
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The binary aluminum scandium clusters AlxScy with x + y = 13: when is the icosahedron retained?
Ngo Tuan Cuong a,
Nguyen Thi Maib,
Nguyen Thanh Tungb,
Ngo Thi Lanbc,
Long Van Duongd,
Minh Tho Nguyene and
Nguyen Minh Tam*fg
a,
Nguyen Thi Maib,
Nguyen Thanh Tungb,
Ngo Thi Lanbc,
Long Van Duongd,
Minh Tho Nguyene and
Nguyen Minh Tam*fg
aFaculty of Chemistry, Center for Computational Science, Hanoi National University of Education, Hanoi, Vietnam
bInstitute of Materials Science, Graduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc, Hanoi Vietnam
cDepartment of Physics and Technology, Thai Nguyen University of Science, Thai Nguyen, Vietnam
dInstitute for Computational Science and Technology (ICST), Quang Trung Software City, Ho Chi Minh City, Vietnam
eDepartment of Chemistry, KU Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium
fComputational Chemistry Research Group, Ton Duc Thang University, Ho Chi Minh City, Vietnam. E-mail: nguyenminhtam@tdtu.edu.vn
gFaculty of Applied Sciences, Ton Duc Thang University, Ho Chi Minh City, Vietnam
First published on 16th December 2021
Abstract
Geometrical and electronic structures of the 13-atom clusters AlxScy with x + y = 13, as well as their thermodynamic stabilities were investigated using DFT calculations. Both anionic and neutral isomers of AlxScy were found to retain an icosahedral shape of both Al13 and Sc13 systems in which an Al atom occupies the endohedral central position of the icosahedral cage, irrespective of the number of Al atoms present. Such a phenomenon occurs to maximize the number of stronger Al–Al and Sc–Al bonds instead of the weaker Sc–Sc bonds. NBO analyses were applied to examine their electron configurations and rationalize the large number of open shells and thereby high multiplicities of the mixed clusters having more than three Sc atoms. The SOMOs are the molecular orbitals belonged to the irreducible representations of the symmetry point group of the clusters studied, rather than to the cluster electron shells. Evaluation of the average binding energies showed that the thermodynamic stability of AlxScy clusters is insignificantly altered as the number y goes from 0 to 7 and then steadily decreases when y attains the 7–13 range. Increase of the Sc atom number also reduces the electron affinities of the binary AlxScy clusters, and thus they gradually lose the superhalogen characteristics with respect to the pure Al13.
1. Introduction
During the last four decades, a very large number of both experimental and theoretical studies on atomic clusters were reported with the aim to understand their novel physical and chemical properties as well as to emphasize their abilities to be used for new promising technological applications.1 The atomic clusters possessing high symmetry geometries have often attracted more attention, in part due to the fact that they are expected to have an enhanced stability along with appealing features. In particular they can be considered as building blocks for assembled nanostructured materials.1,2 Especially, many investigations found that various clusters formed by 13 atoms, including both homonuclear and heteronuclear derivatives, exist in an icosahedral shape and have interesting physico-chemical properties that could lead to significant applications.3 For instance, to adjust the overall valence state and thereby the chemical behavior of the silicon doped aluminum Al12Si icosahedron, it is possible to substitute the Si atom by another dopant such as B, P and Ca, to form the superhalogen Al12B, super alkali metal Al12P, and superchalcogen Al12Ca, respectively.4 Of the main group element clusters, only the aluminum clusters at this size, Al13, in both neutral and anionic states, are icosahedra formed by 13 homonuclear atoms,5–7 that are one of the most well-known and inspiring example for superatomic clusters.1 The anion Al13− exhibits 40 valence electrons in a closed shell structure and thus emerges as a magic cluster with an enhanced thermochemical stability. For its part, the neutral Al13, having one valence electron less than the anion Al13−, has a very high electron affinity exceeding those of halogen atoms and has been regarded as a superhalogen.8,9 Stimulated by the existence of the icosahedral Al13, many similar structures were found by doping of various hetero-atoms into aluminum hosts.3 A previous study using photoelectron spectroscopy combined with theoretical calculations pointed out that an icosahedral anion Al12Li− can be composed by replacing a surface Al atom of the icosahedron Al13− by a Li atom.10 For the doped aluminum Al12X2−, while the beryllium, X = Be, sits at the center of an aluminum icosahedral cage due to its small atomic radius, the remaining dopants with X being an alkaline earth metal having larger atomic radius, favor its attachment outside at the surface.11–13 Although the aluminum doped boron clusters B12Al favor a quasi-planar shape,14 it was also found that other dopants which belong to the same group IIIA as aluminum including B and Ga, can substitute the Al atom(s) at the center of icosahedral cage Al13.15,16 An investigation of the cationic clusters Al12X+, with X being a tetravalent atom including C, Si, Ge, Sn, and Pb, showed that except for the low symmetry structure of Al12C+, the remaining structures of Al12X+ prefer an icosahedral shape. The Si and Ge atoms are encapsulated at the central position of the icosahedra Al12Si+ and Al12Ge+, respectively, whereas for the Al12Sn+ and Al12Pb+, the dopant substitutes an aluminum atom on a vertex of the icosahedral framework.17 Moreover, a recent study on singly and doubly silicon doped aluminum clusters reported that both neutral and cationic states of Al11Si2 keep on favoring the icosahedral shape with one Si dopant embedded at the central position, whereas the remaining Si atom substitutes an external Al position of the icosahedron Al13.18 Similarly, another study of Al12X clusters at both neutral and cationic states, with dopant X being a pentavalent atom including P, As, Sb and Bi, also showed that the P atom prefers to be at the center of the Al12 icosahedron whereas the rest of the dopants favor occupancy of a vertex site due to their larger size.19Previous studies on transition metal doped aluminum clusters also reported that an icosahedral structure can be composed by 12 aluminum atoms plus a transition metal dopant such as Co, Ni, Cu, and Zn. The atomic radius of the latter seems small enough to allow its position at the center of such a cage.10,11,20–22 A combined theoretical and experimental study on AlnV clusters23 showed that the anionic Al12V− prefers a distorted icosahedral shape in which the vanadium atom occupies a convex capped site. Interestingly, Kumar and Kawazoe carried out a study using density functional theory (DFT) calculations on a doping of a Cu atom into Al12 and discovered a perfect icosahedral Cu@Al12 possessing a high spin state with a 3 μB magnetic moment.22 A subsequent theoretical study found that the Al12Cu could play as a stable building block to form ionic salts, as shown from stable dimers, trimers, and tetramers of the Al12CuM3 complex.24
On the other hand, it appears that an icosahedral structure based on the elements of the main group IVA can be also obtained upon doping of an impurity atom onto a 12 tetravalent atom system. A theoretical study indicated that the most stable isomers of the Ge12Mx clusters, with M = Li, Na, Be, Mg, B and Al with x going from −1 to +1 and each containing 50 valence electrons, prefer a high symmetry icosahedron.25 Particularly for the anion Ge12Li−, the lithium dopant was found at the central position of the icosahedral cage instead of on its surface as in the case of Al12Li−.25 Goicoechea and McGrady carried out a theoretical investigation of endohedrally transition metal doped silicon MSi12 and germanium MGe12 clusters, and found that while MSi12 have a hexagonal prism or a bicapped pentagonal prism shape, some MGe12 with M being Cr, Mn, Fe, Cu, Zn, Ag, and Cd, tend to favor an icosahedral form.26 Similarly, many icosahedra bearing an endohedrally doped metal such as tin MSn12 and lead MPb12 were also found from both theoretical and experimental approaches.27–31
Previous investigations also demonstrated that many pure clusters formed by 13 transition metal atoms in both neutral and ionic states exist in ideal or distorted icosahedral shape, including the first-row transition metal clusters such as Sc13, Ti13, V13, Cr13, Mn13 and Fe13 and heavier transition metal clusters such as Y13, Hg13, Zr13, Lu13 and Hf13.32–34 A characteristic difference of the latter from the main group element clusters is that most of the transition metal icosahedra possess very high total spin and thereby large magnetic moments, due to their unpaired nd electrons. Along with the homonuclear icosahedra, it was found that many doped transition metal clusters favor an icosahedral shape. Datta et al. reported from DFT calculations that the most stable isomers of vanadium doped cobalt clusters Co13−mVm, with m = 1–4, adopt an icosahedral geometry, unlike the hexagonal symmetry preference of the pure Co13 clusters.35 Other theoretical studies also demonstrated that an icosahedral 13-atom structure can be formed by endohedrally doping a transition metal atom into a coinage metal cluster Cu12 and Ag12.36–39 In particular, doping of a Cr atom into Cu12 as well as Ag12 was shown to form a Kondo-like system that enhances the thermodynamic stability of both resulting CrCu12 and CrAg12 icosahedra but quenches the large magnetic moment of the dopant simultaneously.37,39
Of the transition metal clusters possessing icosahedral geometry, the scandium-based clusters could deserve more attention because of their formal similarities with aluminum-based clusters in terms of valence electrons. As a matter of fact, the scandium element belongs to group IIIB and also has 3 valence electrons (3d14s2). Additionally, both Sc13 and Al13 are characterized by a perfect icosahedral structure. However, unlike the anion Al13− having a closed shell electronic structure and the neutral Al13 exhibiting a doublet ground state, both anionic and neutral states of Sc13 are known to exist in very high spin ground states, with the total spin magnetic moments of 18 and 19 μB, respectively.32,40 A previous theoretical investigation of the singly aluminum doped scandium clusters ScnAl, with n = 1–8, 12, showed that the ScnAl isomers prefer an Al substitution at a Sc position of a structure of the corresponding Scn+1 size. In fact, the resulting doped cluster Sc12Al exists in a high spin state along with an icosahedral shape in which the Al dopant is put into the center.41 Surprisingly, despite such a formal association between Sc and Al, an understanding of the geometric structure and electronic properties of the mixed Sc–Al clusters is still limited; they have not much been investigated so far. In this context, we set out to perform a detailed and systematic investigation on the binary Al–Sc clusters AlxScy with x + y = 13, at both anionic and neutral states using DFT method, with the purpose of gaining deeper insights into the structural and electronic features of these intriguing systems. A question of interest is as to whether the icosahedral geometry, ideal or distorted, remains predominant following atomic mixture, and the identity of the central atom.
2. Computational methods
All standard electronic structure calculations in this study are carried out using the Gaussian 09 package.42 The possible isomers of each cluster are searched for using different approaches. In the first step, a stochastic genetic algorithm43 is used to generate the possible structures of each size AlxScy with x + y = 13. This algorithm has been used in previous studies and shown to be highly efficient for systems containing different components.18,44–46 Another way of generating the initial isomers is a manual substitution by a Sc-atom at all Al positions of the well-known icosahedron Al13 to generate the Al12Sc structures, and successively for the following sizes with more Sc atoms. The search also includes the shapes of other 13-atom clusters that have previously been reported. Geometry optimizations are first carried out to generate the first initial set of isomers using the popular hybrid B3LYP functional in combination with the small LANL2DZ basis set.47 The local energy minima identified with relative energies of <5 eV with respect to the corresponding lowest-lying isomer of each size are then reoptimized at various spin states using the same functional but with the larger 6-311+G(d) basis set.48 Harmonic vibrational frequencies are subsequently calculated at this level to confirm the identity of the true local minima obtained, as well as to evaluate their zero-point energy corrections.The lower-lying isomers of the neutral 13-atom clusters AlxScy that have 39 valence electrons each, are then obtained from the corresponding anionic isomers upon removal of an electron and then geometrically optimized at different spin states. Moreover, a natural bond orbital (NBO) analysis is performed to examine the electronic configuration and thereby to rationalize the chemical bonding and magnetic properties of the clusters considered by using the NBO 3.1 program implemented in Gaussian 09. The total and local electron densities are defined as the difference between the numbers of spin-up and spin-down electrons occupying the molecular/atomic orbitals of the cluster/atom.
3. Results and discussion
3.1. Lower-lying isomers of binary Al–Sc clusters in both anionic and neutral states
Since there is a large number of local minima located on the potential energy surface of each size considered in different spin states, we only present here the lowest-lying isomers whose relative energies are close to the corresponding most stable structure, being <1 eV in relative energy. The shapes of AlxScy equilibrium structures in both anionic and neutral states, their spin states, and their relative energies obtained at the B3LYP/6-311+G(d)+ZPE level are shown in Fig. 1–3.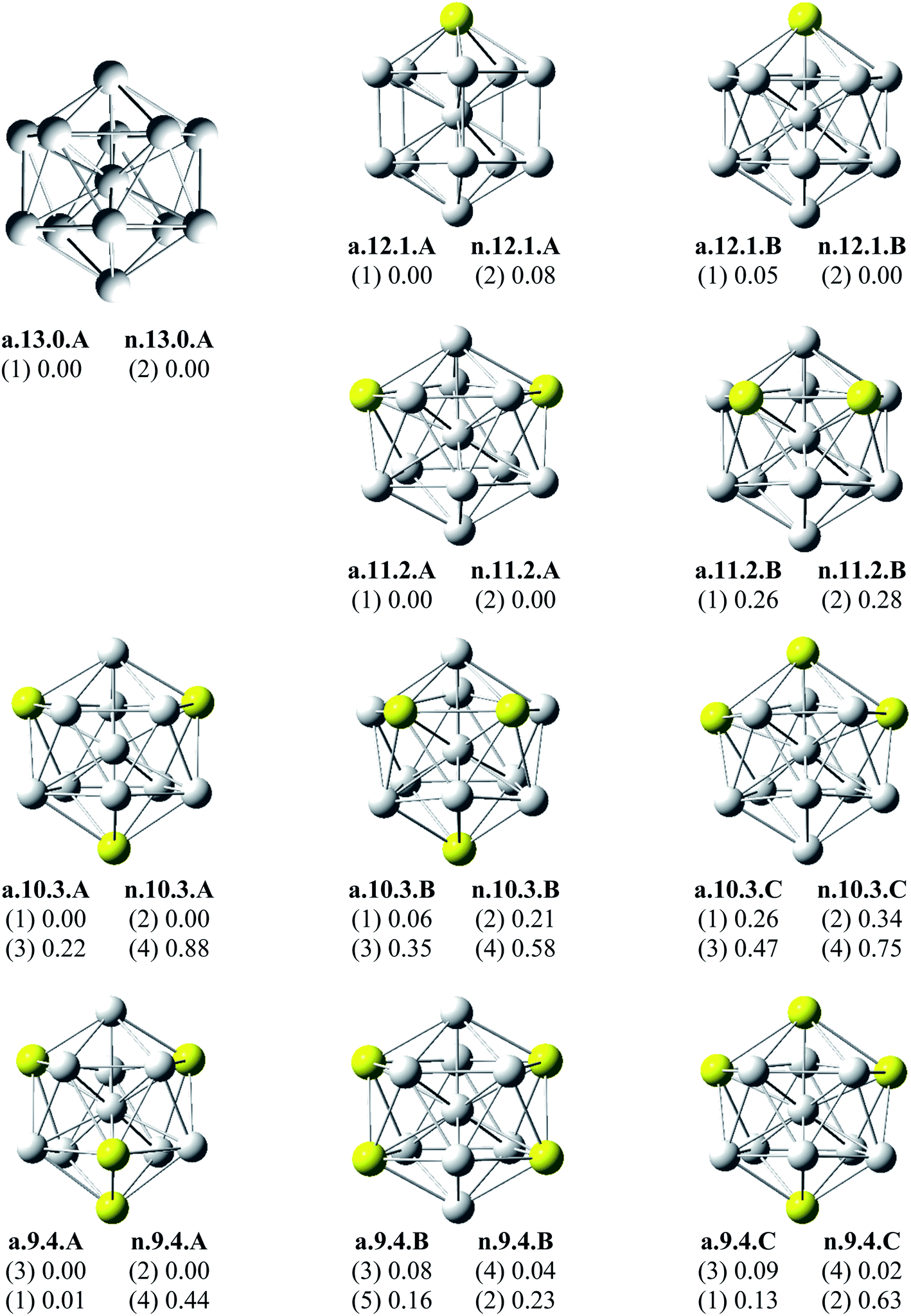 | ||
| Fig. 1 Geometry, relative energy (with ZPE corrections), and spin state (in the bracket) of the most stable isomers AlxScy (x + y = 13 with y = 0–4) using (U)B3LYP/6-311+G(d) optimizations. | ||
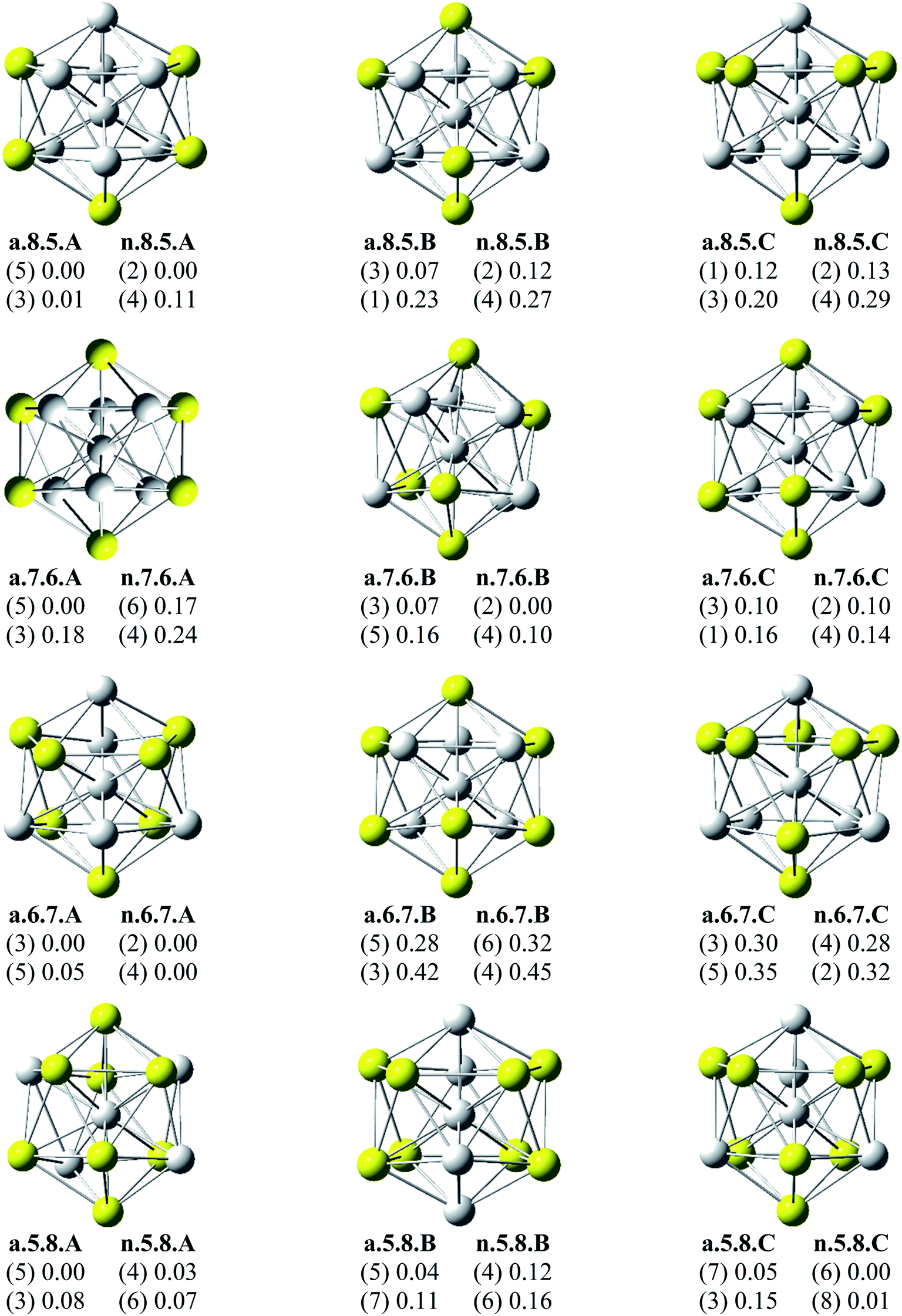 | ||
| Fig. 2 Geometry, relative energy (with ZPE corrections), and spin state (in the bracket) of the most stable isomers AlxScy (x + y = 13 with y = 5–8) using (U)B3LYP/6-311+G(d) optimizations. | ||
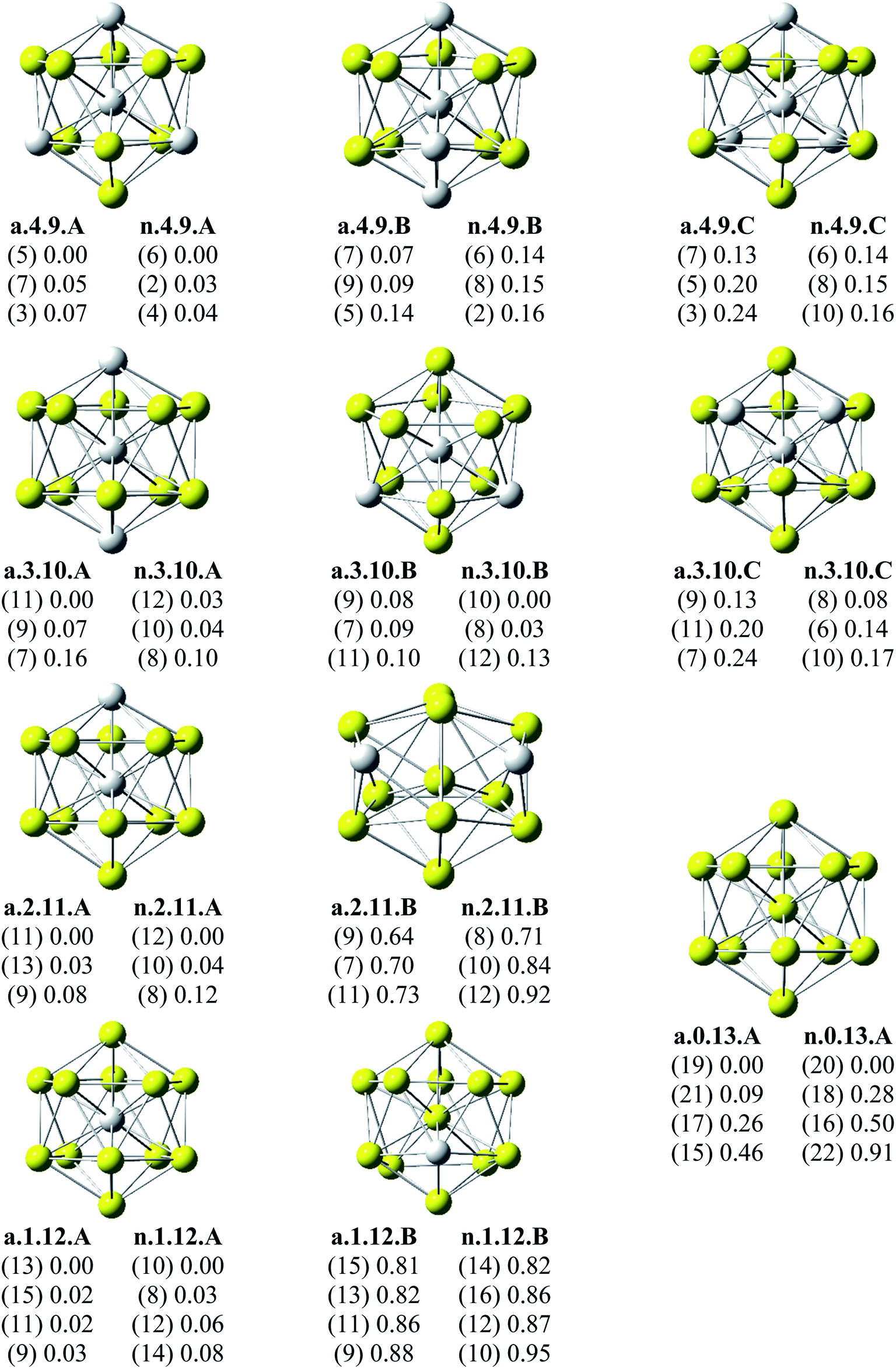 | ||
| Fig. 3 Geometry, relative energy (with ZPE corrections), and spin state (in the bracket) of the most stable isomers AlxScy (x + y = 13 with y = 9–13) using (U)B3LYP/6-311+G(d) optimizations. | ||
As for a convention, the S.x.y.Z label is used to denote the isomers in which S = a and n stand for an anionic state and its corresponding neutral with a similar geometrical shape, respectively, x being the number of Al atoms and y the number of Sc atoms, and Z = A, B, C… referring to the isomers with an increasing relative energy. Accordingly, the notation a.x.y.A invariably refers to the most stable anionic isomer (A) of the a.x.y series, and the n.x.y.A to its corresponding neutral.
Calculated results reveal an interesting discovery about geometrical features. All the lowest-lying AlxScy isomers in both anionic and neutral states, irrespective of the number x of Al atoms, have an icosahedral shape in which an Al atom is invariably situated at the cage center, whereas the remaining Al and Sc atoms form the corresponding icosahedron in different positions.
For the size x = 12 Al12Sc−, a.12.1.A, formed by capping the Sc atom on a vertex of the bicapped pentagonal prism framework, is only 0.05 eV lower in energy than the icosahedral structure a.12.1.B in which the Sc atom is placed on a surface position of the icosahedron. However, such a relative energy gap is too small to be meaningful, and therefore both isomers can be considered as energetically degenerate. DFT results also point out some energetic degeneracies with small energy gaps of <0.1 eV for most of the (x, y) combination of the series of AlxScy with y > 2. These isomers have an icosahedral framework with different positions of Sc atoms on the surface and in different spin states. As stated above, a remarkable feature of AlxScy structures is that an Al atom is consistently found at the center regardless of the number of Al atoms. The fact that the Sc atoms occupy surface positions of the icosahedron can be understood by the smaller atomic radius of aluminum which favors the Al atom to occupy a position inside the cage. This is in agreement with a previous experimental study on the argon physisorption ability of the first row transition metals,49 showing that the transition metal doped clusters AlnTM+ are able to attach one argon atom up to a critical cluster size ncrit, with ncrit = 16 for TM = V, Cr and ncrit = 19–21 for TM = Ti, and undergo a geometrical transition in going from exohedrally to endohedrally doped clusters in which the transition metal atom becomes, from ncrit, located inside an aluminum cage. Furthermore, this feature can be rationalized on the basis of the strength of the homo- and heteronuclear bonds between Al and Sc atoms. According to DFT calculations, the bond energies of the Al2, AlSc and Sc2 dimers, also obtained at the B3LYP/6-311+G(d) level, amount to 1.2, 0.6, and 0.5 eV, respectively. Although bond energies of the dimers are expected to differ from the energies of corresponding bonds in the clusters, this result shows that in the cluster the bond between two Al atoms is much stronger than the Al–Sc and Sc–Sc bonds. In a AlxScy cage structure both Al and Sc atoms are arranged in such a way of favoring formation of a maximum number of strong bonds, along with a minimum number of weak bonds. Accordingly, the Al atom thus prefers to occupy the icosahedral center in order to maximize the possible number of Al–Al and Al–Sc bonds.
3.2. Electron configuration and multiplicity
Along with a consistent possession of an icosahedral shape, another typical feature is that the AlxScy ground electronic states are associated with high number of unpaired electrons. The search for the most stable isomers is carried out for all plausible electron spin state for each cluster composition, and the multiplicity of AlxScy tends to gradually increase with an increasing number of Sc atoms. For a small number of Sc atoms, y = 0–3, the most stable structures of Al12Sc, Al11Sc2 and Al10Sc3 and their anions keep the lowest spin states alike the Al13 cluster, that are singlet state for species having an even number of electrons and doublet state for those having an odd number of electrons. A competition between the low and high spin states begins to occur at the size of four Sc atoms, namely the Al9Sc4. For y = 10–13, both anionic and neutral AlxScy structures are more stable at very high spin state in which each has nine or more unpaired electrons, approaching that of the pure scandium Sc13. Indeed, the most stable isomer of the latter has the highest spin states with 18 and 19 open shells for both anionic and neutral states, respectively. Fig. 4 displays the variation of the multiplicity of the binary clusters considered.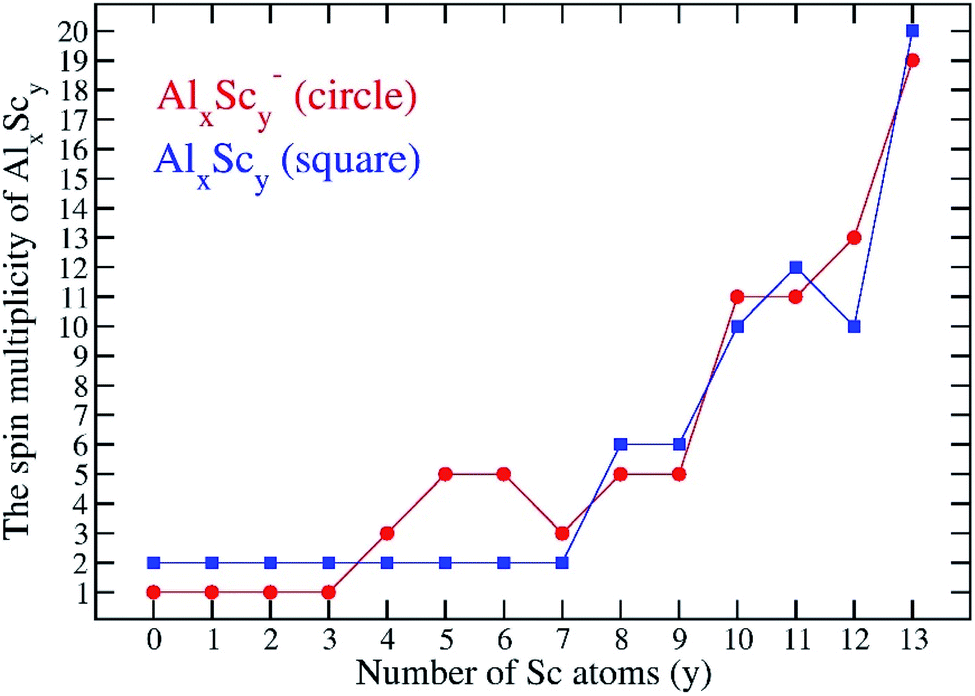 | ||
| Fig. 4 Spin multiplicities of the most stable isomers of the binary AlxScy clusters considered. | ||
In order to rationalize the high spin states of AlxScy clusters and obtain a deeper understanding of their electronic structures, we now analyze the natural bond orbitals of the anions.
Each 13-atom AlxScy− anion has 40 valence electrons, as each of the Al and Sc atoms has three valence electrons. For Al13−, all of its 40 valence electrons contribute to the shell molecular orbitals resulting in a closed electron shell of [1S21P61D102S22P61F14], without any unpaired electron, as shown in Fig. 5. Similarly, the Al12Sc−, Al11Sc2−, and Al10Sc3− anions also have the same closed electron configuration of [1S21P61D102S22P61F14] without open shell. The clusters having up to three Sc atoms do not have any Sc–Sc bond for the reason described above, as their geometrical structures seen in Fig. 1. In these cases, the isolation of Sc atoms around an Al environment results in a quenching of its spin upon formation of the Al–Sc bonds. In order to understand how the AlxScy clusters come to possess high multiplicity upon increase of the Sc atom number going from four up to thirteen, we would start examining the electron configuration of the pure Sc13 cluster which is characterized by the highest spin multiplicity as a special reference case, and the other binary clusters with a reduced number of Sc atoms, going down to Al9Sc4, are subsequently examined.
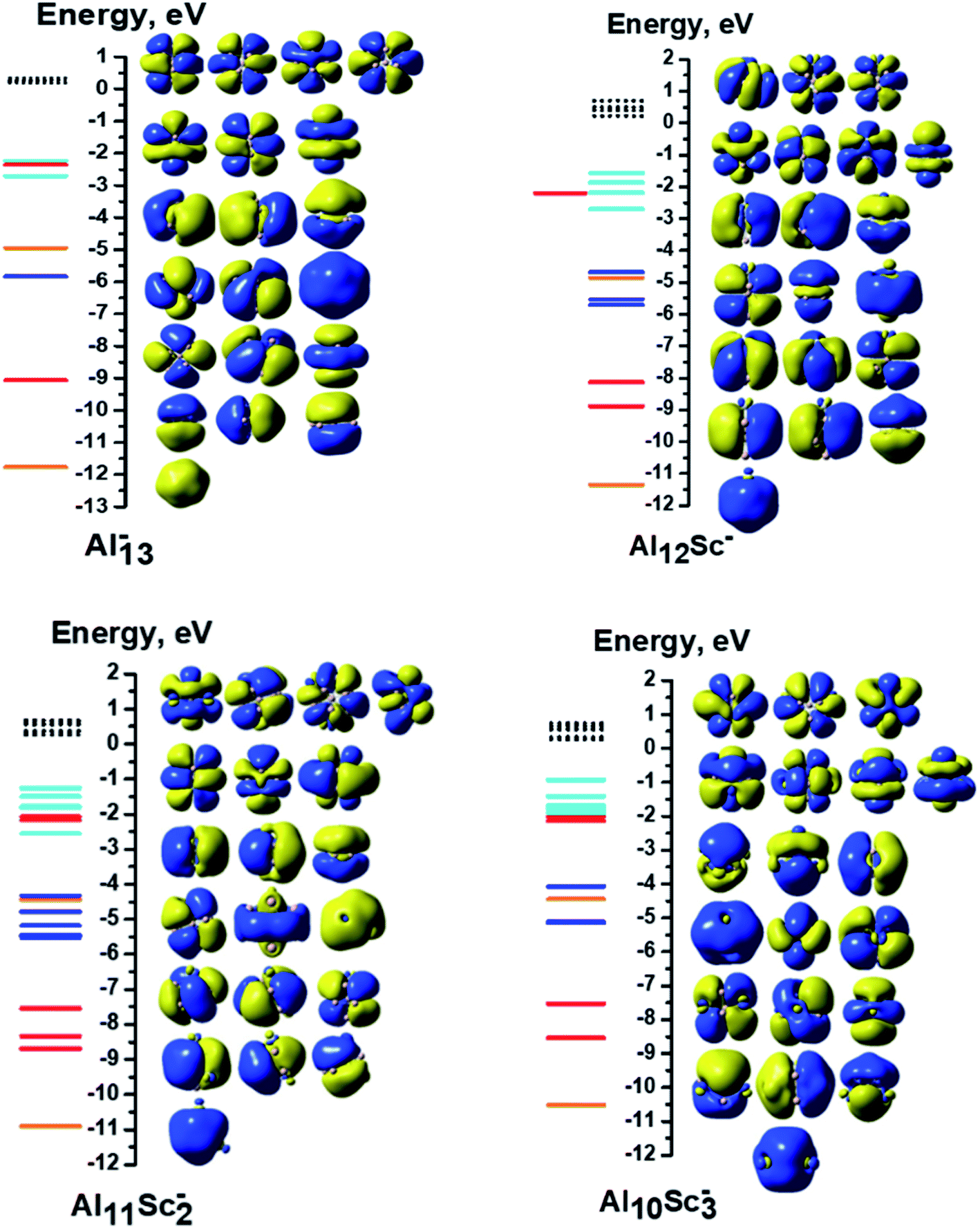 | ||
| Fig. 5 MO interaction diagrams of the anions AlxScy− (x + y = 13 with y = 0–3) with shapes of the delocalized orbitals and the localized 3d Sc atomic orbitals. The orange, red, blue, cyan lines represent orbitals corresponding to the 1S, 1P, 1D and 1F cluster shells, respectively. The green lines represent the localized 3d AO(Sc)s. The filled lines stand for occupied orbitals and the dashed lines denote the unoccupied ones. | ||
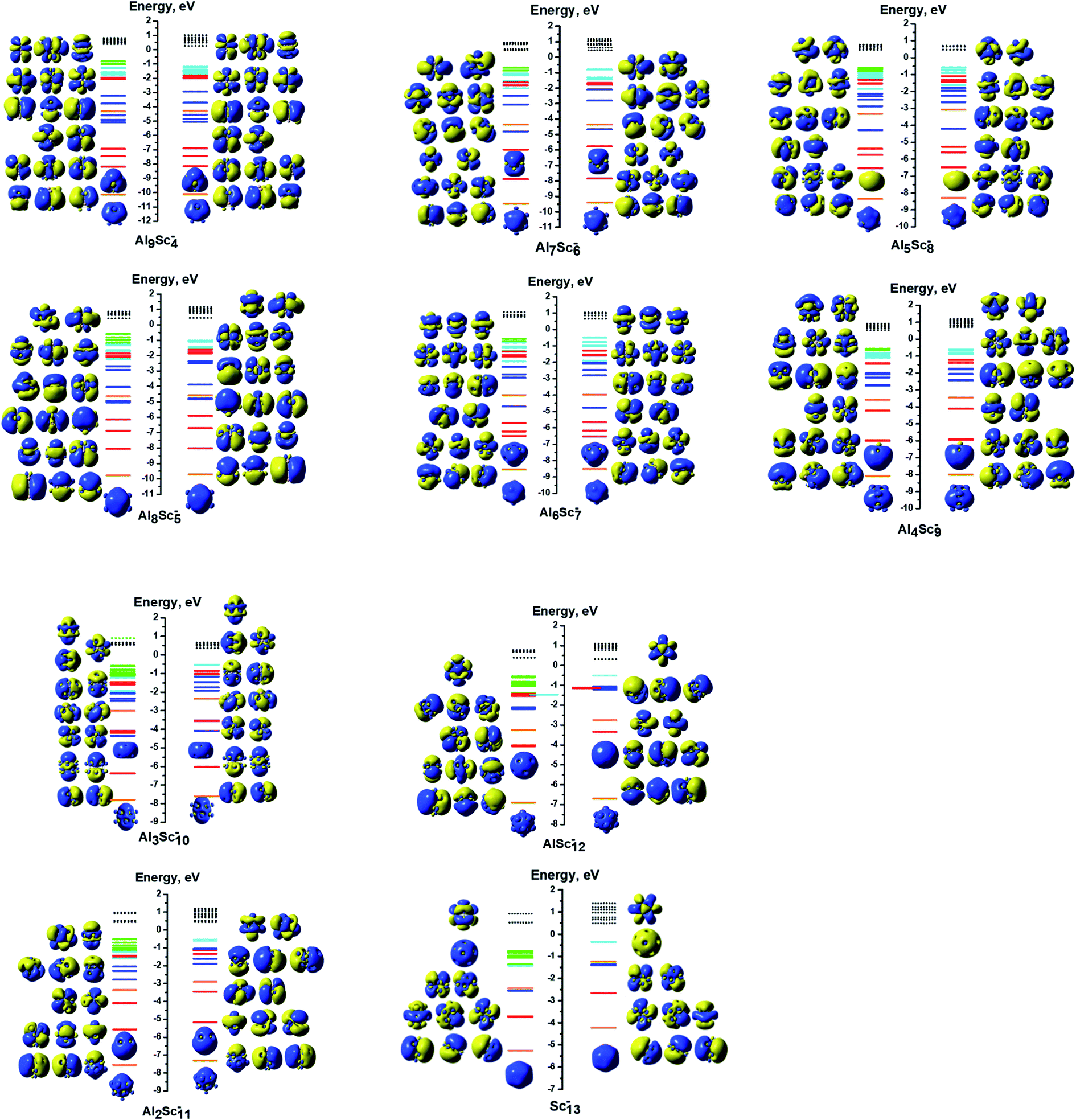 | ||
| Fig. 6 MO interaction diagrams of the anions AlxScy− (x + y = 13 with y = 4–13) with shapes of the delocalized orbitals and the localized 3d Sc atomic orbitals. The orange, red, blue, cyan lines represent orbitals corresponding to the 1S, 1P, 1D and 1F cluster shells, respectively. The green lines represent the localized 3d AO(Sc)s. The filled lines stand for occupied orbitals and the dashed lines denote the unoccupied ones. | ||
| Clusters | Sc | Al | |||||
|---|---|---|---|---|---|---|---|
| 4s | 4p | 3d | 4d | 5d | 3s | 3p | |
| Sc13− | 0.99 | 1.77 | 14.81 | 0.25 | 0.00 | — | — |
| AlSc12− | −0.05 | 0.01 | 11.84 | 0.13 | 0.00 | −0.02 | −0.01 |
| Al2Sc11− | 0.04 | 0.44 | 9.20 | 0.11 | 0.00 | 0.00 | 0.15 |
| Al3Sc10− | −0.04 | 0.31 | 9.23 | 0.19 | 0.00 | 0.00 | 0.34 |
| Al4Sc9− | 0.12 | 0.32 | 3.65 | 0.06 | 0.00 | −0.02 | −0.2 |
| Al5Sc8− | 0.03 | 0.25 | 3.55 | 0.06 | 0.00 | 0.02 | 0.05 |
| Al6Sc7− | −0.06 | 0.12 | 2.37 | 0.05 | 0.00 | −0.12 | −0.41 |
| Al7Sc6− | −0.14 | 0.10 | 3.97 | 0.10 | 0.00 | −0.10 | 0.03 |
| Al8Sc5− | 0.05 | 0.33 | 2.99 | 0.07 | 0.00 | 0.09 | 0.42 |
| Al9Sc4− | 0.27 | 0.23 | 1.24 | 0.02 | 0.00 | 0.08 | 0.12 |
| No. | Al9Sc4− | Al8Sc5− | Al7Sc6− | Al6Sc7− | Al5Sc8− | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| a The atom locates at center of icosahedral cage. | ||||||||||
| 1 | Al | 0.1 | Al | 0.1 | Al | 0.0 | Ala | 0.0 | Al | 0.1 |
| 2 | Ala | 0.0 | Ala | 0.0 | Al | 0.0 | Al | 0.0 | Al | 0.1 |
| 3 | Al | 0.1 | Al | 0.1 | Ala | 0.0 | Al | 0.0 | Al | 0.0 |
| 4 | Al | 0.1 | Al | 0.1 | Al | 0.0 | Al | −0.1 | Ala | 0.0 |
| 5 | Al | 0.1 | Al | 0.1 | Al | 0.0 | Al | −0.2 | Al | 0.0 |
| 6 | Al | 0.0 | Al | 0.1 | Al | 0.0 | Al | −0.2 | Sc | 0.4 |
| 7 | Al | 0.0 | Al | 0.1 | Al | 0.0 | Sc | 0.2 | Sc | 0.4 |
| 8 | Al | 0.0 | Al | 0.0 | Sc | 0.7 | Sc | 0.5 | Sc | 0.6 |
| 9 | Al | 0.0 | Sc | 0.8 | Sc | 0.7 | Sc | 0.6 | Sc | 0.3 |
| 10 | Sc | 0.8 | Sc | 0.8 | Sc | 0.7 | Sc | 0.5 | Sc | 0.7 |
| 11 | Sc | 0.0 | Sc | 0.7 | Sc | 0.7 | Sc | 0.2 | Sc | 0.6 |
| 12 | Sc | 0.8 | Sc | 0.7 | Sc | 0.7 | Sc | 0.0 | Sc | 0.7 |
| 13 | Sc | 0.0 | Sc | 0.4 | Sc | 0.7 | Sc | 0.6 | Sc | 0.3 |
| Total | 2.0 | Total | 4.0 | Total | 4.0 | Total | 2.0 | Total | 4.0 | |
| No. | Al4Sc9− | Al3Sc10− | Al2Sc11− | AlSc12− | Sc13− | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Al | 0.0 | Al | 0.2 | Ala | 0.0 | Ala | 0.0 | Sc | 1.4 |
| 2 | Ala | 0.0 | Ala | 0.0 | Al | 0.2 | Sc | 1.0 | Sc | 1.5 |
| 3 | Al | 0.0 | Al | 0.2 | Sc | 0.9 | Sc | 1.0 | Sc | 1.4 |
| 4 | Al | 0.0 | Sc | 0.9 | Sc | 0.8 | Sc | 1.0 | Sc | 1.4 |
| 5 | Sc | 0.3 | Sc | 1.1 | Sc | 0.8 | Sc | 1.0 | Sc | 1.5 |
| 6 | Sc | 0.3 | Sc | 0.9 | Sc | 1.0 | Sc | 1.0 | Sc | 1.5 |
| 7 | Sc | 0.1 | Sc | 1.0 | Sc | 1.0 | Sc | 1.0 | Sc | 1.4 |
| 8 | Sc | 0.6 | Sc | 1.1 | Sc | 0.9 | Sc | 1.0 | Sc | 1.5 |
| 9 | Sc | 0.7 | Sc | 0.9 | Sc | 0.9 | Sc | 1.0 | Sca | 0.5 |
| 10 | Sc | 0.6 | Sc | 1.0 | Sc | 0.9 | Sc | 1.0 | Sc | 1.4 |
| 11 | Sc | 0.5 | Sc | 1.0 | Sc | 0.9 | Sc | 1.0 | Sc | 1.4 |
| 12 | Sc | 0.5 | Sc | 0.9 | Sc | 0.9 | Sc | 1.0 | Sc | 1.5 |
| 13 | Sc | 0.8 | Sc | 1.0 | Sc | 0.9 | Sc | 1.0 | Sc | 1.5 |
| Total | 4.0 | Total | 10 | Total | 10 | Total | 12 | Total | 18 | |
Moreover, an observation of the shapes of 18 SOMOs points out that they are neither the shell orbitals (such as 1F, 2D…) of the Sc13− cluster nor the individual AOs of any Sc atom but rather they are the molecular orbitals that belong to the A, E and T irreducible representations of a T point group. Accordingly, the electron configuration of the Sc13− cluster can best be written as [1S21P61D102S21F23dE23dT33dA13dE23dA13dT33dA13dA13dE23dE2]. In this electron configuration, the notations of 3dA, 3dE and 3dT represent the SOMOs that are formed from combinations of 3d atomic orbitals of Sc atoms and belong to the A, E and T irreducible representations of T point group, respectively.
A similar phenomenon occurs in the remaining AlxScy− sizes with a decreasing Al atom number. On average, each dAO(Sc) of AlSc12− contains 0.2 unpaired electron, proportionately 1.0 unpaired electron found on each Sc atom. Thus, those twelve spatial orbitals are no longer individual 3d orbitals of any single Sc atom, but rather their combination creates molecular orbitals that belong to the A, E and T irreducible representations of the T point group. Similar to the previous case, the electron configuration of the Al1Sc12− cluster can be written as [1S21P61D102S22P61F23dE23dA13dE23dA13dE23dE23dE2].
Similarly, the electron configuration based on the symmetry point group and spin multiplicity of the remaining anions can concisely be expressed in the same way as follows:
The Al3Sc10− cluster (C5v, 11-et). Each of the Sc atoms contains one unpaired electron and 30 valence electrons are paired in the shell orbitals of the electron configuration [1S21P61D102S22P61F43dE23dE23dA13dE23dE23dA1].
The Al4Sc9− cluster (C3v, quintet). Its electron configuration can be written as [1S21P61D102S22P61F103dE23dA13dA1].
The Al5Sc8− cluster (C2, quintet). The ordering of its valence electron filling is [1S21P61D102S22P61F103dA13dB13dA13dB1].
The Al6Sc7− cluster (Cs, triplet). The remaining 38 valence electrons are coupled in pairs in the electron shells of [1S21P61D102S22P61F123dA′′13dA′1].
The Al7Sc6− cluster (C3v, quintet). The filling of valence electrons of this cluster is [1S21P61D102S22P61F103dA13dE23dA1].
The Al8Sc5− cluster (Cs, quintet) has an electron configuration of [1S21P61D102S22P61F103dA′′13dA′′13dA′13dA′1].
The Al9Sc4− cluster (C2v, triplet). The 38 paired valence electrons fill the shell orbitals resulting in an electron configuration of [1S21P61D102S22P61F123dA113dA11].
The symmetries, spin multiplicities, and electron configurations of all anionic AlxScy− clusters are also summarized in Table 4. In general, in the open shell systems of AlxScy−, with y = 4–13, the unpaired electrons are mostly distributed on the Sc atoms located at the cage vertexes. Moreover, the spatial orbitals of the SOMOs are neither the shell molecular orbitals nor the individual AOs of any Sc atoms of the AlxScy− clusters, but rather the MOs that belong to the irreducible representations of the corresponding point group, depending on the geometrical symmetry of the cluster considered.
As shown in Table 4, the calculation results show that all the d electrons of Sc atoms take part in cluster bonding in the Al12Sc−, Al11Sc2−, and Al10Sc3− clusters while the AlxScy− clusters contain unpaired d electrons when x is larger than three. In the clusters Al12Sc−, Al11Sc2−, and Al10Sc3− the Sc atoms are far apart from each other, their d electrons rather participate in formation of bonds with the Al atoms. For the AlxScy− (x > 3) clusters, the Sc atoms are located next to each other (see Fig. 1 and 2) and their 3d unpaired electrons are not bonded but having parallel spins that create a magnetism.
In the cluster Al9Sc4−, each of the Sc(10) and Sc(12) atoms, which are next to each other, has 0.8 unpaired electrons and these unpaired electrons mainly belong to their d AOs (see Tables 1 and 2). Therefore, it can be concluded that they do not use their single electron d AOs to form bonds with each other.
In the cluster Al8Sc5−, the Sc(9), Sc(10) atoms are the neighbors of Sc(12) and Sc(11) ones, respectively, at a distance of 2.970 Å, 0.148 Å closer than the distance to the Sc(13) atom. Tables 1 and 2 show that each of the Sc(9), Sc(10), Sc(11) and Sc(12) atoms have ∼0.75 single electrons that mainly belong to their d AOs. Therefore, we can also conclude that these Sc atoms do not use their unpaired d electron to form bonds with each other.
In the Al7Sc6, the six Sc atoms next to each other forming a ring with equal Sc–Sc distance of 3.043 Å. Tables 1 and 2 show that each Sc atom has ∼0.7 unpaired electrons and these unpaired electrons mainly belong to their d AOs.
In the case of Al6Sc7−, Al5Sc8−, we observe the same phenomenon as in previous clusters having four or more Sc atoms. Although the number of Sc atoms increases, there are still enough Al atoms in the alternative positions of the Sc atoms and their magnetism does not increase. A leap in the magnetic property is observed in subsequent clusters from Al3Sc10−, Al2Sc11− to Al1Sc12−, in which the number of Al atoms becomes sufficiently small that all the Sc atoms are located in positions next to each other with similar distance and do not use their d AOs for bonding to each other. Therefore, they exhibit 10, 10 and 12 unpaired electrons, respectively.
A special case involves the Sc13− where no Al atom is present and all Sc atoms do not use their d AOs, and some of their s and p AOs for bonding. As a result, the cluster is exceptionally magnetic, bearing up to 18 unpaired electrons.
3.3. Thermodynamic stability
The inherent thermodynamic stability of the 13-atom clusters AlxScy are now evaluated through the examination of average binding energies (Eb) which can conventionally be defined in eqn (1) and (2):| Eb(AlxScy−) = [(x − 1)E(Al) + E(Al−) + yE(Sc) − E(AlxScy−)]/13 | (1) |
| Eb(AlxScy) = [xE(Al) + yE(Sc) − E(AlxScy)]/13 | (2) |
Particularly for the anionic Sc13−, the Eb can be defined by eqn (3):
| Eb(Sc13−) = [12E(Sc) + E(Sc−) − E(Sc13−)]/13 | (3) |
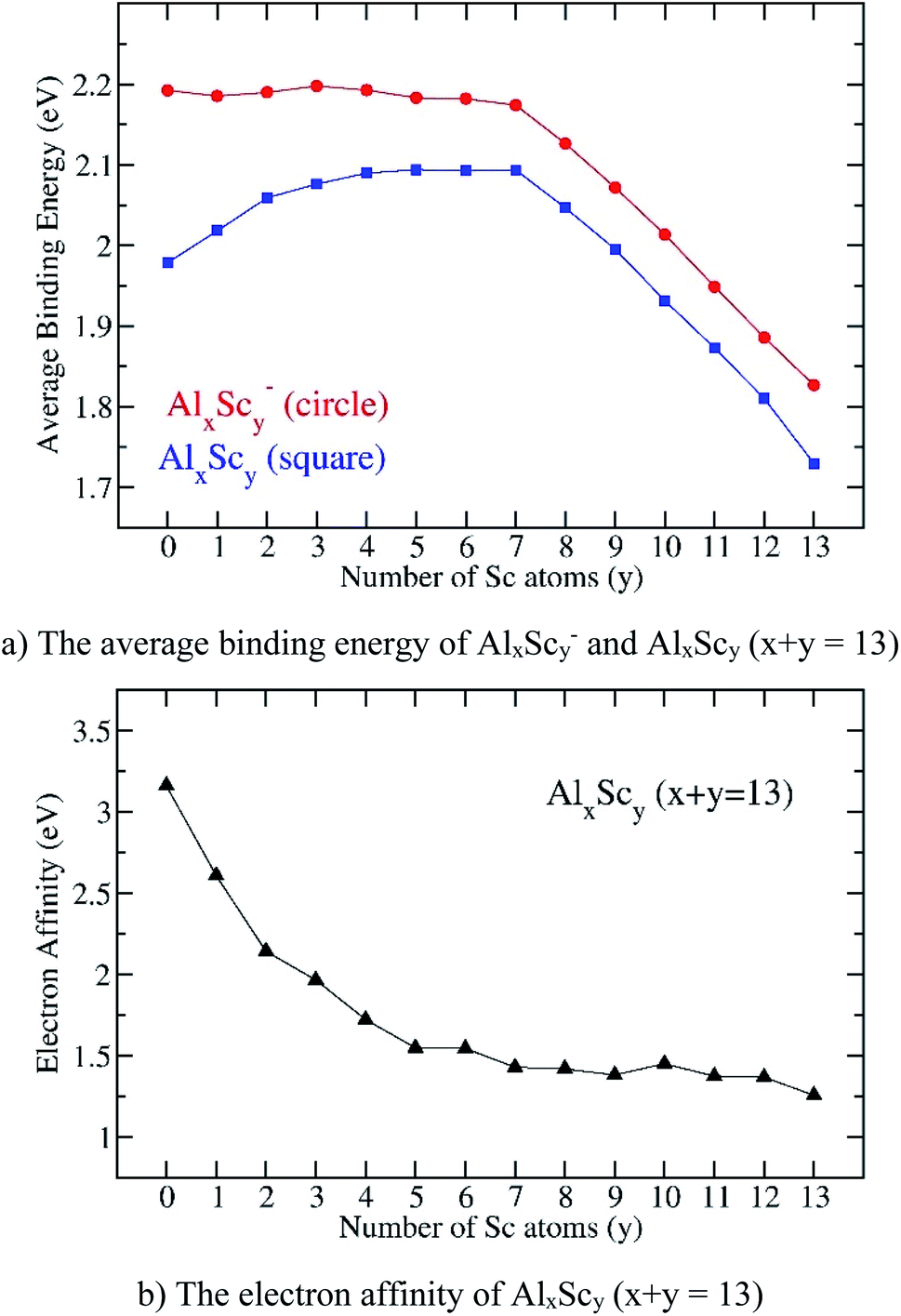 | ||
| Fig. 7 Evolution of (a) the average binding energy and (b) electron affinity of the AlxScy clusters considered in their ground state. Values are obtained from (U)B3LYP/6-311+G(d)+ZPE computations. | ||
There is no significant change of Eb values of the anions AlxScy− whereas those values of the neutral structures increase when y goes from 0 to 4. For y = 4–7, the Eb values of the anions slightly decrease whereas those of neutrals continue to increase, even slightly. However, when the Sc atom number is greater than seven, the Eb of both anionic and neutral states steadily decrease, and attains the lowest value at y = 13, corresponding to the value of the pure scandium cluster Sc13. In order to interpret such a trend, the total Al–Al, Al–Sc and Sc–Sc bond order within each cluster are analyzed using NBO calculations.
The bond order for each of the bonds connecting two atoms, including Al–Al, Al–Sc, and Sc–Sc in the clusters can be calculated as half of the difference between the electron occupancies in the corresponding bonding and anti-bonding orbitals. The total bond order for each of the bonds involved are thus summed up in the total Al–Al, Al–Sc and Sc–Sc bond orders. The values of bond orders, NBO charges of the central atom of the icosahedral cage are listed in the Table 3. In going from Al13− to Al7Sc6−, the Al–Al bond order decreases, whereas the Al–Sc and Sc–Sc bond orders increase, and the total bond order of each cluster remains relatively high. Therefore, the energy needed to break all the bonds in a cluster to form the constituent atoms is high as compared to that in other clusters. It is worth noting again that the Al–Al bond is markedly stronger than those made between Sc and Sc (cf. above). Nevertheless, the Sc–Sc bond order in the Sc2 dimer amounts to 2.3, being much larger as compared to that of the Al–Al dimer.52 From the size Al6Sc7− onward to AlSc12− and finally Sc13−, the Al–Al bond order of AlxScy is going close to zero, and this makes their average binding energies much lower as compared to those the AlxScy clusters having y < 7. Consequently, the average binding energy tends to decrease from Al6Sc7− to Al1Sc12− and to Sc13− and this could attribute to a substantial decrease in the Al–Sc bond order in these sizes.
| Cluster | Summation of all Al–Al bond order | Summation of all Al–Sc bond order | Summation of all Sc–Sc bond order | Total bond order | Charge of center atoma | Charge of the remain cage |
|---|---|---|---|---|---|---|
| a Except for Sc13−, all remaining clusters have Al atom located at the central position of the icosahedral cage. | ||||||
| Al13− | 7.3 | 0.0 | 0.0 | 7.3 | −1.7 | +0.7 |
| Al12Sc1− | 6.2 | 3.5 | 0.0 | 9.7 | −1.1 | +0.1 |
| Al11Sc2− | 3.6 | 7.1 | 0.0 | 10.7 | −0.7 | −0.3 |
| Al10Sc3− | 1.6 | 10.0 | 0.0 | 11.6 | −0.4 | −0.6 |
| Al9Sc4− | 2.5 | 4.6 | 0.8 | 7.8 | −0.1 | −0.9 |
| Al8Sc5− | 2.5 | 4.0 | 1.8 | 8.3 | 0.1 | −1.1 |
| Al7Sc6− | 1.8 | 5.2 | 2.5 | 9.6 | 0.4 | −1.4 |
| Al6Sc7− | 0.0 | 5.6 | 2.5 | 8.1 | 0.6 | −1.6 |
| Al5Sc8− | 0.0 | 3.2 | 4.3 | 7.6 | 0.7 | −1.7 |
| Al4Sc9− | 0.0 | 4.1 | 4.3 | 8.4 | 0.7 | −1.7 |
| Al3Sc10− | 0.1 | 0.2 | 5.6 | 6.0 | 0.8 | −1.8 |
| Al2Sc11− | 0.2 | 0.5 | 6.3 | 7.0 | 0.7 | −1.7 |
| Al1Sc12− | 0.0 | 0.0 | 8.7 | 8.7 | 0.7 | −1.7 |
| Sc13− | 0.0 | 0.0 | 8.9 | 8.9 | −3.7 | +2.7 |
| Cluster | Point group | Spin multiplicity | Order of valence electron filling |
|---|---|---|---|
| Al13− | Ih | 1 | 1S21P61D102S22P61F14 |
| Al12Sc− | C5V | 1 | 1S21P61D102S22P61F14 |
| Al11Sc2− | C2V | 1 | 1S21P61D102S22P61F14 |
| Al10Sc3− | C3V | 1 | 1S21P61D102S22P61F14 |
| Al9Sc4− | C2V | 3 | 1S21P61D102S22P61F123dA113dA11 |
| Al8Sc5− | CS | 5 | 1S21P61D102S22P61F103dA′′13dA′′13dA′13dA′1 |
| Al7Sc6− | C3V | 5 | 1S21P61D102S22P61F103dA13dE23dA1 |
| Al6Sc7− | CS | 3 | 1S21P61D102S22P61F123dA′′13dA′1 |
| Al5Sc8− | C2 | 5 | 1S21P61D102S22P61F103dA13dB13dA13dB1 |
| Al4Sc9− | C3V | 5 | 1S21P61D102S22P61F103dE23dA13dA1 |
| Al3Sc10− | C5V | 11 | 1S21P61D102S22P61F43dE23dE23dA13dE23dE23dA1 |
| Al2Sc11− | C5V | 11 | 1S21P61D102S22P61F43dE23dA13dE23dE23dE23dA1 |
| Al1Sc12− | T | 13 | 1S21P61D102S22P61F23dE23dA13dE23dA13dE23dE23dE2 |
| Sc13− | T | 19 | 1S21P61D102S21F23dE23dT33dA13dE23dA13dT33dA13dA13dE23dE2 |
Fig. 7a also shows the Eb values of all anionic AlxScy− clusters that are obviously higher than those of the neutral counterparts, and this proves that the neutral tends to receive an electron to form the more stable anionic state. This feature can be observed more closely by examining the computed electron affinities shown in Fig. 7b. Starting from the superhalogen Al13 with a very large electron affinity, EA(Al13) = 3.2 eV, the substitution of one to five Al atoms in Al13 by a corresponding number of Sc atoms rapidly reduces this parameter, down to a value of 1.6 eV at Al8Sc5. When the Sc atom number increases from 5 to 13, the electron affinity turns out to slightly decrease and takes the lowest value of 1.3 eV at the pure Sc13. Such a reduction is no doubt due to the low electron affinity of the transition metal atom. Thus, substitution of Al atoms in Al13 by Sc atoms makes the binary clusters losing their superhalogen characteristic.
4. Concluding remarks
In the present theoretical study, the geometrical and electronic structures of the 13-atom clusters AlxScy, with x + y = 13, were investigated using DFT calculations. Geometry optimizations remarkably pointed out that all the most stable isomers of AlxScy, in both anionic and neutral states, retain the icosahedral shape in which the Al atom is favored to occupy the central location, irrespective of the Al atom number. The Sc atoms are consistently located at the vertexes of an icosahedral cage. The icosahedral shape is thus retained, even with some slight geometric distortions. The perfect icosahedral shape is kept only for the Al13, Sc13 and AlSc12 sizes.The electron configurations of the clusters considered, in their ground state, have been established and rationalized along with the spin multiplicities. NBO analyses revealed that the unpaired electrons are mostly distributed on the Sc atoms and the SOMOs are the molecular orbitals belonging to the irreducible representations of the symmetry point group of the corresponding cluster. Thermodynamic stabilities were also examined through the average binding energy per atom in each size. The stable geometrical structure, the unpaired electron and thereby multiplicity, and the average binding energy follow some clear trends as follows:
(i) The scandium atoms prefer to be located on exohedral sites of icosahedron nearby the aluminum atoms in order to maximize the number of stronger Al–Al and Sc–Al bonds at the expense of the weaker Sc–Sc bonds;
(ii) The AlxScy clusters are stable at low multiplicity when the Sc atom number goes from 0 to 3, whereas a high spin state predominates as the Sc atom number increases from 4 to 13, and
(iii) The cluster stability insignificantly changes from the superhalogen Al13 to the Al6Sc7 and then regularly decreases from the size Al6Sc7 to the pure Sc13. Moreover, substitution of Al atoms in Al13 by Sc atoms results in a loss of the superhalogen characteristics, in which the electron affinities of the binary AlxScy superatoms are reduced with respect to that of the pure Al13 cluster.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 103.01-2019.372.References
- P. Jena and Q. Sun, Chem. Rev., 2018, 118, 5755–5870 CrossRef CAS PubMed.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS PubMed.
- J. Zhao, Q. Du, S. Zhou and V. Kumar, Chem. Rev., 2020, 120, 9021–9163 CrossRef CAS PubMed.
- L. Wang, J. Zhao, Z. Zhou, S. B. Zhang and Z. Chen, J. Comput. Chem., 2009, 30, 2509–2514 CrossRef CAS PubMed.
- B. K. Rao and P. Jena, J. Chem. Phys., 1999, 111, 1890–1904 CrossRef CAS.
- R. Fournier, J. Chem. Theory Comput., 2007, 3, 921–929 CrossRef CAS PubMed.
- L. Cândido, J. N. T. Rabelo, J. L. F. Da Silva and G.-Q. Hai, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 245404 CrossRef.
- X. Li, H. Wu, X.-B. Wang and L.-S. Wang, Phys. Rev. Lett., 1998, 81, 1909–1912 CrossRef CAS.
- D. E. Bergeron, A. W. Castleman, T. Morisato and S. N. Khanna, Science, 2004, 304, 84 CrossRef CAS PubMed.
- R. Pal, L.-F. Cui, S. Bulusu, H.-J. Zhai, L.-S. Wang and X. C. Zeng, J. Chem. Phys., 2008, 128, 024305 CrossRef CAS PubMed.
- E. Jimenez-Izal, D. Moreno, J. M. Mercero, J. M. Matxain, M. Audiffred, G. Merino and J. M. Ugalde, J. Phys. Chem. A, 2014, 118, 4309–4314 CrossRef CAS PubMed.
- W.-M. Sun, D. Wu, X.-H. Li, Y. Li, J.-H. Chen, C.-Y. Li, J.-Y. Liu and Z.-R. Li, J. Phys. Chem. C, 2016, 120, 2464–2471 CrossRef CAS.
- X. Xing, J. Wang, X. Kuang, X. Xia, C. Lu and G. Maroulis, Phys. Chem. Chem. Phys., 2016, 18, 26177–26183 RSC.
- S. Jin, B. Chen, X. Kuang, C. Lu, W. Sun, X. Xia and G. L. Gutsev, J. Phys. Chem. C, 2019, 123, 6276–6283 CrossRef CAS.
- J. J. Castro, J. R. Soto and B. Molina, AIP Conf. Proc., 2012, 1420, 145–150 CrossRef.
- J. C. Smith, A. C. Reber, S. N. Khanna and A. W. Castleman, J. Phys. Chem. A, 2014, 118, 8485–8492 CrossRef CAS PubMed.
- G. Chen and Y. Kawazoe, J. Chem. Phys., 2007, 126, 014703 CrossRef CAS PubMed.
- N. M. Tam, L. V. Duong, N. T. Cuong and M. T. Nguyen, RSC Adv., 2019, 9, 27208–27223 RSC.
- C. Wu, P. Lu, Z. Yu, L. Ding, Y. Liu and L. Han, J. Comput. Theor. Nanosci., 2013, 10, 1055–1060 CrossRef CAS.
- M. Wang, X. Huang, Z. Du and Y. Li, Chem. Phys. Lett., 2009, 480, 258–264 CrossRef CAS.
- Y. Hua, Y. Liu, G. Jiang, J. Du and J. Chen, J. Phys. Chem. A, 2013, 117, 2590–2597 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 115405 CrossRef.
- X. Xia, Z.-G. Zhang, H.-G. Xu, X. Xu, X. Kuang, C. Lu and W. Zheng, J. Phys. Chem. C, 2019, 123, 1931–1938 CrossRef CAS.
- J. U. Reveles, T. Baruah and R. R. Zope, J. Phys. Chem. C, 2015, 119, 5129–5137 CrossRef CAS.
- T. B. Tai and M. T. Nguyen, Chem. Phys. Lett., 2010, 492, 290–296 CrossRef CAS.
- J. M. Goicoechea and J. E. McGrady, Dalton Trans., 2015, 44, 6755–6766 RSC.
- V. Kumar and Y. Kawazoe, Appl. Phys. Lett., 2002, 80, 859–861 CrossRef CAS.
- S. Neukermans, E. Janssens, Z. F. Chen, R. E. Silverans, P. v. R. Schleyer and P. Lievens, Phys. Rev. Lett., 2004, 92, 163401 CrossRef CAS PubMed.
- L.-F. Cui, X. Huang, L.-M. Wang, J. Li and L.-S. Wang, Angew. Chem., Int. Ed., 2007, 46, 742–745 CrossRef CAS PubMed.
- Y.-J. Bai, H.-Y. Cheng, H.-Q. Sun, N. Xu and K.-M. Deng, Phys. B, 2011, 406, 3781–3787 CrossRef CAS.
- T. B. Tai, N. M. Tam and M. T. Nguyen, Chem. Phys., 2011, 388, 1–8 CrossRef CAS.
- G. L. Gutsev, C. W. Weatherford, K. G. Belay, B. R. Ramachandran and P. Jena, J. Chem. Phys., 2013, 138, 164303 CrossRef CAS PubMed.
- M. J. Piotrowski, P. Piquini and J. L. F. Da Silva, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 155446 CrossRef.
- J. T. A. Gilmour and N. Gaston, Phys. Chem. Chem. Phys., 2020, 22, 4051–4058 RSC.
- S. Datta, M. Kabir, T. Saha-Dasgupta and A. Mookerjee, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 085418 CrossRef.
- M. Zhang, X.-Y. Gu, W.-L. Zhang, L.-N. Zhao, L.-M. He and Y.-H. Luo, Phys. B, 2010, 405, 642–648 CrossRef CAS.
- H. T. Pham, N. T. Cuong, N. M. Tam and N. T. Tung, J. Phys. Chem. A, 2016, 120, 7335–7343 CrossRef CAS PubMed.
- R. Xiong, D. Die, L. Xiao, Y.-G. Xu and X.-Y. Shen, Nanoscale Res. Lett., 2017, 12, 625 CrossRef PubMed.
- N. T. Mai, N. T. Lan, N. T. Cuong, N. M. Tam, S. T. Ngo, T. T. Phung, N. V. Dang and N. T. Tung, ACS Omega, 2021, 6(31), 20341–20350 CrossRef CAS PubMed.
- J. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 155422 CrossRef.
- F.-Y. Tian, Q. Jing and Y.-X. Wang, Phys. Rev. A: At., Mol., Opt. Phys., 2008, 77, 013202 CrossRef.
- M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, et al., Gaussian 09 Revision: D.01, 2009 Search PubMed.
- H. T. Pham, L. V. Duong, B. Q. Pham and M. T. Nguyen, Chem. Phys. Lett., 2013, 577, 32–37 CrossRef CAS.
- H. T. Pham, N. T. Cuong, N. M. Tam, V. D. Lam and N. T. Tung, Chem. Phys. Lett., 2016, 643, 77–83 CrossRef CAS.
- N. M. Tam, L. V. Duong, H. T. Pham, M. T. Nguyen and M. P. Pham-Ho, Phys. Chem. Chem. Phys., 2019, 21, 8365–8375 RSC.
- H. T. Pham, C.-T. D. Phan, M. T. Nguyen and N. M. Tam, RSC Adv., 2020, 10, 19781–19789 RSC.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- P. J. Hay, J. Chem. Phys., 1977, 66, 4377–4384 CrossRef CAS.
- S. M. Lang, P. Claes, S. Neukermans and E. Janssens, J. Am. Soc. Mass Spectrom., 2011, 22, 1508 CrossRef CAS PubMed.
- M. Scheer, R. C. Bilodeau, J. Thøgersen and H. K. Haugen, Phys. Rev. A: At., Mol., Opt. Phys., 1998, 57, R1493–R1496 CrossRef.
- C. S. Feigerle, Z. Herman and W. C. Lineberger, J. Electron Spectrosc. Relat. Phenom., 1981, 23, 441–450 CrossRef CAS.
- T. Chen and T. A. Manz, RSC Adv., 2019, 9, 17072–17092 RSC.
| This journal is © The Royal Society of Chemistry 2021 |