
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3-Methoxybutan-2-one as a sustainable bio-based alternative to chlorinated solvents†
Saimeng Jin‡
 a,
Fergal P. Byrnea,
James H. Clarka,
Con Robert McElroya,
Alex Quinna,
James Sherwooda and
Andrew J. Hunt*b
a,
Fergal P. Byrnea,
James H. Clarka,
Con Robert McElroya,
Alex Quinna,
James Sherwooda and
Andrew J. Hunt*b
aGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, York, YO10 5DD, UK
bMaterials Chemistry Research Center, Department of Chemistry and Center of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail: andrew@kku.ac.th
First published on 10th December 2021
Abstract
Methylation of acetoin with dimethyl carbonate was performed in a sustainable one-step process, with improved process mass intensity (PMI) and atom economy compared to previously published methods. The resulting product, 3-methoxybutan-2-one (MO) was successfully evaluated as a bio-based solvent, while both Kamlet–Taft solvatochromic parameters and Hansen solubility parameters demonstrate its potential viability in the substitution of chlorinated solvents. MO exhibited a low peroxide forming potential and a negative Ames mutagenicity test and was successfully used as a solvent in a Friedel–Crafts acylation (79% yield compared to 77% in dichloromethane) and for N-alkylations. MO is a renewable oxygenated solvent, with the potential ability to substitute carcinogenic halogenated solvents in some applications.
Introduction
Solvents are ubiquitous in the chemical industry and in laboratories as reaction or separation media.1,2 Many of the most common solvents are toxic or pose hazards to the environment, such as chloroform,3 and dichloromethane (DCM).4 Strict regulations, including Registration, Evaluation, Authorisation and restriction of Chemicals (REACH) in the EU, are preventing further use of the most hazardous substances, including several solvents.5Different approaches have been put forward for the development of replacement solvents that combine in silico methods with laboratory-based testing.6–8 The substitution of solvents is typically guided with polarity scales. Kamlet–Taft (KT) solvatochromic parameters are widely used for understanding the nature and strength of solvent interactions with solutes.9 They consist of three individual parameters: α (hydrogen bond donating ability),10 β (hydrogen bond accepting ability),11 and π* (a combination of dipolarity and polarisability).12 Plotting β versus π* on a solvent map allows easy visualisation of the polarity of aprotic solvents (Fig. 1).13
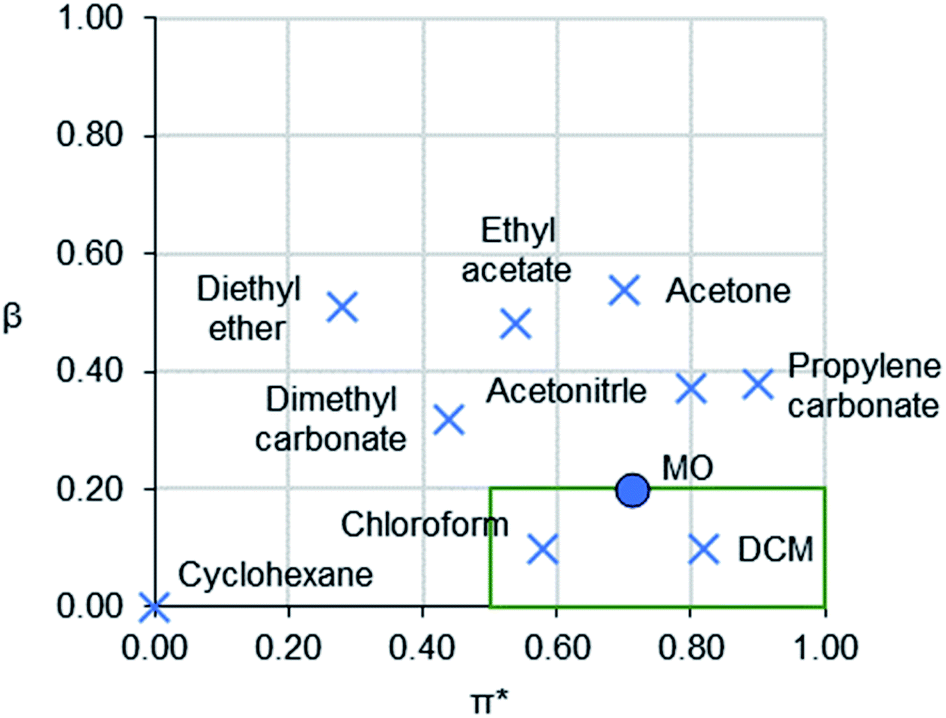 | ||
| Fig. 1 A KT solvent map of 3-methoxybutan-2-one (MO) and selected traditional aprotic solvents for comparison. Chlorinated solvents typically occupy the boxed region. | ||
To the best of the authors' knowledge, there are no prior examples of bio-based, room-temperature aprotic liquids with medium to high polarity (0.50 ≤ π* ≤ 1.00) and low basicity (β ≤ 0.20) (i.e. the lower right corner of the solvent map in Fig. 1). Conventional solvents with this polarity are dichloromethane (DCM), chloroform, nitromethane and nitrobenzene. These solvents are characterised by environmental, health and safety hazards such as reproductive toxicity, mutagenicity, suspected carcinogenicity, and ozone-depletion.3,4 Thus, finding a bio-based solvent that has similar physical properties to these solvents yet without the same degree of hazards is of significant importance.
Most common solvents are made from non-renewable resources,2 yet biomass feedstocks offer access to chemical functionalities not easily attainable from petrochemicals. This means novel structures can be synthesised while avoiding the chemical functionality of conventional solvents associated with health and environmental hazards.14 Agricultural and food wastes are an ideal feedstock to produce renewable platform chemicals due to their global availability and low cost. Acetoin (3-hydroxybutan-2-one) is one such compound present in a variety of fresh foods.15–18 Acetoin can also be produced on an industrial scale from various types of biomass.19–22
Herein, for the first time the one-step solvent-free synthesis of 3-methoxybutan-2-one (MO) from acetoin and dimethyl carbonate catalysed by p-toluenesulfonic acid (PTSA) is reported (Scheme 1). Dimethyl carbonate is a potentially bio-based, biodegradable, and non-toxic methylating agent to replace the highly toxic dimethyl sulphate and methyl iodide.23 The solvent properties of MO have been investigated against traditional solvents, and representative Friedel–Crafts acylation and Menshutkin reactions performed.
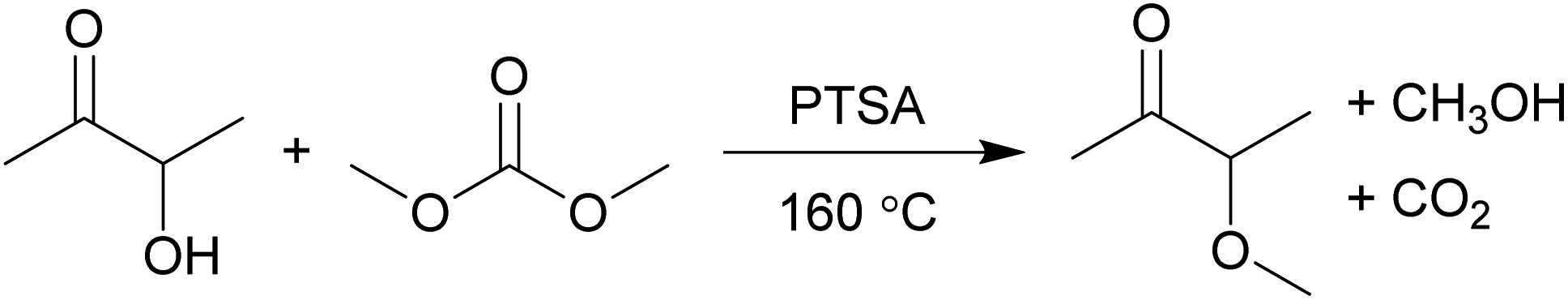 | ||
| Scheme 1 The synthesis of the new bio-based solvent MO from acetoin by dimethyl carbonate chemistry. | ||
Materials and methods
Materials
Acetoin ≥95%, dimethyl carbonate 99%, p-toluenesulphonic acid monohydrate 98.5%, 4-methoxyacetophenone 99%, anhydrous anisole 99.7%, acetic anhydride ≥99%, anhydrous acetonitrile 99.8%, anhydrous cyclohexane 99.5%, anhydrous diethyl ether ≥99%, anhydrous propylene carbonate 99.7%, iron(III) chloride 97%, anhydrous ethyl acetate 99.8%, anhydrous magnesium sulphate 99.5%, Nile red ≥98%, 4-nitroaniline ≥99%, 4-nitroanisole analytical standard, 4-nitrophenol spectrophotometric grade, 1,2-dimethylimidazole 98%, 1-bromodecane 98%, methanol 99.9%, dichloromethane 99.8%, acetone 99.9%, chloroform-d (CDCl3, 99.8% D), chloroform 99.9%, dimethyl sulfoxide 99.9% were purchased from Sigma-Aldrich. Ames MPF 98/100 kits, 2-nitrofluorene and 4-nitroquinoline-N-oxide were purchased from Xenometrix. TA98 and TA100 were stored at −70 °C. QUANTOFIX® Peroxide 100 was purchased from Macherey-Nagel. N,N-Diethyl-4-nitroaniline was purchased from VWR.Catalyst screening for synthesis of 3-methoxybutan-2-one (MO)
Into each pot of a 6 well multipoint high-pressure reactor was added acetoin (0.4405 g, 5 mmol), dimethyl carbonate (0.8418 mL, 10 mmol), catalyst (5 mol%) and a magnetic stirrer. The reaction vessel was sealed, each well isolated and heated to 160 °C for 1 hour and allowed to cool to room temperature before being characterised using GC and 1H NMR.Large scale synthesis and isolation of (MO)
Into a high-pressure reactor was added acetoin (100 g, 1.09 mol), p-toluenesulphonic acid monohydrate (10.4 g, 5 mol%), dimethyl carbonate (92 mL, 1.09 mol) and a magnetic stirring bar. The air-tight high-pressure reactor was stirred with heating up to 160 °C for 2 hours by a magnetic hot plate (Heidolph, MR Hei-Tec), and then immediately cooled in an ice-bath. No water was observed in MO after synthesis and methanol was removed by evaporation. The crude reaction mixture was filtered and gently rectified by a Vigreux distillation column to give 3-methoxybutan-2-one (MO) as a colourless liquid (85% isolated yield, 99% purity by GC). 1H NMR (400 MHz, CDCl3): δ = 1.27 (d, J = 6.96 Hz, 3H), 2.14 (s, 3H), 3.33 (s, 3H), 3.68 (q, J = 6.60 Hz, 1H) ppm. GC-MS (relative intensity, 70 eV) m/z: 102 (M+), 59 (100), 43.Synthesis of 4-methoxyacetophenone (1) from anisole and acetic anhydride
To a 3 mL sample bottle was placed anisole (5.4 μL, 0.05 mmol), iron(III) chloride (8.1 mg, 0.05 mmol), acetic anhydride (4.7 μL, 0.05 mmol), and 0.5 mL of the chosen solvent. The sample bottle was sealed for 2 hours and stirred by a roller (Stuart, roller mixer SRT6) at room temperature. Each experiment was repeated at least twice. After 2 hours an aliquot of the reaction was taken and analysed by GC and GC-MS using the comparison to commercial 4-methoxyacetophenone (1). GC-MS (relative intensity, 70 eV) m/z: 150 (M+), 135, 107, 92, 77, 64, 63, 62, 51, 50, 43 (100), 42.Synthesis of 1-decyl-2,3-dimethylimidazolium bromide (2) from 1,2-dimethylimidazole and 1-bromodecane
To a solution of 1,2-dimethylimidazole (0.288 g, 3.00 mmol) preheated to 50 °C in 3 mL MO was placed 1-bromodecane (0.69 mL, 3.33 mmol). The reaction rate was monitored by 1H NMR spectroscopy based on the method provided in the literature, a representative 1H NMR spectra is presented in the ESI.† 24Synthesis of 1-(3,4-dimethylcyclohex-3-enyl) ethanone from 2,3-dimethylbuta-1,3-diene (diene) and 3-buten-2-one
To a 3 cm3 sample bottle was placed 0.036 mmol anhydrous ytterbium(III) chloride, 0.5 mL MO solvent, 0.358 mmol 3-buten-2-one and 0.358 mmol diene. The argon gas protected sample bottle remained sealed for 16 hours and was stirred by a roller throughout (Stuart, roller mixer SRT6) at room temperature. Each experiment was repeated at least twice. After 16 hours, the sample was removed and analysed by GC and GC-MS. Following filtration of the reaction mixture, 1-(3,4-dimethylcyclohex-3-enyl) ethanone (DE) was isolated by distillation, and its GC, GC-MS, 1H NMR and 13C NMR were all analysed. (3): 1H NMR (400 MHz, CDCl3): δ = 2.59–2.48 (m, 1H), 2.14 (s, 3H), 2.12–1.85 (m, 5H), 1.60 (s, 3H), 1.58 (s, 3H), 1.55–1.43 (m, 1H) ppm. 13C NMR (400 MHz, CDCl3): δ = 18.8, 19.0, 25.3, 27.9, 31.2, 33.0, 48.2, 123.9, 125.3, 211.8 ppm. GC-MS (relative intensity, 70 eV) m/z: 152 (M+), 137, 119, 109 (100), 107, 95, 93, 91, 81, 79, 77, 67, 55, 43.GC analysis
During the experiments, an Agilent 6890N gas chromatography with a flame ionisation detector (GC-FID) was employed for the analysis of the corresponding reaction results. The GC-FID had a ZB5HT capillary column (30 m × 250 μm × 0.25 μm nominal, max temperature 400 °C) at 20.2 psi constant pressure. The carrier gas utilised in the GC-FID was helium with flow rate at 2.0 mL min−1 in constant flow mode. The split ratio was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The GC method was used as following: the starting oven temperature was held at 30 °C for 3 minutes. After that, the temperature was then increasing by 4 °C per minute to 110 °C. And then the temperature was increasing by 30 °C per minute to 300 °C and maintained at 300 °C for 1 minute. The temperature of injector and flame ionisation detector were held at 300 °C and 340 °C, respectively. Each of these GC samples consisted of 30 mg product mixture and 1.5 mL dichloromethane (DCM) as GC solvent.
:1. The GC method was used as following: the starting oven temperature was held at 30 °C for 3 minutes. After that, the temperature was then increasing by 4 °C per minute to 110 °C. And then the temperature was increasing by 30 °C per minute to 300 °C and maintained at 300 °C for 1 minute. The temperature of injector and flame ionisation detector were held at 300 °C and 340 °C, respectively. Each of these GC samples consisted of 30 mg product mixture and 1.5 mL dichloromethane (DCM) as GC solvent.
GC-MS analysis
Gas chromatography-mass spectrometry (GC-MS) was carried out on a PerkinElmer Clarus 500 GC along with a Clarus 560 S quadrapole mass spectrometer, installed with a DB5HT capillary column (30 m × 250 μm × 0.25 μm nominal, max temperature 430 °C). The carrier gas was helium with flow rate at 1.0 mL min−1, and the split ratio used was 10:1. The injector temperature was set at 330 °C. The initial oven temperature was 30 °C and held for 3 minutes. Initial ramp rate was 4 °C min−1 to 110 °C, followed by an increase in rate to 30 °C min−1 to 300 °C, at which it was held at 300 °C for 1 minute. The Clarus 500 quadrapole mass spectrum was conducted in electron ionisation (EI) mode at 70 eV with the source temperature and the quadrapole both at 300 °C. The data was collected by the PerkinElmer enhanced TurboMass (Ver. 5.4.2) chemical software. All GC-MS samples consisted of 30 mg product mixture and 1.5 mL DCM as solvent.
1H NMR spectroscopic analysis
The 1H NMR spectrum results of samples in this research were recorded by a JEOL JNM-ECS 400 MHz spectrometer. For preparation, 100 mg sample from the experiment was dissolved in 1 mL chloroform-d. 16 scans were utilised for the 1H NMR testing. The data of 1H NMR was processed and analysed by ACD/NMR Processor Academic Edition software (Ver. 12.01).Kamlet–Taft solvatochromic parameters of MO
The Kamlet–Taft (KT) solvatochromic parameters of MO were determined based on the ultraviolet-visible (UV-vis.) spectra (tested on JENWAY, 6705 UV/Vis spectrophotometer) of different dyes probes: Nile red (NR), 4-nitroaniline (NA), N,N-diethyl-4-nitroaniline (NN), 4-nitroanisole (NS) and 4-nitrophenol (NP) dissolved in MO in quartz cuvettes at 25 °C. NA/NN were used to measure β1 and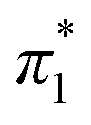 , while NS/NP were utilised to measure β2 and
, while NS/NP were utilised to measure β2 and 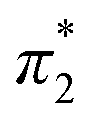 . When the νmax of these dyes was confirmed, the individual KT parameters α, β1 and
. When the νmax of these dyes was confirmed, the individual KT parameters α, β1 and 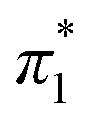 were calculated using eqn (1)–(3) below (where νmax-NR, νmax-NA and νmax-NN stand for the wavenumbers at maximum absorbance on the UV-vis. Spectra for NR, NA and NN, respectively). β2 and
were calculated using eqn (1)–(3) below (where νmax-NR, νmax-NA and νmax-NN stand for the wavenumbers at maximum absorbance on the UV-vis. Spectra for NR, NA and NN, respectively). β2 and 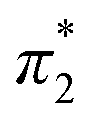 were calculated based on literature.11,12 β and π* of MO are the average value of NA/NN system and NS/NP system.
were calculated based on literature.11,12 β and π* of MO are the average value of NA/NN system and NS/NP system.
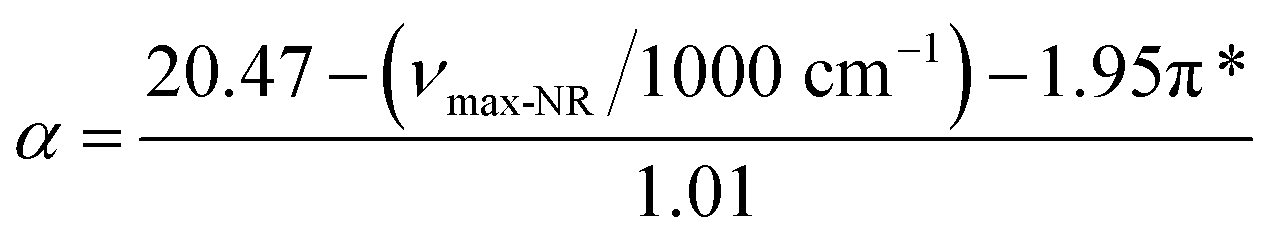 | (1) |
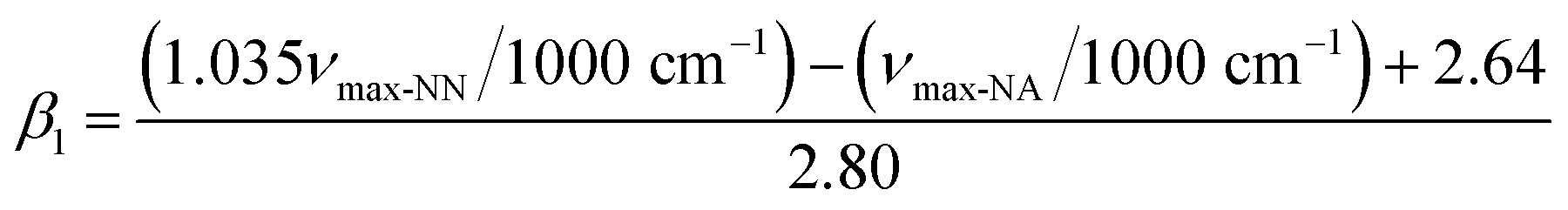 | (2) |
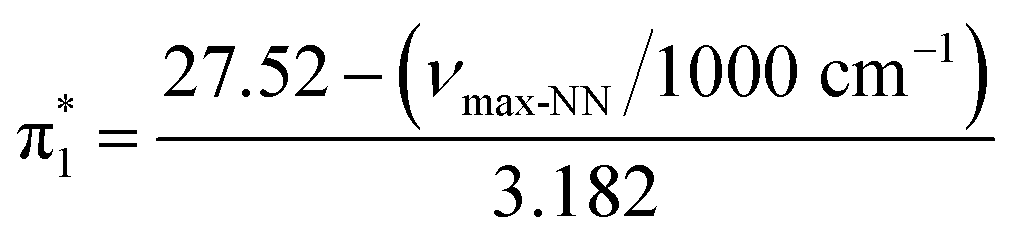 | (3) |
Ames test for MO
Mutated Salmonella typhimurium TA98 (hisD3052) and TA100 (hisG46) were used for testing. TA98 and TA100 were utilised for the detection of frameshift mutations and base substitution mutations respectively. Dimethyl sulfoxide (DMSO) was used as a solvent (negative) control. A mixture of 2-nitrofluorene (2-NF) and 4-nitroquinoline-N-oxide (4-NQO) was used as the positive control. This Ames test did not use S9 microsomal activation. The experimental procedure was based on manufacturer's guidelines as following: TA98 and TA100 were tested in 6 different concentrations (0.16 mg mL−1, 0.31 mg mL−1, 0.63 mg mL−1, 1.25 mg mL−1, 2.5 mg mL−1, 5 mg mL−1) of MO, as well as a positive (concentration of 2 μg mL−1 for 2-NF and 0.1 μg mL−1 for 4-NQO) and a negative (DMSO solvent) control, in a medium including sufficient histidine for 90 minutes in order to conduct about two cell divisions. After exposure, the cultures were diluted in histidine-free pH indicator medium and then aliquoted into 48 wells of a 384-well plate (each concentration was carried out in triplicate). After 48 hours at 37 °C, the wells containing mutated bacteria changed in colour from purple to yellow (reversed to His+) since pH of the medium was decreased by the metabolism of the His+ strains. The number of wells, in which the colour changed, was counted manually for each dose, to obtain the average value. After testing, no mutagenicity for MO was observed for both TA98 and TA100 strains since no obvious dose effect was exhibited.Results and discussion
Catalyst screen
To obtain the desired target compound. MO, methylation of acetoin is required. Dimethyl carbonate (DMC) has been identified as non-toxic, renewable and safe methylating agent of both alcohols,25 and phenols.26 In these examples the catalyst in question was a potassium or calcium carbonate, i.e. base mediated, as with the vast majority of the literature.27 However acid catalysed reactions have also recently been shown to be effective in DMC chemistry.28 As such a number of Lewis and Brønsted acid catalysts were investigated for methylation of MO, with calcium carbonate as a baseline and no catalyst as a control. Results are reported in Table 1. Several heterogeneous catalysts (including zeolites, acidic clays and acid resins) were also screened, but they were found to have little or no conversion under conventional heating conditions (Table S2, ESI†). Several heterogeneous catalysts demonstrated some promise under microwave heating and future work will focus on utilising these routes in flow for developing industrially relevant processes (Table S3, ESI†).| Entry | Catalyst | MO yield (NMR) |
|---|---|---|
| a Acetoin (5 mmol), dimethyl carbonate (10 mmol), acid catalyst (5 mol%), 160 °C for 1 hour. | ||
| 1 | H2SO4 | 85% |
| 2 | p-TSA | 76% |
| 3 | AlCl3 | 11% |
| 4 | FeCl3 | 22% |
| 5 | CaCO3 | 0% |
| 6 | None | 0% |
A low 2:1 DMC:acetoin ratio was selected to allow ease of following the reaction via NMR without the need for work up which would result in loss of volatile product. The catalyst screen reviled that the favoured catalyst in the literature, calcium carbonate (entry 5), did not result in any methylation of acetoin occurring at all, although carboxymethylation did take place. Both Lewis acids showed some yield towards the desired product (entries 3 and 4). The best yield observed was using concentrated sulphuric acid (entry 1) and the NMR of this product looks very clean. However, GC-FID data shows a significant number of more volatile components in the system, most likely because of various aldol condensations. PTSA (entry 2) gave a good yield after 1 hour, with products exhibiting lower boiling points visible by GC-FID. As such, PTSA was selected as the best catalyst going forward. In addition, PTSA has previously been highlighted as a green organic acid catalyst.29 No reaction was observed in the control (entry 6).
Large-scale synthesis of MO from acetoin
The one-step solvent-free large-scale synthesis of MO from acetoin and dimethyl carbonate was achieved with >95% conversion (as determined by 1H NMR spectroscopy), catalysed by 0.05 equivalents of PTSA at 160 °C. Isolation and further purification of MO was achieved by distillation, resulting in an isolated yield of 85%. The reaction was then assessed using the CHEM21 metrics toolkit (Table 2).30 The reaction mass efficiency (RME) of the process is low because of dimethyl carbonate being applied in excess as a solvent as well as a reactant, while the atom economy is limited by the stoichiometric production of methanol and CO2. However, a comparison with the literature shows several benefits of these reaction conditions. The only previously published synthetic route to MO from acetoin was for the purpose of synthesising an intermediate and not to produce a solvent.31 The use of iodomethane as the methylating agent lowers the atom economy due to its relatively high molecular weight (Table 2).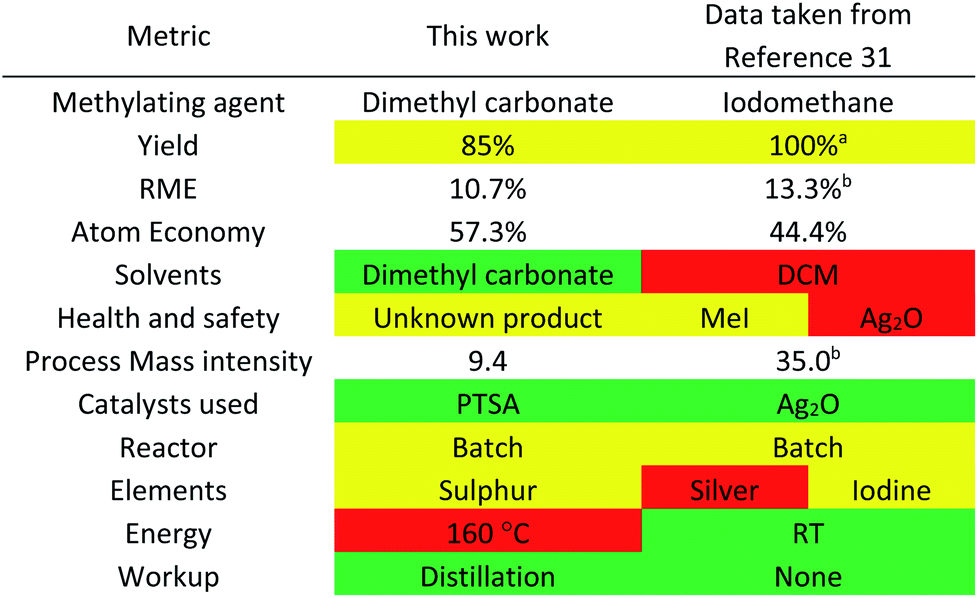
The process mass intensity (PMI) of the literature method is also worse compared to the dimethyl carbonate route proposed in this current work due to an excess of iodomethane and dichloromethane as solvent. A catalyst was employed, silver oxide, which contains a critical element as well as being very toxic to aquatic life. However, the reaction does occur readily at room temperature when using the more reactive methylating agent.
Hansen solubility parameters
Hansen solubility parameters (HSPs) are a tool to determine the solvency power of solvents (Hansen, 2007).33Consisting of dispersion forces (δD), dipole forces (δP) and hydrogen bonding forces (δH), the three HSPs can be used as coordinates to establish the position of a solvent in three-dimensional Hansen space. The HSP distance (Ra) between two compounds is defined in eqn (4).
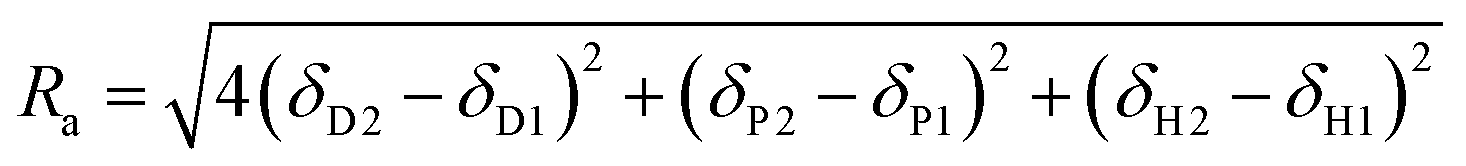 | (4) |
The original purpose of calculating Ra values was to predict the suitability of a particular solvent towards a substrate. It can also be used to establish the similarity between two solvents. Hence the smaller the Ra value between two solvents is, the more similar their solubility properties are predicted to be.
Hansen Solubility Parameters in Practice (HSPiP, 5th edition, version 5.0.03) was used to calculate the HSPs of MO and compare it to other solvents (Fig. 2).34 It can be observed that MO is more similar to DCM within the Hansen space than structurally similar solvents such as acetone (Table 3 and Fig. 2).
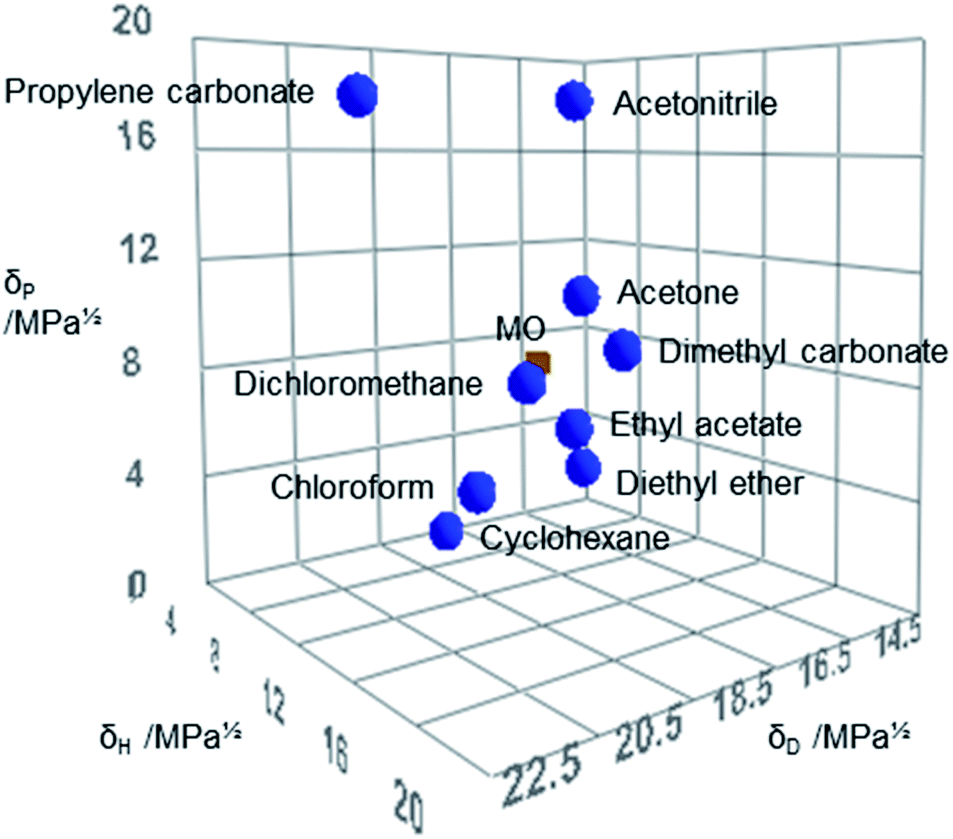 | ||
| Fig. 2 The position of MO (red) and selected traditional aprotic solvents (blue) in Hansen space constructed with HSPiP. | ||
| MO | DCM | Chloroform | Ethyl acetate | Diethyl ether | Acetonitrile | Acetone | Cyclohexane | Dimethyl carbonate | Propylene carbonate | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated with HSPiP (version 5.0.03).b Average value of 4-nitrophenol/4-nitroanisole and 4-nitroaniline/N,N-diethyl-4-nitroaniline systems.c Hansen radius to MO.d Measured by DSC.e Ref. 39.f Ref. 36.g Ref. 33.h Ref. 40.i Ref. 41.j Ref. 42.k Ref. 43.l Ref. 44.m Ref. 45.n Ref. 11.o Ref. 46.p Ref. 47.q Ref. 48.r Ref. 49. | ||||||||||
| Mol. wt | 102.1a | 84.9e | 119.4e | 88.1e | 74.1e | 41.1e | 58.1e | 84.2e | 90.1a | 102.1e |
| α | 0.33 | 0.13f | 0.20f | 0.00n,o | 0.00n,o | 0.35p | 0.20p | 0.00j | 0.00j | 0.00q |
| β | 0.20b | 0.10f | 0.10f | 0.48n,o | 0.51 | 0.37p | 0.54q | 0.00j | 0.32j | 0.38q |
| π* | 0.71b | 0.82f | 0.58f | 0.54n,o | 0.28n,o | 0.80p | 0.70p | 0.00j | 0.44j | 0.90q |
| δD/MPa0.5 | 16.1a | 18.2g | 17.8g | 15.8g | 14.5g | 15.3g | 15.5g | 16.8g | 15.5g | 20.0g |
| δP/MPa0.5 | 7.7a | 6.3g | 3.1g | 5.3g | 2.9g | 18.0g | 10.4g | 0.0g | 3.9g | 18.0g |
| δH/MPa0.5 | 5.5a | 6.1g | 5.7g | 7.2g | 5.1g | 6.1g | 7.0g | 0.2g | 9.7g | 4.1g |
| Rac | 0.00 | 4.57 | 5.97 | 3.25 | 6.03 | 10.14 | 3.07 | 9.70 | 5.99 | 12.76 |
| b.p./°C | 112 | 40e | 61e | 77e | 34e | 82e | 56e | 81e | 90k | 242e |
| m.p./°C | <−90d | −95e | −64e | −84e | −116e | −44e | −95e | 7e | 5l | −55e |
| ρ/g mL−1 at 298 K | 0.90 | 1.32e | 1.48e | 0.89e | 0.71e | 0.78e | 0.78e | 0.77e | 1.06k | 1.20e |
| Viscosity/cP at 293 K | 0.48 | 0.44h | 0.5 to 6r | 0.45e | 0.24e | 0.36h | 0.33i | 0.98e | 0.62m | 2.76e |
Kamlet–Taft solvatochromic parameters
While HSP describes the solvency power of solvents, KT parameters can be correlated to dynamic chemical phenomena, such as reaction rates and equilibria.The Kamlet–Taft (KT) solvatochromic parameters of MO were obtained experimentally via spectroscopic methods (Table 3 and Fig. 2). MO has an equivalent dipolarity (π*) to other ketones such as acetone, but a reduced capability to accept hydrogen bonds (β) more akin with chlorinated solvents. This is a significant result since the replacement of hazardous medium–high polarity and low basicity solvents is a challenge.35 The weak hydrogen bond accepting ability of MO is surprising, but a similar effect is exhibited by multifunctional alcohols and ethers, where diols are poorer hydrogen bond acceptors than monoalcohols, and 1,2-dimethoxyethane has a lower β value than diethyl ether for example.36 In the formation of a hydrogen bond, the available electron density is reduced by the inductive effect of a neighbouring electronegative functional group. Yet the alignment of the polarised bonds maximises the dipole of the MO molecule, resulting in a reasonably high π* value.
Solvents derived from bio-based platform molecules will not provide an exact drop-in replacement for a solvent like DCM, because organohalides have unique properties that oxygenated compounds cannot replicate. A carbon–chlorine bond results in an outfacing antibonding pi orbital (belonging to the chlorine atom) along the axis of the covalent bond.37 This electron deficient ‘crown’ defines the nature of intermolecular interactions with chlorinated solvents. Halogen bonding becomes possible, but at the expense of hydrogen bonding (as represented by β). The high electronegativity of chlorine polarises the carbon–hydrogen bonds in DCM and chloroform resulting in mild acidity (see α values in Table 3). These phenomena result in a strong complementarity between chlorinated solvents and oxygenated substrates, further justifying the use of DCM and chloroform in extractions.38 As a ketone, MO, can undergo keto–enol tautomerisation and provide weak hydrogen bond donating ability.
Physical properties and hazards
Table 3 summarises the properties of MO compared to a range of traditional aprotic solvents. The melting point (m.p.) of MO was measured by differential scanning calorimetry (DSC) and was found to be below −90 °C. This is comparable to most established solvents and allows MO to be used as a solvent at low temperatures. The boiling point (b.p.) of MO was measured as 112 °C by refluxing at atmospheric pressure. Although the boiling point is higher than typical chlorinated solvents it is significantly lower than many other bio-based solvents. For reaction chemistry, the reasonably high boiling point makes it attractive for some applications, for instance where 1,2-dichloroethane or chlorobenzene is currently used.35 The viscosity of MO is 0.48 cP at 293 K, and its density was experimentally determined to be 0.90 g mL−1 at 298 K, typical of conventional solvents. Unlike the chlorinated solvents, MO is water miscible, and unsuitable for aqueous-organic separations.As MO contains ether functionality, it can potentially form explosive peroxide compounds by reacting with O2.50 A preliminary test to monitor the formation of peroxides in MO was conducted using peroxide test strips (Macherey-Nagel, QUANTOFIX® Peroxide-100). After 210 days of testing in the absence of antioxidants or stabilisers, the total peroxide concentration in MO was less than 1 mg L−1. This demonstrates that despite containing an ether functional group, thus far MO has demonstrated a low risk of forming hazardous peroxides.
It is important that new substances intended to replace toxic solvents do not themselves possess high toxicity. The benefits of the Ames test as a first test of toxicity have recently been demonstrated.28 The Ames test uses bacterial strains to detect mutagenicity, which has been shown to correlate with carcinogenicity.51–53 The Ames test is quick, with results obtained after four days, and importantly, is relatively low cost. As such, it is a useful test for the early screening of new molecules, and a first step towards regulatory approval. In this work, a preliminary mutagenicity test of MO was conducted using the Ames test, in which it did not exhibit any mutagenicity. Although this result is promising at this early stage of development, a full set of toxicity tests will be required before commercial scale use and production of this solvent can be considered.
Unlike halogenated hydrocarbon solvents, MO contains only carbon, hydrogen and oxygen atoms and therefore will not form highly toxic compounds such as phosgene, an oxidation product of DCM observed in combustion, or deplete the ozone layer,54 adding to its green credentials. Its higher boiling point compared to DCM implies a lower vapour pressure and less risk of exposure and environmental release.
Friedel–Crafts acylation
The Friedel–Crafts acylation reaction is an electrophilic aromatic substitution reaction, often undertaken in a chlorinated hydrocarbon solvent, such as DCM.55–57The commercially important, Lewis acid catalysed (FeCl3) synthesis of 4-methoxyacetophenone (1) from anisole and acetic anhydride was used to assess the suitability of MO for the substitution of traditional organic solvents (Scheme 2).
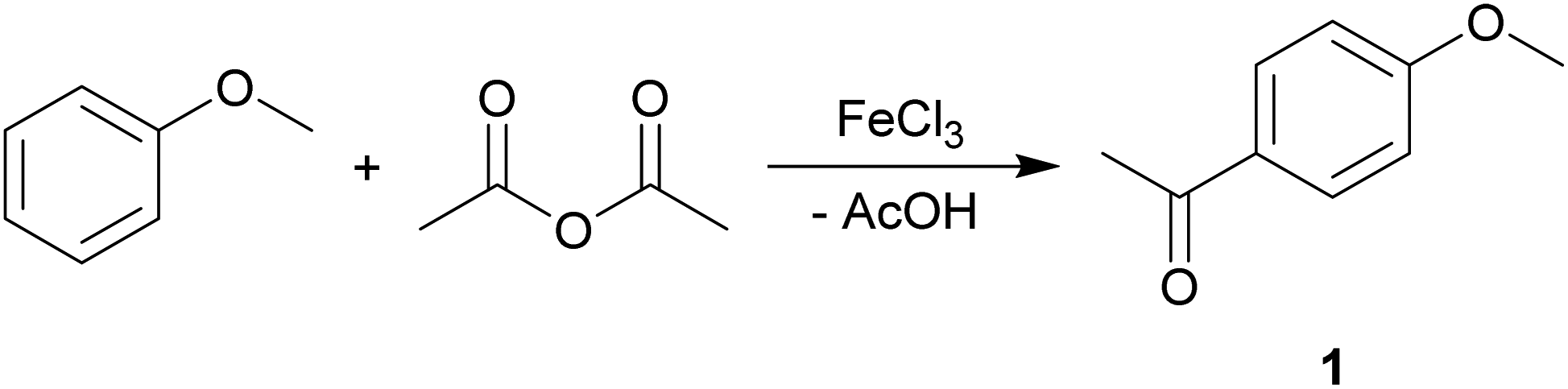 | ||
| Scheme 2 The Friedel–Crafts acylation of anisole to synthesise 1. | ||
It was found that conversions in MO (79% yield) matched DCM (77%) and exceeded chloroform (30%). In fact, MO exhibited the highest yield of a variety of traditional solvents (Fig. 3). This demonstrates the potential of MO to be a high-performance solvent in organic synthesis.
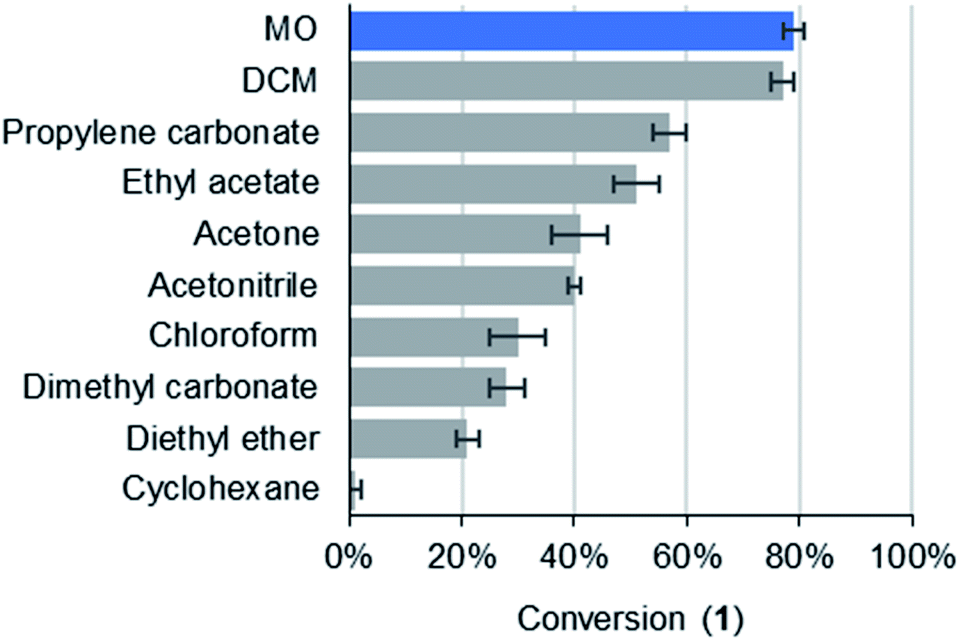 | ||
| Fig. 3 Conversions to 1 at room temperature after 2 hours (>99% selectivity towards 1 was observed in all solvents). | ||
Menschutkin reaction
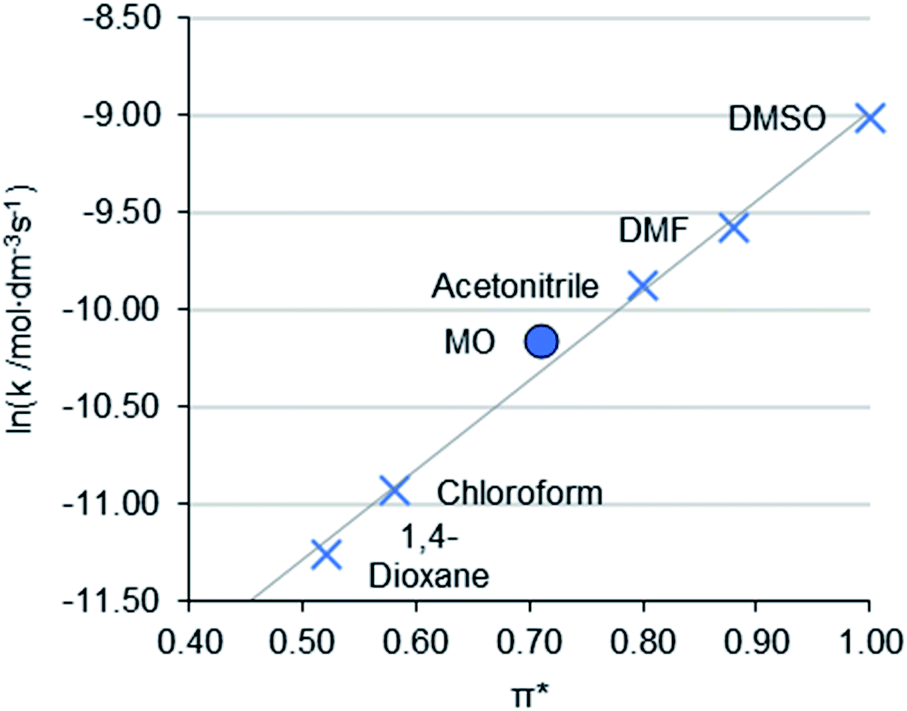
The Menschutkin reaction is an alkylation reaction, used as an important method of preparing imidazolium ionic liquids (Scheme 3).58 The rate constants of 1-decyl-2,3-dimethylimidazolium bromide (2) formation in conventional solvents was obtained previously by Sherwood et al.24 MO performed moderately well under the same conditions, with the rate of reaction (lnk = −10.16 mol L−1 s−1) correlating to the π* value of the solvent (Fig. 4). A 1H NMR spectrum of a representative Menschutkin reaction is presented in the ESI.†
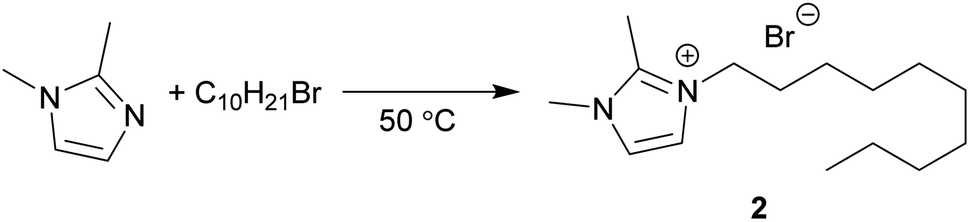 | ||
| Scheme 3 The synthesis of 2 from 1,2-dimethylimidazole and 1-bromodecane. | ||
 | ||
| Fig. 4 Conversions to 2 at room temperature after 2 hours (>99% selectivity towards 2 was observed in all solvents). | ||
Diels–Alder reaction
In this study, the performances of MO and other traditional solvents were evaluated for the Diels–Alder reaction of 2,3-dimethylbuta-1,3-diene (diene) and 3-buten-2-one to form 1-(3,4-dimethylcyclohex-3-enyl) ethanone (3) (Scheme 4).59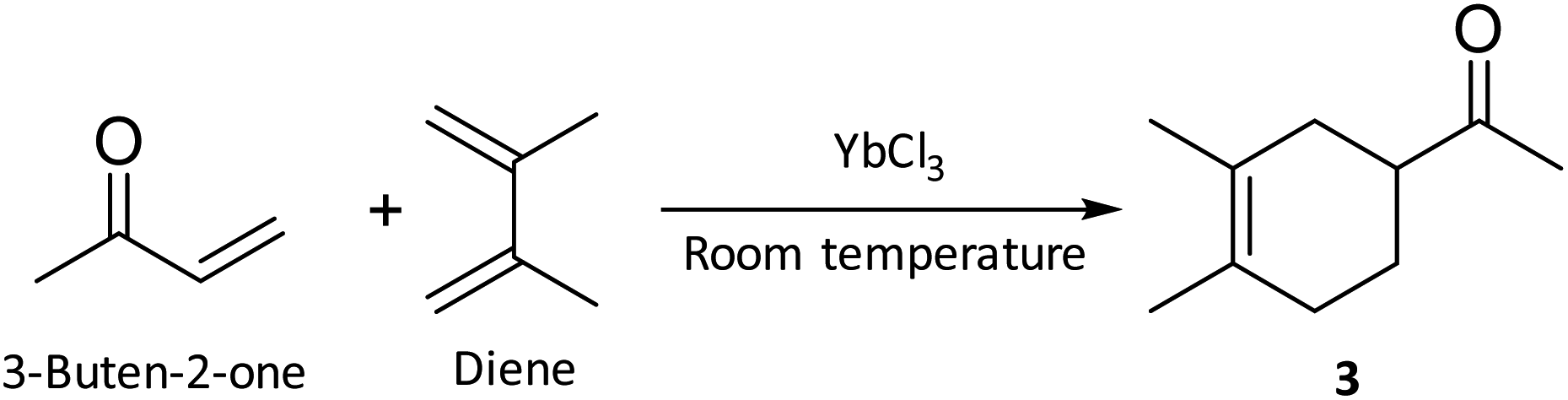 | ||
| Scheme 4 Diels–Alder synthesis of 3 from 3-buten-2-one and diene catalysed by ytterbium(III) chloride. | ||
It can be clearly observed in Fig. 5 that MO was not an effective solvent in the Diels–Alder reaction for the formation of 3. This is potentially down to MO forming a complex with the Lewis acid ytterbium(III) chloride, thus hindering its catalytic performance.
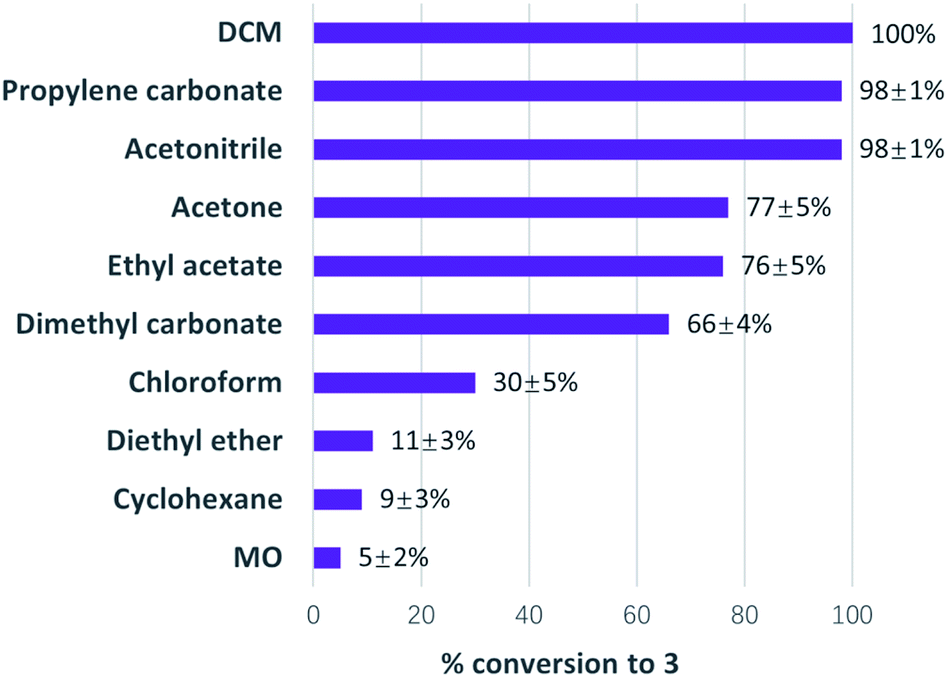 | ||
| Fig. 5 Conversions to 3 from 3-buten-2-one and diene in various solvents catalysed by ytterbium(III) chloride via Diels–Alder reaction; selectivity >99% towards 3 for all the solvents investigated in the research. | ||
Such results highlight some of the limitations of MO, where the activity of the carbonyl limits its application, e.g. systems which might promote imine/enamine formation or aldol chemistry. In addition, MO is water miscible, which limits it suitability for organic-aqueous workup and having a b.p. of 112 °C, increases energy consumption when removing by distillation. The synthesis of MO in flow and utilising heterogeneous catalysts would be vital for this solvent to be employed at scale. Future investigations will also focus on expanding substrate scope and examine the workup or product isolation steps of synthesis, which are also important in evaluating a solvents performance.
MO is a renewable solvent, which represent one of the first oxygenated compound that demonstrates promise in substitution of carcinogenic halogenated solvents in some applications.
Conclusions
3-Methoxybutan-2-one (MO) has been synthesised in a one-step, solvent-free process in yields greater than 85% at 100 g scale, from the platform molecule acetoin, which can be derived from waste biomass. The synthetic methodology utilised in this study avoided the use (or generation) of toxic substances and critical elements. MO is an oxygenated, bio-based solvent with uniquely low basicity and medium–high polarity. MO demonstrated no mutagenicity in preliminary Ames tests, while peroxide testing highlighted only limited formation after 210 days. Solvent properties including the Kamlet–Taft polarity scales and Hansen solubility parameters indicate that 3-methoxybutan-2-one is a new bio-based candidate suitable for the substitution of traditional polar solvents with low basicity including halogenated solvents such as chloroform or DCM. It has also been shown that MO is able to support acylation and alkylation reactions as a solvent. It is envisaged that bio-based molecules such as MO will aid in the replacement of those traditional solvents now deemed to be unacceptable by legislation such as REACH. The utilisation of agricultural residues and food wastes for the production of chemicals including solvents also drives the bio-economy and can increase the sustainability of processes or products as part of a circular economy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Andrew Hunt would like to acknowledge the financial support of Materials Chemistry Research Center (Khon Kaen University), Thailand Research Fund (RSA6280031) and Khon Kaen University. Financial support from the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation is gratefully acknowledged. The authors would like to thank the Biorenewables Development Centre (BDC) for their assistance with the Ames test. The authors also thank Dr Duncan J. Macquarrie (University of York) and Dr Thomas J. Farmer (University of York) for their beneficial suggestions during this research.Notes and references
- C. Reichardt, Org. Process Res. Dev., 2007, 11, 105–113 CrossRef CAS.
- F. M. Kerton and R. Marriott, Alternative Solvents for Green Chemistry, Royal Society of Chemistry, 2013 Search PubMed.
- Chloroform - Substance Information - ECHA, https://echa.europa.eu/substance-information/-/substanceinfo/100.000.603, accessed 25th July 2020.
- Dichloromethane - Substance Information - ECHA, https://echa.europa.eu/substance-information/-/substanceinfo/100.000.763, accessed 25th July 2020.
- ECHA, Candidate List of substances of very high concern for Authorisation (published in accordance with Article 59(10) of the REACH Regulation), 2019, available at https://echa.europa.eu/candidate-list-table (accessed 25th July 2020) Search PubMed.
- S. Jonuzaj, J. Cui and C. S. Adjiman, Comput. Chem. Eng., 2019, 130, 106518 CrossRef CAS.
- J. Sherwood, J. Granelli, C. R. McElroy and J. H. Clark, Molecules, 2019, 24, 2209 CrossRef CAS.
- F. P. Byrne, B. Forier, G. Bossaert, C. Hoebers, T. J. Farmer and A. J. Hunt, Green Chem., 2018, 20, 4003–4011 RSC.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS.
- R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 2886–2894 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud and R. W. Taft, J. Am. Chem. Soc., 1977, 99, 6027–6038 CrossRef CAS.
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- S. Jin, F. Byrne, C. R. McElroy, J. Sherwood, J. H. Clark and A. J. Hunt, Faraday Discuss., 2017, 202, 157–173 RSC.
- J. F. Guymon and E. A. Crowell, Am. J. Enol. Vitic., 1965, 16, 85–91 CAS.
- Y. Xu, W. Fan and M. C. Qian, J. Agric. Food Chem., 2007, 55, 3051–3057 CrossRef CAS PubMed.
- B. S. Radovic, M. Careri, A. Mangia, M. Musci, M. Gerboles and E. Anklam, Food Chem., 2001, 72, 511–520 CrossRef CAS.
- R. G. Buttery, L. C. Ling and D. J. Stern, J. Agric. Food Chem., 1997, 45, 837–843 CrossRef CAS.
- W. Hummel, M. R. Kula and F. Boermann,US Pat., US5164314 A, 1992 Search PubMed.
- J. Liu, X. Zhao, J. Zhang, Y. Tian, Y. Han and L. Han, US Pat., US20080182306 A1, 2008 Search PubMed.
- Z. J. Xiao, P. H. Liu, J. Y. Qin and P. Xu, Appl. Microbiol. Biotechnol., 2007, 74, 61–68 CrossRef CAS.
- L. Zhang, Q. Liu, Y. Ge, L. Li, C. Gao, P. Xu and C. Ma, Green Chem., 2016, 18, 1560–1570 RSC.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS.
- J. Sherwood, M. D. Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- M. Selva, V. Benedeta and M. Fabris, Green Chem., 2012, 14, 188–200 RSC.
- N. I. Prakoso, N. V. Nugroho and D. Rubiyanto, AIP Conf. Proc., 2020, 2229, 030001, DOI:10.1063/5.0002543.
- G. Fiorani, A. Perosa and M. Selva, Green Chem., 2018, 20, 288–322 RSC.
- S. Jin, A. J. Hunt, J. H. Clark and C. R. McElroy, Green Chem., 2016, 18, 5839–5844 RSC.
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC.
- C. R. McElroy, A. Constantinou, L. C. Jones, L. Summerton and J. H. Clark, Green Chem., 2015, 17, 3111–3121 RSC.
- N. Oku, K. Takada, R. W. Fuller, J. A. Wilson, M. L. Peach, L. K. Pannell, J. B. McMahon and K. R. Gustafson, J. Am. Chem. Soc., 2010, 132, 10278–10285 CrossRef CAS.
- D. G. H. Crout and S. M. Morrey, J. Chem. Soc., Perkin Trans., 1983, 1, 2435–2440 RSC.
- C. M. Hansen, Hansen Solubility Parameters: A User's Handbook, CRC Press, 2nd edn, 2007 Search PubMed.
- C. M. Hansen and S. Abbott, Hansen Solubility Parameters in Practice, Hansen-Solubility, 2008 Search PubMed.
- J. Sherwood, Angew. Chem., Int. Ed., 2018, 57, 14286–14290 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, G. Priimagi, A. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- E. Stefanis and C. Panayiotou, Int. J. Pharm., 2012, 426, 29–43 CrossRef CAS.
- J. A. Riddick, W. B. Bunger and T. K. Sakano, Organic Solvents, Wiley-Interscience, New York, 4th edn, 1986 Search PubMed.
- I. H. M. V. Stokkum, T. Scherer, A. M. Brouwer and J. W. Verhoeven, J. Phys. Chem., 1994, 98, 852–866 CrossRef.
- V. H. Staudinger and H. Jörder, J. prakt. Chem., 1942, 160, 166–175 CrossRef.
- J. Sherwood, PhD thesis, University of York, 2013.
- M. Fukano, H. Matsuda, K. Kurihara and K. Ochi, J. Chem. Eng. Data, 2006, 51, 1458–1463 CrossRef CAS.
- H. Matsuda, H. Kimura, Y. Nagano, K. Kurihara, K. Tochigi and K. Ochi, J. Chem. Eng. Data, 2011, 56, 1500–1505 CrossRef CAS.
- F. Chen, Z. Yang, Z. Chen, J. Hu, C. Chen and J. Cai, J. Mol. Liq., 2015, 209, 683–692 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- L. Crowhurst, P. R. Mawdsley, J. M. Perez-Arlandis, P. A. Saltera and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- H. L. Parker, J. Sherwood, A. J. Hunt and J. H. Clark, ACS Sustainable Chem. Eng., 2014, 2, 1739–1742 CrossRef CAS.
- J. C. Prager, Environmental Contaminant Reference Databook, Van Nostrand Reinhold, New York, NY 1995, Vol. 1, p. 453 Search PubMed.
- D. E. Clark, Chem. Health Saf., 2001, 8, 12–22 CrossRef CAS.
- B. N. Ames, F. D. Lee and W. E. Durston, Proc. Natl. Acad. Sci., 1973, 70, 782–786 CrossRef CAS.
- A. J. F. Griffiths, J. H. Miller, D. T. Suzuki, R. C. Lewontin and W. M. Gelbart, Introduction to Genetic Analysis, W. H. Freeman, New York, 7th edn, 2000 Search PubMed.
- J. McCann, E. Choi, E. Yamasaki and B. N. Ames, Proc. Natl. Acad. Sci., 1975, 72, 5135–5139 CrossRef CAS.
- M. J. Molina and F. S. Rowland, Nature, 1974, 249, 810–812 CrossRef CAS.
- V. Rozenberg, T. Danilova, E. Sergeeva, E. Vorontsov, Z. Starikova, K. Lysenko and Y. Belokon’, Eur. J. Org. Chem., 2000, 2000, 3295–3303 CrossRef.
- S. Dahmen and S. Bräse, Tetrahedron: Asymmetry, 2001, 12, 2845–2850 CrossRef CAS.
- M. S. Shchepinov, V. A. Korshun, R. D. Egeland and E. M. Southern, Tetrahedron Lett., 2000, 41, 4943–4948 CrossRef CAS.
- J. C. Schleicher and A. M. Scurto, Green Chem., 2009, 11, 694–703 RSC.
- X. Fang, B. P. Warner and J. G. Watkin, Synth. Commun., 2000, 30, 2669–2676 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07322b |
| ‡ Current address: School of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, People's Republic of China. |
| This journal is © The Royal Society of Chemistry 2021 |