
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrediction of flavor and retention index for compounds in beer depending on molecular structure using a machine learning method†
Yu-Tang Wangbc,
Zhao-Xia Yanga,
Zan-Hao Piaobc,
Xiao-Juan Xubc,
Jun-Hong Yu*a and
Ying-Hua Zhang *bc
*bc
aState Key Laboratory of Biological Fermentation Engineering of Beer, Tsingtao Brewery Co., Ltd, Qingdao, 266061, Shandong, China. E-mail: yujh@tsingtao.com.cn
bDepartment of Food Science, Northeast Agricultural University, Harbin 150030, PR China
cKey Laboratory of Dairy Science, Ministry of Education, Northeast Agricultural University, China. E-mail: yinghuazhang@neau.edu.cn
First published on 17th November 2021
Abstract
In order to make a preliminary prediction of flavor and retention index (RI) for compounds in beer, this work applied the machine learning method to modeling depending on molecular structure. Towards this goal, the flavor compounds in beer from existing literature were collected. The database was classified into four groups as aromatic, bitter, sulfury, and others. The RI values on a non-polar SE-30 column and a polar Carbowax 20M column from the National Institute of Standards Technology (NIST) were investigated. The structures were converted to molecular descriptors calculated by molecular operating environment (MOE), ChemoPy and Mordred, respectively. By combining the pretreatment of the descriptors, machine learning models, including support vector machine (SVM), random forest (RF) and k-nearest neighbour (kNN) were utilized for beer flavor models. Principal component regression (PCR), random forest regression (RFR) and partial least squares (PLS) regression were employed to predict the RI. The accuracy of the test set was obtained by SVM, RF, and kNN. Among them, the combination of descriptors calculated by Mordred and RF model afforded the highest accuracy of 0.686. R2 of the optimal regression model achieved 0.96. The results indicated that the models can be used to predict the flavor of a specific compound in beer and its RI value.
1. Introduction
Flavor is the soul of beer. The identification of a flavor compound in beer and understanding its flavor have always been the core and difficulty of beer flavor research. Numerous studies have focused on the flavor compounds in beer. However, they individually measured what flavors some typical compounds showed. Further sorting and analyzing the published data of a large number of beer flavor substances will provide new information for the description of product flavor according to the classification of molecular properties. This study aimed at filling this gap by exploring the relationship between beer flavor and molecular structure. It therefore provided a starting point for developing a tool for prediction of flavor and retention index for compounds in beer.There are kinds of flavor compounds in beer, involving alcohols, esters, fatty acids, phenol, sulfur compounds, etc.1 Possessing dramatic chemical diversity, the analysis of flavor compounds seems difficult. Fortunately, machine learning is an excellent choice to analyze the large amounts of data. Richter et al. used SVM classifiers to predict the 275 asparagus samples from six countries of origin, with an accuracy of 0.970.2 Dagan-Wiener et al. gathered 691 bitter molecules and non-bitter molecules from database together with published literature to create positive set and negative set, respectively. They correctly classified beyond 80% of the compounds based on decision trees machine learning algorithm.3 Similarly, composed a dataset including 707 bitterants and 592 non-bitterants, Zheng et al. built the bitter/bitterless classification models and the accuracy of SVM model was 0.918.4 The previous studies demonstrated that the suitable model can be a potential method to classify compounds on the basis of molecular structures. Our study extended this approach to the flavor compound in beer.
To determine compounds that are responsible for the flavor of beer, the crucial step is the identification of the odor-active compound. Proposed by Kováts in 1958,5 retention indices (RI) are independent from the experimental conditions, except for the temperature and the polarity of stationary phases. Therefore, RI as a useful parameter is applied for the purpose of identification by researchers. In the study of Neiens et al., the structure assignment of each odor-active compound in beer was based on the comparison of RI values as well as its mass spectrum obtained by GC-MS.6 Because it would make mistake only based on mass spectrum, when structurally related compounds that provide similar mass spectra, such as isomeric compounds.7 In the same way, volatile compounds were identified by mass spectral matching with National Institute of Standards Technology (NIST) and confirmed by RI values.8–10 Some libraries cover the RI information of diverse stationary phases for a huge amount of registered compounds. In some cases, compounds are not registered in chemical libraries. A suitable alternative to deal with these obstacles is the quantitative structure–retention relationship (QSRR), which integrates experimental RI data and various molecular descriptors of the identified compounds in order to obtain models for prediction of RI values for compounds without experimental data. Numerous researchers have reported good correlation of QSRR. For example, Rojas et al. developed chemoinformatic modeling of volatile organic compounds of different samples of peppers based on QSRR of 273 identified compounds. The coefficient of determination and root mean square deviation for predictions were 0.915 and 55.4, respectively.11 Veenaas et al. implemented partial least squares (PLS) to predict RI values and the average deviation between the predicted and the experimental value was 5%.12 Therefore, the QSRR methodology was employed to obtain more RI of flavor compounds in beer and more accurate identification.
Given these premises, this work explored the machine learning models to predict the flavor and RI for compounds in beer. The data was collected from previous literature and FlavorDB.13 There are vast number of words used to describe the flavor. The Flavor Wheel, consisted of a set of agreed flavor terminology, solved the arguments of flavor expression.14 Beer has been attracting consumers due to desirable aroma and mildly bitter taste. The bitter taste of beer plays an important role in consumers expect and enjoy to a varying degree during consumption.15,16 Sulfury also has a significant impact on beer flavor and consumers liking.17 According to the Flavor Wheel and taking these important flavors into account, the collected compounds were separated into four flavors as aromatic, bitter, sulfury, and others. The RI data was collected from NIST.18 The structures were converted to molecular descriptors calculated by molecular operating environment (MOE), ChemoPy and Mordred, respectively. By combining the pre-treatment of the descriptors, machine learning models, including Support Vector Machine (SVM), Random Forest (RF), k-Nearest Neighbour (kNN), and Partial Least Squares (PLS) regression were utilized to predict the flavor and RI value of beer compound. New ideas were provided for recognizing the beer flavor compound depending on the molecular structure. As a result, a promising and rapid tool based on machine learning method has been developed to research the beer flavor compound.
2. Material and methods
2.1 Data collection and screening
The flavor compounds in beer were collected from various literature and published database. In order to guarantee a meaningful chemical space for training and evaluating the machine learning models, the duplicate molecules were removed. To further reduce noise, peptides, salt ions and molecules with less than 3 atoms were removed. Thus, 301 molecules were retained in the flavor compounds data set, and they were classified into four groups according to the Flavor Wheel, as aromatic, bitter, sulfury, and others. The simplified molecular input line entry system (SMILES) strings and Chemical Abstract Services numbers (CAS) were obtained for each compound from PubChem (https://pubchem.ncbi.nlm.nih.gov). Then used the Chemical Identifier Resolver (https://cactus.nci.nih.gov/chemical/structure) to check the SMILES. The RI values were searched from NIST. To guarantee the homogeneity and thus comparability of the RI values, only target Kováts RI values on standard non-polar SE-30 and polar Carbowax 20M were considered. For RI values obtained under homogeneous conditions for the same compound, the average value was considered.2.2 Molecular descriptors calculation
The molecular descriptors are used as the structural representation of molecules in order to develop models. Descriptors are the final result of a logical and mathematical procedure that transforms chemical information encoded within a symbolic representation of a molecule into a numerical quantity or into the result of some standardized experiments.19 Two- and three-dimensional molecular descriptors were calculated in three different platforms as MOE, ChemoPy and Mordred.MOE is a commercial software released by Chemical Computing Group (CCG) that can calculate 206 two-dimensional descriptors.20 The 2D molecular descriptors are the numerical properties evaluated from the connection tables. MOE represents a molecule by physical properties, subdivided surface areas, atom counts, bond counts, Kier and Hall connectivity and κshape indices, adjacency and distance matrix descriptors containing BCUT and GCUT descriptors, pharmacophore feature descriptors, and partial charge descriptors (PEOE descriptors).21
ChemoPy is a freely available, open-source python package named chemoinformatics in python.22 It can generate common structural and physicochemical descriptors including constitutional descriptors, topological descriptors, connectivity indices, charge descriptors, molecular property, etc.
Mordred is a developed descriptor-calculation software application that can calculate more than 1800 two- and three-dimensional descriptors.23 The 2D descriptors include adjacency matrix, aromatic, atom counts, auto correlation, carbon types, etc. A demonstration server is available at http://mordred.phs.osaka-u.ac.jp. The SMILES can be uploaded from the front page.
2.3 Molecular descriptors pre-processing
The molecular descriptors were imported into R (version 3.6.0). In some situations, the data generating mechanism can create predictors that only have a single unique value (i.e. a “zero-variance predictor”), this may cause the model to crash or the fit to be unstable. Similarly, predictors might have only a handful of unique values that occur with very low frequencies, these “near-zero-variance” predictors may need to be identified and eliminated prior to modelling. Models may benefit from reducing the level of correlation between the predictors. So the “caret” package was used to remove the descriptors that had zero- and near zero-variance and cut off high correlated descriptors.24 The variables were scaled to have standard deviation one and mean zero, in order to make the variables comparable.2.4 Dimensionality reduction
Principal component analysis (PCA) is a technique for reducing the dimensionality of large dataset, increasing interpretability, minimizing information loss by creating new uncorrelated variables that successively maximize variance. Every principal component can be expressed as a combination of one or more existing variables. All principal components are orthogonal to each other, and each one captures some amount of variance in the data.25 PCA was implemented using “FactoMineR” package for the analysis and “factoextra” package for visualization.26 The principal components (PCs) extract was implemented using the function “prcomp” that built in R “stats” package.2.5 Development of flavor model
All individuals were divided into training set and test set according to stratified sampling. The training set, representing 75% of the total number of compounds, was used to develop the models via SVM, RF, and kNN algorithm. The remaining 25% data was assigned to the test set and used to validate the models. 10-fold cross validation was performed to objectively evaluate the robustness and validity of models. The data set is split into 10 mutuality exclusive subsets of similar size. Then reserve one subset and train the model on all other subsets, test the model on the reserved subset and record the prediction error. This process was repeated until each of the 10 subsets has served as the test set.SVM is a machine learning technique used for classification tasks. It was originally developed by Cherkassky,27 which is a supervised machine learning method based on the statistical learning theory. The basic idea of SVM is to transform the input vector into a high-dimensional Hilbert space and seek a separating hyperplane in this space. It targets on minimizing the structural risk and uses kernel function to tackle nonlinearly separable problem. The free R package “e1071” is used to construct a SVM with “kernel” function.28
RF algorithm is one of the most common and powerful machine learning techniques, which is applied to decision trees.29 RF was implemented using the “randomForest” package.30 The “ntree” values were tested from 300 to 700 with 200 intervals, while “mtry” was tested from 15 to 25 with 5 intervals. A grid search was used to select the optimal number “ntree” and “mtry” of predictor variables randomly sampled as candidates at each split, and fit the final best random forest model that explains the best our data.
kNN algorithm predicts the outcome of a new observation by comparing it to k similar cases in the training data set.31 kNN was implemented using the function “class” from the R package. The best k was selected using a grid search from k = 2 to 10.
2.6 Development of RI model
Regression models were built for analysis of RI according to the GC information of beer flavor compounds. RF can be used for both classification that is predicting a categorical variable and regression that is predicting a continuous variable.30 Random forest regression (RFR) was used to develop models for RI values prediction. Principal component regression (PCR) applies principal component analysis on the data set to summarize the original predictor variables into a few new variables as principal components (PCs), which are linear combination of the original data. These PCs are used to build the linear regression model.32 The number of principal components, to incorporate in the model, is chosen by cross-validation. PLS is an alternative to PCR, which identifies new principal components related to the outcome, that summarizes the original predictors. These components are used to fit the regression model. PLS was implemented using the “pls” package.33 10-folds cross validation was applied in this modelling procedure. After the model was established, the test set was then analyzed in order to estimate the predictive capability of the established models, to minimize the risk of overfitting.GC-MS analyses were performed on Shimadzu nexis gc2030 gas chromatograph for five compounds (hexanoic acid ethyl ester, phenylethyl alcohol, ethyl caprylate, ethyl caprate, and ethyl laurate) to verify the prediction of models.
2.7 Evaluation metrics
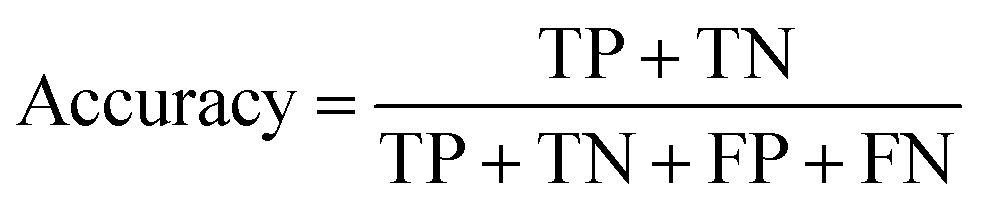
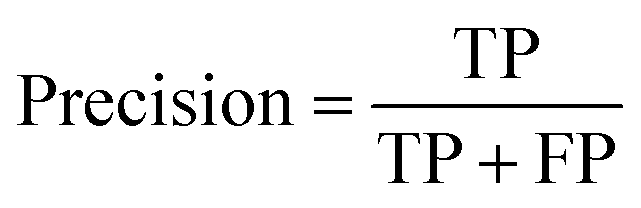
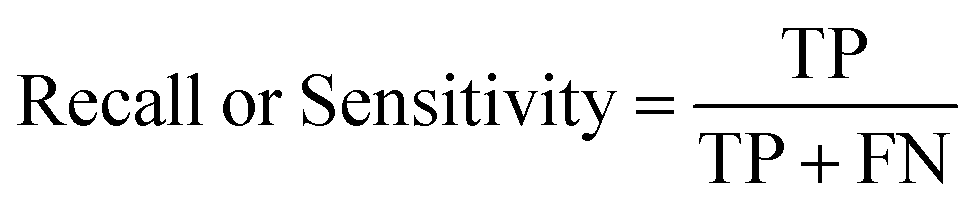
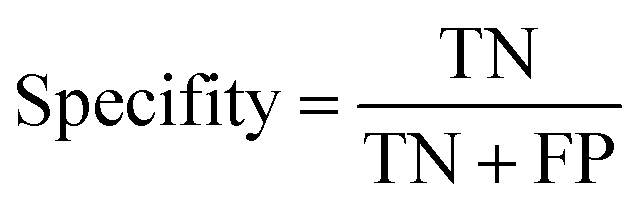
The ROC curve (receiver operating characteristics curve) is a graphical measure for assessing the performance or the accuracy of a classifier, which corresponds to the total proportion of correctly classified observations. The test will be declared positive when the corresponding predicted probability, returned by the classifier algorithm above a fixed threshold. This threshold is generally set to 0.5, which corresponds to the random guessing probability. The ROC curve is typically used to plot the true positive rate (or sensitivity on y-axis) against the false positive rate (or “1-specificity” on x-axis) at all possible probability cutoffs. This shows the trade off between the rate at which correctly predict something with the rate of incorrectly predicting something. The Area Under the Curve (AUC) summarizes the overall performance of the classifier, over all possible probability cutoffs. It represents the ability of a classification algorithm to distinguish positives from negatives. AUC is calculated by taking the average of precision across all recall values corresponding to different thresholds. It is a relevant measure when there is class imbalance in the data set. These can be performed using “ggplot2” package.
where Xobs is the observed known values of the outcome, Xpred is the predicted value by the model, n is the number of samples in the test set.
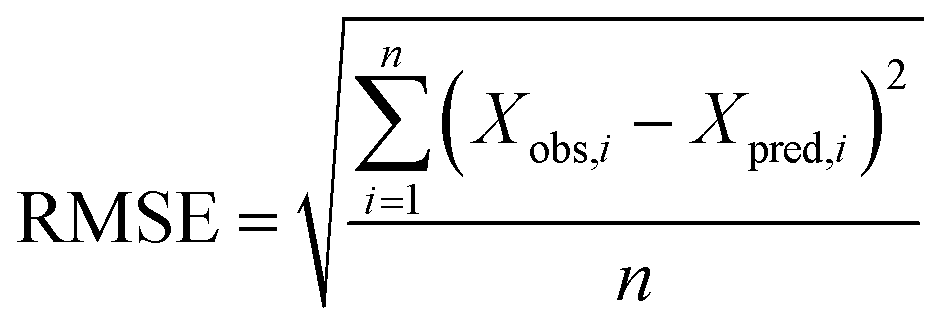
3. Results and discussion
3.1 Beer flavor database
In this study, information of flavor compounds in beer was collected from a wide variety of resources ranging from database to literature. Molecules, which exact information of taste was either unavailable or conflicting, were removed. The collected dataset consisted of 301 molecules. The database was classified into four groups of 139 aromatic, 62 bitter, 38 sulfury, and 62 others. A beer flavor database was built that contained beer flavor ID, chemical name, SMILES, CAS, FlavorDB ID, etc. The beer flavor database is available at http://ficbf.neau.edu.cn/. A brief summary of the datasets is given in Table 1. The GC information from NIST was curated. The largest amount of retention index data can be obtained for non-polar SE-30 column and polar Carbowax 20M column as the most commonly used columns for the analysis of flavor substances. 75 RI data on non-polar SE-30 column and 72 on polar Carbowax 20M column was obtained in the present study.| Flavor | Number of molecules | Reference |
|---|---|---|
| FlavorDB | ||
| Aromatic | 139 | Coelho et al.1 |
| Gonzalez et al.34 | ||
| Ochiai et al.35 | ||
| Lehnert et al.36 | ||
| Bitter | 62 | Irwin et al.37 |
| Kaneda et al.38 | ||
| Verstrepen et al.39 | ||
| Shen et al.40 | ||
| Sulfury | 38 | Intelmann et al.15 |
| Sanekata et al.41 | ||
| Bettenhausen et al.42 | ||
| Neiens et al.6 | ||
| Other | 62 | Dresel et al.43 |
| Pires et al.44 | ||
| Sigler et al.45 |
3.2 Molecular descriptors
MOE, ChemoPy and Mordred calculated 206, 628, and 1610 descriptors, respectively. They held hundreds of variables that contained redundant and co-linear information. The redundant descriptors that had zero- and near zero-variance were removed, and high correlated descriptors were cut off. The choice of molecular descriptors plays a key role in the performance of machine learning models, the PCA algorithm was implemented to select the features such that the features are orthogonal to each other and capture the maximum variance of the data. As it can be seen from Fig. 1, the first two PCs were able to capture 76.7%, 72.4% and 60.8% of the total variance in all the descriptor sets, respectively. Variables that were correlated with PC1 and PC2 were the most important in explaining the variability in the data set. Positively correlated variables were grouped together. Negatively correlated variables were positioned on opposed quadrants. The contributions of variables in accounting for the variability in PC1 and PC2 were expressed in percentage. The larger the value of the contribution, the more the variable contributes to the component.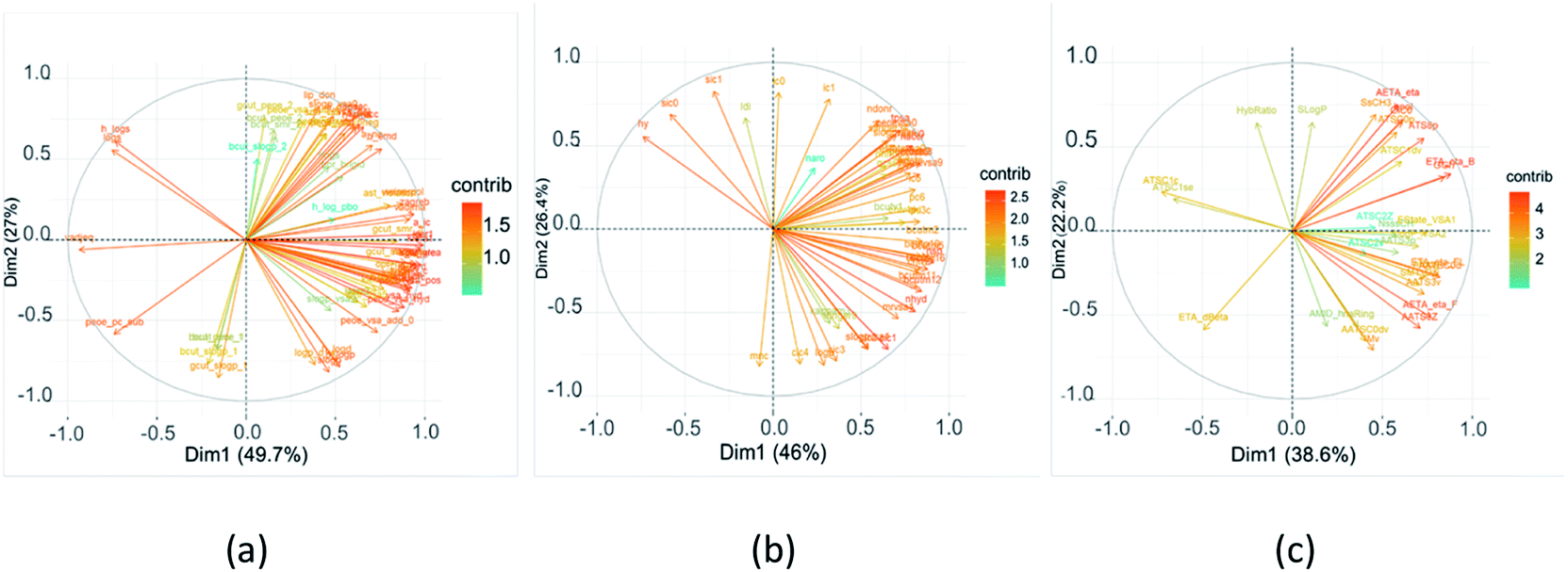 | ||
| Fig. 1 Variable correlation plot of flavor compounds shows contribution rate of each variable to the principal component (a) MOE, (b) ChemoPy, (c) Mordred. | ||
Each PC accounts for consecutively decreasing the amount of data variance, which results in the compression of significant data into a few PC variables. The correlation coefficient between each PC and the original variable is called loading. As it can be seen from Fig. 2, the absolute values of loading factors corresponding to PC1 and PC2 were more than zero. Thus, the selection of the descriptors was rational.
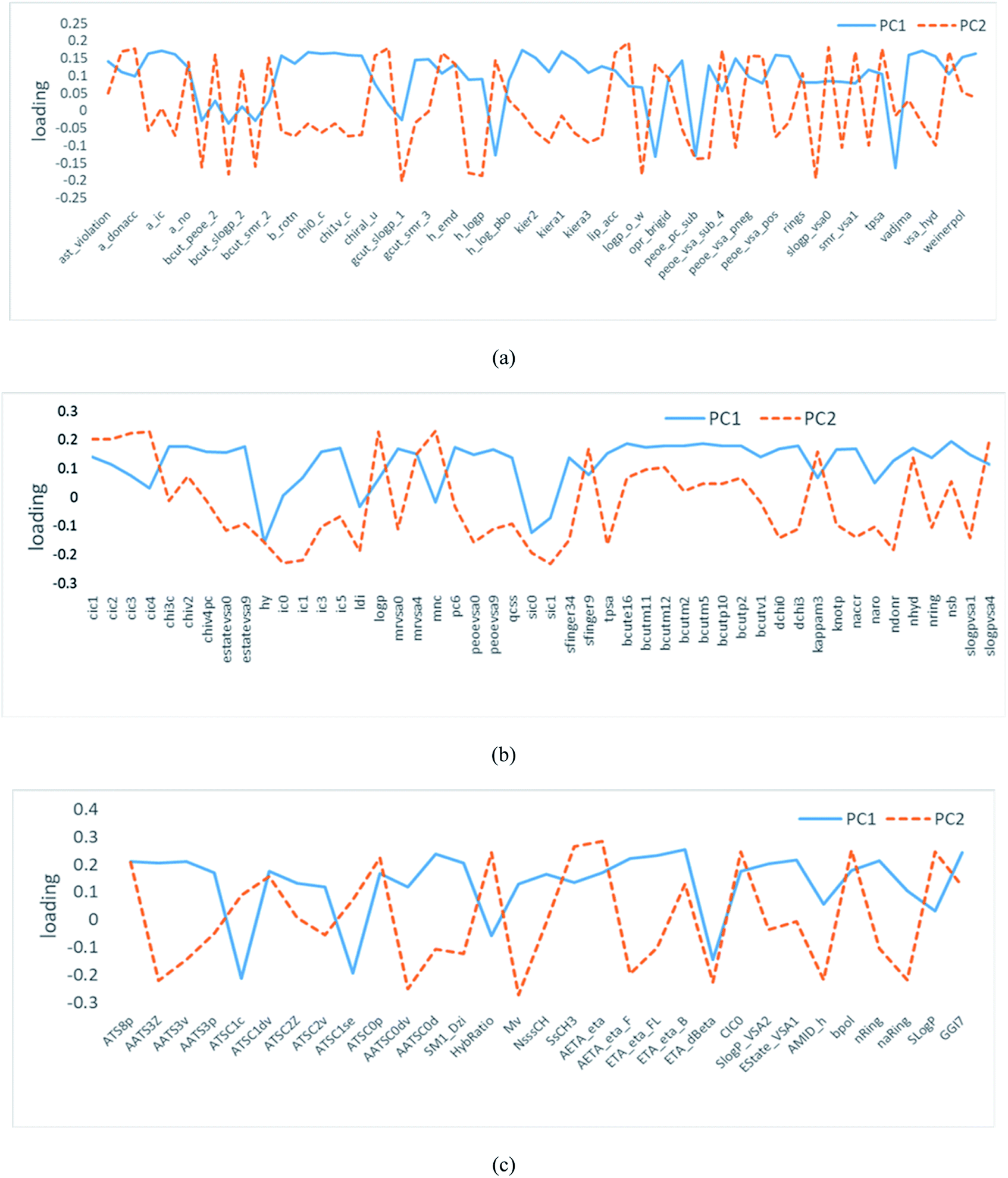 | ||
| Fig. 2 Loading profiles for the PCs of flavor compounds based on the descriptors calculated by (a) MOE, (b) ChemoPy, (c) Mordred. | ||
3.3 Models for flavor
For the discrimination of different flavor on the basis of descriptors, the data set was submitted for interpretation with the use of the machine learning techniques. Machine learning techniques as the classifiers, including SVM, RF, and kNN were used in the study. The most important standard to evaluate the classification model is the prediction accuracy of test set. Fig. 3 shows the accuracy of the models. The box chart represents the distribution of data, and the thick line in the middle represents the median. In this study, the model is trained through 10-fold cross validation. The standard accuracy of model performance evaluation is expressed by the average value of 10-fold cross validation results. The accuracy of RF was higher than SVM and kNN. The mean accuracy was over 0.60, of which the RF model with the Mordred descriptors had the highest mean accuracy of 0.686. With the continuous growth of the beer market, beer manufacturers have been working hard to provide consumers with beer with abundant flavor. Previous research developed artificial intelligence models based on aroma profiles, chemometrics, and chemical fingerprinting to assess beers.46 At the same time, researchers continue to improve the methods of separating and identifying beer flavor substances. However, these methods either detect several flavor substances, or detect some types of flavor substances. Each research result is independent.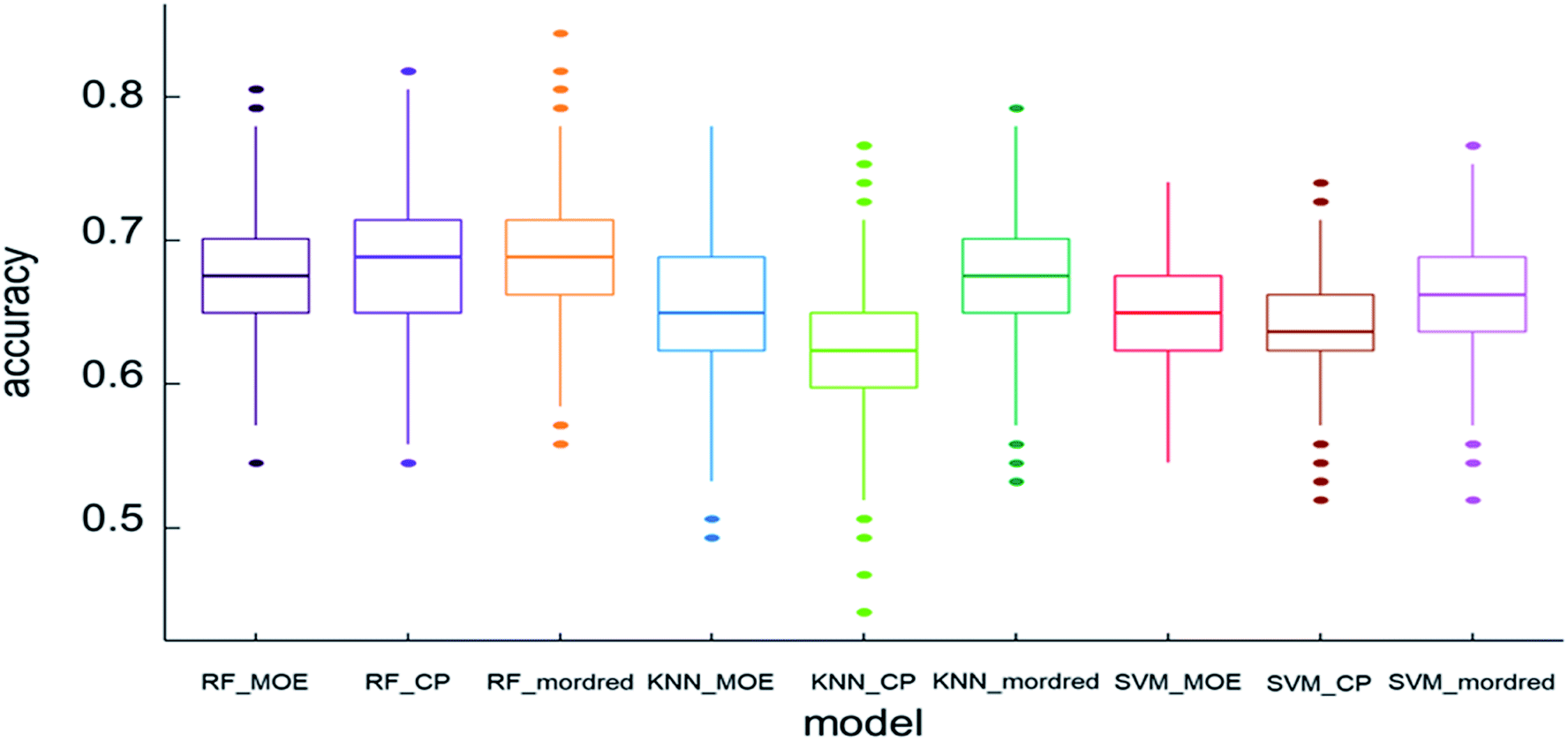 | ||
| Fig. 3 The accuracy comparisons of 9 models. | ||
In order to avoid contingency of evaluation metrics on specific thresholds, models were evaluated using threshold-independent metric AUC additionally. Fig. 4 shows the ROC of 9 models utilizing different algorithms and molecular descriptor sets. The AUC of RF was higher than SVM and kNN. On the whole, RF was found to give the best performance. As a result, a beer flavor prediction tool based on RF and Mordred descriptors was released. The complex types and large quantities of beer flavor substances lead to chemical diversity, which seems difficult to carry out systematic analysis. The principle of machine learning is that the property of compounds is related to their molecular structure. Molecular structure information is encoded by molecular descriptors. With molecular descriptors as independent variables and property as dependent variables, the mathematical relationship between descriptors and property of known compounds is established by mathematical statistical method to predict the activities of unknown compounds. In this study, the structural parameters of beer flavor substances are independent variables and their flavor is dependent variables. Based on machine learning method, the relationship between the structure of flavor substances and flavor is established, and the flavor prediction model is established, so that the flavor can be predicted according to the structure of new flavor substances.
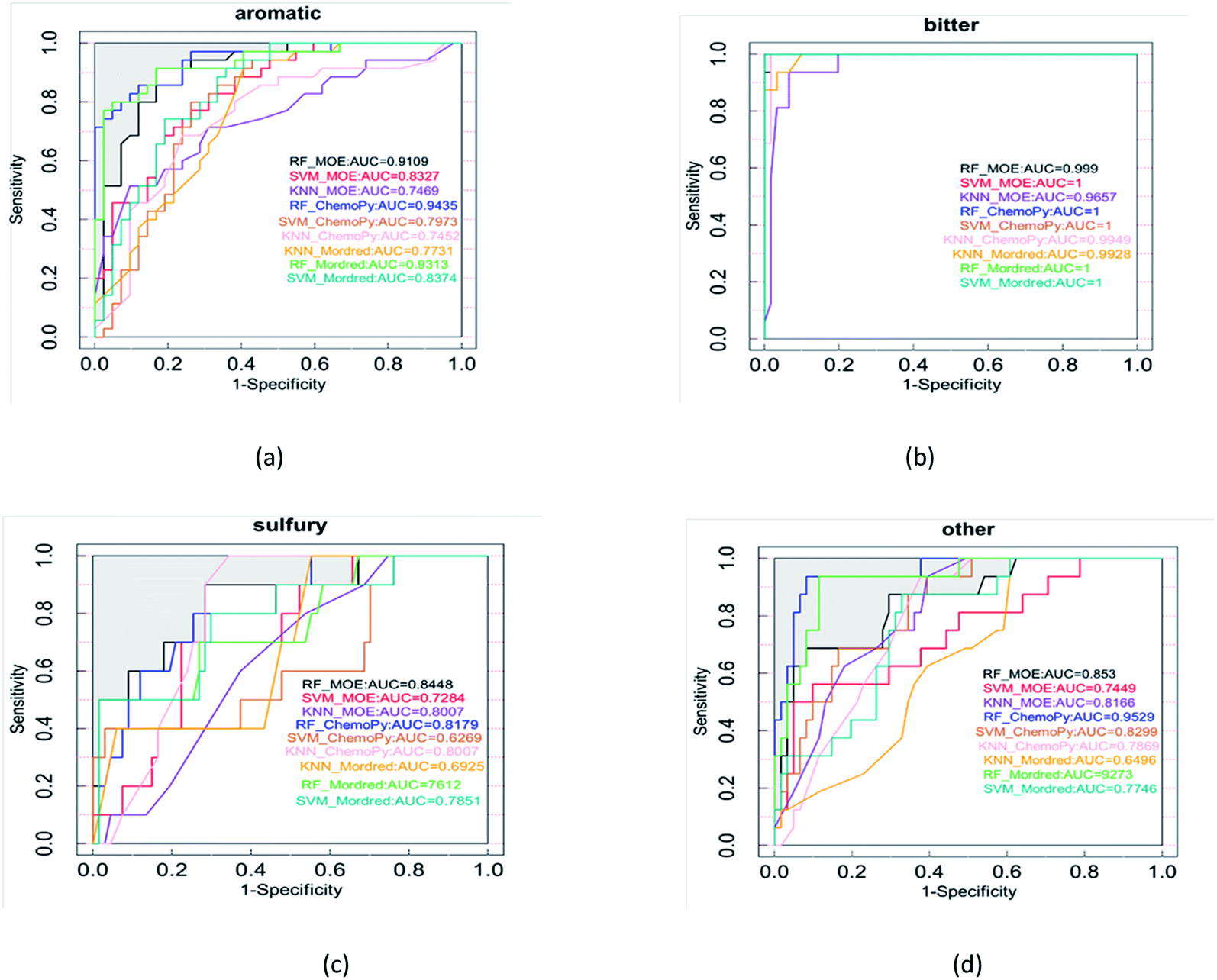 | ||
| Fig. 4 The AUC values of each flavor calculated under the RF, SVM, and kNN models include (a) aromatic, (b) bitter, (c) sulfury, (d) other. | ||
3.4 Models for RI
RFR, PCR, and PLS algorithms were used to develop models to predict for RI values. Fig. 5 displays experimental versus predicted RI values for SE-30 column and Carbowax 20M column, respectively. The models created scatter plots with the regression line in blue and the perfect fit in red. The data sets of retention index on non-polar SE-30 stationary phase and polar Carbowax 20M stationary phase were divided into training set and test set respectively, in which the training set is used to establish the model and the test set is used to verify the model. In order to ensure the significance of data set segmentation, 12 RI on non-polar SE-30 stationary phase are extracted as test set, and the rest as training set by random sampling method. 15 RI on polar Carbowax 20M stationary phase were selected as the test set and the rest as the training set. The confidence interval reflects the uncertainty around the mean predictions. The grey band in figures displays the 95% confidence intervals. The performance of model was evaluated by R2 and RMSE. R2 represented the correlation between the experimental values and the predicted values. The higher the R2, the better the model. While RMSE represented the average difference between the experimental values in the test set and the predicted values by the model. The lower the RMSE, the better the model. As can be seen, the R2 values ranged from 0.89 to 0.96, and RMSE ranged from 0.03 to 0.06. For SE-30 column, the RFR model was found to be marginally better than PLS and outperform PCR. High R2 values, 0.96 for RFR and 0.95 for PLS, indicated strong correlation between the experimental and predicted values on the test sets. Predictions of RI values were very close to the experimental values, as indicated by the low RMSE values obtained. For Carbowax 20M column, the PLS model was better than RFR and PCR. A possible reason for poor performance of PCR is that there is no guarantee that the selected PCs are associated with the outcome. The selection of PCs to incorporate in the model is not supervised by the outcome variable.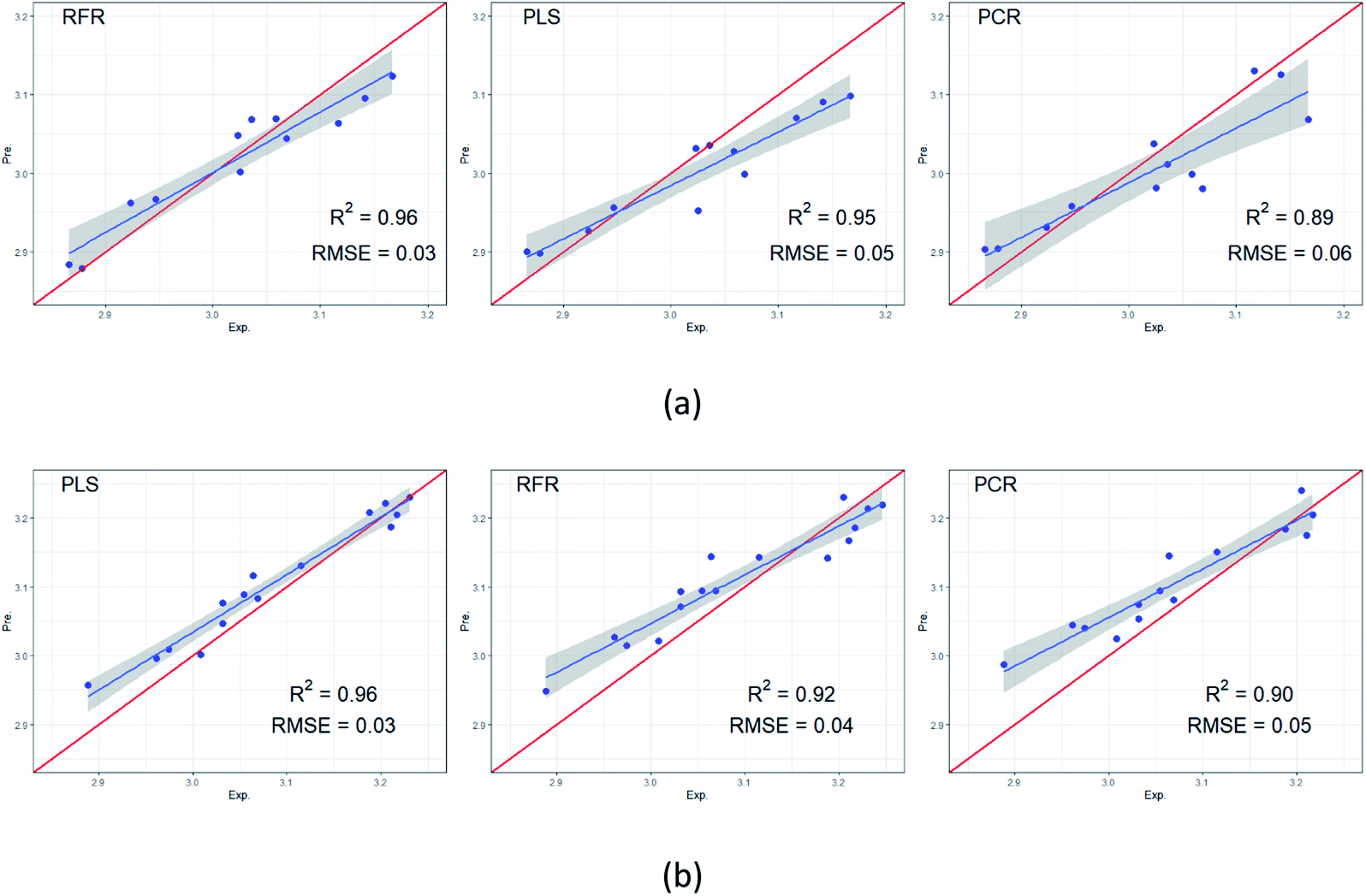 | ||
| Fig. 5 Experimental versus predicted retention indices for (a) non-polar SE-30 column and (b) polar Carbowax 20M column. | ||
Using the same experimental testing protocol, the RI values of five compounds (hexanoic acid ethyl ester, phenylethyl alcohol, ethyl caprylate, ethyl caprate, and ethyl laurate) were calculated, which was used to compare to the prediction results in the present study. Relative errors of prediction and experiment range from −0.21% to 0.48%. Therefore, RI prediction models established in this study were experimentally validated.
Differently, PLS uses a supervised dimension reduction strategy to identify PCs that summarize the original variable and that are related to the outcome. Overall, PLS had the best performance for predict RI values, so that it can be applied to compounds with unknown RI values. Thus, it provided another evidence for identification of compounds besides mass spectrum.
4. Conclusions
In this study, the beer flavor database (BeerFlavorDB) was established. Based on BeerFlavorDB, the most relevant molecular descriptors calculated by MOE, ChemoPy and Mordred were used. Beer flavor prediction models were trained by using SVM, RF and kNN algorithms. Beer flavor models trained using open source Mordred molecular descriptors and RF algorithm afforded the highest accuracy of 0.686. The RI models were developed with RFR, PCR and PLS algorithms for SE-30 and Carbowax 20M column, respectively. The regression results showed that PLS had the best performance for predict RI values. Considering the variability of beer flavor compounds, it appears that larger training set is necessary to achieve more accurate calibrations for determination of flavors. This study provided a starting point for developing a tool for prediction of flavor and retention index for compound in beer.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
This work was funded by Open Research Fund of State Key Laboratory of Biological Fermentation Engineering of Beer, funder grant No. M/Z-01-11-04-2-10F03. The authors wish to thank the anonymous reviewers and editors for their valuable advice.References
- E. Coelho, J. Magalhães, F. B. Pereira, F. Macieira, L. Domingues and J. M. Oliveira, LWT, 2019, 108, 129–136 CrossRef CAS.
- B. Richter, M. Rurik, S. Gurk, O. Kohlbacher and M. Fischer, Food Control, 2019, 104, 318–325 CrossRef CAS.
- A. Dagan-Wiener, I. Nissim, N. Ben Abu, G. Borgonovo, A. Bassoli and M. Y. Niv, Sci. Rep., 2017, 7, 12074 CrossRef PubMed.
- S. Zheng, M. Jiang, C. Zhao, R. Zhu, Z. Hu, Y. Xu and F. Lin, Front. Chem., 2018, 6, 1–18 CrossRef PubMed.
- E. Kováts, Helv. Chim. Acta, 1958, 41, 1915–1932 CrossRef.
- S. D. Neiens and M. Steinhaus, J. Agric. Food Chem., 2018, 67, 364–371 CrossRef PubMed.
- J. Yan, D. Cao, F. Guo, L. Zhang, M. He, J. Huang, Q. Xu and Y. Liang, J. Chromatogr. A, 2012, 1223, 118–125 CrossRef CAS PubMed.
- W. Zhang and X. Liang, Foods, 2019, 8, 205 CrossRef CAS PubMed.
- C. Qian, W. Quan, C. Li and Z. Xiang, Microchem. J., 2019, 149, 104064 CrossRef CAS.
- X. Zhao, H. Wu, J. Wei and M. Yang, Ind. Crops Prod., 2019, 130, 137–145 CrossRef CAS.
- C. Rojas, P. R. Duchowicz and E. A. Castro, J. Food Sci., 2019, 84, 770–781 CrossRef CAS PubMed.
- C. Veenaas, A. Linusson and P. Haglund, Anal. Bioanal. Chem., 2018, 410, 7931–7941 CrossRef CAS PubMed.
- N. Garg, A. Sethupathy, R. Tuwani, R. NK, S. Dokania, A. Iyer, A. Gupta, S. Agrawal, N. Singh, S. Shukla, K. Kathuria, R. Badhwar, R. Kanji, A. Jain, A. Kaur, R. Nagpal and G. Bagler, Nucleic Acids Res., 2018, 46, D1210–D1216 CrossRef CAS PubMed.
- M. C. Meilgaard, D. S. Reid and K. A. Wyborski, J. Am. Soc. Brew. Chem., 1982, 40, 119–128 CrossRef CAS.
- D. Intelmann, G. Kummerlà We, G. Haseleu, N. Desmer, K. Schulze, R. Frà Hlich, O. Frank, B. Luy and T. Hofmann, Chem.–Eur. J., 2009, 15, 13047–13058 CrossRef CAS PubMed.
- O. Oladokun, S. James, T. Cowley, F. Dehrmann, K. Smart, J. Hort and D. Cook, Food Chem., 2017, 230, 215–224 CrossRef CAS PubMed.
- S. Landaud, S. Helinck and P. Bonnarme, Appl. Microbiol. Biotechnol., 2008, 77, 1191–1205 CrossRef CAS PubMed.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69 Search PubMed.
- M. C. Hutter, ChemMedChem, 2010, 5, 306–307 CrossRef CAS.
- CCG, 2015.
- S. Das, P. Roy, M. A. Islam, A. Saha and A. Mukherjee, Chem. Pharm. Bull., 2013, 61, 125–133 CrossRef CAS PubMed.
- D. Cao, Q. Xu, Q. Hu and Y. Liang, Bioinformatics, 2013, 29, 1092–1094 CrossRef CAS PubMed.
- H. Moriwaki, Y. Tian, N. Kawashita and T. Takagi, J. Cheminf., 2018, 10, 4 Search PubMed.
- M. Kuhn, J. Stat. Software, 2008, 28 Search PubMed.
- I. T. Jolliffe and J. Cadima, Philos. Trans.: Math., Phys. Eng. Sci., 2016, 374, 20150202 Search PubMed.
- S. Lê, J. Josse and F. Husson, J. Stat. Software, 2008, 25, 1–18 Search PubMed.
- V. Cherkassky, IEEE Trans. Neural Network., 1997, 8 DOI:10.1080/00401706.1996.10484565.
- R. Zuo and E. J. M. Carranza, Comput. Geosci., 2011, 37, 1967–1975 CrossRef CAS.
- L. Breiman, Mach. Learn., 2001, 45, 5–32 CrossRef.
- A. Liaw and M. Wiener, Forest, 2001, 23 Search PubMed.
- Z. Zhang, Ann. Transl. Med., 2016, 4, 218 CrossRef PubMed.
- P. T. Reiss and R. T. Ogden, J. Am. Stat. Assoc., 2007, 102, 984–996 CrossRef CAS.
- B. Mevik and R. Wehrens, J. Stat. Software, 2007, 18, 1–23 CrossRef.
- C. Gonzalez Viejo, S. Fuentes, D. D. Torrico, A. Godbole and F. R. Dunshea, Food Chem., 2019, 293, 479–485 CrossRef CAS PubMed.
- N. Ochiai, K. Sasamoto, S. Daishima, A. Heiden and A. Hoffmann, J. Chromatogr. A, 2003, 986, 101–110 CrossRef CAS PubMed.
- R. Lehnert, M. Kuřec, T. Brányik and J. A. Teixeira, J. Am. Soc. Brew. Chem., 2008, 66, 233–238 CrossRef CAS.
- A. J. Irwin, R. L. Barker and P. Pipasts, J. Am. Soc. Brew. Chem., 1991, 40, 140–149 CrossRef.
- H. Kaneda, Y. Kano, T. Sekine, S. Ishii, K. Takahashi and S. Koshino, J. Ferment. Bioeng., 1992, 73, 456–460 CrossRef CAS.
- S. M. G. Saerens, F. Delvaux, K. J. Verstrepen, P. Van Dijck, J. M. Thevelein and F. R. Delvaux, Appl. Environ. Microbiol., 2008, 74, 454–461 CrossRef CAS PubMed.
- S. Geng, Z. Jiang, H. Ma, Y. Wang, B. Liu and G. Liang, Food Chem., 2020, 312, 126066 CrossRef CAS PubMed.
- A. Sanekata, A. Tanigawa, K. Takoi, Y. Nakayama and Y. Tsuchiya, J. Agric. Food Chem., 2018, 66, 12285–12295 CrossRef CAS PubMed.
- H. M. Bettenhausen, L. Barr, C. D. Broeckling, J. M. Chaparro, C. Holbrook, D. Sedin and A. L. Heuberger, Food Res. Int., 2018, 113, 487–504 CrossRef CAS PubMed.
- M. Dresel, A. Dunkel and T. Hofmann, J. Agric. Food Chem., 2015, 63, 3402–3418 CrossRef CAS PubMed.
- E. J. Pires, J. A. Teixeira, T. Brányik and A. A. Vicente, Appl. Microbiol. Biotechnol., 2014, 98, 1937–1949 CrossRef CAS PubMed.
- K. Sigler, D. Matoulková, M. Dienstbier and P. Gabriel, Appl. Microbiol. Biotechnol., 2009, 82, 1027–1035 CrossRef CAS PubMed.
- G. V. Claudia and F. Sigfredo, Fermentation, 2020, 6 DOI:10.3390/fermentation6040104.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06551c |
| This journal is © The Royal Society of Chemistry 2021 |