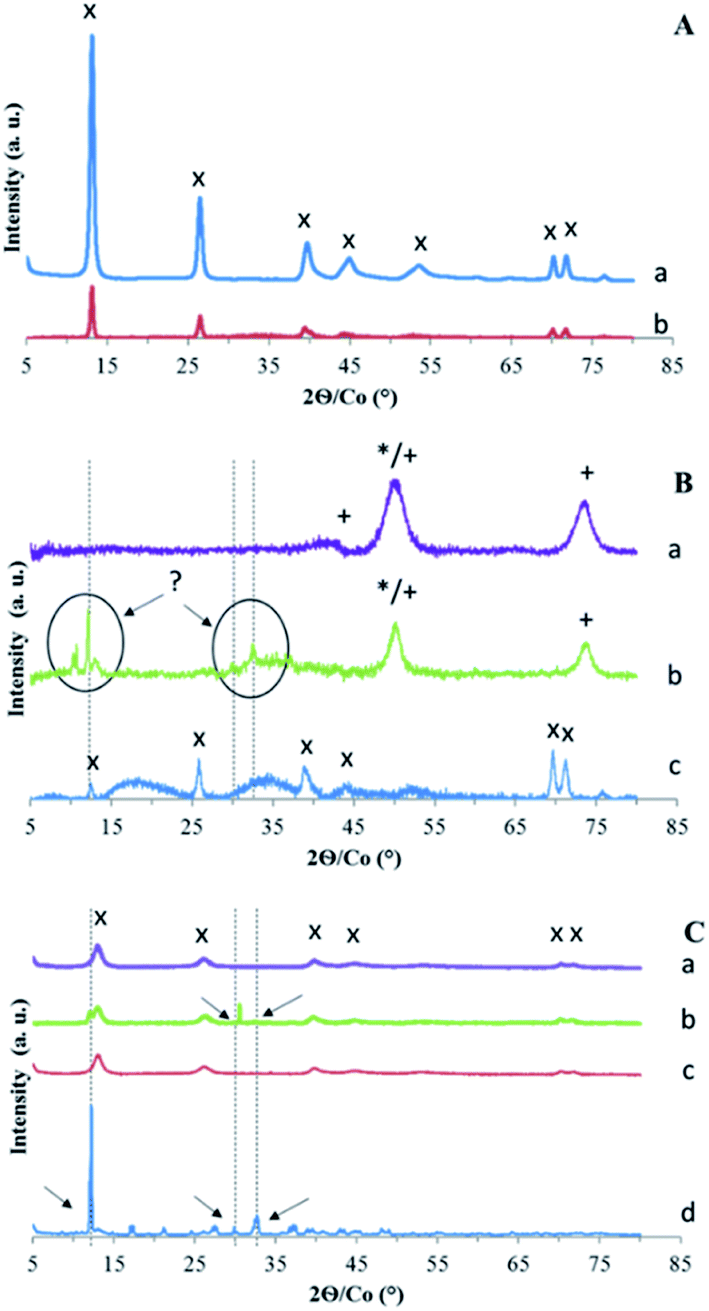 | ||
| Fig. 3 X-ray diffraction patterns of (A) LDH Mg–Fe–CO3 (a) before and (b) after sorption of Ta. (B) (a) CLDH Mg–Fe, (b) after its sorption of Ta and (c) after its reconstruction by CO32−. (C) CLDH Mg–Fe (a) reconstructed by Cl− and (b) its sorption of Ta, (c) reconstructed by NO3− and (d) its sorption of Ta. Identified phases are MgO (*), MgFe2O4 (+) and LDH structure (×). The other peaks correspond to unidentified Ta precipitate (see text). | ||
According to the literature, the exchange of the carbonate anion by another anion with a different size leads to a change of the distance between the layers observed by a shift of peak (00l).18,44 If 14% of the AEC were balanced with Ta6O198−, it would lead to the presence of a small shifted peak for (00l). Thus, the diffractogram indicates that Ta is not intercalated into the LDH structure, maybe due to its too large size, and its uptake could be due to its adsorption.
To evaluate this possibility, the zeta potential of particles has been measured for LDH Mg–Fe–CO3 before and after sorption of Ta. This method allows to probe the evolution of the surface potential, and the adsorption of a POM lead to highly negative values.45 On the contrary, a pure anion exchange does not influence the zeta potential.23 The variation of zeta potential is shown in Fig. 4: the surface of LDH before the sorption of Ta is positively charged up to pH 11. Above this value, the ionic strength of the solution increases, which would complicate the interpretation of the results. In contrast, for the LDH sample after sorption of Ta, the surface is positively charged only for the lowest pH values, while above pH 7, the zeta potential levels off at low negative values. This low isoelectric point would be consistent with the adsorption of tantalum species.
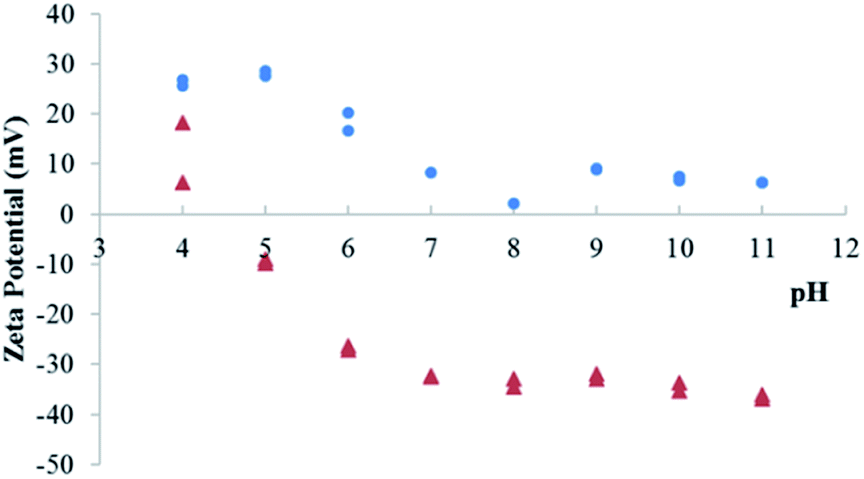 | ||
| Fig. 4 Zeta potential of LDH Mg–Fe–CO3 suspensions before (●) and after (▲) sorption of Ta. | ||
To complete the interpretation of the sorption mechanism for this series samples, the nitrogen adsorption–desorption isotherms of Mg–Fe LDH before and after sorption of Ta was done. The BJH plots show an average pore size of 27–30 nm and BET plots show a surface area of 66 m2 g−1 for both materials. It appears that the adsorption of Ta on the surface does not lead to any change on the material geometry.
The second sample series using calcined LDH have been first characterized by XRD (Fig. 3B). CLDH is found to consist in a mixture of metal oxide MgO and MgFe2O4 as expected.36,37 After contact with a solution of POTa, XRD patterns (Fig. 3B) do not shown that CLDH has been reconstructed, but is characterized by the appearance of new unidentified peaks. Then, in order to validate that CLDH is able to reconstruct in the presence of anions in solution the same protocol was done with a solution which contains sodium carbonate. In this case, XRD patterns show its reconstruction with the characteristic peaks of LDH (Fig. 3B). In conclusion, this analyses rule out the sorption of POTa by reconstruction. To go further in the understanding of the extraction mechanism of CLDH, microscopic observations was done for the 3 cases: CLDH, reconstructed CLDH by CO32− and CLDH after Ta extraction (Fig. 5). The observations confirmed the peculiarity of this latter sample, where some white crystals cover the surface of LDH particles. These could be a precipitate on the surface, resulting of the reaction of POTa with a soluble phase formed during calcination. Indeed, the high solubility of MgO is known35 and magnesium cations could precipitate in presence of Ta species.
 | ||
| Fig. 5 Microscopy images of (a) CLDH, (b) CLDH after recontraction with CO32−, (c) CLDH after extraction of Ta, (d) CLDH reconstructed with Cl− (LDH Mg–Fe–Cl–) following by the extraction of Ta and (e) CLDH reconstructed with NO3− (LDH Mg–Fe– NO3−) following by the extraction of Ta. | ||
The last series of samples corresponds to the reconstruction of LDH with Cl− or NO3− as counter anion. The diffractograms (Fig. 3C) show the characteristic peaks of the initial LDH structure in reconstructed samples. After contact with a POTa solution, new peaks are present, shown by arrows. For MgFe–Cl, it consists mainly in a peak at 30.5° and a shoulder at 12.7°. For Mg–Fe–NO3, the bands of the LDH structure become very weak, with several new narrow peaks.
The observation by optical microscopy, Fig. 5, validates the presence of second phases (white patches) which would be consistent with a precipitate. This can be explained by the fact that the reconstruction of LDH Mg–Fe–Cl or NO3 leads to the formation of a structure less stable that the initial Mg–Fe–CO3 and its contact with a solution containing POTa would lead to the formation of a precipitation of a magnesium (polyoxo)tantalate.
Validation step: precipitation of magnesium (polyoxo)tantalate
The new phase shown by XRD and optical microscopy would be formed by the presence of POTa and soluble Mg2+. Unfortunately, such a structure is not documented, and the XRD patterns do not correspond to a reference in the database for Ta, Mg, O and H composition compound. However, the solubilization of MgO is likely since MgO, identified in CLDH, has a very high solubility. It is higher than those of most divalent oxide such as Zn < Co ≅ Ni < Mn < Mg.46,47In order to validate this hypothesis, a POTa solution was mixed either with MgO powder or with MgCl2 followed by the measurement of Ta concentration as a function of time.
Fig. 6 shows the percentage of Ta measured in solution as a function of time in the case of presence of MgCl2 or MgO and for a blank solution that corresponds to a solution of POTa alone. In both cases, as soon as they are brought into contact, a white precipitate is observed and after 60 min, Ta is no longer detected in solution. However, in the case of presence of POTa alone in solution the concentration of Ta measured remains the same without any variation. The solid recovered in both cases was analysed by XRD. Fig. 7 represents its diffractogram, where peaks similar to the ones observed in the case of CLDH-Ta, Mg–Fe–Cl–Ta or Mg–Fe–NO3–Ta are found. Its structure does not have equivalence in the XRD database. In the literature, some studies on the synthesis of Mg–Ta based compounds for various applications exist but none has led to a similar crystalline structure.48,49 The identification of crystalline structure is in progress but is out of the scope of this work.
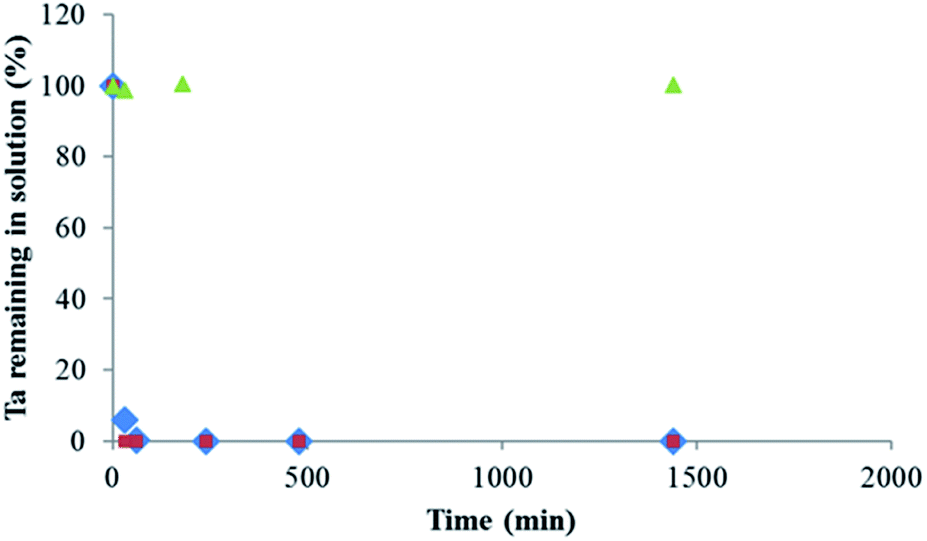 | ||
| Fig. 6 Percentage of Ta measured in solution as function of time after adding (♦) MgCl2 or (●) MgO, and (▲) blank, for solutions containing initially soluble POTa, [Ta]0 = 300 mg L−1. | ||
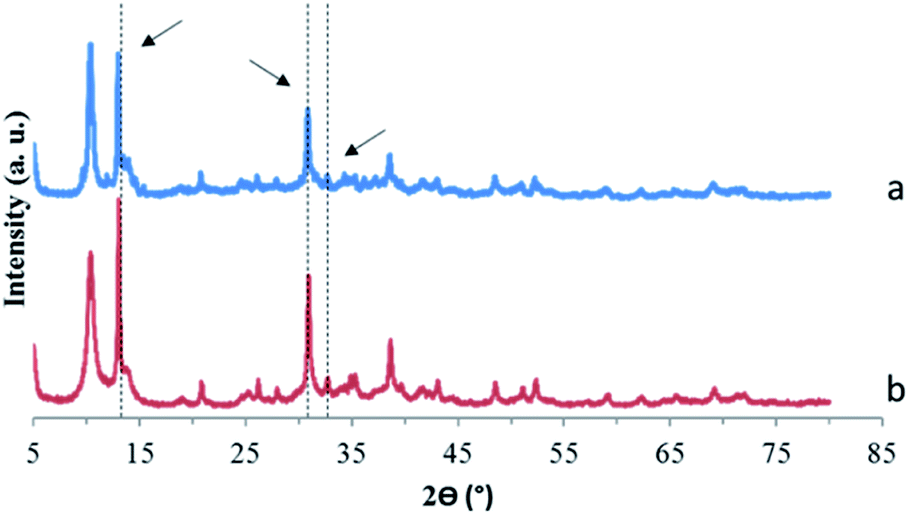 | ||
| Fig. 7 X-ray diffraction patterns of precipitates recovered after adding (a) MgO or (b) MgCl2 to a solution containing soluble POTa. The arrows correspond to unidentified peaks observed in Fig. 3. | ||
Moreover, it can be observed that the intensity of unidentified peaks appearing after sorption of Ta on CLDH, Mg–Fe–Cl, Mg–Fe–NO3 and MgO/MgCl2 depends on the sample. Thus, several phases can be responsible to Ta precipitation. Different reasons can be given: first, the kinetics of precipitation is very different between MgO/MgCl2 and LDH samples, related to the different solubilities. In addition, the germination step to form the precipitate can be influenced by the type of solid present in the system. Moreover, for the peaks present at the same angle but with different intensities, a preferential orientation during the preparation of the sample cannot be excluded.
Conclusions
The possibility of extraction of Ta(V) by LDH was studied in an alkaline medium corresponding to the solubility range of this refractory metal. In these conditions, it forms highly charged POMs anions such as H2Ta6O196−, HTa6O197− or Ta6O198−. The high pH is a constraint to choose the LDH, which should be stable. Thus, Mg–Fe–CO3 LDH was used in this study, since the presence of the ferric ions increases its stability in comparison on more usual Mg–Al LDH. We have therefore synthesized an LDH of formula Mg0.168Fe0.056(CO3)0.049(OH)0.407·0.199H2O in order to evaluate the possibility of the exchange of carbonate ions by POM Ta, like previously observed with other POMs (V, Mo or W). Nevertheless, the results indicate that the contact of LDH with POM Ta solution results in an uptake of 100 ± 5 mg g−1 of Ta, which is only 14% of its anionic exchange capacity. Moreover, characterizations by XRD and zeta potential suggested that Ta was adsorbed. Other protocols were then followed to promote the POM Ta uptake inside the solid layer structure and consequently increase the sorption capacity: a calcination of the LDH at 400 °C was done in order to reconstruct the calcined LDH in a POTa solution, and the reconstruction of Mg–Fe–Cl and Mg–Fe–NO3 was done to prepare Mg–Fe LDH with better leaving species. Higher uptakes result from these modifications, but we conclude that the leading mechanism is the precipitation of a magnesium tantalate, whose structure is neither known nor referenced. Despite the sorption mechanism is not the one expected, given the high extraction capacity, the simplicity of the process, and the expected selectivity (Mg salts are generally soluble), this approach deserves a further study in order to validate its selectivity and efficiency on the one hand and on the other hand to identify the chemical structure of the precipitate. This would allow to optimize a hydrometallurgical process based on this step, followed by the magnesium POTa treatment to obtain tantalum oxide or metal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
PSL Valorisation is thanked for the funding of the project Lix_LDH. Thomas Degabriel, Aurélie Makolana and Julie Lion (IRCP) are thanked for the preliminary experiments on Ta/LDH system.Notes and references
- J. Ma, X. Guo, H. Xue, K. Pan, C. Liu and H. Pang, Chem. Eng. J., 2020, 390, 122428 CrossRef.
- European Commission, 2020, En ligne, available, https://ec.europa.eu/info/index_en, accès le 2021.
- L. M. Kinsman, R. A. M. Crevecoeur, M. Singh, B. Ngwenya, C. A. Morrison and J. B. Love, Metals, 2020, 10, 346 CrossRef CAS.
- K. Mineta and T. Okabe, J. Phys. Chem. Solids, 2005, 66, 318–321 CrossRef CAS.
- Z. Sun, Y. Xiao, H. Agterhuis, J. Sietsma and Y. Yang, J. Cleaner Prod., 2016, 112, 2977–2987 CrossRef.
- O. Ayanda and F. Adekola, J. Miner. Mater. Charact. Eng., 2011, 10, 245–256 Search PubMed.
- T. Fujita, H. Ono, G. Dodbiba and K. Yamaguch, Waste Manage., 2014, 34, 1264–1273 CrossRef CAS PubMed.
- D. Bose, Miner. Process. Extr. Metall. Rev., 2010, 10, 217–237 CrossRef.
- B. Niu, Z. Chen and Z. Xu, J. Cleaner Prod., 2017, 166, 512–518 CrossRef CAS.
- I. Gaballah, E. Allaih and M. Djona, Metall. Mater. Trans. B, 1997, 28, 360 Search PubMed.
- M. Ungerer, C. Van Sittert, D. Van der Westhuizen and H. Krieg, Comput. Theor. Chem., 2016, 1090, 112–119 CrossRef CAS.
- M. Ungerer, D. Van der Westhuizen, H. Krieg and C. Van Sittert, J. Mol. Liq., 2019, 288, 111056 CrossRef CAS.
- O. Sanda and E. Taiwo, Hydrometallurgy, 2012, 127–128, 168–171 CrossRef CAS.
- G. Deblonde, A. Chagnes, S. Bélair and G. Cote, Hydrometallurgy, 2015, 156, 99–106 CrossRef CAS.
- G. Deblonde, D. Bengio, D. Beltrami, S. Bélair, G. Cote and A. Chagnes, Sep. Purif. Technol., 2019, 215, 634–643 CrossRef CAS.
- G. Arana, N. Etxebarria, L. Fernandez and J. Madariaga, J. Solution Chem., 1995, 24, 611–621 CrossRef CAS.
- L. Zhang and Z. Chen, Int. J. Energy Res., 2020, 1–31 Search PubMed.
- K. Goh, T. Lim and Z. Dong, Water Res., 2008, 42, 1343–1368 CrossRef CAS PubMed.
- S. Lee, K. Jung, J. Choi and Y. Lee, Chem. Eng. J., 2019, 368, 914–923 CrossRef CAS.
- M. Doeuff, T. Kwon and T. Pinnavaia, Synth. Met., 1989, 34, 609–615 CrossRef CAS.
- A. Davantès and G. Lefèvre, J. Phys. Chem. A, 2013, 117, 12922–12929 CrossRef PubMed.
- A. Davantès, D. Costa and G. Lefèvre, J. Phys. Chem. C, 2015, 119, 12356–12364 CrossRef.
- G. Lefèvre, J. Lion and A. Makolana, Sep. Sci. Technol., 2018, 1505911 Search PubMed.
- D. Bish, Bone Miner., 1980, 103, 170–175 CAS.
- F. Cavani, F. Trifir and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- S. Samuei, F. Rad and Z. Rezvani, Appl. Clay Sci., 2020, 184, 105388 CrossRef CAS.
- O. Mrozek, P. Ecorchard, P. Vomacka, J. Ederer, D. Smrzova, M. Slusna, A. Machalkova, M. Nevoralova and H. Benes, Appl. Clay Sci., 2019, 169, 1–9 CrossRef CAS.
- C. Peng, J. Dai, J. Yu and J. Yin, AIP Adv., 2015, 5, 057138 CrossRef.
- Y. Guo, Z. Gong, C. Li, B. Gao, P. Li, X. Wang, B. Zhang and X. Li, Chem. Eng. J., 2020, 392, 123682 CrossRef CAS.
- R. Goswami, P. Sengupta, K. Bhattacharyya and D. Dutta, Appl. Clay Sci., 1998, 13, 21–34 CrossRef.
- G. Mascolo and M. Mascolo, Microporous Mesoporous Mater., 2015, 214, 246–248 CrossRef CAS.
- B. Prasad, P. Kamath and K. Vijayamohanan, Langmuir, 2011, 27, 13539–13543 CrossRef CAS PubMed.
- S. Miyata, Anion-Exchange Properties of Hydrotalcite-Like Compounds, Clays Clay Miner., 1983, 31, 305 CrossRef CAS.
- Y. Israeli, C. T. Gueho, J. P. Besse, J. P. Morel and N. M. Desrosiers, J. Chem. Soc., Dalton Trans., 2000, 791 RSC.
- J. W. Boclair and P. S. Braterman, Chem. Mater., 1999, 11, 289 Search PubMed.
- N.-H. Abdelkader, A. Bentouami, Z. Derriche, N. Bettahar and L. De Menorval, Chem. Eng. J., 2011, 169, 231–238 CrossRef.
- F. Jiao, L. Shuai, J. Yu, X. Jiang, X. Chen and S. Du, Trans. Nonferrous Met. Soc. China, 2014, 24, 3971–3978 CrossRef CAS.
- P. Abramov, A. Abramov, E. Peresypkina, A. Gushchin, S. Adonin and M. Sokolov, J. Struct. Chem., 2011, 52, 1012–1017 CrossRef CAS.
- M. Filella, Earth-Sci. Rev., 2017, 173, 122–140 CrossRef CAS.
- J. Das, D. Das, G. Dash and K. Parida, J. Colloid Interface Sci., 2002, 251, 26–32 CrossRef CAS PubMed.
- C. Tiberg, C. Sjöstedt and J. Gustafsson, Chemosphere, 2018, 196, 556–565 CrossRef CAS PubMed.
- K. Bourikas, J. Vakros, C. Kordulis and A. Lycourghiotis, J. Phys. Chem. B, 2003, 107, 9441–9451 CrossRef CAS.
- E. Peltier, R. Allada, A. Navrotsky and D. Sparks, Clays Clay Miner., 2006, 54, 153–164 CrossRef CAS.
- F. Cavani, A. Trifirb and F. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- K. Morimoto, S. Anraku, J. Hoshino, T. Yoneda and T. Sato, Journal Colloid Interface Sciences, 2012, 384, 99–104 CrossRef CAS PubMed.
- J. Park, D. Kim and J. Park, J. Am. Ceram. Soc., 2015, 98, 1974–1981 CrossRef CAS.
- L. Bhattacharya and E. Elzinga, Soil Syst., 2018, 2, 20 CrossRef CAS.
- A. Vahid, P. Hodgson and Y. Li, Mater. Sci. Eng., A, 2017, 685, 349–357 CrossRef CAS.
- H. Nakao, A. Shirakawa, K. Ueda, A. Kaminskii, S. Kuretake, N. Tanaka, Y. Kintaka and T. Yanagitani, Opt. Mater., 2013, 35, 700–703 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07383d |
| This journal is © The Royal Society of Chemistry 2021 |