
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in the transition-metal-free synthesis of quinoxalines
Biplob Borah
 and
L. Raju Chowhan
*
and
L. Raju Chowhan
*
School of Applied Material Sciences, Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar-382030, India. E-mail: rchowhan@cug.ac.in
First published on 19th November 2021
Abstract
Quinoxalines, also known as benzo[a]pyrazines, constitute an important class of nitrogen-containing heterocyclic compounds as a result of their widespread prevalence in natural products, biologically active synthetic drug candidates, and optoelectronic materials. Owing to their importance and chemists' ever-increasing imagination of new transformations of these products, tremendous efforts have been dedicated to finding more efficient approaches toward the synthesis of quinoxaline rings. The last decades have witnessed a marvellous outburst in modifying organic synthetic methods to create them sustainable for the betterment of our environment. The exploitation of transition-metal-free catalysis in organic synthesis leads to a new frontier to access biologically active heterocycles and provides an alternative method from the perspective of green and sustainable chemistry. Despite notable developments achieved in transition-metal catalyzed synthesis, the high cost involved in the preparation of the catalyst, toxicity, and difficulty in removing it from the final products constitute disadvantageous effects on the atom economy and eco-friendly nature of the transformation. In this review article, we have summarized the recent progress achieved in the synthesis of quinoxalines under transition-metal-free conditions and cover the reports from 2015 to date. This aspect is presented alongside the mechanistic rationalization and limitations of the reaction methodologies. The scopes of future developments are also highlighted.
 Biplob Borah | Biplob Borah was born in 1995 in Garukhunda, a small village in the Nagaon district of Assam, India. He had graduated BSc in Chemistry from Nowgong College (Gauhati University), Assam in 2017 and received his Master's degree in Industrial Chemistry from the Central University of Gujarat, India in 2019. Currently, he was joined as a PhD scholar in the School of Applied Material Sciences at the Central University of Gujarat under the guidance of Dr L. Raju Chowhan. His research interest includes organocatalysis, multicomponent reactions (MCRs), green chemistry, synthesis of medicinally privileged heterocycles, and hybrid molecules in an aqueous medium. |
 L. Raju Chowhan | Dr L. Raju Chowhan obtained his BSc degree from Osmania University, Hyderabad in 2005, and Master's degree in Chemical Science from Hyderabad Central University, Hyderabad, in 2007, and received his PhD from CSIR-Indian Institute of Chemical Technology, in association with Hyderabad Central University. He joined as an Assistant Professor in the Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar on 5th September 2012. His research interests include stereoselective synthesis of natural products, development of novel methodologies for asymmetric synthesis, multicomponent reactions, and 1,3-dipolar cycloaddition reactions. |
1. Introduction
Nitrogen-containing heterocycles, of natural and synthetic origin, represent a huge family of indispensable structural motifs and have attracted enormous attention in synthetic and medicinal chemistry as well as in material sciences owing to their wide-ranging chemical landscape and prolific bioactivity profiles.1 Among various nitrogen heterocycles, quinoxalines and their derivatives have been considered as versatile and privileged structural scaffolds in synthetic and medicinal chemistry.2 These well-established nitrogen-containing bicyclic heterocycles mainly consist of a benzene ring fused with a pyrazine ring in their structure. Quinoxalines structural motifs are widely distributed in the architecture of numerous natural products,3 pharmaceutical agents,4 agrochemicals,5 pharmacologically active synthetic compounds.6 They have been found to possess a diverse range of biological activities including antidiabetic,7 antimicrobial,8 anticancer,9 antituberculosis,10 antibacterial,11 anti-inflammatories,12 anti-HIV,13 antiviral,14 PI3Kγ inhibitors,15 cyclooxygenase 2 (COX-2) inhibitors,16 antimalarial,17 anti-dengue,18 antioxidant,19 anticonvulsant,20 antidepressant,21 antitumors,22 antiplasmodial,23 antileishmanial activity,24 and antiprotozoal activity.25Several natural products that featured quinoxaline rings as an integral part are depicted in Fig. 1A. The natural product echinomycin (A) isolated from Streptomyces echinatus sp. was established for the first time in 1957.26 They have been found to possess anti-bacterial activity against Gram-negative organisms and have good anti-tumour activity. Another natural product triostin A (B) was isolated from Streptomycesaureus S-2-210 and has antitumor and antibacterial activity.27 Lumiphenazines (C) possesses anticancer activity was isolated from Streptomyces sp. IFM 11204.28 The natural product quinoxapeptin B (D) isolated from Betula papyrifera has been known for its anti-HIV activity.29 Quinoxalines are the building blocks of vitamin B2. Two marine alkaloids baimantuoluoamide B (F) and hunanamycin A (G) were isolated from alkaloidal fraction Datura metel L and Bacillus hunanensis.30
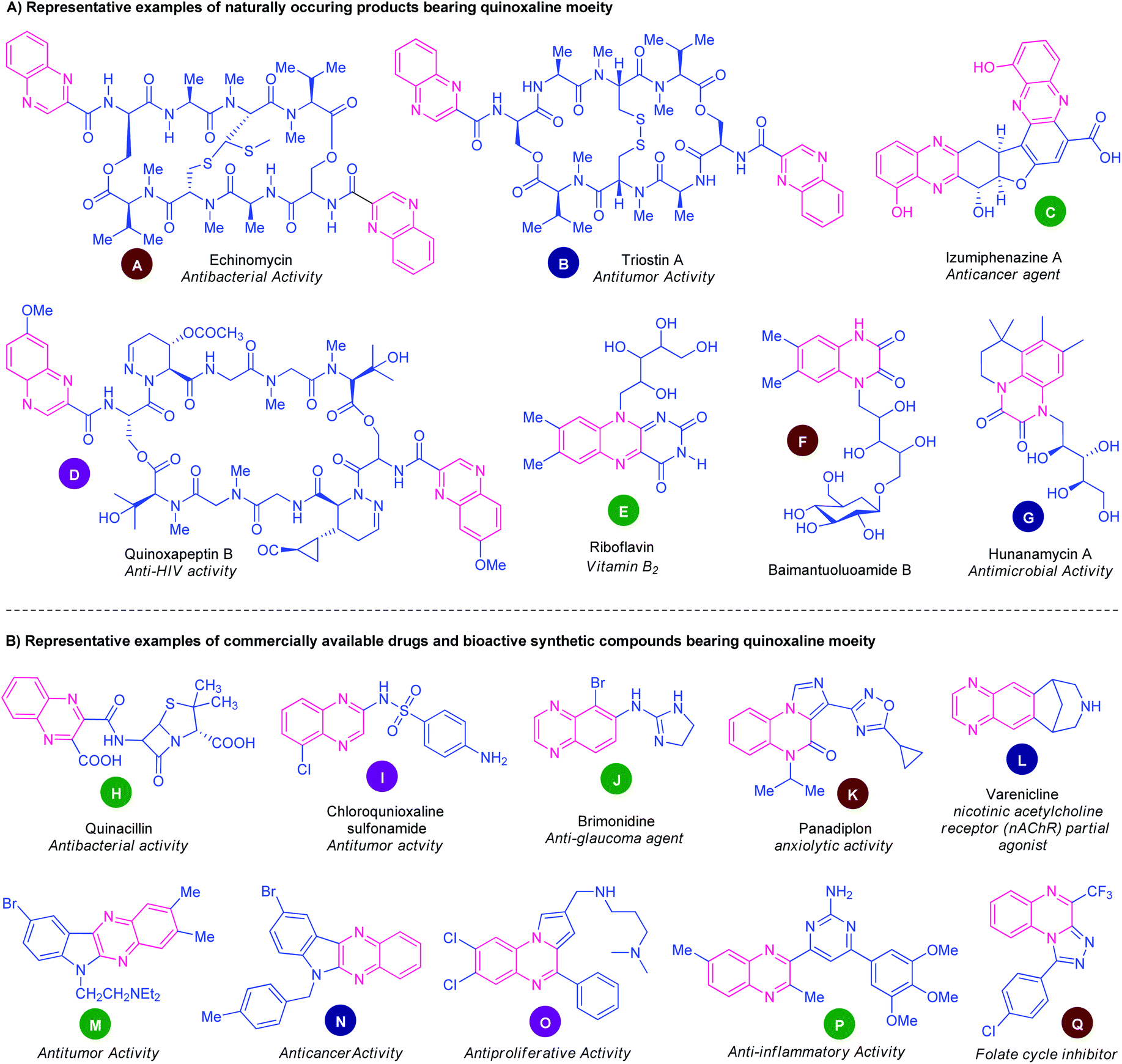 | ||
| Fig. 1 Representative examples of natural products, drugs, and bioactive synthetic compounds containing quinoxaline ring. | ||
Besides these natural products, non-natural quinoxalines displayed prominent biological activities. In this pursuit, several quinoxaline-containing drugs and synthetic compounds of pharmacological profiles have been discovered (Fig. 1B). Quinacillin (H) is a quinoxaline-based semisynthetic penicillinase-resistant penicillin and has antibacterial activity.31 Chloroquinoxaline sulfonamide (I) is a topoisomerase II α/β-poison and is active against murine and human solid tumours.32 Brimonidine (J) is a commercially available α2 adrenergic agonist used to treat open-angle glaucoma, ocular hypertension, and rosacea.33 Panadiplon (K) acts as a high-affinity GABAA receptor partial agonist with anxiolytic activity.34 Varenicline (L) is a quinoxaline-based α4β2 nicotinic receptor partial agonist drug available in the market and is used for smoking cessation.35 Still, there are lots of quinoxaline derivatives possessing different biological activities which are under the stage of preclinical or clinical development. Several synthetic quinoxaline-based compounds M, N, O, P, and Q exhibited potent pharmacological activities such as antitumor,36 anticancer,37 antiproliferative,38 anti-inflammatory,39 and folate cycle inhibitors40 respectively.
Despite these wide-ranging biological activities, a diverse molecular structure that featured the quinoxaline framework has been established as potential fragments in diverse areas of materials science. The applications included organic photovoltaic devices,41 organic semiconductors,42 electroluminescent materials,43 fluorescent probes,44 organic light-emitting diodes (OLEDs),45 organic solar cells,46 sensitizers for dye-sensitized solar cells,47 polymer light-emitting diodes (PLEDs),48 and fuel cells.49
Considering the wide-ranging application and tremendous chemical landscape, a great deal of attention has been subjected toward the synthesis of quinoxalines in the last decades. However, most of the synthetic strategy utilizes hazardous reagents, toxic volatile solvents, harsh reaction condition, and high energy inputs that adversely affects the overall chemical process as well as the environment from being making eco-friendly and sustainable nature.
Despite, transition metal catalyst has been proven as a powerful catalyst and successfully employed in many organic synthesis for the diverse construction of valuable structural frameworks,50 they possess many limitations and drawbacks in terms of synthetic efficiency and green chemistry point of view. The high cost involved in the preparation of transition metal catalyst alongside the requirement of non-commercial supporting ligands marks major limitations of transition metal catalysis. In addition, they are very sensitive to air and moisture. The removal of transition metal catalysts from a reaction mixture which is particularly crucial to the pharmaceutical industry often becomes a formidable challenge and is very expensive. The requirements of co-catalysts and additives in some cases for enhancing the efficacy and reactivity of the transition metal-catalyzed transformation once again point towards the failure of green and more sustainable synthesis. Consequently, the occurrence of transition metal catalyst even at the lowest loading corresponds to disadvantageous effects on the eco-friendly and environmentally friendly nature of the chemical process.51 Intriguingly, the development of a chemical process that employed alternative materials for the synthetic purpose which are not only environmentally friendly but also readily accessible at bulk quantities in anywhere at a very low price, for the construction of structural complexity with high atom- and step-economy by avoiding the requirements of transition metal catalysts, co-catalyst, any additives are highly desired.
Recently, transition-metal-free reactions have been demonstrated as an efficient and environmentally benign strategy toward the synthesis of heterocyclic compounds and have been emerged as a key fascinating area in synthetic organic chemistry.52 They have several advantages in comparison to organic transformations involving transition metal catalysts. Consequently, tremendous growth has been witnessed in the last decade on the transition-metal-free synthesis of quinoxaline and its derivatives. However, to date, no review articles have summarized the development achieved in the synthesis of quinoxalines under transition-metal-free conditions.
Several review articles covered the synthetic strategy of quinoxalines starting from the classical one to non-conventional green ones and compiled them in the last few years.53 Recently, Maikhuri et al., described the recent metal-catalyzed synthesis of quinoxalines.54 Also, Yashwantrao and Saha reviewed the synthesis and reactivity of quinoxaline derivatives.55 The present review article focuses on the recent progress achieved in the synthesis of quinoxalines and their fused structure under transition-metal-free conditions and covers the literature from 2015 to date. Besides, highlighting the so far advances realized in this fascinating area, we also point out the limitations and drawbacks of the strategy, and their possible scope of future developments has also been discussed. On behalf of a clear and understanding overview, the article is organized based on the different types of substrates used rather than compiled in chronological order.
2. Classical methods of quinoxaline synthesis
Several synthetic strategies for the preparation of quinoxaline and its derivatives have existed since the late 1800's. The quinoxaline moiety is generally synthesized by employing classical methods including Körner56 and Hinsberg57 synthesis. Both of these reactions were independently reported in 1884, which mainly involves the condensation of o-phenylenediamine with synthons possessing reactive 1,2-dicarbonyl core (e.g., 1,2-diketones, 1,2-ketoesters, or oxalic acid derivatives).58 A constructive modification of these synthetic methods introduces 1,2-dicarbonyl compound surrogates (e.g., α-haloketones, α-hydroxyketones, epoxides, or alkynes)58 (Fig. 2).Though many of these methods are very attractive in terms of product yield as well as broad functionality, they often utilize various acids, metals, co-catalyst, or reagents that are not environmentally friendly, and produces a large amount of waste as well as hazardous by-products which is difficult to dispose of properly and remove from the reaction mixture and leads to the contamination of the products and the reaction required unusually longer reaction times to complete. Consequently, tremendous growths have been witnessed in the last few years for the metal-free synthesis of quinoxalines, and are compiled in this review.
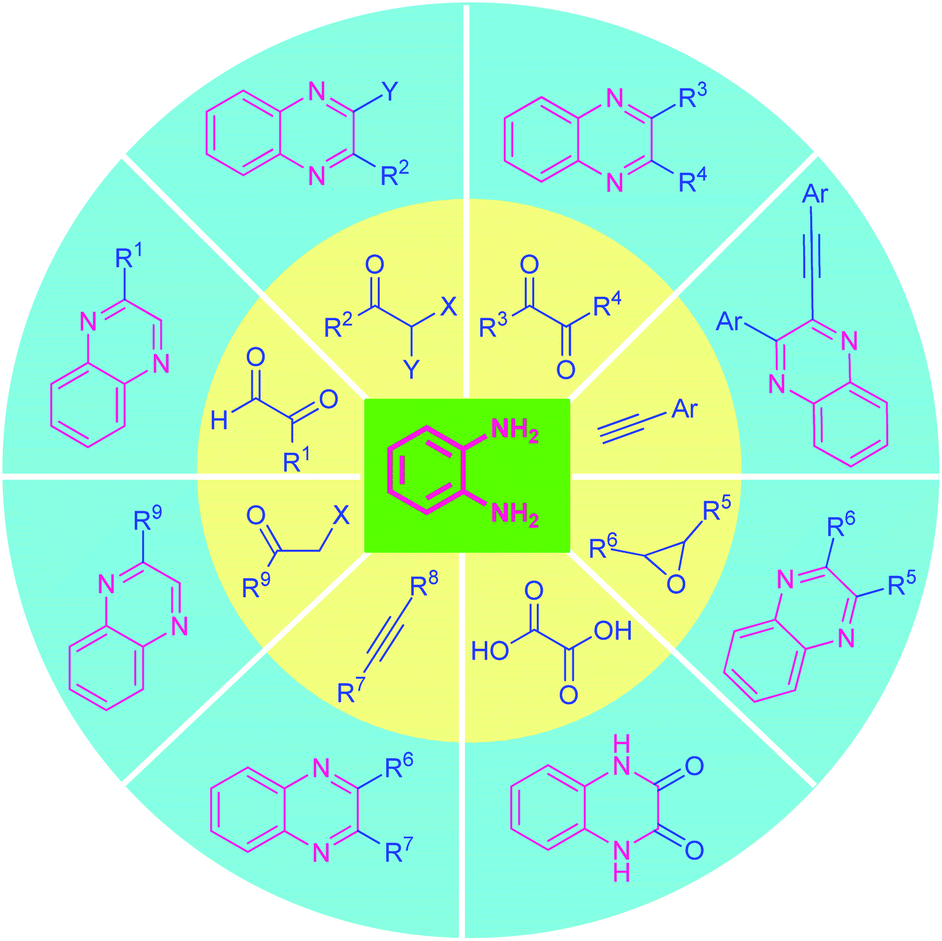 | ||
| Fig. 2 Various classical routes for the synthesis of quinoxalines derivatives. | ||
3. Synthesis of simple quinoxalines based on metal-free two-component reactions
Synthesis of quinoxalines based on a metal-free approach can be accomplished via different reaction types and pathways. Most of them proceed through the modification of classical strategy and newly developed strategy.3.1 From 1,2-dicarbonyl compounds
After the renaissance of organocatalyst, the field of organic synthesis reached an excellent level from the perspective of green and sustainable chemistry. Inspired by the development achieved in the organocatalysis, Fathi and Sardarian demonstrated an efficient organocatalytic approach for the synthesis of various quinoxalines 3 from the reaction of 1,2-diamines 1 and 1,2-carbonyl compounds 2 (Scheme 1).59 With the help of 5 mol% of nitrilotris(methylenephosphonic acid) C-1 as the organocatalyst, the corresponding quinoxaline products have been synthesized in 80–97% yields within a very short reaction time. The reaction condition tolerates a wide variety of substituents on the 1,2-diamine ring as well as on the 1,2-dicarbonyl ring. The efficacy of the protocol was established by demonstrating the recyclability and reusability of the organocatalyst up to several consecutive cycles with a slight change in the product yield.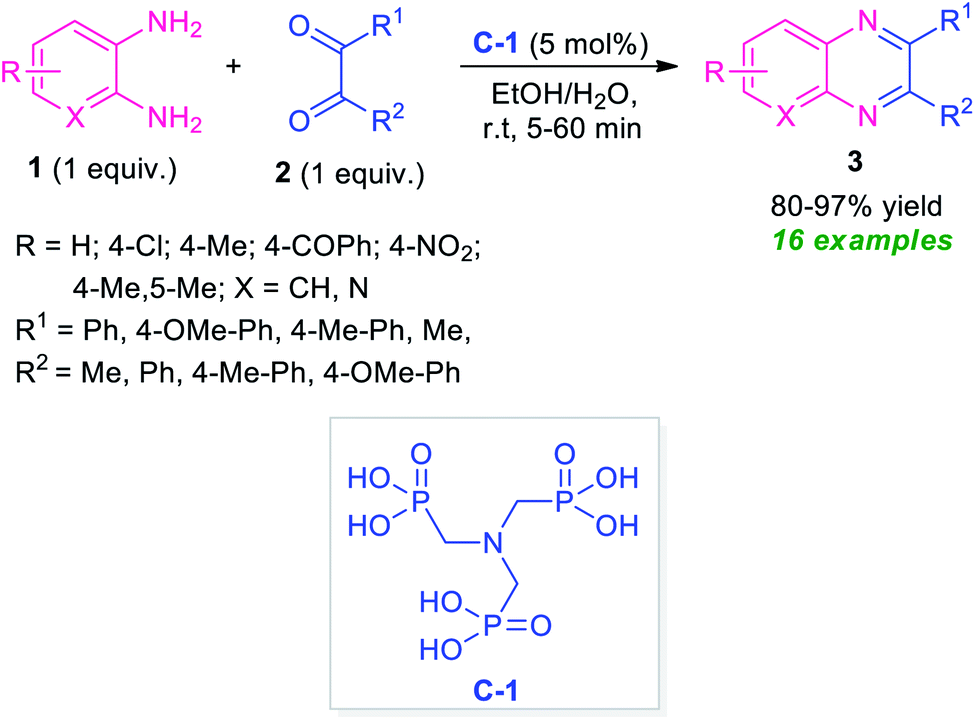 | ||
| Scheme 1 Efficient organocatalytic approach towards the rapid access of quinoxaline derivatives 3. | ||
At the same time, Basu and co-workers disclosed a metal-free one-pot synthesis of quinoxalines 7 from 2-nitroaniline by using graphene oxide (GO) or reduced graphene oxide (rGO) as the carbon catalyst (Scheme 2).60 The reaction proceeded through the initial graphene oxide catalyzed reduction of 2-nitroaniline 4 with hydrazine hydrate and subsequent one-pot tandem reaction with 1,2-dicarbonyl compounds or α-hydroxy ketones 6. Twenty-seven derivatives were synthesized in moderate to excellent yield. The catalyst was easily recovered and reused up to four runs without affecting the product yield.
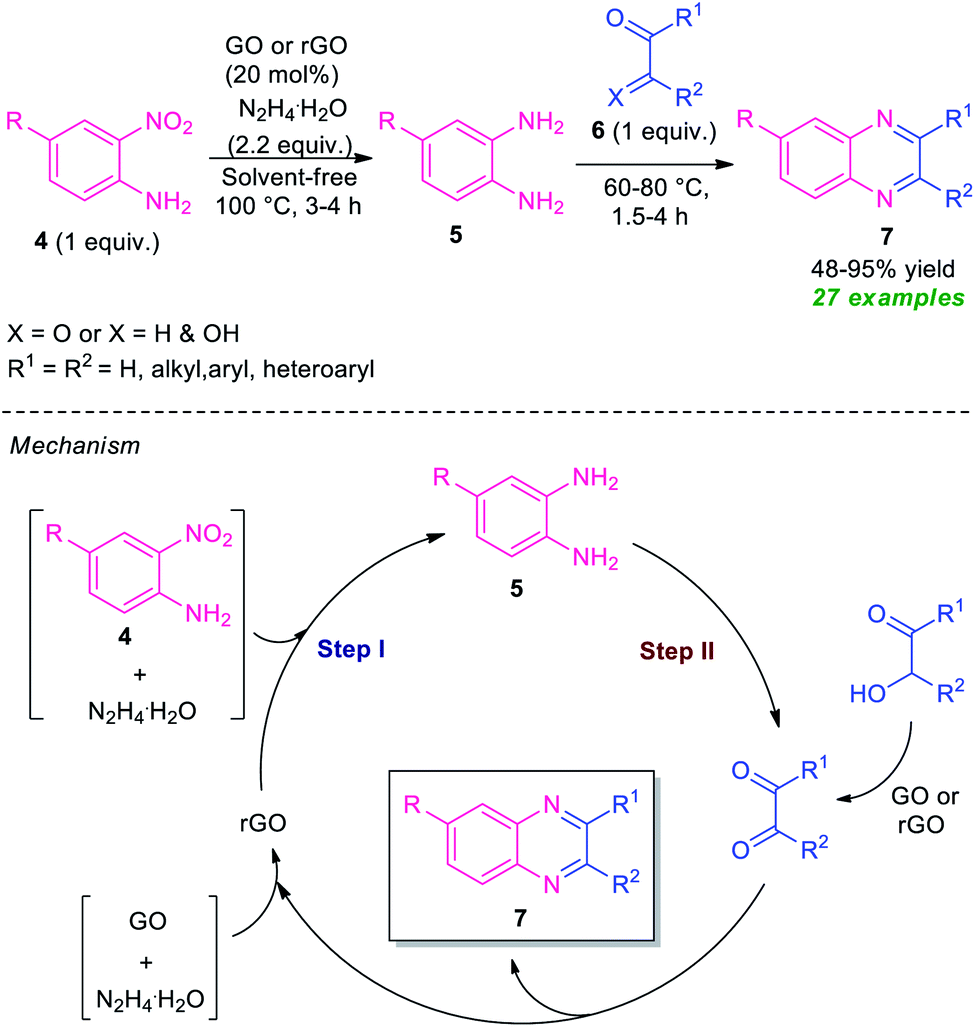 | ||
| Scheme 2 One-pot reduction and subsequent tandem condensation to access diverse quinoxaline derivatives 7. | ||
A mechanism was proposed by the author to explain this transformation which was depicted in Scheme 2. The first step of the reaction involves the reduction of 2-nitroaniline 4 via a four-electron pathway which utilized two-electron reducing agent hydrazine to afford the 1,2-diamine 5 where GO acts as an adsorbent as well as a collector of hydrazines in its surface. In the second step, the catalyst GO or rGO effectively facilitates the condensation of 1,2-dicarbonyls as well as oxidation of α-hydroxyketones to corresponding 1,2-dicarbonyls for subsequent condensation to final products 7.
Treatment of various o-phenylenediamine 8 and 1,2-dicarbonyl compounds 2 was found to proceed under the influence of ammonium bifluoride (NH4HF2) as the catalyst and aqueous ethanol as the solvent system to efficiently provide the corresponding quinoxaline derivatives 9 in 90–98% yields (Scheme 3).61 A variety of alkyl, aryl- and heteroaryl-substituted 1,2-dicarbonyl compounds as well as substituted o-phenylenediamine participate in the reaction smoothly under this mild reaction condition. With unsymmetrical dicarbonyl compounds and o-phenylenediamine, the products were formed regioselectively in excellent yield. The mild reaction condition, simple work-up procedure, low catalyst loading, utilization of easily available starting material, recyclability for further reaction cycles with negligible loss in catalytic property, are some of the key features of this approach.
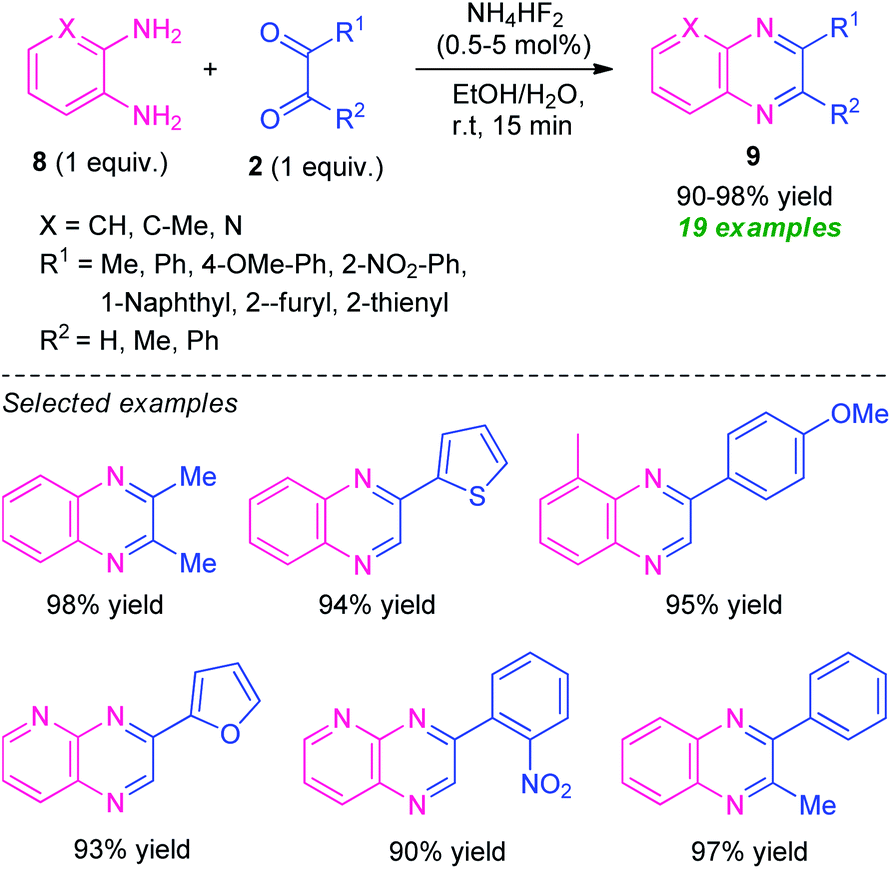 | ||
| Scheme 3 Ammonium bifluoride catalyzed synthesis of various quinoxalines 9. | ||
Recently, the utilization of ionic liquid as a catalyst as well as a solvent system in organic synthesis has attracted much more attention as they provide green alternatives to metal catalysts and hazardous organic solvents.62 In this regard, Moghaddam and Valizadeh designed and prepared ionic liquid 1-methyl-3-(3-trimethoxysilylpropyl) imidazolium hydrogen sulfate functionalized cellulose C-2 as a heterogeneous catalyst, and the catalytic activity was examined in the reaction of diverse o-phenylenediamine 10 with 1,2-dicarbonyl compound 11 (Scheme 4).63 The prepared catalyst C-2 was found to be very effective in catalyzing this condensation reaction to afford the desired quinoxaline products 12 in 78–99% yields. This reaction required only 300 mg of the catalyst and by using water as the green solvent, a total of 13 compounds were synthesized.
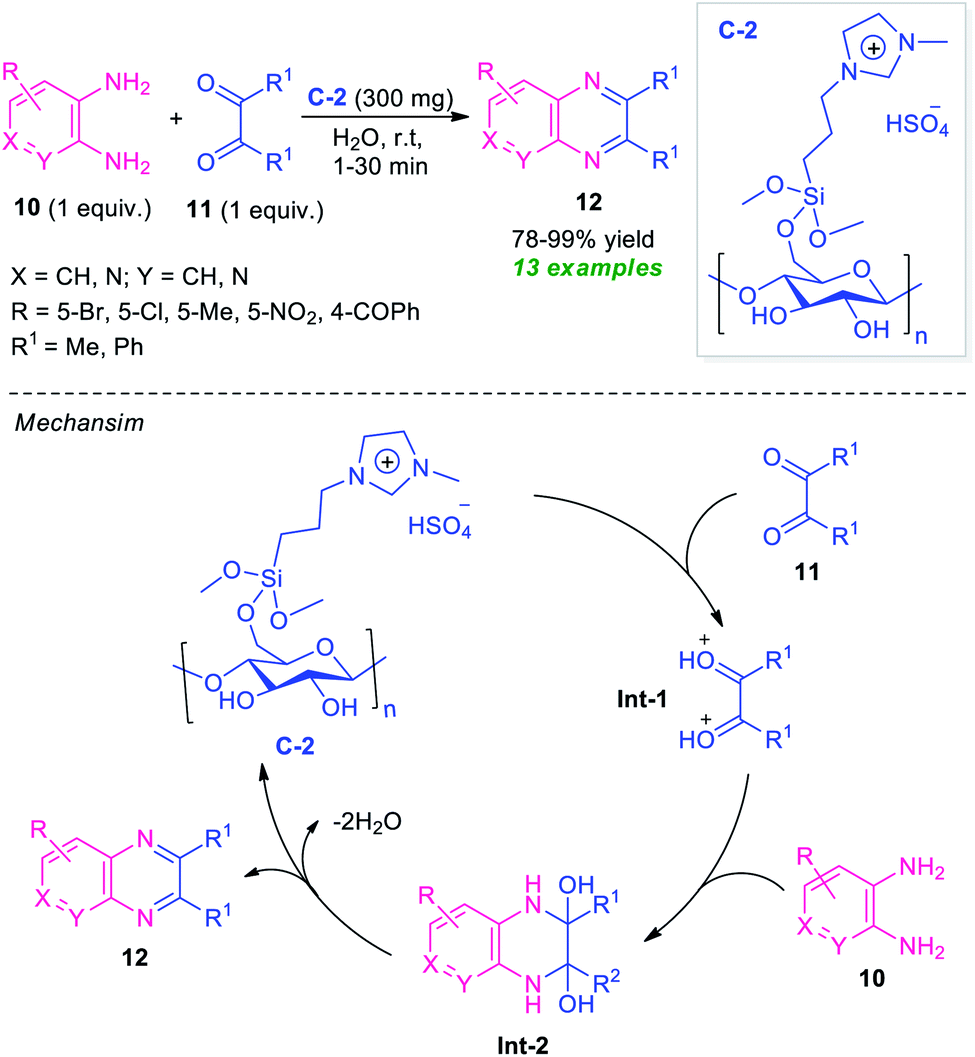 | ||
| Scheme 4 Ionic liquid functionalized cellulose as an efficient catalyst for the rapid access to quinoxalines 12. | ||
The reaction can proceed through the catalytic cycle as depicted in Scheme 4. Initially, the Brønsted acid catalyst C-2 enhances the nucleophilic character of 1,2-carbonyl compounds thereby facilitating the protonation of the carbonyl groups. The resulting intermediate Int-1 experiences a nucleophilic attack from the nitrogen of diamines to deliver the intermediate Int-2, which is followed by dehydration to yield the final product 12.
Considering the importance of quinoxaline rings along with the advantages associated with the transition-metal-free catalysis, several eco-friendly and environmentally benign strategies for the synthesis of quinoxaline derivatives 3 from o-phenylenediamine 1 and 1,2-dicarbonyl compounds 2 has been developed. All the reactions were performed in the presence of different catalytic systems including ionic liquid, bio-based organocatalyst, green catalyst as well as under catalyst-free conditions. The utilization of ethanol, aqueous ethanol, or solvent-free conditions makes the developed methodology green and sustainable. The products were obtained in moderate to excellent yield within a very short reaction time in almost all of the cases. All the reactions required a low amount of catalyst which marks the salient features of the developed methodologies. Broad functional group tolerance, mild reaction condition, recoverability, and reusability of the catalyst without affecting the outcome of the reaction, are some of the advantages of all of the developed strategies (Table 1).64–70
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Loading | Solvent | Condition | Time (min) | Examples | Yield (%) | Ref. |
a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3 ratio is used.b Phospho sulfonic acid.c sulfonated rice husk ash.d Gum arabic.e Time expressed in hours. :1:3 ratio is used.b Phospho sulfonic acid.c sulfonated rice husk ash.d Gum arabic.e Time expressed in hours. |
||||||||
| 1 | DABCO:AcOH:H2Oa |
5 mol% | Solvent-free | 80 °C | 20–45 | 12 | 86–98 | 64 |
| 2 | PSAb | 5 mol% | EtOH | r.t | 10–30 | 15 | 88–95 | 65 |
| 3 | RHA-SO3Hc | 15 mg | Solvent-free | r.t | 5–30 | 11 | 90–98 | 66 |
| 4 | Vitamin C | 11 mol% | EtOH | r.t | 2–100 | 17 | 60–98 | 67 |
| 5 | Sulfated polyborate | 10 wt% | Solvent-free | 100 °C | 3–10 | 18 | 95–99 | 68 |
| 6 | Catalyst-free | — | EtOH | Grinding, r.t | 10–30 | 30 | 80–98 | 69 |
| 7 | GAd | 0.05 g | EtOH:H2O (4:1) |
r.t | 0.6–18e | 11 | 55–98 | 70 |
Although their emergence in organic chemistry in only about the last decades, ultrasound irradiation has been considered as a powerful alternative energy source for the construction of value-added compounds to make them sustainable from the perspective of green chemistry.71
Given the importance of ultrasound irradiation in organic synthesis, Srivastava and co-workers in 2019, demonstrated an ultrasound irradiated catalyst-free protocol for the synthesis of diverse fused quinoxaline derivatives from several substituted o-phenylenediamines by utilizing water as the reaction medium (Scheme 5).72 Treatment of o-phenylenediamine 1 with isatin 13 under this optimized condition found to proceed smoothly to afford indolo[2,3-b]quinoxalines 14 in 87–95% yields; whereas the reaction of diamine 1 with ninhydrin 15 delivered the indeno[1,2-b]quinoxaline derivatives 16 in 92–99% yields within very short reaction time. The scope of the reaction was established by varying different substitutions on the diamine ring and isatin ring. It is interesting to note that, unsubstituted isatin (R1 = H) as well as substituted isatin (R1 = Et, Bn, Pr) were well tolerated by this approach.
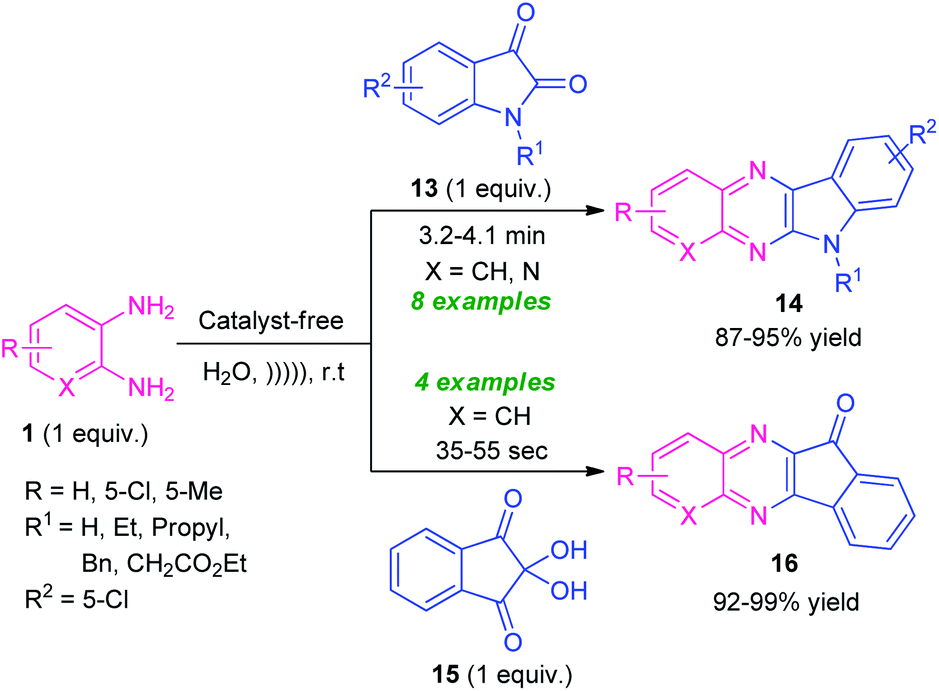 | ||
| Scheme 5 Ultrasound irradiated catalyst-free synthesis of diverse quinoxaline derivatives 14 and 16. | ||
At the same time, Singh and their groups disclosed a visible light promoted organo-photoredox catalyzed chemoselective strategy toward the synthesis of a variety of quinoxalines (Scheme 6).73 With the help of 20 mol% of Rose Bengal C-3 as the organo-photoredox catalyst, the treatment of 1,2-diamines and 1,2-dicarbonyl compounds including substituted benzil 18 or substituted isatin 13 were found to take place at room temperature to afford the desired quinoxaline products 19 and 20 in moderate to excellent yield respectively. The mechanism to explain this reaction has been covered in Scheme 7. Initially, the photoredox catalyst C-3 is excited to its singlet state under visible light irradiation, which can be further excited to the triplet state through intersystem crossing (ISC). Pleasingly, under influence of this triplet state, 1,2-diamine converts into radical-cation Int-3 which reacts with isatin to form intermediate Int-4. Subsequent single electron transfer (SET) from C-3˙− followed by removal of proton results in the formation of intermediate Int-5 from Int-4, which on dehydration afforded the intermediate Int-6. The cyclization of Int-6 leads to the desired products 20.
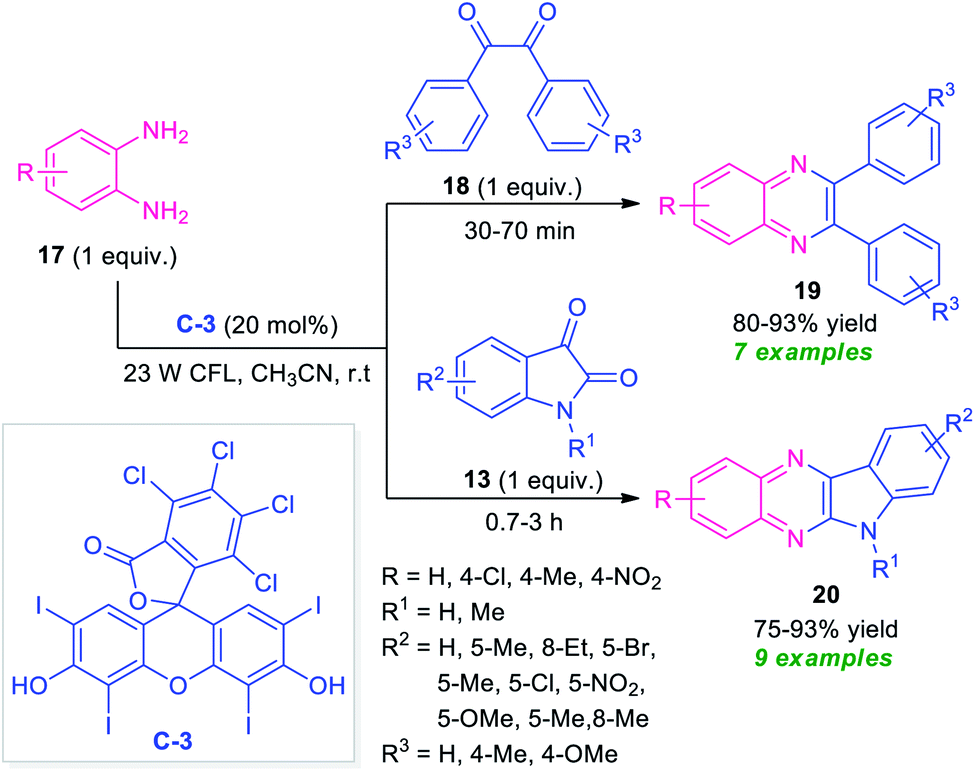 | ||
| Scheme 6 Visible light-mediated Rose Bengal catalyzed synthesis of different quinoxaline derivatives 19 and 20. | ||
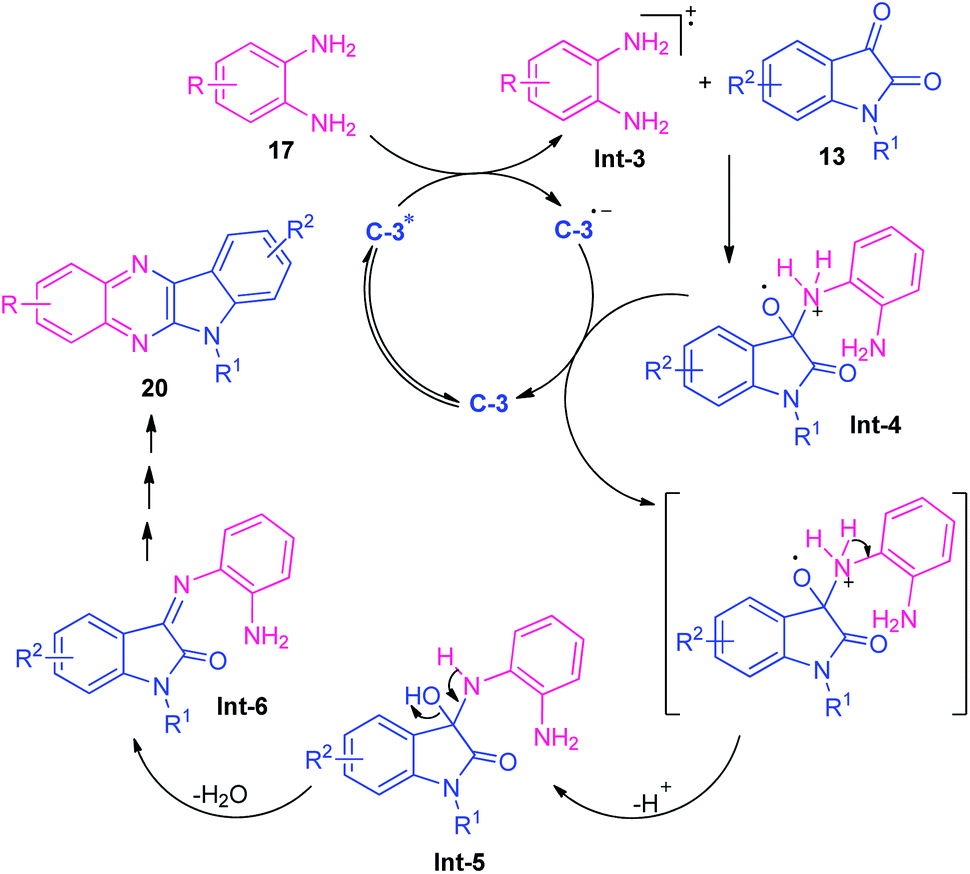 | ||
| Scheme 7 The suggested mechanism to explain the formation of quinoxalines 20. | ||
Bhargava et al., synthesized a series of diverse quinoxaline derivatives 22, 24, 26, and 28 from the equimolar amount of o-phenylenediamine 17 and different types of carbonyl substrates such as acenaphthoquinone 21, substituted bromoacetophenone 23, isatin 25, and benzil 27 by using ionic liquid C-4 as the catalyst as well as reaction medium (Scheme 8).74 Interestingly, the products were obtained in good to excellent yield within a very short reaction time at room temperature in all the cases. The effectiveness of the protocol was established by demonstrating the reactivity of recovered ionic liquids for up to six reaction cycles without loss in its catalytic activity. Also, wide substrates scope, high yields, simple work-up procedure, and short reaction time make this approach very efficient and environmentally benign.
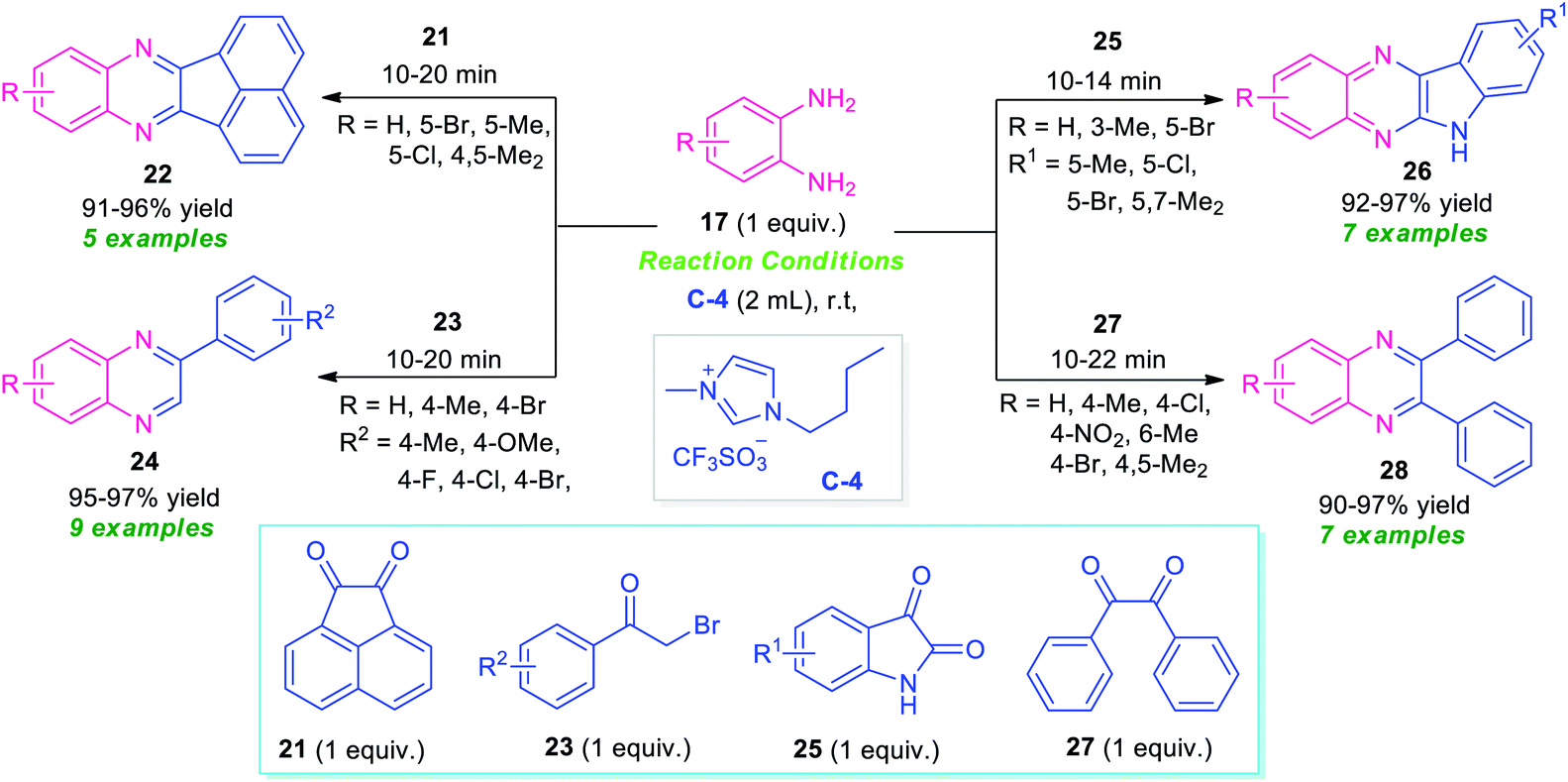 | ||
| Scheme 8 Preparation of library of quinoxaline derivatives in ionic liquid. | ||
Recently, a straightforward organocatalytic strategy for the synthesis of different types of quinoxaline derivatives was developed by Banerjee's group (Scheme 9).75 By using 20 mol% of camphor sulfonic acid as the organocatalyst, various quinoxaline derivatives 29, 30, and 32 derived from easily available starting material o-phenylenediamine 1 and carbonyl substrates acenaphthylene-1,2-dione 21, benzil 27, and phenanthrene-9,10-dione 31, have been obtained in moderate to excellent yield within a very short reaction time. Utilization of commercially available, camphor sulfonic acid as an organocatalyst, aqueous ethanol as the solvent, mild reaction conditions make this protocol environmentally and eco-friendly benign. However, low substrate scopes mark a major limitation of this approach and demand further developments otherwise outstanding work.
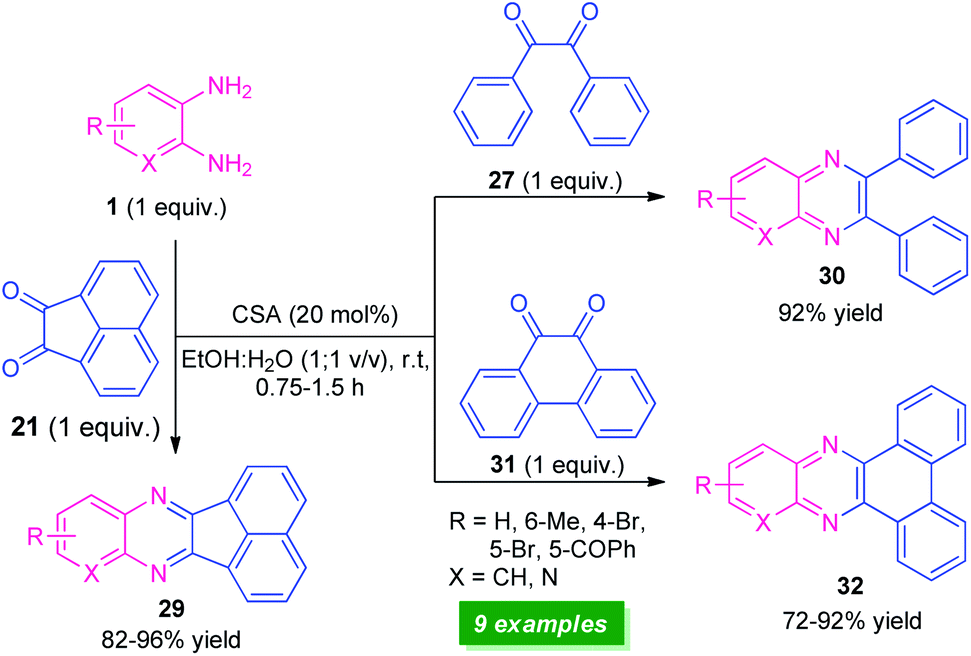 | ||
| Scheme 9 Camphor sulfonic acid-catalyzed synthesis of quinoxalines 29, 30, and 32. | ||
3.2 From α-halo-ketones
Owing to the dual functionality as they contain two electrophilic sites, α-halo-ketones are recently gained as important building blocks for the synthesis of numerous active heterocycles as well as undergo various types of reaction.76 α-Halo-ketones mainly phenacyl bromides were utilized in the synthesis of quinoxaline derivatives.In 2015, a highly efficient and practicable one-pot strategy toward the synthesis of quinoxalines via oxidative cyclization of α-halo ketones and o-phenylenediamine was developed by Nair and their groups (Scheme 10).77 Treatment of several diamines 17 with substituted phenacyl bromide 33 in water at 80 °C afforded the desired quinoxalines 34 in moderate to high yield. This reaction does not require any catalyst as well as co-catalyst, and additives. Phenacyl bromide-bearing electron-donating, as well as electron-withdrawing substituents and symmetrical diamines, smoothly underwent the reaction under this condition. However, when unsymmetrical diamines were employed, the products were formed regioselectively. The overall process can initiate through the nucleophilic attack of diamines 17 with phenacyl bromide 33 to produce the intermediate Int-7 that can then be cyclized into intermediate Int-8. Subsequently, the aromatization of intermediate Int-8 was occurred in air oxidation to afford the final products 34.
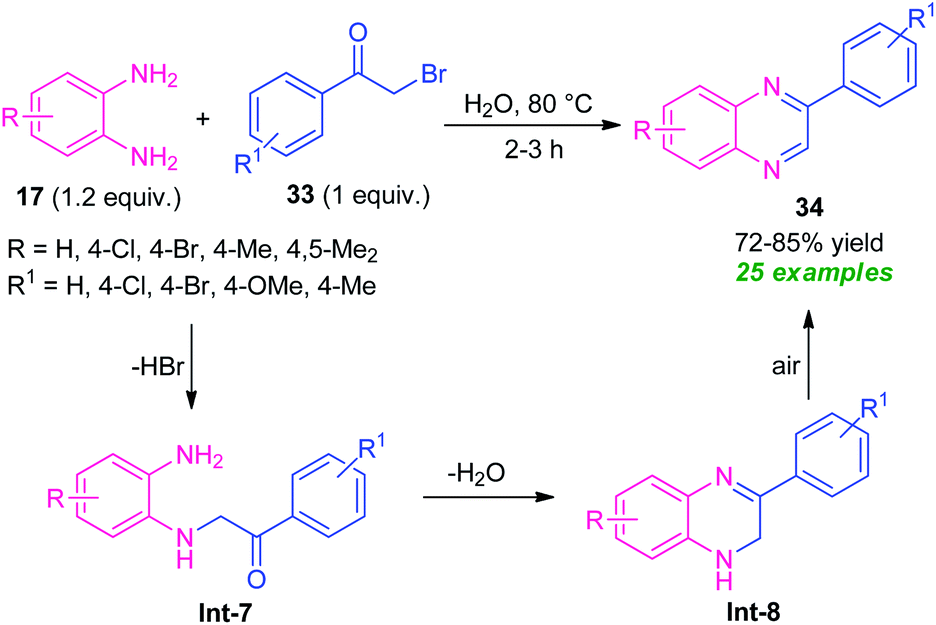 | ||
| Scheme 10 Catalyst-free oxidative cyclization of diamines and phenacyl bromide to access quinoxalines. | ||
Massah et al., in 2017, disclosed a catalyst-free protocol for the synthesis of quinoxaline derivatives 38 from the reaction of o-phenylenediamine 35 and phenacyl bromide 33 under reflux condition using ethanol as the green solvent (Scheme 11).78 The products were obtained in 70–85% yields. The synthesized quinoxaline derivatives were extended to quinoxaline sulfonamides 39 via a two-step reaction. The first step involves the reaction of quinoxaline 36 (R1 = OMe) with ClSO3H to afford the quinoxaline sulfonyl chloride 37. Treatment of 37 with different aryl amines 38 under solvent-free conditions lead to the final quinoxaline sulfonamides 39 via the second steps. The respective products were achieved in moderate to high yield. Most of the synthesized compounds have been established as good antibacterial agents against Staphylococcus spp. and Escherichia coli bacteria.
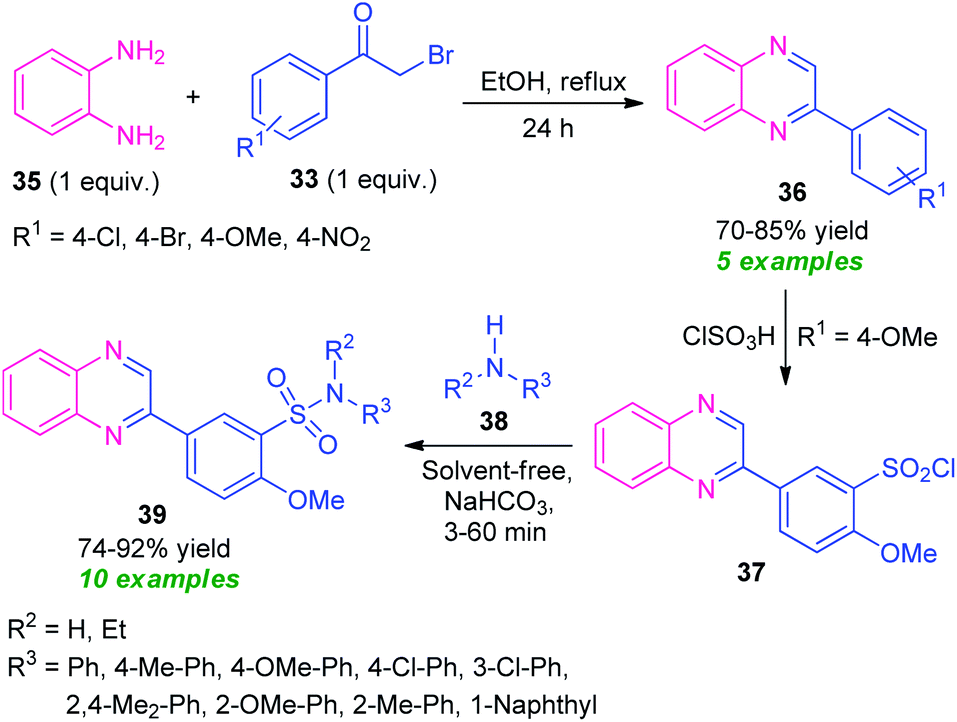 | ||
| Scheme 11 Three-step synthesis of quinoxaline-sulfonamides based on a green catalyst-free strategy. | ||
3.3 From β-ketonitriles
Similar to α-halo-ketones, β-ketonitriles represent an important class of highly reactive precursors for the preparation of diverse biologically active pharmaceuticals and heterocyclic compounds due to the wide functionality of ketone and nitrile groups present on it.79Considering their versatility, along with the prevalence of quinoxaline-1,4-di-N-oxide (QdNO) and N-acylhydrazone in medicinal chemistry, Santos et al., realized a metal-free step-wise approach for the synthesis of quinoxaline hybrid 45 through the Beirut reaction sequence (Scheme 12).80 Initially, the reaction of dioxolan-benzofuroxan 40 with benzoylacetonitrile 41 was successfully carried out via conventional as well as microwave-irradiation to afford the adduct 42 under the influence of either K2CO3 or Et3N as metal-free bases. Although, both the method provides the same amount of yield of adduct 42 (30%), however, microwave technique was found to be very advantageous over conventional one in terms of reaction time. The subsequent deprotection of 42 by acid hydrolysis and in situ condensation of resulting adduct 43 with isoniazid 44 lead to the final quinoxaline-1,4-di-N-oxide-N-acylhydrazone 45 in 66% yields.
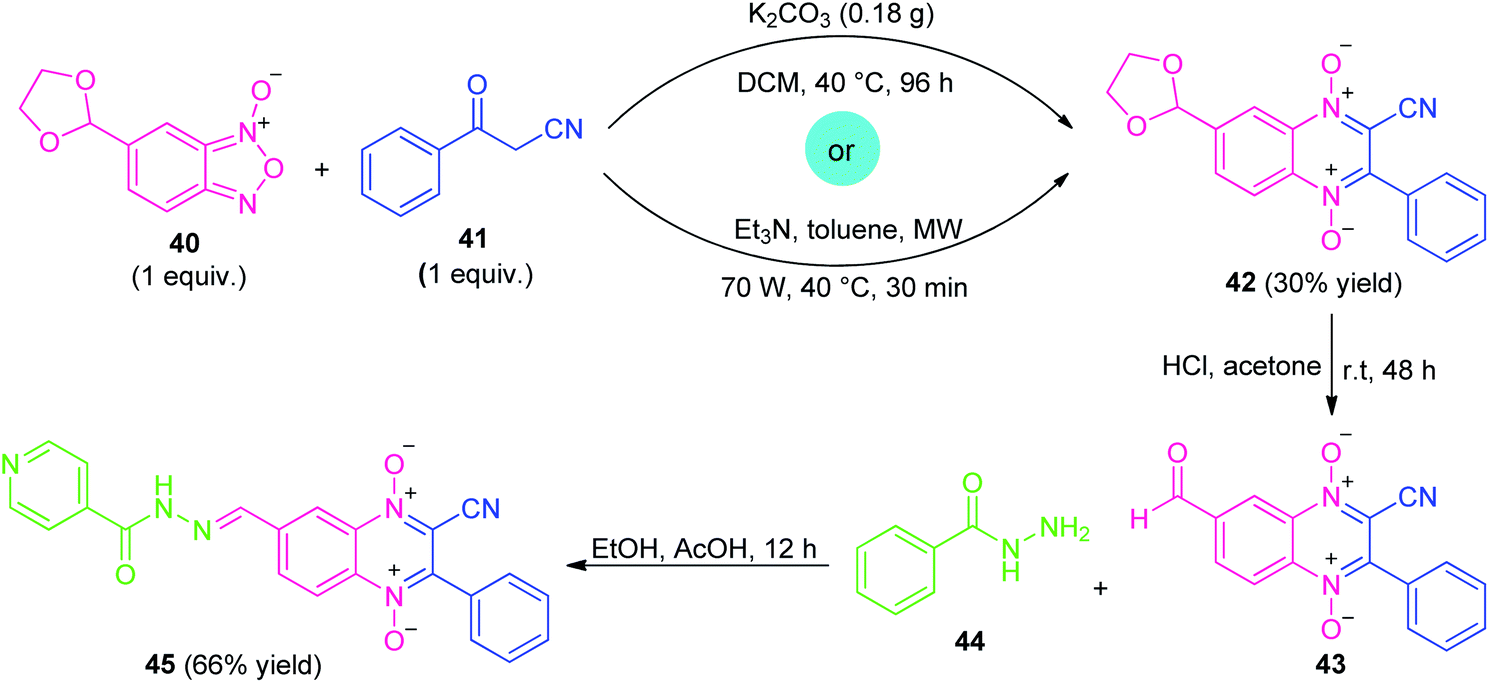 | ||
| Scheme 12 Metal-free conventional as well as the microwave-assisted synthesis of quinoxaline hybrid 45. | ||
A highly efficient visible-light mediated metal-free synthesis of quinoxalines from easily available o-phenylenediamine 17 and benzoylacetonitrile 46 via single electron-transfer and oxidative coupling strategy has been developed by Guo et al., in 2019 (Scheme 13).81 Pleasingly, a total of 10 compounds were synthesized in 50–78% yields by this mild reaction condition at room temperature. A variety of substitutions on the diamine ring as well as on the benzoylacetonitrile were found to effectively work by this approach. The mechanistic pathway behind this transformation involves the reaction of benzoylacetonitrile 46 with singlet oxygen generated from the oxygen under the influence of visible light, to produce a radical intermediate Int-9. The intermediate Int-9 then reacted with HOO radical and deliver the intermediate Int-10 which underwent dehydration to afford the intermediate Int-11.
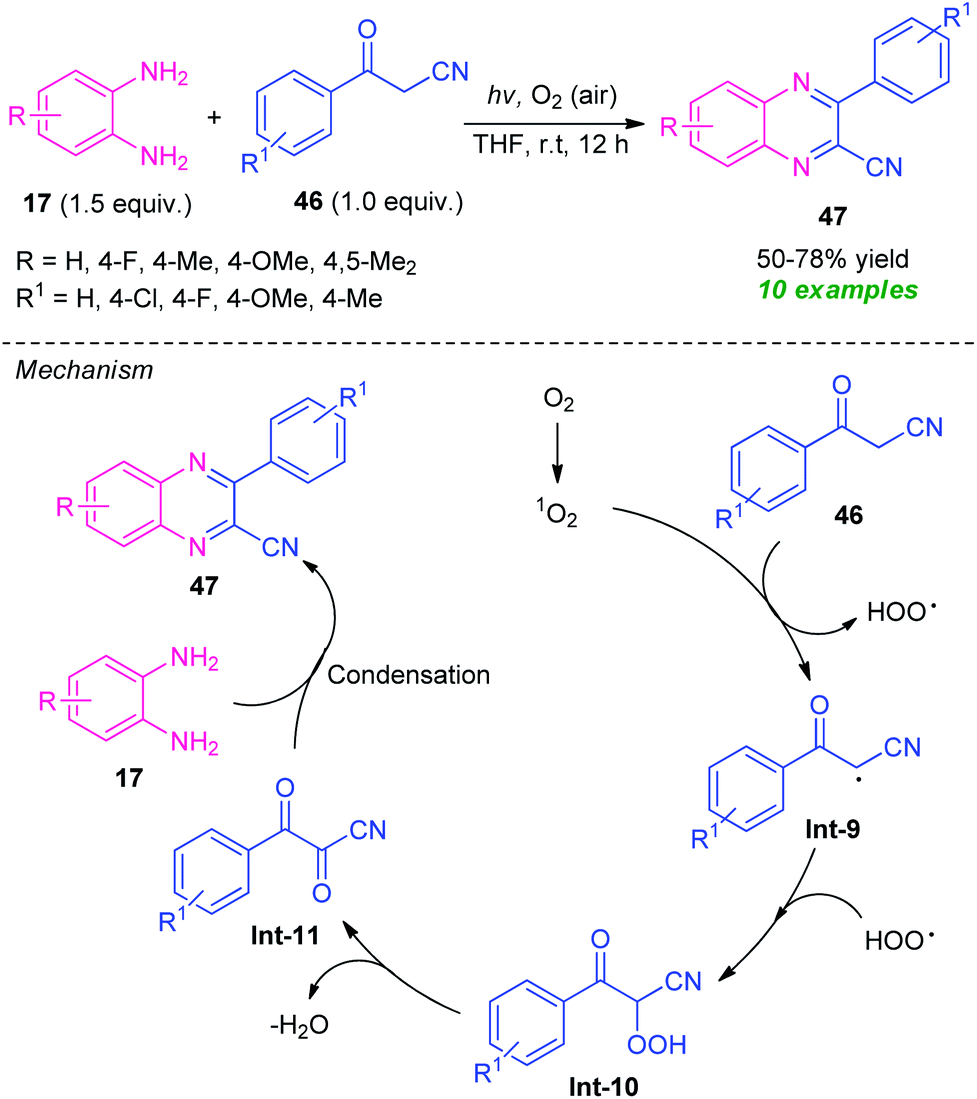 | ||
| Scheme 13 Visible light-induced electron-transfer and oxidative cyclization to access quinoxaline derivatives. | ||
Consequently, condensation of Int-10 with diamine 17 leads to the final product 47.
3.4 From α-hydroxy ketones/diols
Given the importance of α-hydroxy ketones in the compounds of the pharmaceutical interest and optically active value-added compounds, the synthesis and their derivatization into potential lead compounds has received tremendous attention in organic synthesis.82In 2015, Ma and his group demonstrated a straightforward one-pot two-step procedure for the synthesis of quinoxaline derivatives 49 from α-hydroxy ketones 48 (Scheme 14).83 With the help of 20 mol% of I2 as the catalyst and DMSO as the solvent as well as oxidant, the corresponding quinoxaline products 49 have been synthesized in 78–99% yields. Different substituted o-phenylenediamine 17 was found to proceed smoothly under this condition. A total of 23 compounds were synthesized by this method. Broad functional group tolerances, excellent level of yield, metal-free reaction conditions are some of the salient features of this approach. This process starts with the initial oxidation of hydroxy ketones 48 to the desired dicarbonyl compound 11 under the influences of I2 as the oxidant. Then the subsequent condensation of 11 with diamine 17 takes place smoothly, to form the final product 49 under the influence of I2 that acts as Lewis acid in this step. For recycling the catalytic system, HI was oxidized into iodine with DMSO as the oxidant, and then iodine was regenerated for further operation.
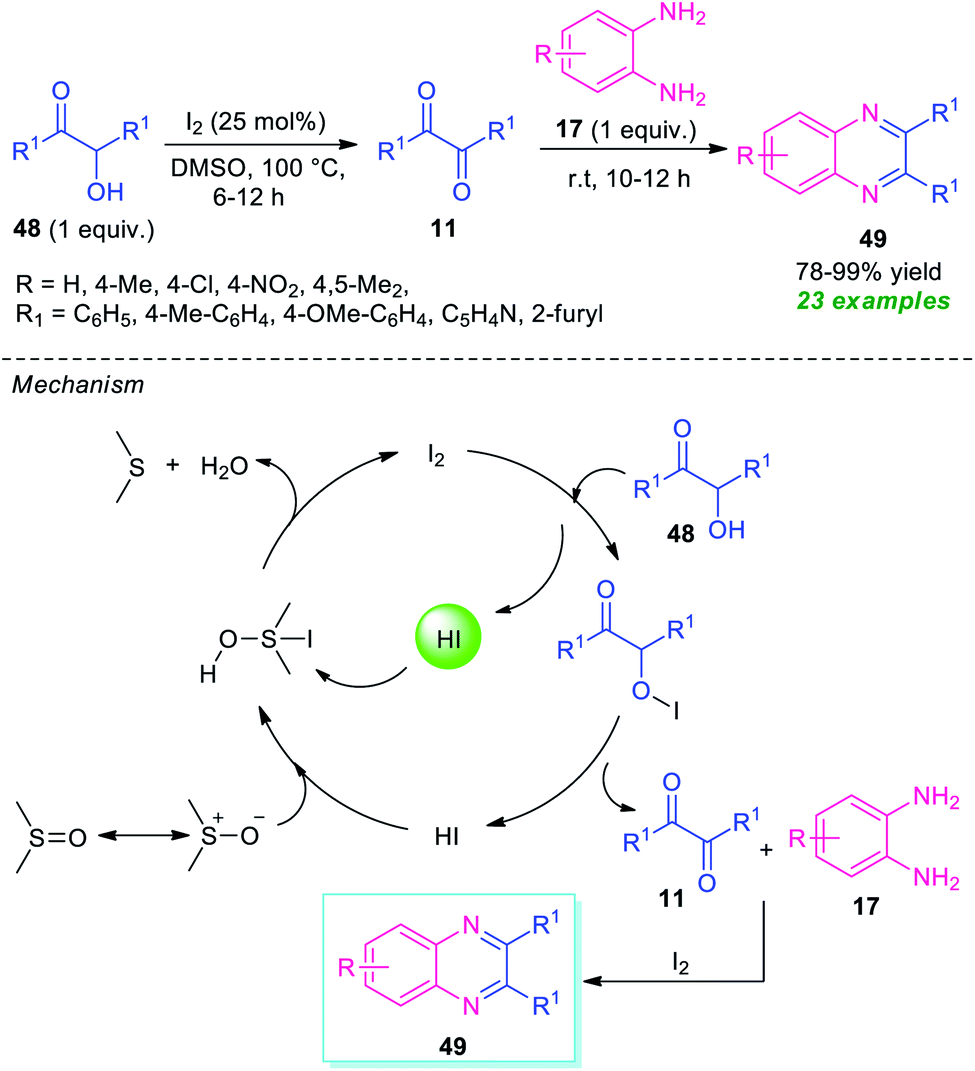 | ||
| Scheme 14 Iodine catalyzed one-pot oxidation/cyclization to access quinoxalines 49. | ||
In 2020, a transition-metal-free highly convenient strategy for the construction of quinoxaline derivatives via redox condensation of 2-nitroamines with α-hydroxy ketone or diols was disclosed by Zhu, Song, and their groups (Scheme 15).84 For optimizing the reaction condition, the reaction was initially carried out under the influence of different bases including NaOH, NaOtBu, KOtBu, KOH as well as in different solvent systems like toluene, o-xylene, dioxane, and also in solvent-free conditions. Among them, the exploitation of NaOH as the base catalyst in toluene was selected as the standard condition. Under this condition, the treatment of 2-nitroamine 50 with diols 51 afforded various quinoxaline derivatives 52 in moderate to excellent yields (46–98%), whereas the reaction of 2-nirtoamine 50 and α-hydroxy ketone 53 delivered the corresponding product 52 in 44–99% yields. By employing both the substrates a total of 55 compounds were has been synthesized.
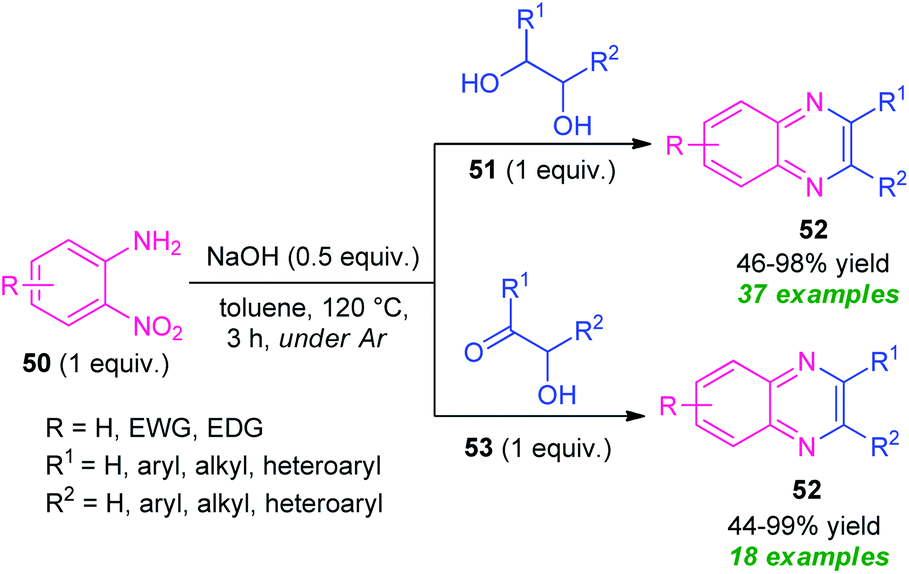 | ||
| Scheme 15 Transition-metal-free redox condensation reaction to access quinoxaline derivatives 52. | ||
Later, Sarma and co-workers disclosed a visible light-assisted catalyst-free green protocol toward the synthesis of a series of quinoxaline derivatives 56 by employing easily accessible o-phenylenediamine 54 and α-hydroxy ketone 55 as the starting material (Scheme 16).85 With the help of tert-Butyl hydroperoxide (TBHP) as the oxidizing agent, the corresponding quinoxaline products 56 were obtained in 78–93% yields. The decomposition of TBHP into its radical was predominately promoted by metal or photocatalysts, or other reagents, however in this reaction, the decomposition of TBHP for the radical formation and its acceleration for the completion of the reaction was achieved simply by visible light irradiation and no extra metal- or photocatalyst is required. The successful utilization of photoinduced catalyst-free protocol in an aqueous medium makes this approach very efficient toward green and sustainable practices.
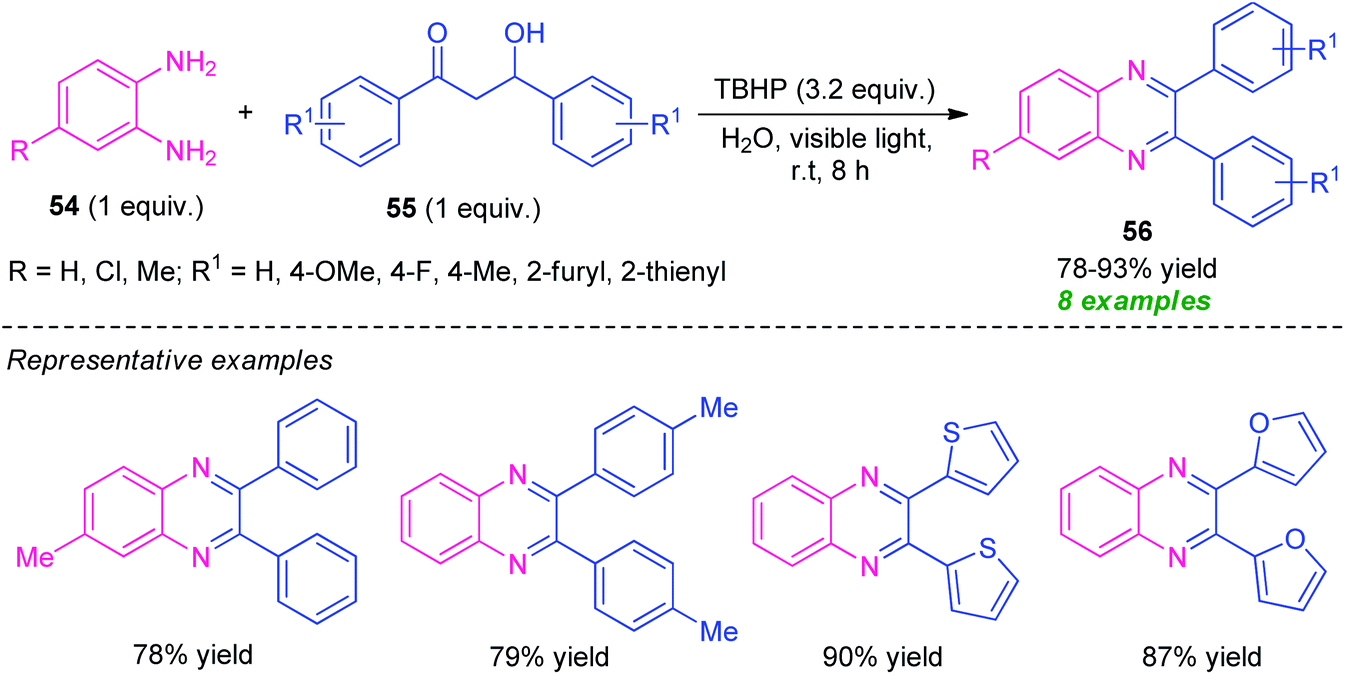 | ||
| Scheme 16 Metal-free visible light-mediated domino synthesis of quinoxalines 56. | ||
3.5 From alkenes
The utilization of alkenes in the metal-free domino synthesis of quinoxaline derivatives has been achieved by Chaskar et al., in 2015 (Scheme 17).86 Using I2 as the catalyst and TBHP as the oxidant in DMSO, the corresponding quinoxaline products 57 derived from various o-phenylenediamine 17 and alkenes 57, were obtained in 31–93% yields. Not only aromatic alkenes but also heteroaromatic alkenes were well tolerated by this atom economic and operationally simple method. A total of 15 compounds were synthesized under this mild reaction condition. Based on a series of experimental results the author proposed a mechanistic pathway for this transformation which involved the oxidation of styrene 57a to the corresponding radical (Int-12) under the influence of I2/TBHP through a radical mechanism as depicted in Scheme 17. Iodination of Int-12 by I2/TBHP afforded the key intermediate Int-13 which undergo Kornblum oxidation to produce phenylglyoxal Int-14. Subsequent treatment with diamine 17, the intermediate Int-14 furnished intermediate Int-15, which on cyclization followed by oxidation provides quinoxaline 58a.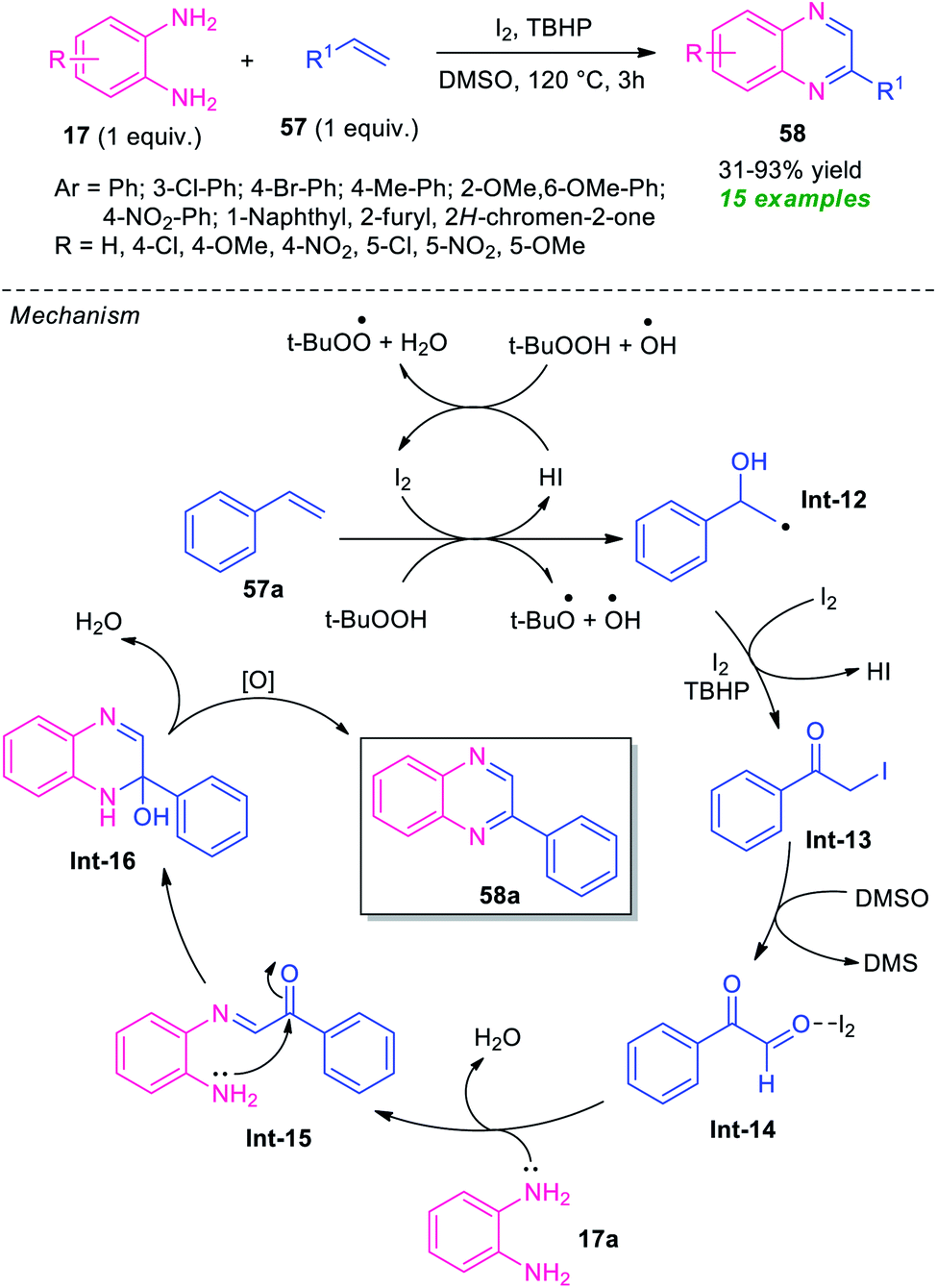 | ||
| Scheme 17 I2-catalyzed domino one-pot atom-economic approach to quinoxaline derivatives 58. | ||
A practical one-pot two-step NBS-promoted efficient synthesis of quinoxalines from the reaction of substituted alkenes 59 and o-phenylenediamines 17 was established (Scheme 18).87 The synthesis involves the initial reaction of alkenes 59 with NBS in aqueous media to deliver the phenacyl bromide 33. Subsequent condensation of 33 with diamine 17.
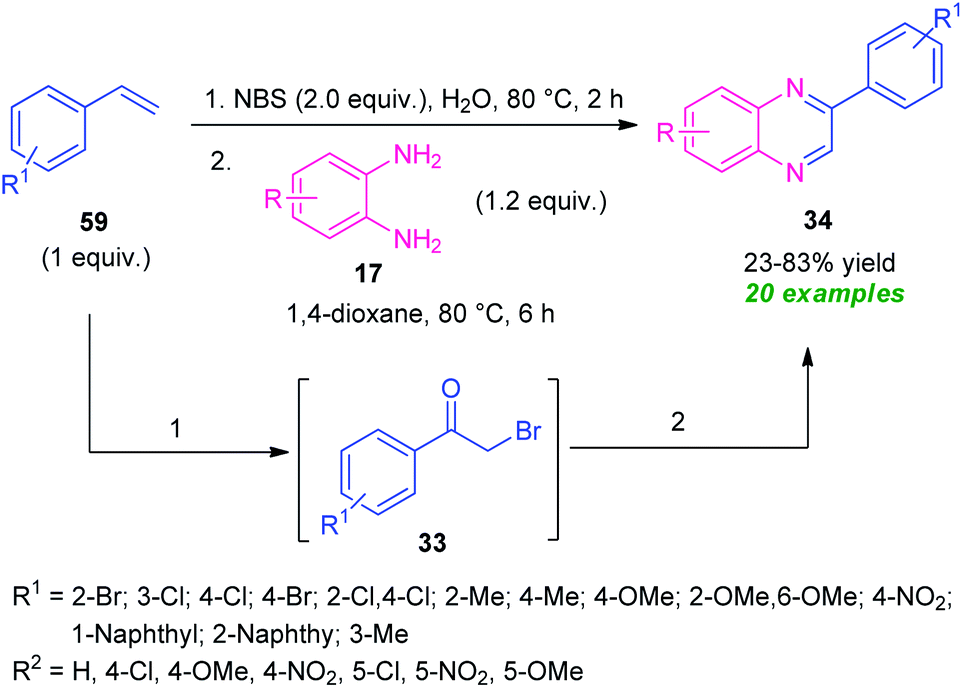 | ||
| Scheme 18 NBS-promoted one-pot two-step green synthesis of quinoxalines 34. | ||
In presence of 1,4-dioxane formed the quinoxaline derivatives 34 in 23–83% yields. A wide variety of substituents present on the alkenes ring smoothly anticipated the reaction under this metal-free approach. Broad functional group tolerance, utilization of inexpensive NBS as bromine source as well as oxidant, water as green solvent, a high level of yield make this approach synthetically as well as environmentally viable.
3.6 From alkynes
In 2016, Cui and his group disclosed a highly convenient one-pot approach for the regiospecific synthesis of quinoxalines 52 from readily available ynones 60 and o-phenylenediamines 17 under metal-free conditions (Scheme 19).88 The reaction proceeded through an intermolecular Michael addition, followed by dehydration and subsequent base-promoted C–α-CH2-extrusion process. A total of 33 quinoxaline derivatives were synthesized by this strategy in 36–95% yields. Various aryl, heteroaryl, and alkyl-substituted ynones were found to be suitable substrates for this reaction. However, when asymmetrical o-phenylenediamine and ynones were employed in the reaction, the products were formed in moderate to good yield with excellent regioselectivity (>20:1). The exploitation of O2 as the environmentally benign oxidant, formation of H2O and CO2 as only the by-products directed this approach toward more environmentally friendly and sustainable.
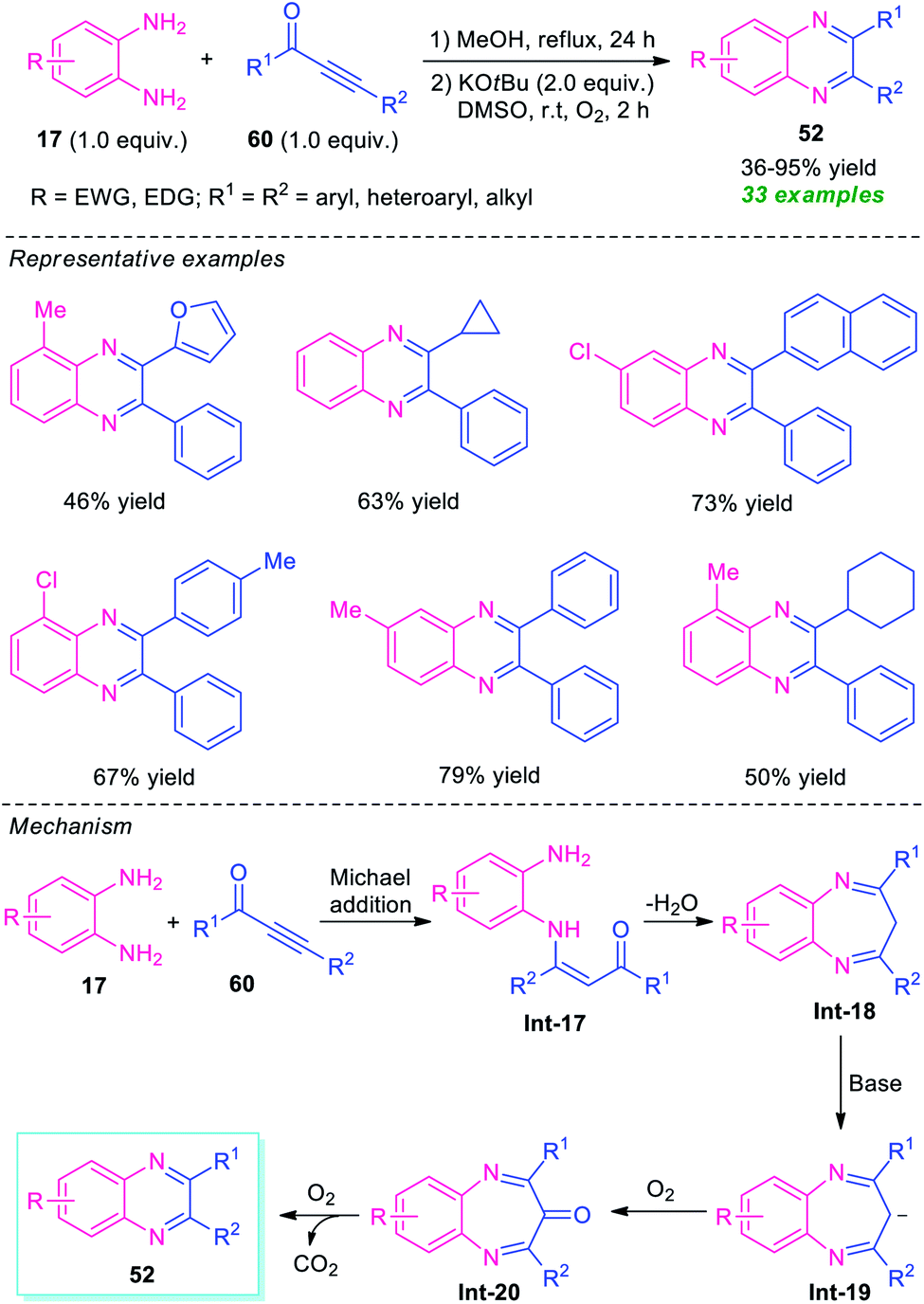 | ||
| Scheme 19 Metal-free one-pot synthesis of quinoxalines via C–α-CH2-extrusion process. | ||
The proposed mechanism for this C–α-CH2-extrusion process involves the initial Michael addition of o-phenylenediamines 17 and ynones 60 to formed intermediate Int-17 regioselectively that can then undergo dehydration as well as condensation to give Int-18. Subsequent deprotonation of Int-18 by KOtBu results in the formation of anion intermediate Int-19 which gets easily oxidized to Int-20 by O2. The final decarbonylation of Int-20 under O2 afforded the products 52.
A metal-free cascade process toward the synthesis of diverse quinoxaline derivatives 61 from alkynes 60 has been developed by Hazarika and Phukan in 2017 (Scheme 20).89 Treatment of alkynes 61 with TsNBr2 (N,N-dibromo-p-toluene sulfonamide) in an aqueous medium first generated the α,α-dibromoketone intermediate which on reaction with 17 under influence of base deliver the desired products 62 in 18–88% yields within one hour. The scope of the reaction was found to be viable to a wide variety of substituents on the phenyl ring of the alkynes as well as diamines. However, the low yield of the products for the reaction with nitro- and chloro-substituted diamines represents a limitation of this approach.
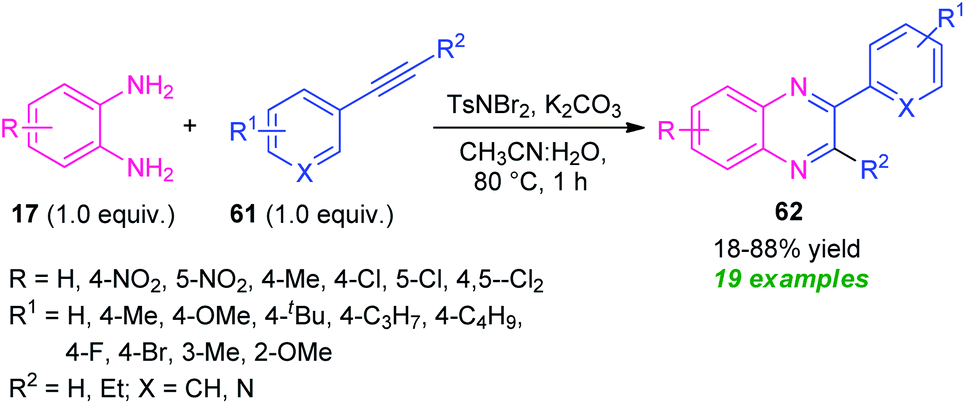 | ||
| Scheme 20 Metal-free domino protocol for the synthesis of quinoxalines via oxidative cyclization. | ||
In 2018, Zhang, Shen, Cao, and their group demonstrated a Michael addition initiated tandem azidation and cycloamination reaction for the construction of diverse fluoroalkylated quinoxalines (Scheme 21).90 Under metal-free conditions, the corresponding quinoxalines derivatives 65 derived from anilines 63 and fluoroalkylated alkynes 64 and TMSN3 as the nitrogen source, were obtained in 31–95% yields. The reactions with ortho-substituted anilines were found to lead to a decrease in the yield of the products as compared to para-substituted anilines. Alternatively, when meta-substituted anilines were employed as the substrates, two regioisomers has been formed in moderate to excellent yield. In this reaction, the utilization of KI as an additive provides the formation of iodine(I) species by reacting with PhI(OAc)2 and thereby promoting the azidation and cycloamination sequence toward the synthesis of the final products.
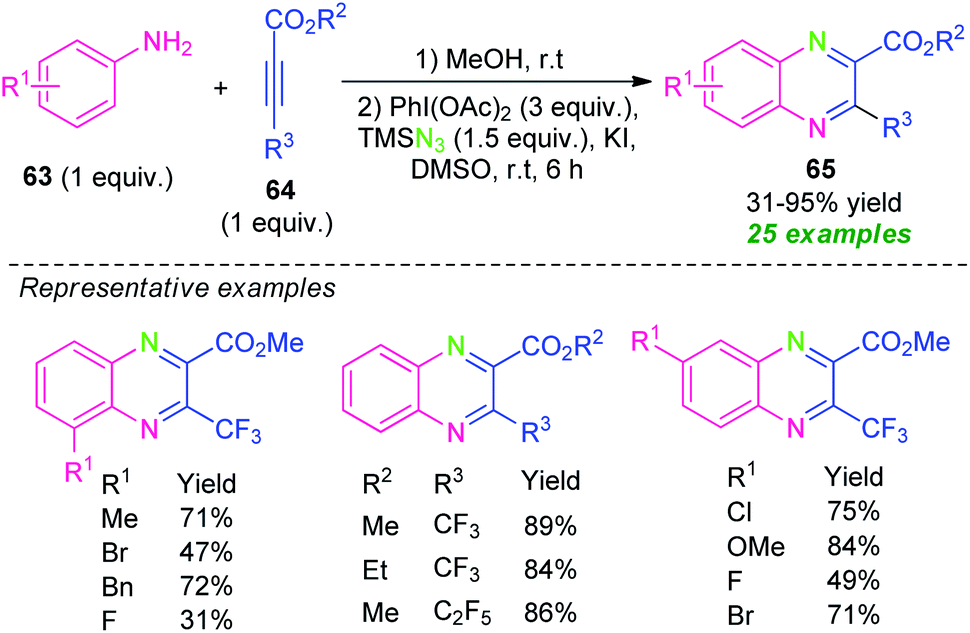 | ||
| Scheme 21 Tandem Michael addition/azidation/cycloamination sequence toward the synthesis of quinoxalines. | ||
3.7 From enaminones
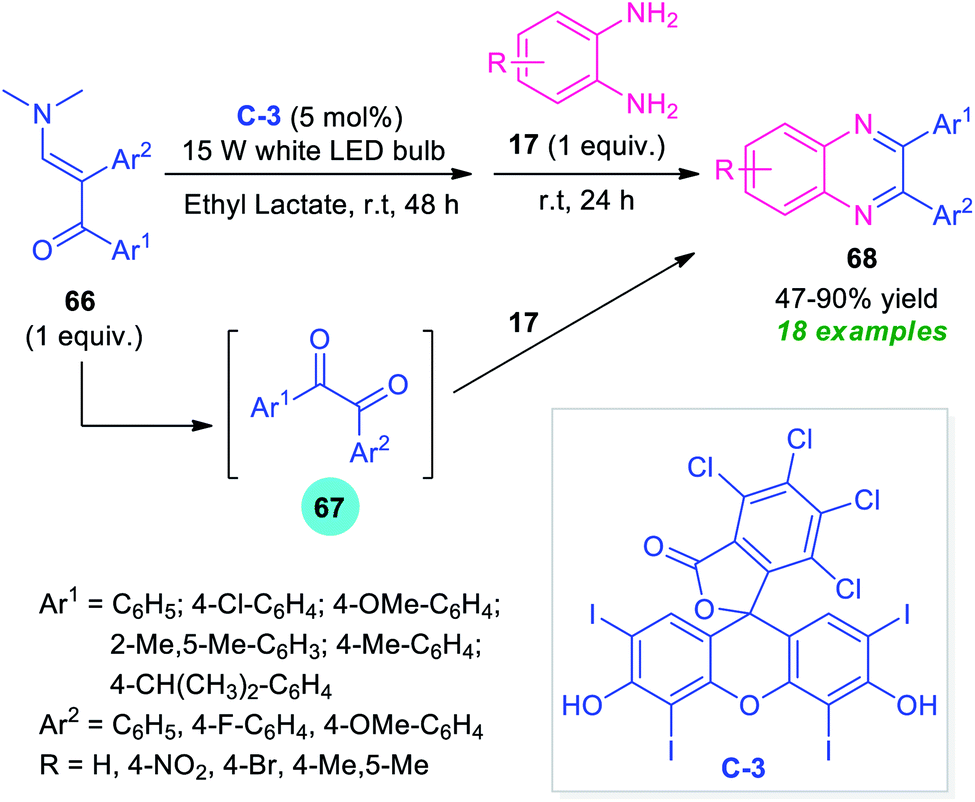
Enaminones are an important class of widely investigated organic compounds in organic chemistry. Due to the highly reactive nature and easy availability, a great variety of organic compounds have been synthesized by making the utilization of enaminones as key building blocks.91By considering this key importance and taking advantage of the high photocatalytic activity of Rose Bengal C-3, Wan, Wen, and co-workers disclosed a one-pot procedure for the synthesis of quinoxalines from enaminones 66 (Scheme 22).92 With the help of 5 mol% of C-3 as the photocatalyst, the visible light-mediated reaction of enaminones 66 afforded the corresponding products 68 in 47–90% yields. The overall process starts with the cleavage of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C of enaminones 66 to furnish the adduct 67 which reacts in situ with o-phenylenediamines 17 under the ambient condition to form the resultant products 68. Broad functional group tolerances, easy accessibility of the starting material, operational simplicity are some of the key advantages of this approach. The low yield of products for the reaction with o-phenylenediamines bearing electron-withdrawing group constitutes a major drawback of this method.
C of enaminones 66 to furnish the adduct 67 which reacts in situ with o-phenylenediamines 17 under the ambient condition to form the resultant products 68. Broad functional group tolerances, easy accessibility of the starting material, operational simplicity are some of the key advantages of this approach. The low yield of products for the reaction with o-phenylenediamines bearing electron-withdrawing group constitutes a major drawback of this method.
 | ||
| Scheme 22 Visible-light induced photocatalytic cleavage of CC of enaminones to access quinoxalines. | ||
3.8 From isonitriles
In 2016, the research group of Ma and Yu developed a visible light-assisted halogen-bond-promoted strategy for the synthesis of quinoxaline derivatives through the double radical isocyanide insertion of o-diisocyanoarenes 69 with perfluoroalkyl iodides 70 (Scheme 23).93 By using this strategy, a series of diverse 2-fluoroalkylated 3-iodoquinoxalines 71 were synthesized in moderate to high yield at room temperature. The overall process can initiate through the formation of halogen-bond adduct Int-21 which under the influence of visible light deliver the fluoroalkyl radical Rf˙, Bn2NH+˙, and iodide anion via a single electron transfer (SET) process. In this reaction, perfluoroalkyl iodides serve as halogen-bond donors and Bn2NH as halogen-bond acceptors. The addition of fluoroalkyl radical Rf˙ to o-diisocyanoarenes 69, followed by radical cyclization produces the radical intermediate Int-24 which then abstracts an iodine atom from 70 to yield the final products 71.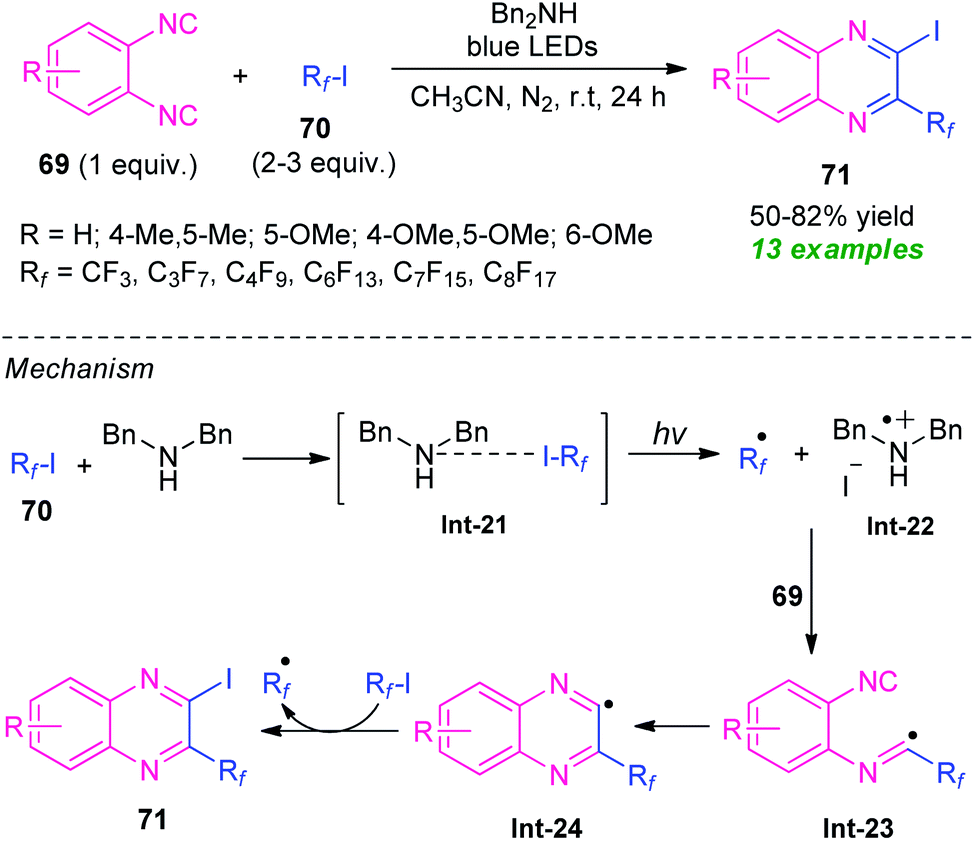 | ||
| Scheme 23 Halogen-bond-promoted construction of various quinoxaline derivatives under visible light irradiation. | ||
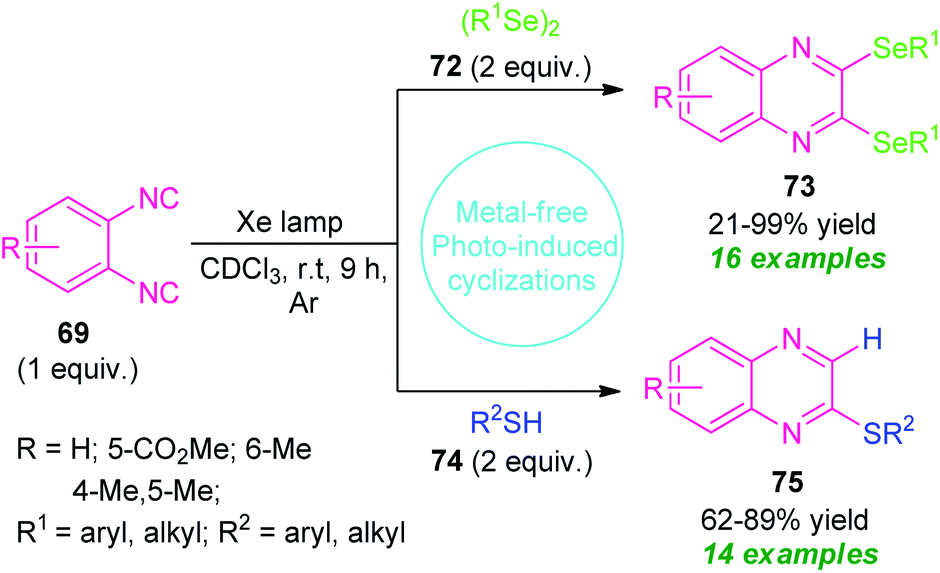
Recently, a metal- and additive-free cyclization sequence under visible-light irradiation has been established for the synthesis of a vast array of quinoxaline derivatives from o-diisocyanoarenes 69 and organic diselenides or thiols (Scheme 24).94 Initial optimization of the reaction condition revealed that utilization of Xe lamp as a light source in presence of CDCl3 as the solvent provides high activity and reactivity of the reaction as compared to high-pressure Hg lamp. By using this condition, a series of 2,3-bis-(selanyl)quinoxalines 73 derived from 69 and 72, were obtained in 21–99% yields. Not only the aryl-substituted selenides but also alkyl-substituted selenides were reacted efficiently under this condition.
 | ||
| Scheme 24 Visible light irradiated construction of different quinoxalines derivatives under metal-free conditions. | ||
Encourage by this result, they further attempted the reaction with 69 and 74 under the same reaction condition. Delightfully, a total of fourteen 2-thiolated quinoxaline compounds 75 were synthesized in moderate to high yield. Unlike diselenides, the reaction condition also tolerates several alkyl and aryl-substituted thiols. But unfortunately, thiols bearing unprotected alcohol groups, amines, and (iPr)3SiSH failed to yield any products which represents a major limitation of this approach (Scheme 24).
3.9 From oximes
A tandem one-pot protocol for the construction of functionalized quinoxalines from easily accessible arylamines 63 and β-keto oximes 76 has been developed by Pan, Mo, Su, and co-workers in 2017 (Scheme 25).95 The reaction sequence involves the initial condensation of amines 63 with oximes 76 as the nitrogen source and subsequent metal-free N-arylation of key intermediate Int-25 under Lewis acid catalysis was found to lead to the desired products 77 in 42–82% yields. The effectiveness of the developed protocol was established by its applicability in the gram-scale synthesis and other useful transformations. A wide range of substrate scopes, exploitation of the synthesized compounds in useful synthetic modifications, mild reaction conditions, water as only the by-products, systematic design of the metal-free sequence makes this protocol synthetically valuable and environmentally benign.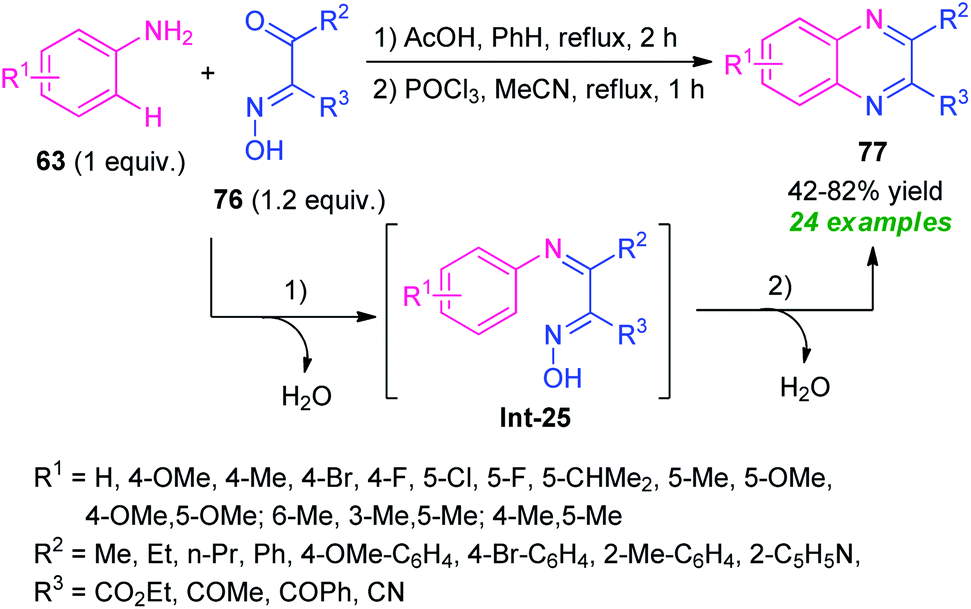 | ||
| Scheme 25 One-pot tandem cyclization/metal-free N-arylation toward the synthesis of functionalized quinoxalines. | ||
4. Synthesis of fused quinoxalines based on metal-free two-component reactions
4.1 Synthesis of pyrrolo/indolo[1,2-a]quinoxalines
Among various fused quinoxaline derivatives, pyrrolo/indolo[1,2-a]quinoxalines as the most important and well-established nitrogen-containing heterocycles play a vital role in medicinal and pharmaceutical chemistry due to their remarkable pharmacological profile.96 In particular, the presence of this moiety in many products found to exhibit reverse transcriptase inhibitor (R), antileishmanial activity (S), protein kinase CK2 inhibitor (T), anti-HIV activity (U), antifungal activity (V), and anticancer activity (W) (Fig. 3).97 Owing to these tremendous properties, significant efforts have been demonstrated toward their synthesis in the last decades.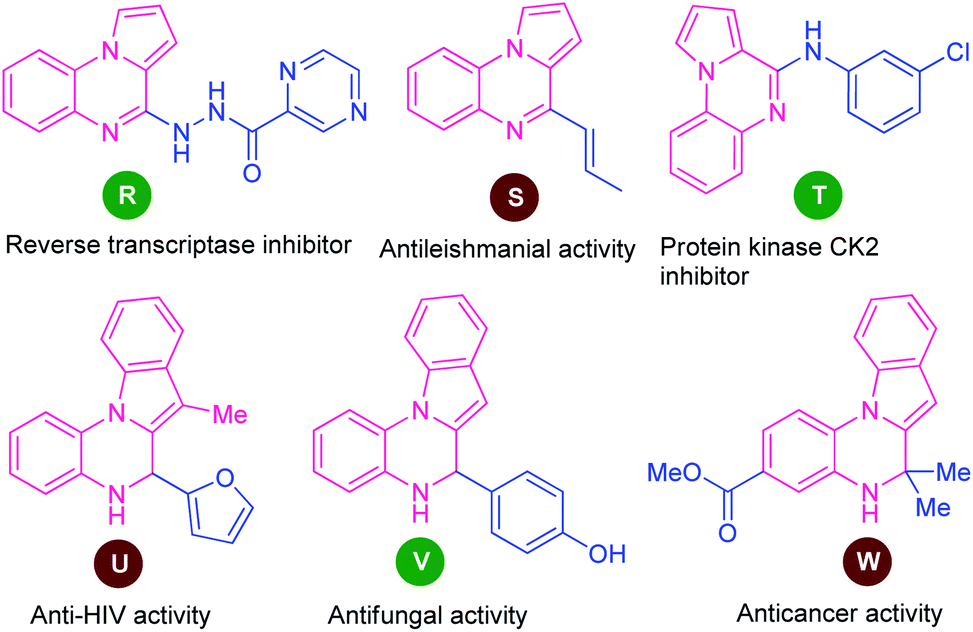 | ||
| Fig. 3 Biologically active various pyrrolo/indolo[1,2-a]quinoxalines derivatives. | ||
In this perspective, Ma and co-workers in 2016, developed a highly efficient one-pot domino strategy by employing 2-(1H-pyrrol-1-yl)aniline 78 and 1,3-dicarbonyl compounds 79 as the easily available starting material (Scheme 26).99 With transition metal-free condition, a variety of pyrrolo[1,2-a]quinoxalines 80 in 54–96% yields has been synthesized utilizing TsOH·H2O as the organocatalyst. The reaction condition was found to be appropriate for both β-diketones 79 (R3 = aryl, alkyl; R4 = aryl, alkyl) and β-keto esters 79 (R3 = aryl, alkyl; R4 = OEt). However, the reaction with diketones 79 (R3 = R4 = t-Bu) failed to yield any product which represents the limitation of this approach.
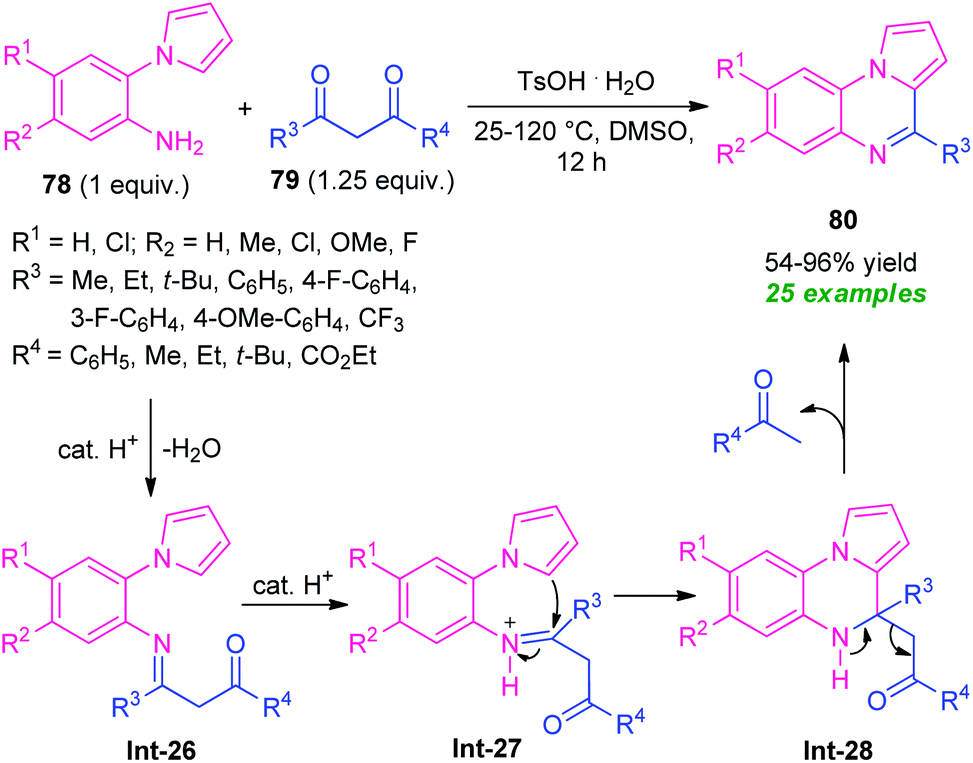 | ||
| Scheme 26 Brønsted acid-catalyzed one-pot domino synthesis of pyrrolo[1,2-a]quinoxalines. | ||
The requirement of a longer reaction time also points toward the drawback of this method. This transformation can proceed via the initial formation of the imine intermediate Int-26, which then abstracts a proton from the catalyst to form Int-27. Subsequently, an intramolecular cyclization of intermediate Int-27 followed by cleavage of C–C bond in intermediate Int-28 form the final product 80.
One year later, the same research group disclosed a novel one-pot green strategy for the synthesis of pyrrolo/indolo[1,2-a]quinoxalines by employing DMSO as reactant as well as the solvent system in metal-free condition (Scheme 27).100 With the help of AcOH acid as the catalyst, treatment of dimethyl sulfoxide 81 with 2-(1H-indol-1-yl)aniline 82 was found to take place at 120 °C to deliver indolo[1,2-a]quinoxalines 83 in 12–93% yields while using 2-(1H-pyrrol-1-yl)aniline 83 instead of 82 under the same reaction condition provided pyrrolo[1,2-a]quinoxalines 85 in 44–94% yields. A wide variety of electron-rich, as well as electron-poor substituents present on 82 and 84, were well tolerated by this condition. However, the low yield of the products 83 (12% yield) for the reaction with 2-(1H-indol-1-yl)aniline 82 (R1 = H, R2 = H; 3-Me is absent in indole ring)and no reaction for 3-methyl-2-(1H-pyrrol-1-yl)aniline 84 (R1 = R2 = H; X = C–Me), constitutes a major limitation of this approach. In addition, the strategy offers several advantages including mild reaction conditions, metal-free, utilization of oxygen as cheap and green oxidant, etc.
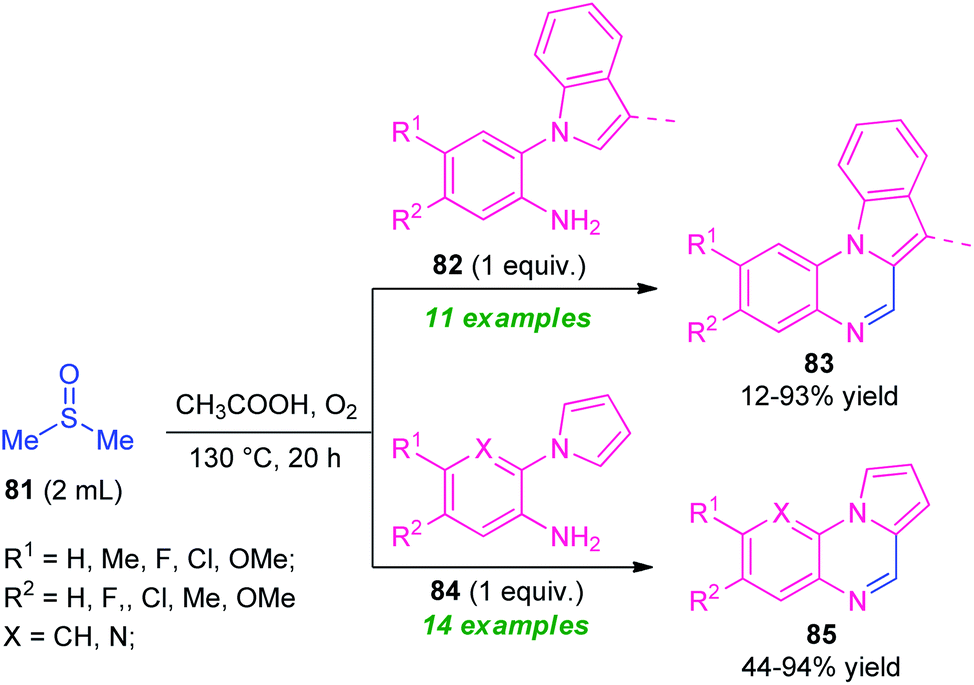 | ||
| Scheme 27 Versatile activity of DMSO in the synthesis of quinoxaline. | ||
In 2019, Patel et al. reported an efficient acetic acid-catalyzed synthesis of pyrrolo[1,2-a]quinoxalines 87 via a Pictet–Spengler type reaction of 2-(1H-pyrrol-1-yl)aniline 78 to aldehyde 86 (Scheme 28).101 With only 10 mol% of AcOH acid as the catalyst, a total of 16 compounds were synthesized in 82–89% yields. Pleasingly, various substituted and unsubstituted 2-(1H-pyrrol-1-yl)aniline 78 underwent the reaction smoothly to provide the desired product efficiently. While, the reaction with CF3 and CO2Me substituted 78 (R1 = H, R2 = CF3, CO2Me) failed to yield any product which draws a shortcoming of this approach. Similarly, aryl, as well as heteroaryl substituted aldehydes, were efficiently worked under this condition, except for electron-withdrawing substituents present on the meta-position of the aryl ring of aldehyde (R3 = 3-NO2-C6H4, 3-Cl-C6H4, 3-Br-C6H4) which affected the outcome of the reaction. Despite these limitations, the broad functional group tolerance, easily accessible starting material, are some advantages of this protocol. In addition, this reaction doesn't require any extra oxidant or additives, and the air was used as a green oxidant.
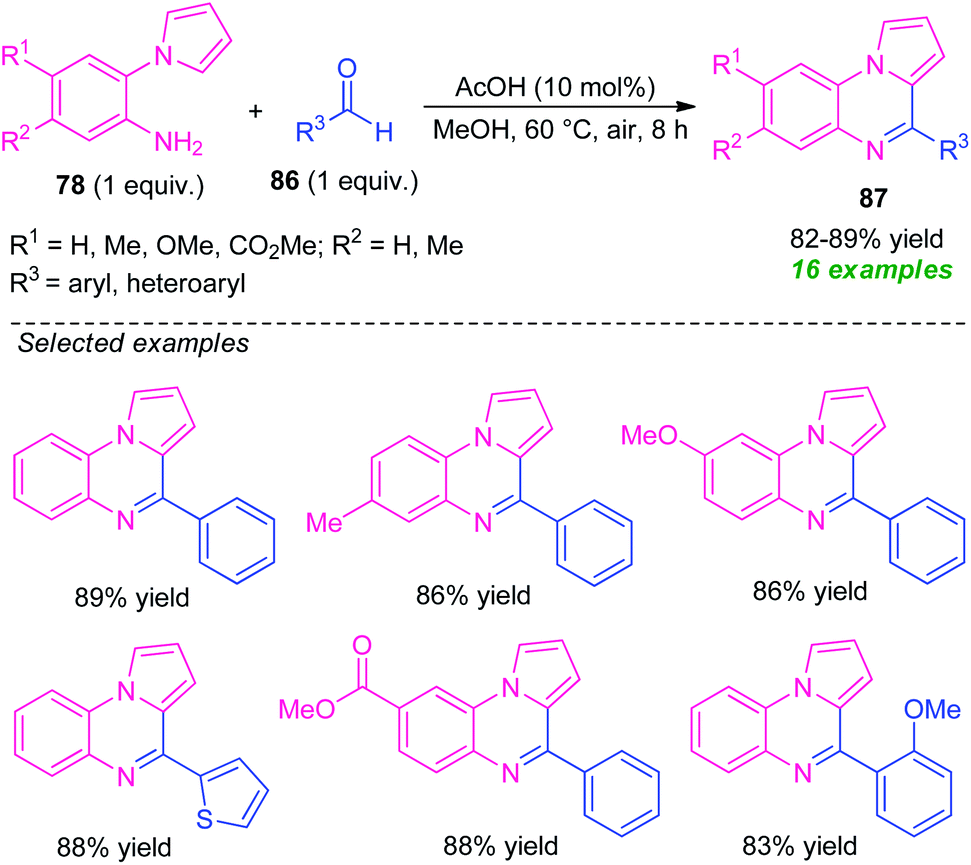 | ||
| Scheme 28 Acetic acid-catalyzed efficient synthesis of pyrrolo[1,2-a]quinoxalines through Pictet–Spengler reaction. | ||
In 2020, Jung and their groups demonstrated α-hydroxy acid as an aldehyde surrogate for the construction of pyrrolo[1,2-a]quinoxalines based on a metal-free approach (Scheme 29).102 The synthesis involves the reaction of 2-(1H-pyrrol-1-yl)aniline 78 with α-hydroxy acid 88 in presence of TBHP (tert-butyl hydrogen peroxide) as the oxidant, leading to the desired products 89 in 38–76% yields. The key step behind this reaction is the in situ formation of aldehydes from α-hydroxy acid 88 and then condensation with 78, followed by intramolecular cyclization and subsequent oxidation step. Although diverse substitutions on the 2-(1H-pyrrol-1-yl)aniline ring and α-hydroxy acid could be tolerable, the low yield of the products, as well as the longer reaction time, constitutes a major drawback of this approach other than outstanding work.
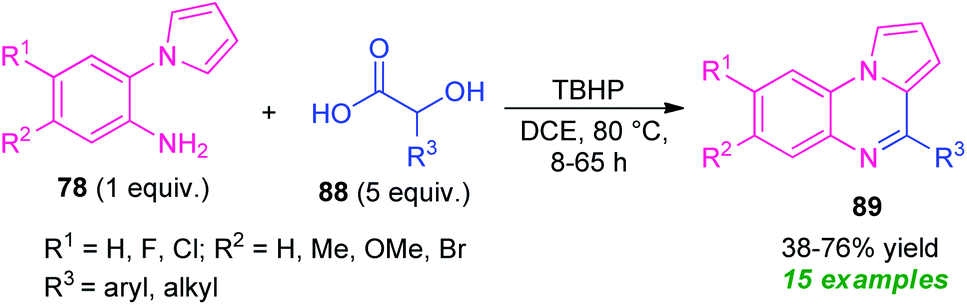 | ||
| Scheme 29 TBHP catalyzed metal-free synthesis of quinoxalines based on in situ generated aldehydes. | ||
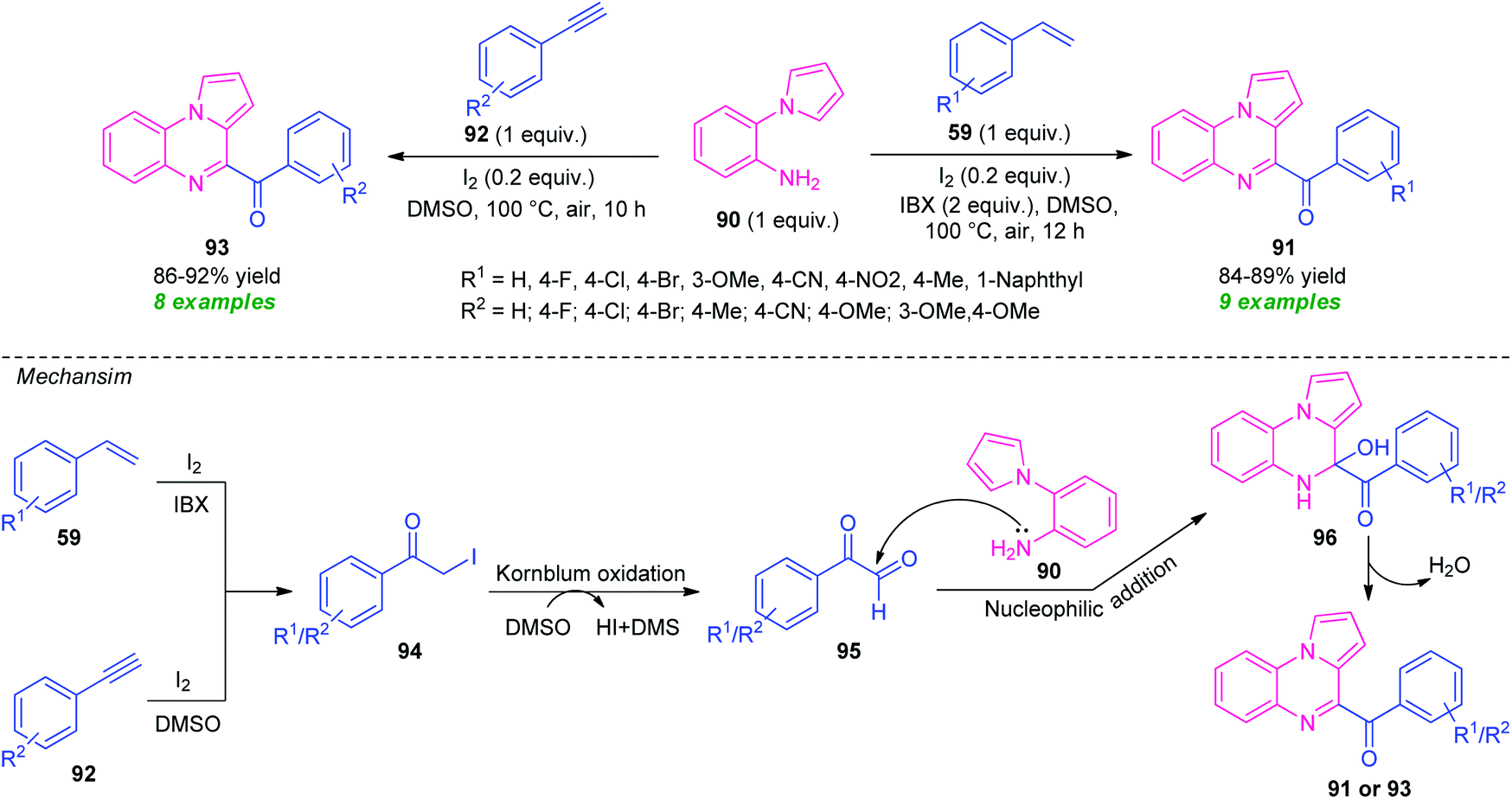 | ||
| Scheme 30 Metal-free diversity-oriented one-pot synthesis of fused quinoxaline derivatives from alkenes and alkynes. | ||
Initially, intermediate 94 was formed in situ either from 59 or from 92, which undergo Kornblum oxidation with DMSO to form an intermediate 95. A subsequent reaction of 95 with 90, followed by dehydration afforded the final products 91 or 93.
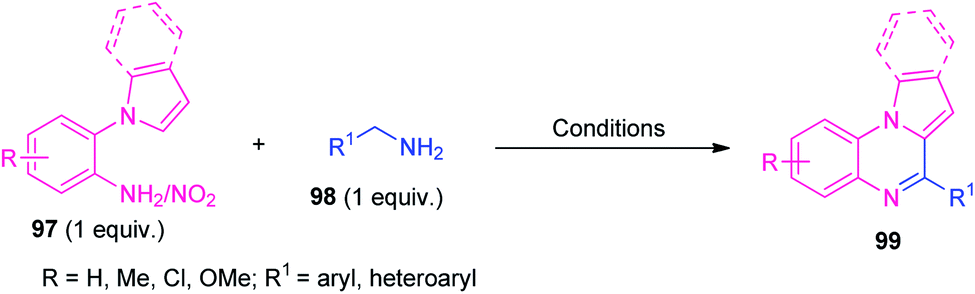
In 2015, Zhai et al., and Jayaprakash et al., independently reported two highly efficient metal-free one-pot strategies toward the synthesis of pyrrolo/indolo[1,2-a]quinoxalines 99 from 1-(2-aminoaryl)pyrrole/indole 97 and amines 98 (R1 = aryl) by employing molecular iodine as the catalyst as well as the promoter respectively. In 2019, direct one-pot access to several pyrrolo/indolo[1,2-a]quinoxalines 99 under metal-free green reaction conditions from 1-(2-amino/nitroaryl)pyrrole/indole 97 and amine 98 (R1 = aryl/alkyl/heteroaryl) were reported separately by Pardeshi, Chaskar, Patil and their groups and Wang et al. In all the cases, the corresponding products were formed in good to excellent yield (Table 2).105–109 Broad functional group tolerances, eco-friendly as well as environmentally friendly nature, operational simplicity, metal-free, additive-free, redox reagent-free, toxic-free, gram-scale synthesis, are some of the advantages of all of the reported methods. Although all the reported methodology has several significant advantages, they also suffer some limitations including longer reaction time, low yield of the products, higher energy source; products were isolated by conventional purification techniques that utilize volatile solvents and are also involved in the waste of solvents. Therefore, attention has also needed to be paid to these reported methodologies, to solve the concerns raised.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Catalyst/promoter | Loading | Oxidant | Solvent | Temperature | Time (h) | Examples | Yield (%) | Ref. |
| a 1,2-Dinitrobenzene.b Activated carbon. | |||||||||
| 1 | I2 | 5 mol% | O2 | o-Xylene | 140 °C | 12 | 27 | 79–95 | 105 |
| 2 | I2 | 2 equiv. | — | MeCN | 80 °C | 5–8 | 22 | 82–95 | 106 |
| 3 | — | — | K2S2O8 | PEG-400 | r.t | 8 | 25 | 65–92 | 107 |
| 4 | — | — | 1,2-DNBa | Diglyme | 130 °C | 9 | 25 | 62–89 | 108 |
| 9 | Acb | 15 mol% | — | H2O | 140 °C | 20 | 11 | 41–99 | 109 |
Another successful example for the synthesis of fused quinoxalines from amines under transition-metal-free conditions has been realized by Ma and co-workers (Scheme 31).110 With the help of (NH4)2S2O8 as the oxidant, the pyrrolo/indolo[1,2-a]quinoxaline products 102, derived from 1-(2-aminoaryl)pyrrole/indole 100 and α-amino acid 101, were obtained in poor to high yield. The optimized reaction conditions were found to be very compatible for a wide range of substituted 1-(2-aminoaryl)pyrrole/indole 100 possessing electron-deficient and electron-rich groups as well as for a variety of short- and long-chain amino acids. However, the reaction with methyl, tert-butyl, and cyclohexyl substituted amino acids (R1 = Me, t-Bu, cyclohexyl) gave only 26%, trace, and 31% yield of the products respectively. This is presumably due to involved steric hindrance that affects the outcome of this approach. Therefore, the development of a novel protocol that provides a clean pathway for expanding the hindered substrates scopes is highly desired.
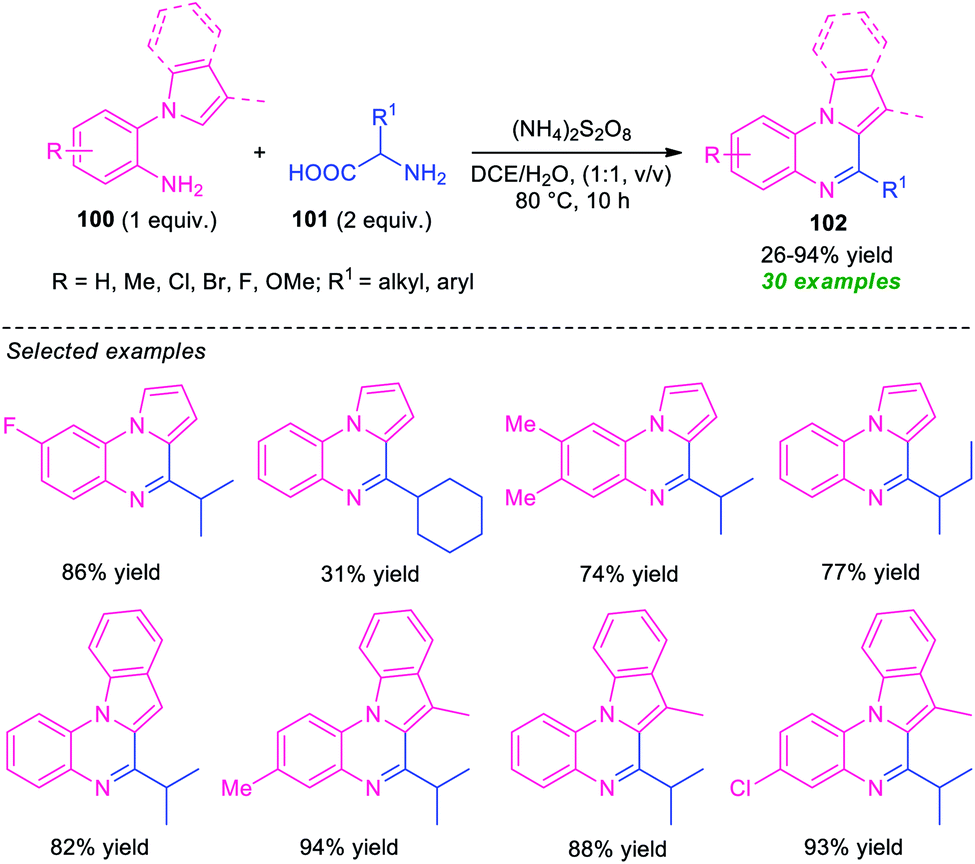 | ||
| Scheme 31 Transition-metal-free synthesis of fused quinoxalines from α-amino acid. | ||
4.2 Synthesis of imidazo[1,5-a]- and imidazo[1,2-a]quinoxalines
Unlike pyrrolo/indolo[1,2-a]quinoxalines, the derivatives of imidazoquinoxalines including imidazo[1,5-a]quinoxalines and imidazo[1,2-a]quinoxalines have extensively been studied and attracted considerable attention in medicinal and organic chemistry due to their tremendous biological activity such as anticancer activity, antimicrobial activity, antiallergenic activity, anticonvulsant activity, and kinase inhibitory activity.111 Because of this importance, significant efforts have been made for their synthesis.112In 2015, Ma and co-workers developed a metal-free one-pot cascade coupling process for the construction of imidazo[1,5-a]quinoxalines 105 (Scheme 32).113 By using 20 mol% of molecular iodine as the catalyst and DMSO as the oxidant as well as solvent, the reaction of 2-(1H-imidazol-1-yl)aniline 103 and ketones 104 were found to proceed smoothly under nitrogen atmosphere to afford the desired products 105 in good to high yield. Aryl-substituted ketones as well as substituted 2-(1H-imidazol-1-yl)aniline efficiently participated in the reaction under this condition. The advantages of this approach included mild reaction conditions, eco-compatible, readily accessible starting material, etc. However, the low substrate scopes alongside high catalyst loading, long reaction time constitutes a major limitation of this approach and needs ample attention to extended the substrate scopes of this approach.
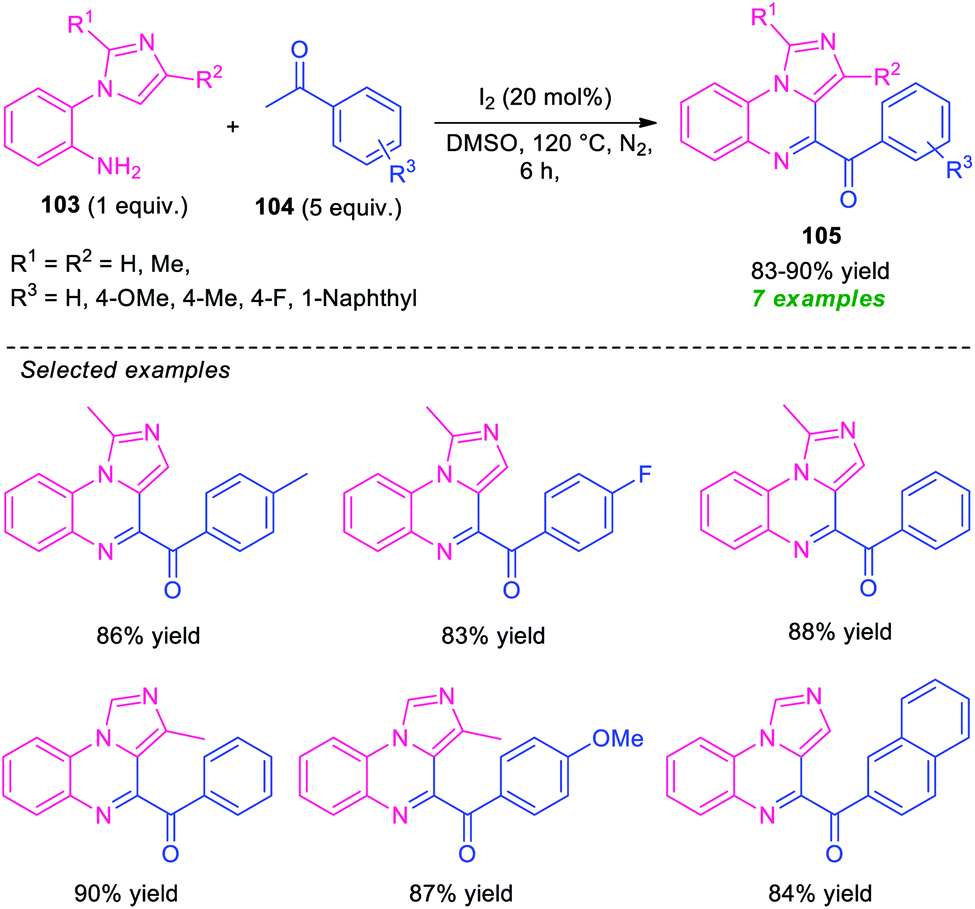 | ||
| Scheme 32 Metal-free iodine catalyzed one-pot synthesis of imidazo[1,5-a]quinoxalines 105. | ||
Highly regioselective synthesis of imidazo[1,5-a]quinoxalines via an unconventional Pictet–Spengler reaction was reported by Chung, Sun, and their group in 2016 (Scheme 33).114 Treatment of ionic liquid supported 2-(1H-imidazol-1-yl)aniline 106 with ketones 107 in presence of TFA under microwave condition at 130 °C, afforded ionic liquid immobilized imidazo[1,5-a]quinoxalines 108 within 20 minutes. Subsequently, cleavage of ionic liquid from 108 was accomplished by KCN in MeOH at room temperature to furnish the final products 109 in moderate to high yield. The reaction condition was found to be compatible with a broad range of aryl, heteroaryl, and alkyl-substituted ketones. However, when the same reaction was carried out with aryl aldehydes instead of ketones, aryl derivatives of imidazo[1,5-a]quinoxalines bearing ionic liquids were formed, but on treating with KCN for removal of ionic liquids, only cleavage of C(3a)-C(4) bond was observed rather than getting the final products. Pleasingly, modify the reaction condition from microwave to reflux in CH3CN rather than toluene, aliphatic aldehydes found to underwent the reaction smoothly providing the final imidazo[1,5-a]quinoxalines 112 in 76–80% yields. However, narrow substrate scopes represent a limitation of this approach.
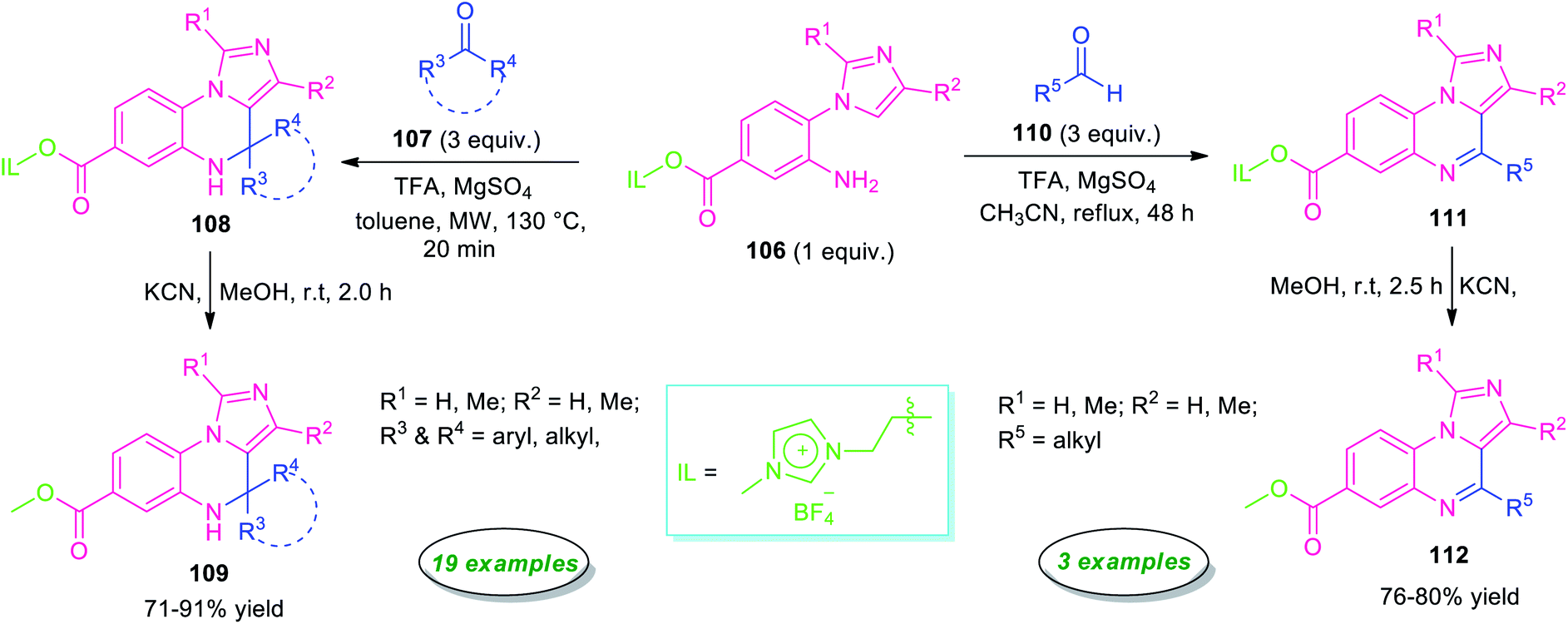 | ||
| Scheme 33 Regioselective metal-free synthesis of imidazo[1,5-a]quinoxalines from ionic liquid supported 2-(1H-imidazol-1-yl)aniline. | ||
As a continuation of their outstanding work on the synthesis of pyrrolo/indolo[1,2-a]quinoxalines described in Scheme 27, Xie and co-workers also disclosed the synthesis of imidazo[1,5-a]quinoxalines 113 by employing metal-free reaction condition (Scheme 34).100 With the help of DMSO as the solvent cum reagent, the corresponding imidazo[1,5-a]quinoxalines 113 derived from 1-(2-aminophenyl)imidazole 103, has been achieved in 18–82% yields. However, narrow substrates scopes and low yield of the products call for further development of this approach.
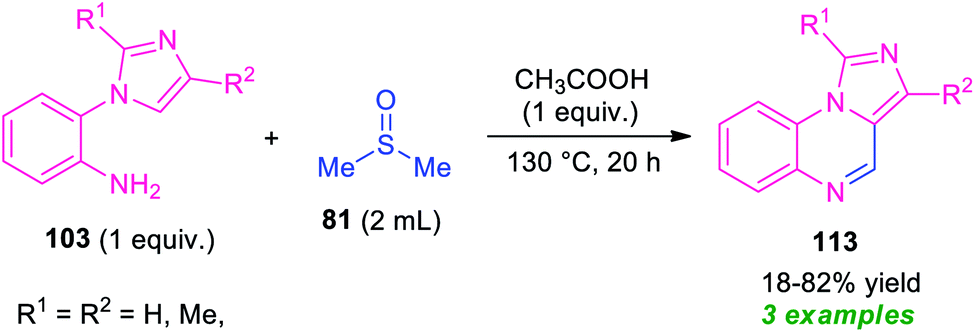 | ||
| Scheme 34 Synthesis of imidazo[1,5-a]quinoxalines 113 by using DMSO as solvent cum reagent in metal-free condition. | ||
In 2018, Kumar et al. introduced the microwave technique as a powerful green energy source for the synthesis of diverse imidazo[1,2-a]quinoxalines via a 6-endo-trig cyclization of 5-amino-1-(2-aminophenyl)-imidazole-4-carbonitriles 114 with carbonyl compounds (Scheme 35).115 By using p-TsOH as a metal-free catalyst, treatment of the equimolar amount of aldehyde 115 with 114 performed in an open condenser under microwave heating at 80 °C, provides imidazo[1,2-a]quinoxalines 116 in 84–89% yields. While the same reaction, when carried out with 2 equivalents of 115 and 1 equivalent of 114, the products 117 were formed in 75–93% yields. Conversely, the reaction of an equimolar amount of ketone 118 and 114 performed in a sealed tube under microwave irradiation, afforded dihydroimidazo[1,2-a]quinoxalines 119 in 74–95% yields, whereas, employing 2 equivalents of 118 under the same reaction condition deliver products 120 in moderate to good yield. The utilization of microwave techniques not only reduces the reaction time in both cases but also provides a clean pathway for these reactions and improves the yield of the products as compared to the conventional method.
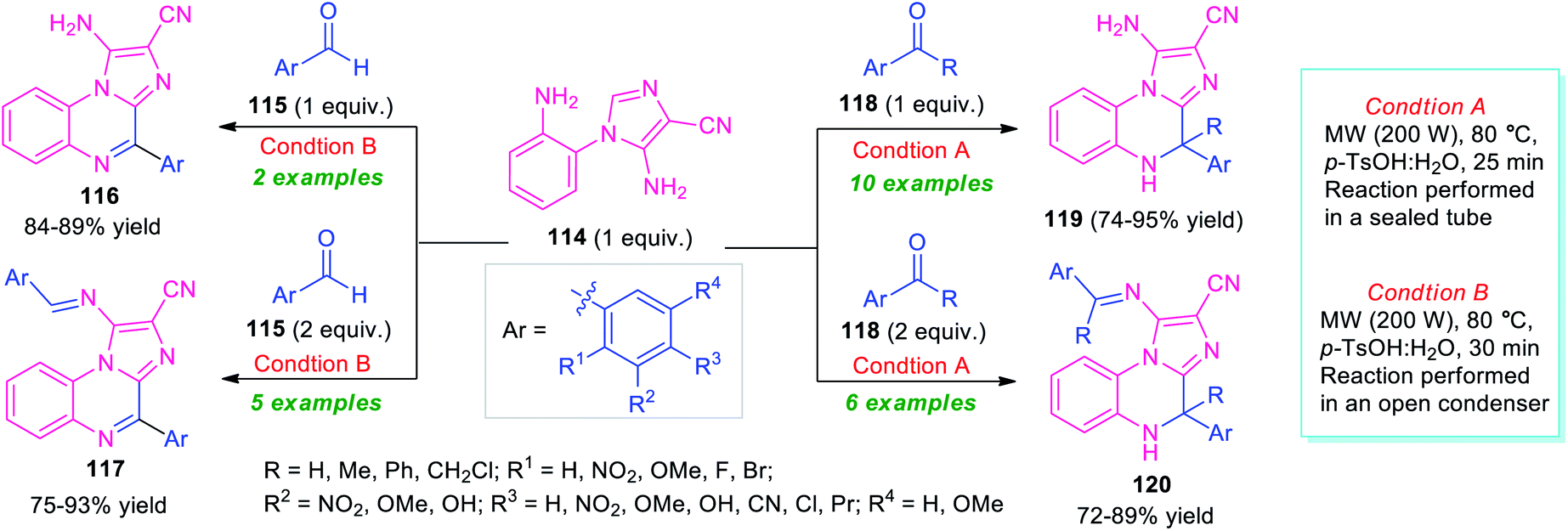 | ||
| Scheme 35 Metal-free microwave-assisted one-pot preparation of library of imidazo[1,2-a]quinoxalines. | ||
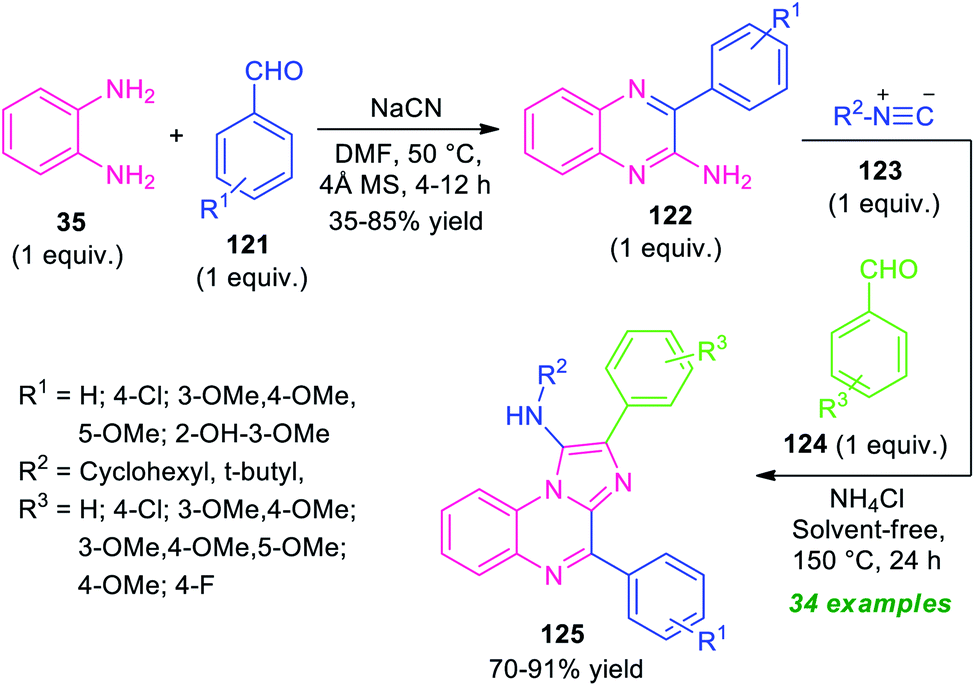
A step-wise synthetic route for imidazo[1,2-a]quinoxalines 125 has been realized recently by Amini and his groups (Scheme 36).116 In the initial step, the reaction of o-phenylenediamine 35 with aldehydes 121 was carried out by using NaCN as a catalyst in DMF at 50 °C. This step requires 4 Å molecular sieves as the additives. The resulting amino-substituted quinoxalines 122 were formed in 35–85% yields within 4–12 hours. The second step involves a multicomponent reaction of an equimolar amount of 122 with isocyanides 123 and aldehydes 124 using ammonium chloride as the catalyst in solvent-free condition at 150 °C for 24 hours. The corresponding imidazo[1,2-a]qunioxaline derivatives 125 were formed in moderate to good yield. A variety of aryl aldehydes possessing different electron-withdrawing as well as electron-donating substituents well worked by this stepwise protocol. Broad functional group tolerances, solvent-free conditions, metal-free nature, a high level of yield are some of the salient features of this approach.
 | ||
| Scheme 36 Two-step synthesis of imidazo[1,2-a]quinoxalines in metal-free condition. | ||
4.3 Synthesis of other fused quinoxalines
A metal-free tandem cyclization approach to access other fused quinoxalines, namely isoindolo[2,1-a]quinoxalines 127 has been demonstrated by Das et al., in 2019 (Scheme 37).117 To find out the optimum condition, the reaction of ninhydrin 126 bearing phenolic adducts, with o-phenylenediamine 35 was initially performed in the presence of different bases including Et3N, piperazine, pyridine, Et2NH, and solvent system like ethylene glycol, water, MeOH, EtOH, glycerol. From these experiments, it was concluded that the utilization of Et3N in ethylene glycol provides easy access to the products within a very short reaction time. The optimum reaction condition was found to be compatible for a wide range of substituted phenolic ninhydrin adducts 126 that smoothly underwent the reaction to deliver the isoindolo[2,1-a]quinoxaline products 127 in 63–78% yields. Most of the synthesized compounds were found to have a fluorescent activity which remarks outstanding achievements of this work.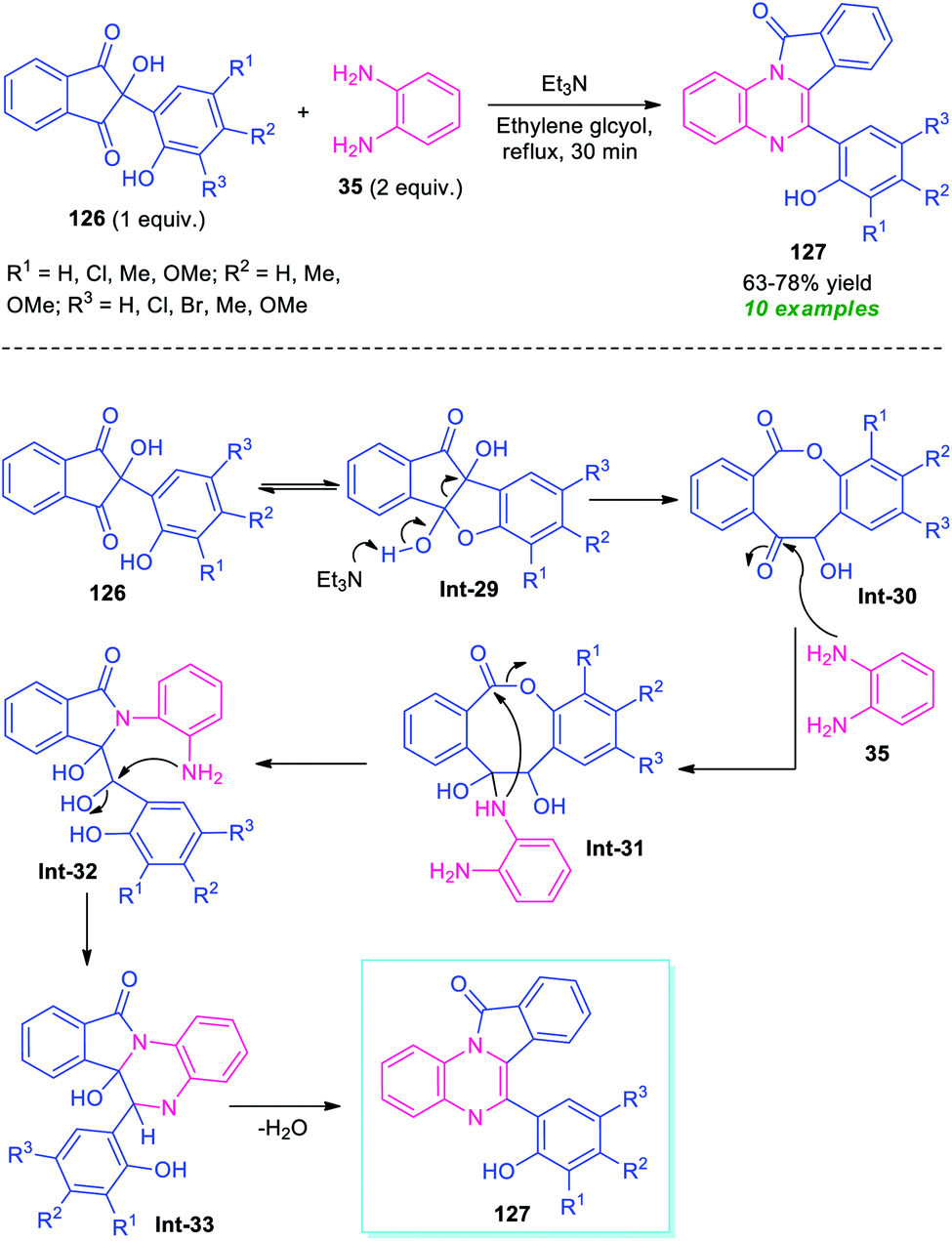 | ||
| Scheme 37 Metal-free base promoted tandem cyclization approach toward the rapid access of fused quinoxalines. | ||
A mechanistic investigation suggests the initial formation of 8-membered lactone intermediate Int-30 from Et3N catalyzed cleavage of C–C bond of Int-29. The lactone Int-30 then experiences nucleophilic attack from –NH of 35 to form an intermediate Int-31, which provides intermediate Int-32 via an intramolecular nucleophilic attack of –NH to CO moiety and subsequent breaking of C–O bond. Consequently, the intramolecular cyclization of (Int-30), followed by elimination of water leads to the final products 127 (Scheme 37).117
5. Synthesis of simple quinoxalines based on metal-free multicomponent reactions
The development of an efficient concise route that provides easy access to highly functionalized and value-added compounds by fulfilling the criteria of green and sustainable chemistry has emerged as a most significant goal and formidable challenge for the synthetic chemist. Compared to synthetic step-wise process which deals with a number of limitations in the product development as well as in consistent with green chemistry principles, multicomponent reactions (MCRs) has recently come to receive widespread global acceptances as the most valuable choice of synthetic routes in industry and academia.118–121 Multicomponent reactions (MCRs) provide a clean and safe platform for multiple-bond forming events to occur in a single one-pot operation from more than two starting materials without separation and purification of the intermediates. Consequently, this reaction increases the product development rates by shortening the reaction steps, and also by reducing waste formation, utilizing inexpensive easily accessible reagents and reactants, etc.As a consequence of these tremendous academic, economic and ecological interests, an isocyanide-based multicomponent approach122 for the synthesis of highly functionalized quinoxalines has been realized by Rouhani and Ramazani in 2018 (Scheme 38).123
 | ||
| Scheme 38 Ultrasound-assisted isocyanide-based multicomponent approach for the synthesis of quinoxalines 129. | ||
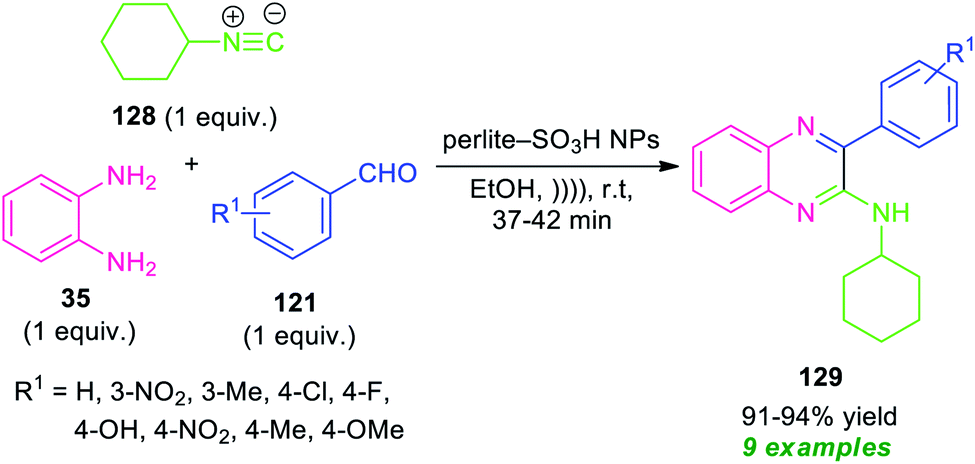
By using perlite-SO3H nanoparticles as the catalyst, the three-component reaction of o-phenylenediamine 35, aldehydes 121, and cyclohexyl isocyanide 128 under ultrasonic irradiation were found to take place smoothly within a very short reaction time to provide the easy access of corresponding quinoxaline derivatives 129 in 91–94% yields. A variety of electron-withdrawing, as well as electron-donating groups present on the aryl ring of aldehydes, were well tolerated by this methodology. The exploitation of ultrasonic technology provides a clean synthesis of the desired quinoxaline products within a very short reaction time as compared to the conventional method which takes lots of time for the completion of the reaction.
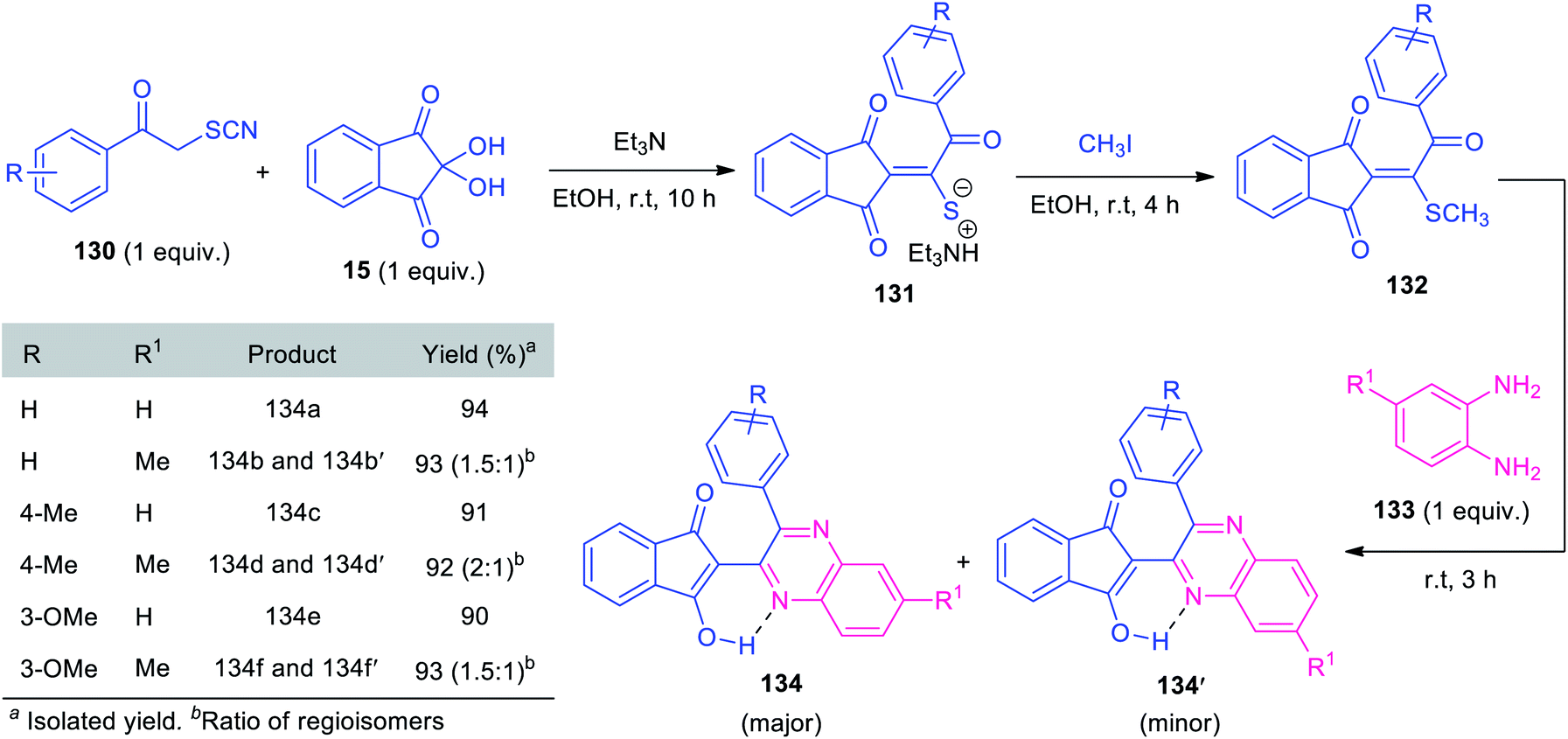
In 2019, Alizadeh and co-workers synthesized triethylammonium thiolate salts 131 based on a formal [3 + 2] cycloaddition, followed by subsequent ring-opening reaction of phenacyl thiocyanate 130 with ninhydrin 15 and has been demonstrated as an efficient reagent for the synthesis of quinoxalines. Under metal-free reaction conditions, a one-pot sequential three-component reaction of triethylammonium thiolate salts 131, with methyl iodide and o-phenylenediamines 133 was found to proceed smoothly to deliver several quinoxalines 134 bearing indandione framework, in high yields (Scheme 39).124 Compound 131 containing various electron-rich groups efficiently participated in this reaction. Similarly, simple o-phenylenediamine 133a (R1 = H) selectively afforded 134 as the major products. However, when methyl-substituted diamine 133b (R1 = Me) was employed as the substrates, two regioisomeric products (134 and 134′) were achieved. Despite the remarkable advantages including mild condition, high yield, operational simplicity, and eco-benign, the limited substrate scope represents a limitation of this approach and calls for further developments.
 | ||
| Scheme 39 Application of triethylammonium thiolate salts as efficient reagents for the multicomponent synthesis of quinoxalines 134. | ||
6. Synthesis of fused quinoxalines based on metal-free multicomponent reactions
A novel one-pot three-component domino reaction between o-phenylenediamine 1, 2-hydroxynaphthalene-1,4-dione 135, and tetracyanoethylene 137 using pyridine as a catalyst in ethanol at room temperature was presented by Mohebat and co-authors for the efficient synthesis of pyrano-fused quinoxalines 138 (Scheme 40).125 The resulting quinoxalines 138 were obtained in good to high yields. The initial step involving the reaction of 1 with 135 takes place within 5–10 minutes under reflux condition in ethanol to form benzo[a]pyrano[2,3-c]phenazine 136, which on treating with 137 under the influence of 10 mol% of pyridine afford the final products 138. The products were simply isolated by recrystallization techniques that do not require further conventional purification procedures. Although the presented approach provides several key features like short reaction time, cost-effectiveness, operation simplicity, however, the narrow substrate scopes again constitute a limitation toward this method and calls for further development.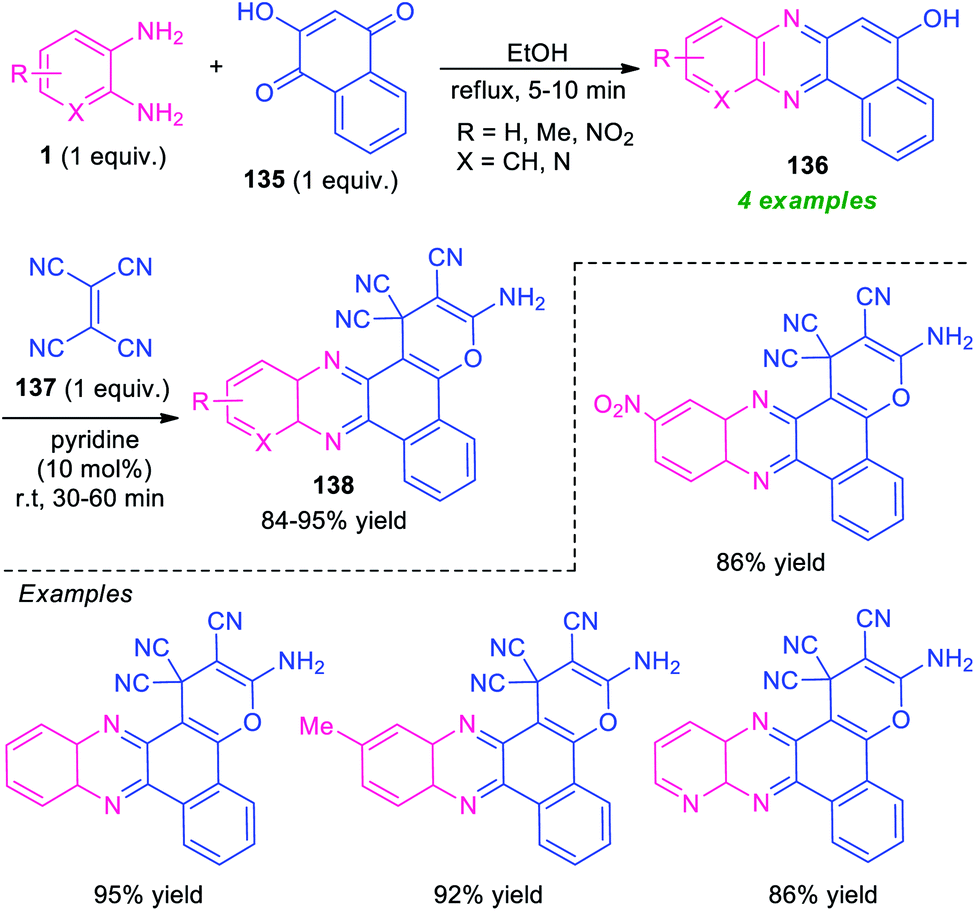 | ||
| Scheme 40 One-pot two-step three-component synthesis of pyran-fused quinoxalines. | ||
For the construction of diverse annulated quinoxalines, ninhydrin was employed as a versatile and easily accessible starting material. In this regard, Maghsoodlou and co-workers in 2017, disclosed a one-pot multicomponent domino protocol under the metal-free condition to access a variety of spiro-furan-indeno[1,2-b]quinoxalines in 85–92% yields by employing ninhydrin 15 as the starting material, alongside o-phenylenediamine 1, and dialkyl acetylene dicarboxylate 139 (Scheme 41).126 The reaction was performed in presence of 5 mol% of triphenylphosphine (PPh3) in CH2Cl2 at ambient temperature that could be completed within 6–8 hours. In this transformation, PPh3 reacts with 139 to produce 1,3-dipolar intermediate Int-34, which eventually attacks the CO of spiro-indeno[1,2-b]quinoxaline Int-35 formed in situ from the condensation of 15 and 1. The resulting zwitterionic intermediate Int-36 underwent intramolecular cyclization to form an intermediate Int-37, which is followed by subsequent rearrangement to yield the desired products 140.
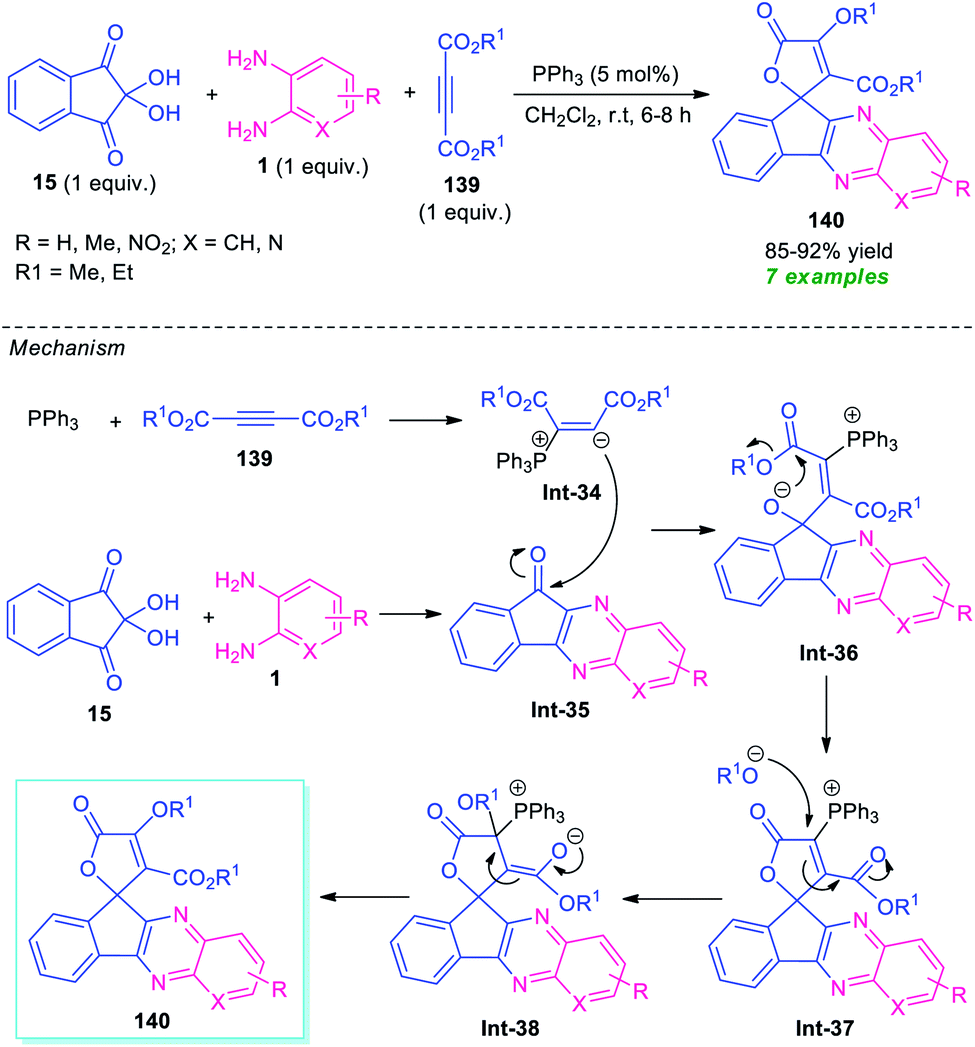 | ||
| Scheme 41 PPh3 catalyzed one-pot domino reaction to access spiro-fused quinoxaline 140. | ||
Around the same time, Heravi and Norouzy showed the successful application of ninhydrin in the multicomponent reactions for the synthesis of a library of fused quinoxaline derivatives by employing ultrasonic techniques as a non-conventional strategy (Scheme 42).127 With the catalyst-free condition, treatment of ninhydrin 15 with o-phenylenediamine 35, active methylene compound 141, and amines 142 or 144 in presence of trifluoroethanol (TFE) under ultrasound irradiation at room temperature provides easy access of spiro[benzo[g]quinoline-4,10′-indeno[2,3-b]quinoxaline] 143 and spiro[benzo[h]quinoline-4,10′-indeno[2,3-b]quinoxaline] 145 in high yield respectively. On the other hand, the same reaction condition was found to be very suitable for various CH-activated acidic compounds 146 that react with ninhydrin 15, o-phenylenediamine 35, and active methylene compound 141 to form various spiro-fused quinoxalines 147 in good to excellent yield. All the tested CH-activated acidic compounds efficiently worked well under this condition. A comparative study for both conventional and ultrasonic methods revealed that the conventional method utilized a higher amount of energy, as well as a long reaction time as compared to ultrasonic conditions. Conversely, the exploitation of ultrasound not only shortened the reaction time but also improves the yield of the products.
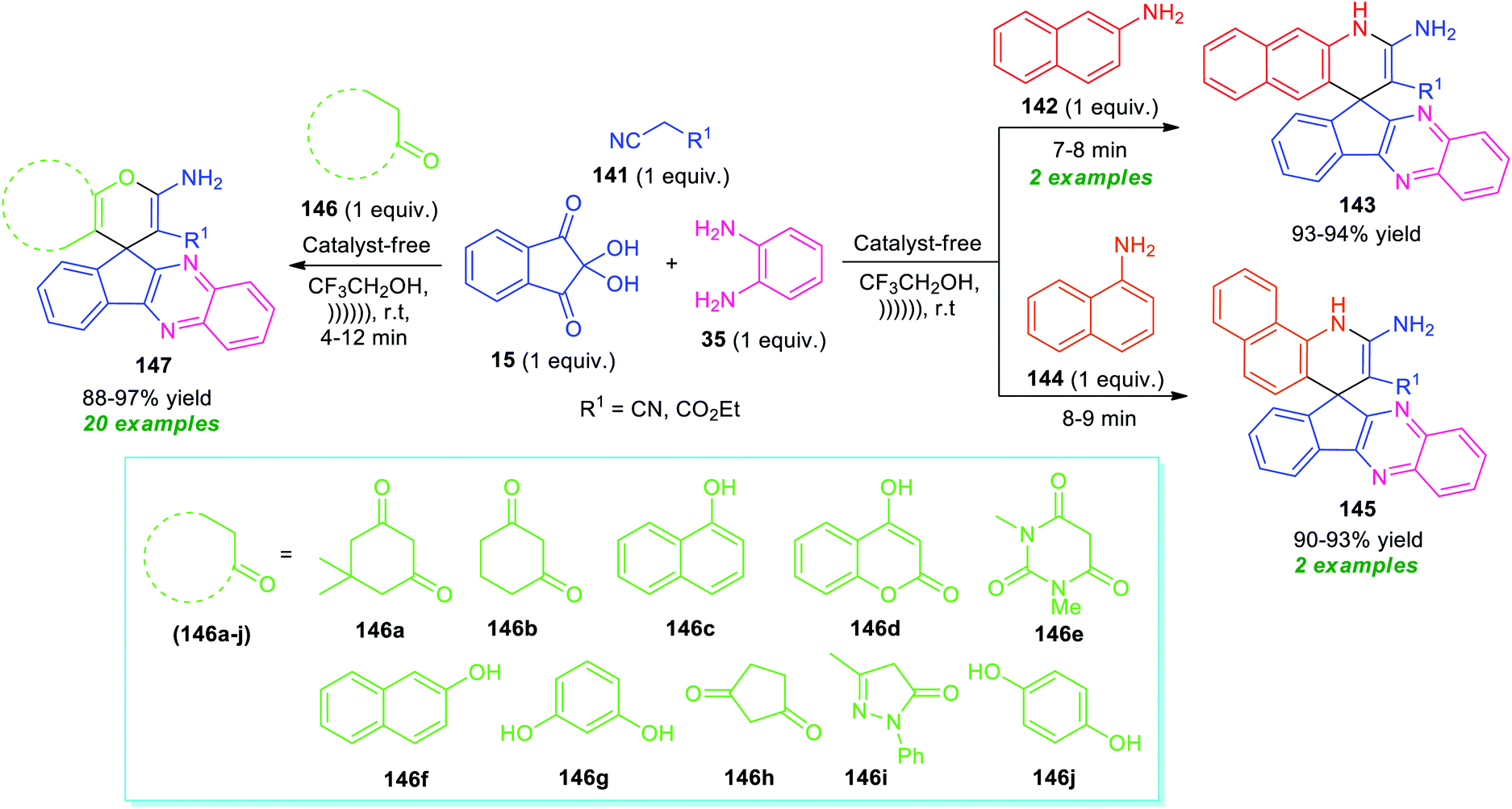 | ||
| Scheme 42 Ultrasound irradiated one-pot construction of several fused quinoxaline derivatives under metal-free condition. | ||
Unlike ultrasonic techniques,128 the attractiveness of microwaves129 for the synthesis of promising molecular structures has recently gained favour due to its remarkable advantages associated with the synthetic practitioner like mild-condition, reduces reaction times, enhances product yield, and provides high purity of products as well as green chemistry point of view.
In 2017, Kantam, Trivedi, and co-authors introduced a microwave technique to develop a convenient strategy for the four-component synthesis of spiro-indeno[1,2-b]quinoxaline-11,3′-pyrrolizines derivative 150 based on an azomethine ylide 1,3-dipole mediated [3 + 2]-cycloaddition reaction (Scheme 43).130 The synthesis starts with the treatment of ninhydrin 15, o-phenylenediamine 35, L-proline 148, and β-nitrostyrene 149 in microwave heating at 80 °C in ethanol, to form the corresponding products 150 as single regio- and diastereomer in good to high yield. Not only aryl-substituted β-nitrostyrene bearing various electron-rich and electron-poor substituents but also heteroaryl as well as alkyl-substituted β-nitrostyrene were well tolerated by this method. The use of conventional heating conditions instead of microwave techniques required a longer reaction time for the completion of the reaction and also a slightly lower yield of the product was observed. The exploitation of microwaves provides a clean reaction profile, shortened the reaction time, and increases the product yield. Other salient features of this approach included broad functionality, eco-friendly, and environmentally benign nature.
 | ||
| Scheme 43 Microwave-assisted regio- and stereoselective one-pot four-component synthesis of spiro-fused quinoxalines. | ||
To rationalize the stereochemistry of this regioselective dipolar cycloaddition of azomethine ylide Int-41 generated from 15′, 35, and 148 to a dipolarphile 149a, two transition states were proposed, TS-A and TS-B based on steric considerations. As shown in Scheme 43, only the endo-product 150 was formed. This is presumably due to the thermodynamically more stable TS-B, while the exo-product 150′ was not formed due to more steric repulsion (TS-A).
An acid-free Ugi-deprotection-cyclization (UDC) approach toward the synthesis of quinolinone-fused quinoxalines has been demonstrated by Xu and co-workers (Scheme 44).131 The overall process starts with the initial post-Ugi three-component reactions of 2-oxo-2-phenylacetaldehyde 151, N-Boc-protected o-phenylenediamine 152, and isocyanide 123 under the influence of phenyl phosphonic acid (PPOA) as the catalyst in MeOH at room temperature. The resulting Ugi product 153, on treating with 10 mol% of TFA in 1,2-dichloroethane (DCE) under microwave heating condition at 110 °C, afforded the adduct 154 via deprotection and cyclization sequence. Subsequently, a nucleophilic substitution reaction of 154 was occurred in presence of Cs2CO3 in DMF in microwave condition at 150 °C to furnish the final products 155. This multi-component reaction provides a total of six compounds in 37–51% yields from different substituted o-phenylenediamine, aryl as well as alkyl-substituted isocyanide and 2-oxo-2-phenylacetaldehyde.
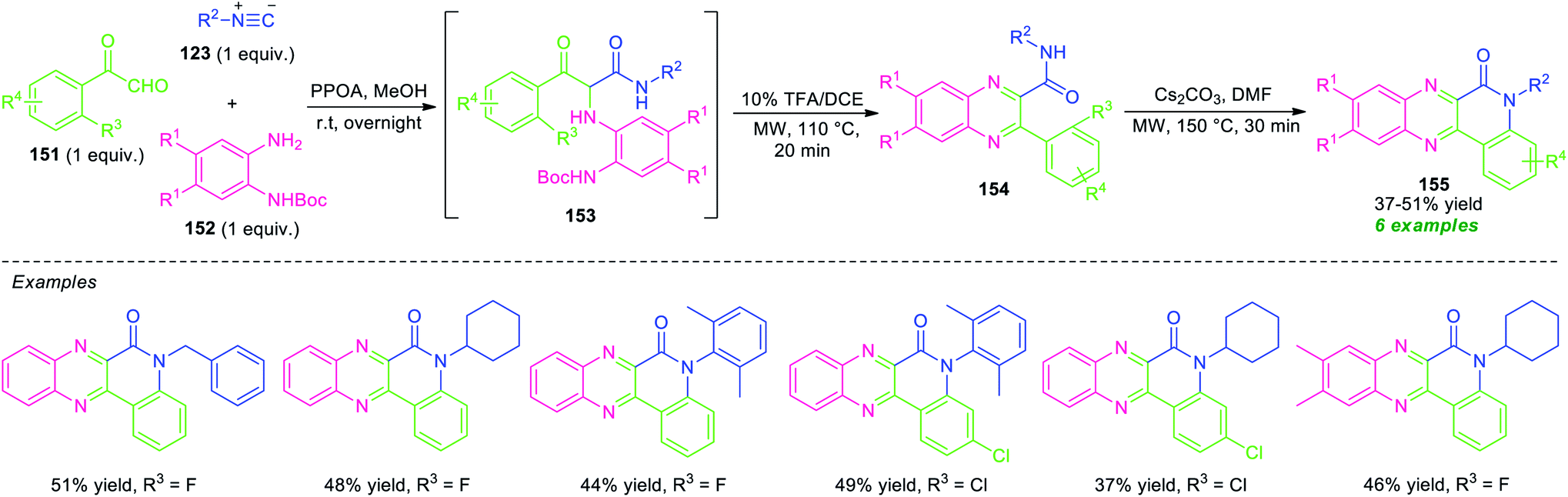 | ||
| Scheme 44 Acid-less Ugi-deprotection-cyclization-substitution sequence for the synthesis of quinolinone-fused quinoxalines. | ||
In 2018, Chowhan et al. disclosed a catalyst-free four-component reaction of ninhydrin 15, o-phenylenediamine 5, 3-methyl-4-nitro-5-alkenylisoxazoles 156, and benzylamine 157 under reflux condition in methanol for 3 hours to provide a series of spiro-indeno[1,2-b]quinoxaline-pyrrolidines 158 (Scheme 45).132 This multicomponent approach proceeds via an in situ generated azomethine ylide 1,3-dipole mediated [3 + 2] cycloaddition reaction. The resultant products were achieved in moderate to high yield with outstanding diastereoselectivity. A variety of differently substituted aryl and heteroaryl rings of 156, as well as unsubstituted or methylated diamines 5, were well tolerated by this method. However, nitro-substituted diamines failed to yield any product. This is presumably due to the electron-poor nature of the nitro group that makes the substrate labile.
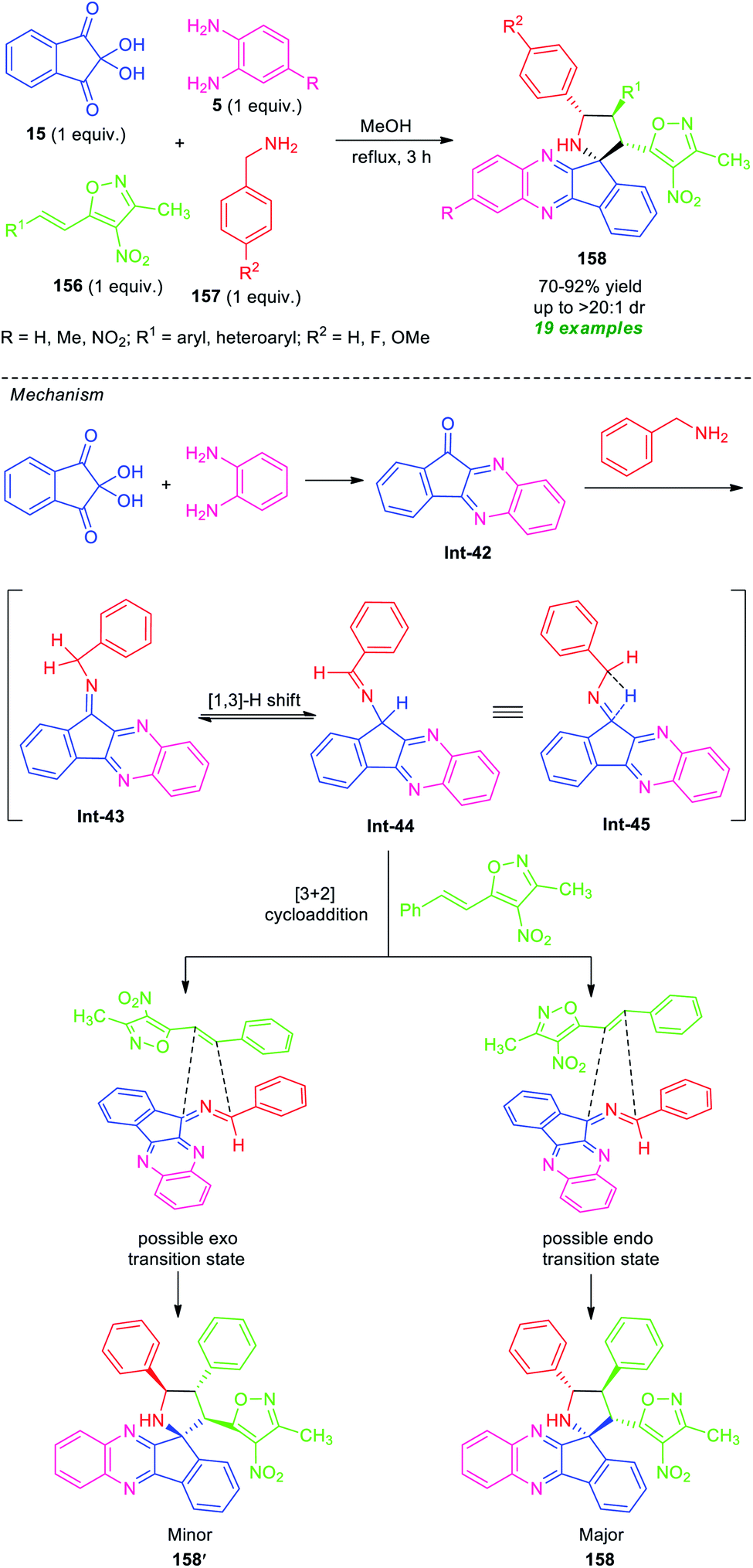 | ||
| Scheme 45 Catalyst-free regio- and diastereoselective one-pot synthesis of spiro-fused quinoxalines. | ||
Based on a series of observations, a mechanism and transition state to account for the stereochemistry were proposed by the authors. Initial dehydration reaction between ninhydrin, o-phenylenediamine, benzylamine provided the isomeric intermediate Int-43 and Int-44 which can be equilibrium to intermediate Int-45. This azomethine ylide type intermediate Int-45, then stereoselectively undergoes [3 + 2] cycloaddition with phenyl-isoxazole via two transition states. The endo isomer 158 was formed as the major isomer over exo product 158′. This can be attributed based on the fact that in 158, the 3-methyl-4-nitroisoxazoles core is positioned closer to the dipolar region that is opposite in 158′.132
Recently, a transition-metal-free one-pot domino process for the synthesis of diverse pyrrolo/indolo[1,2-a]quinoxaline derivatives has been realized by Mandal and Pramanik (Scheme 46).133 By using 50 mol% of p-TsOH as the catalyst, a three-component reaction of N-(2-aminophenyl)pyrroles/indoles 159, with various cyclic 1,2-dicarbonyl compounds 160 or 163 and alcohols 161 or aliphatic amines 166 were performed in open-air heating condition. The reactions provide a variety of different fused quinoxaline derivatives 162, 164, and 167 in moderate to good yields. In these reactions, aliphatic amines and alcohols were acted both as solvent as well as the reactant. To explore the generality of the developed protocol, different N-(2-aminophenyl)pyrroles/indoles 159 and cyclic 1, 2-dicarbonyl compounds bearing electron-withdrawing as well as electron-donating groups, and amines or aliphatic alcohols were employed. All the substrates were found to be well tolerated. However, tertiary alcohols (tert-butyl alcohol and 2,2,2-trifluoroethanol) failed to yield any products by this method. This is presumably due to the steric crowding and reduced nucleophilicity of the free hydroxyl group. The wide substrate scopes, mild reaction condition, metal- and oxidant-free, air as green oxidant are some of the key features of this approach.
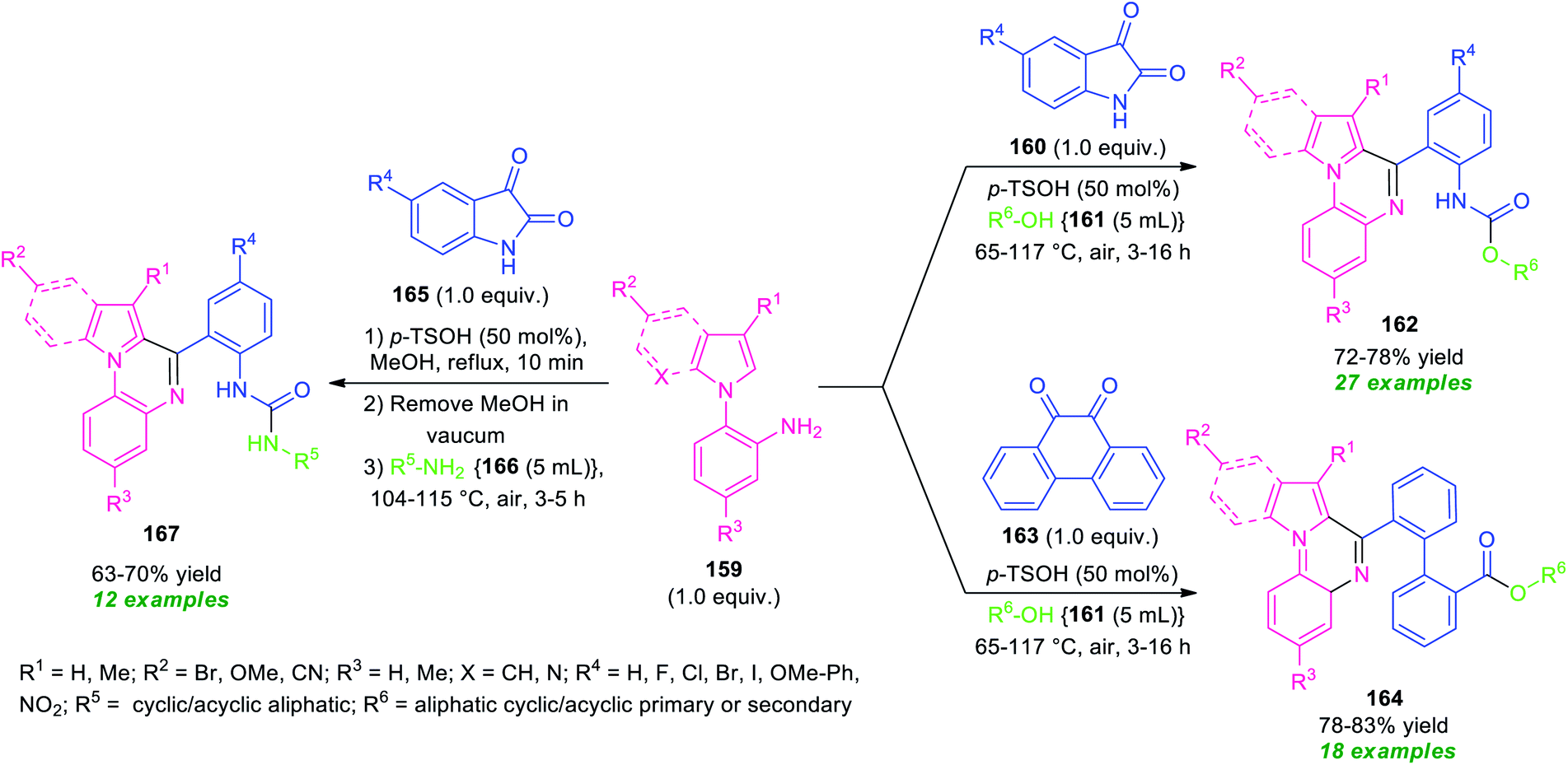 | ||
| Scheme 46 Metal-free solvent-mediated diversity-oriented one-pot domino synthesis of various fused quinoxalines. | ||
7. Conclusion and future perspective
Considering the great importance of the quinoxaline framework in many branches of chemistry including natural products chemistry, drug discovery, medicinal chemistry, material science, and agrochemical area, tremendous efforts have been dedicated toward their synthesis. Consequently, many more useful and efficient synthetic protocol has been developed. Synthetic modifications of the classical methods for the preparation of numerous quinoxalines have also emerged. However, most of them are not clearly focused on the development of an efficient route that not only enhances the yield of the product but also reduces the cost of the reaction, provides a clean reaction profile, atom-economic, and utilized an environmentally benign pathway. Therefore, the development of novel synthetic strategies that directs the efficient synthesis of quinoxalines inconsistent with the green and sustainable chemistry principles to make a toxic and waste-free nature are highly desired.In this pursuit, the last few years have witnessed special attention in the metal-free synthesis of quinoxalines by employing various non-metal inorganic and organic catalysts, ionic liquids, organocatalysts, molecular iodine, catalyst-free condition, green catalyst, visible-light-induced and non-conventional strategies like microwaves, and ultrasound irradiation. The exploitation of these reagents and techniques certainly makes these protocols environmentally as well as eco-friendly benign compared to those transition-metal-catalyzed routes. Despite, notable developments achieved in the transition-metal catalyzed synthesis, high cost involved in the preparation of the catalyst, toxicity, and difficulty in removing it from the final products constitutes disadvantageous effects on the atom economy and eco-friendly nature of the transformation.
In this review article, we have summarized the recent progress achieved in the synthesis of quinoxalines and various fused quinoxalines by employing two-component and multicomponent reactions under metal-free conditions and cover the reports from 2015 to date. This aspect is presented alongside the mechanistic rationalization and limitation of the reaction methodologies. The scopes of future developments are also highlighted.
As illustrated through the review, it is clear that these protocols have significant advantages such as a high level of yield, broad functional group tolerance, mild reaction condition, cost-effectiveness, etc. Other key features included the utilization of water as green solvents over other toxic and volatile solvents due to its remarkable properties like wide abundance, non-toxic, and inexpensive nature. Besides these, solvent-less protocols are also developed under metal-free conditions. In addition to these, the utilization of microwaves and ultrasonic techniques shortened the reaction times as well as enhanced the reactivity and selectivity of the reaction, and provides an excellent yield of the quinoxaline products.
On the other hand, the combination of the metal-free condition along with multicomponent reactions opened a new gateway for the easy access of highly functionalized quinoxalines.
Despite, notable advancement achieved in the metal-free synthesis of quinoxalines, limited substrate scopes, requirements of higher energy source, low yield of the products, longer reaction time, and high catalyst loading, constitutes the major limitations of some so far developed protocols. Therefore, ample attention needs to be paid in the upcoming days for broadening the substrate scopes with excellent yields by employing mild pathways which are safe, eco-benign, atom-economic, and provide scale-up synthesis in low catalyst loading or catalyst and metal-free condition for application to the industrial area. Also, the biological as well as material science applications of novel quinoxaline derivatives need to be screened and should be strengthened. We hope, the present review article will help researchers working on this fascinating area for the design and developments of more precise synthetic routes for the construction of quinoxalines and for further outstanding creation of a highly functionalized novel quinoxaline framework that would find immense application in the field of medicinal chemistry and optoelectronic materials.
Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
The author thank Central University of Gujarat, Gandhinagar, India, and Prof. Rama Shanker Dubey, Vice-Chancellor, the Central University of Gujarat for the encouragement and continuous support. BB thanks UGC-India for the Non-NET fellowship.Notes and references
- (a) J. A. Joule, Adv. Heterocycl. Chem., 2016, 119, 81–106 CrossRef CAS; (b) C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075–3081 CrossRef CAS; (c) N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed; (d) A. Chugh, A. Kumar, A. Verma, S. Kumar and P. Kumar, Med. Chem. Res., 2020, 29, 1723–1750 CrossRef CAS; (e) G. N. Lipunova, E. V. Nosova, V. N. Charushin and O. N. Chupakhin, Curr. Org. Synth., 2018, 15, 793–814 CrossRef CAS; (f) D. Chen, S. J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565–9578 RSC.
- (a) P. S. Jadhavar, D. Kumar, P. Purohit, B. V. Pipaliya, A. Kumar, S. Bhagat and A. K. Chakraborti, Sustainable approaches towards the synthesis of quinoxalines, in Green Chemistry: Synthesis of Bioactive Heterocycles, Springer, New Delhi, 2014, pp. 37–67 Search PubMed; (b) J. A. Pereira, A. M. Pessoa, M. N. D. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed; (c) A. K. Patidar, M. Jeyakandan, A. K. Mobiya and G. Selvam, Int. J. PharmTech Res., 2011, 3, 386–392 CAS; (d) T. Kaushal, G. Srivastava, A. Sharma and A. S. Negi, Bioorg. Med. Chem., 2019, 27, 16–35 CrossRef CAS PubMed; (e) A. Irfan, S. Ahmad, S. Hussain, F. Batool, H. Riaz, R. Zafar and M. Mojzych, Appl. Sci., 2021, 11, 5702 CrossRef.
- (a) D. G. Martin, S. A. Mizsak, C. Biles, J. C. Stewart, L. Baczynskyj and P. A. Meulman, J. Antibiot., 1975, 28, 332–336 CrossRef CAS PubMed; (b) K. Watanabe, Biosci., Biotechnol., Biochem., 2008, 72, 2491–2506 CrossRef CAS PubMed; (c) M. J. Waring, Echinomycin and related quinoxaline antibiotics, in Molecular Aspects of Anticancer Drug-DNA Interactions, Palgrave, London, 1993, pp. 213–242 Search PubMed; (d) S. Matsuura, J. Antibiot., Ser. A, 1965, 18, 43–46 CAS; (e) Y. Hayakawa, R. Sone, H. Aoki and S. Kimata, J. Antibiot., 2018, 71, 898–901 CrossRef CAS PubMed.
- (a) S. Tariq, K. Somakala and M. Amir, Eur. J. Med. Chem., 2018, 143, 542–557 CrossRef CAS PubMed; (b) M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- (a) C. O. Knowles, Environ. Health Perspect., 1976, 14, 93–102 CrossRef CAS PubMed; (b) T. P. Selby, L. R. Denes, J. J. Kilama and B. K. Smith, Synthesis and Chemistry of Agrochemicals IV, ACS Symp. Ser., 1995, 584, 171–185 CrossRef CAS.
- (a) G. Sakata, K. Makino and Y. Kurasawa, Heterocycles, 1988, 27, 2481–2515 CrossRef CAS; (b) E. Vicente, L. M. Lima, E. Bongard, S. Charnaud, R. Villar, B. Solano and A. Monge, Eur. J. Med. Chem., 2008, 43, 1903–1910 CrossRef CAS PubMed; (c) A. Husain and D. Madhesia, J. Pharm. Res., 2011, 4, 924–929 CAS.
- (a) R. H. Bahekar, M. R. Jain, A. A. Gupta, A. Goel, P. A. Jadav, D. N. Patel and P. R. Patel, Arch. Pharm., 2007, 340, 359–366 CrossRef CAS PubMed; (b) M. Missioui, S. Mortada, W. Guerrab, G. Serdaroğlu, S. Kaya, J. T. Mague and Y. Ramli, J. Mol. Struct., 2021, 1239, 130484 CrossRef CAS; (c) S. A. Shintre, D. Ramjugernath, M. S. Islam, R. Mopuri, C. Mocktar and N. A. Koorbanally, Med. Chem. Res., 2017, 26, 2141–2151 CrossRef CAS.
- (a) M. M. Badran, K. A. Abouzid and M. H. M. Hussein, Arch. Pharmacal Res., 2003, 26, 107–113 CrossRef CAS PubMed; (b) D. P. Singh, S. K. Deivedi, S. R. Hashim and R. G. Singhal, Pharmaceuticals, 2010, 3, 2416–2425 CrossRef CAS PubMed.
- (a) A. C. Pinheiro, T. C. Mendonça Nogueira and M. V. N. de Souza, Anticancer Agents Med. Chem., 2016, 16, 1339–1352 CrossRef CAS PubMed; (b) A. H. Bayoumi, A. H. Ghiaty, S. M. Abd El-Gilil, E. M. Husseiny and M. A. Ebrahim, J. Heterocycl. Chem., 2019, 56, 3215–3235 CrossRef CAS; (c) R. Ingle, R. Marathe, D. Magar, H. M. Patel and S. J. Surana, Eur. J. Med. Chem., 2013, 65, 168–186 CrossRef CAS PubMed.
- (a) Y. Sainz, M. E. Montoya, F. J. Martínez-Crespo, M. A. Ortega, A. L. de Cerain and A. Monge, Arzneimittelforschung, 1999, 49, 55–59 CAS; (b) Y. Pan, P. Li, S. Xie, Y. Tao, D. Chen, M. Dai and Z. Yuan, Bioorg. Med. Chem. Lett., 2016, 26, 4146–4153 CrossRef CAS PubMed.
- (a) J. P. Dirlam, J. E. Presslitz and B. J. Williams, J. Med. Chem., 1983, 26, 1122–1126 CrossRef CAS PubMed; (b) S. Noorulla, N. Sreenivasulu, A. Khan and A. Sayeed, Pharmanest, 2011, 2, 229–238 Search PubMed.
- (a) A. Burguete, E. Pontiki, D. Hadjipavlou-Litina, S. Ancizu, R. Villar, B. Solano and A. Monge, Chem. Biol. Drug Des., 2011, 77, 255–267 CrossRef CAS PubMed; (b) S. Noorulla and N. Sreenivasulu, Int. J. Pharma Sci. Res., 2011, 2, 2337 CAS.
- (a) S. B. Patel, B. D. Patel, C. Pannecouque and H. G. Bhatt, Eur. J. Med. Chem., 2016, 117, 230–240 CrossRef CAS PubMed; (b) L. Fabian, M. T. Porro, N. Gomez, M. Salvatori, G. Turk, D. Estrin and A. Moglioni, Eur. J. Med. Chem., 2020, 188, 111987 CrossRef CAS PubMed.
- M. Montana, V. Montero, O. Khoumeri and P. Vanelle, Molecules, 2020, 25, 2784 CrossRef CAS PubMed.
- (a) P. Wu, Y. Su, X. Liu, L. Zhang, Y. Ye, J. Xu and Y. Hu, Eur. J. Med. Chem., 2011, 46, 5540–5548 CrossRef CAS PubMed; (b) P. Wu, Y. Su, X. Liu, J. Yan, Y. Ye, L. Zhang and Y. Hu, MedChemComm, 2012, 3, 659–662 RSC; (c) P. Wu, Y. Su, X. Liu, B. Yang, Q. He and Y. Hu, Bioorg. Med. Chem., 2012, 20, 2837–2844 CrossRef CAS PubMed.
- (a) S. K. Singh, V. Saibaba, V. Ravikumar, S. V. Rudrawar, P. Daga, C. S. Rao and Y. K. Rao, Bioorg. Med. Chem., 2004, 12, 1881–1893 CrossRef CAS PubMed; (b) S. A. Galal, S. H. Khairat, F. A. Ragab, A. S. Abdelsamie, M. M. Ali, S. M. Soliman and H. I. El Diwani, Eur. J. Med. Chem., 2014, 86, 122–132 CrossRef CAS PubMed.
- (a) J. B. Rangisetty, C. N. V. H. B. Gupta, A. L. Prasad, P. Srinivas, N. Sridhar, P. Parimoo and A. Veeranjaneyulu, J. Pharm. Pharmacol., 2001, 53, 1409–1413 CrossRef CAS PubMed; (b) L. Van Heerden, T. T. Cloete, J. W. Breytenbach, C. de Kock, P. J. Smith, J. C. Breytenbach and D. D. N’Da, Eur. J. Med. Chem., 2012, 55, 335–345 CrossRef CAS PubMed.
- C. H. Tseng, C. R. Han and K. W. Tang, Int. J. Mol. Sci., 2019, 20, 4786 CrossRef CAS PubMed.
- (a) M. M. Hossain, M. H. Muhib, M. R. Mia, S. Kumar and S. A. Wadud, Bangladesh Medical Research Council Bulletin, 2012, 38, 47–50 CrossRef CAS PubMed; (b) M. Missioui, S. Mortada, W. Guerrab, G. Serdaroğlu, S. Kaya, J. T. Mague and Y. Ramli, J. Mol. Struct., 2021, 1239, 130484 CrossRef CAS.
- (a) A. A. Elhelby, R. R. Ayyad and M. F. Zayed, Arzneimittelforschung, 2011, 61, 379–381 CrossRef CAS PubMed; (b) E. Elkaeed, A. Ghiaty, A. El-Morsy, K. El-Gamal and H. Sak, Chem. Sci. Rev. Lett., 2014, 3, 1375–1387 Search PubMed.
- (a) R. Sarges, H. R. Howard, R. G. Browne, L. A. Lebel, P. A. Seymour and B. K. Koe, J. Med. Chem., 1990, 33, 2240–2254 CrossRef CAS PubMed; (b) Y. Q. Fang, H. J. Zhang, Y. Ren and Z. S. Quan, Lat. Am. J. Pharm., 2018, 37, 170–181 CAS.
- M. Montana, F. Mathias, T. Terme and P. Vanelle, Eur. J. Med. Chem., 2019, 163, 136–147 CrossRef CAS PubMed.
- (a) C. Barea, A. Pabón, S. Pérez-Silanes, S. Galiano, G. Gonzalez, A. Monge and I. Aldana, Molecules, 2013, 18, 4718–4727 CrossRef CAS PubMed; (b) A. Gil, A. Pabón, S. Galiano, A. Burguete, S. Pérez-Silanes, E. Deharo and I. Aldana, Molecules, 2014, 19, 2166–2180 CrossRef PubMed.
- (a) C. Barea, A. Pabón, D. Castillo, M. Zimic, M. Quiliano, S. Galiano and I. Aldana, Bioorg. Med. Chem. Lett., 2011, 21, 4498–4502 CrossRef CAS PubMed; (b) J. Cogo, J. Cantizani, I. Cotillo, D. P. Sangi, A. G. Correa, T. Ueda-Nakamura and C. V. Nakamura, Bioorg. Med. Chem., 2018, 26, 4065–4072 CrossRef CAS PubMed.
- X. Hui, J. Desrivot, C. Bories, P. M. Loiseau, X. Franck, R. Hocquemiller and B. Figadere, Synthesis and antiprotozoal activity of some new synthetic substituted quinoxalines, Bioorg. Med. Chem. Lett., 2006, 16, 815–820 CrossRef CAS PubMed.
- R. Corbaz, L. Ettlinger, E. Gäumann, W. K. Schierlein, F. Kradolfer, L. Neipp and H. Zahner, Helv. Chim. Acta, 1957, 40, 199–204 CrossRef CAS.
- (a) J. I. Shōji and K. Katagiri, J. Antibiot., Ser. A, 1961, 14, 335–339 Search PubMed; (b) M. Shin, K. Inouye and H. Otsuka, Bull. Chem. Soc. Jpn., 1984, 57, 2203–2210 CrossRef CAS.
- M. S. Abdelfattah, T. Kazufumi and M. Ishibashi, J. Nat. Prod., 2010, 73, 1999–2002 CrossRef CAS PubMed.
- R. B. Lingham, A. H. Hsu, J. A. O'brien, J. M. Sigmund, M. Sanchez, M. M. Gagliardi and G. M. Salituro, J. Antibiot., 1996, 49, 253–259 CrossRef CAS PubMed.
- (a) B. Y. Yang, Y. G. Xia, Q. H. Wang, D. Q. Dou and H. X. Kuang, Fitoterapia, 2010, 81, 1003–1005 CrossRef CAS PubMed; (b) Y. Hu, K. Wang and J. B. MacMillan, Org. Lett., 2013, 15, 390–393 CrossRef CAS PubMed.
- H. C. Richards, J. R. Housley and D. F. Spooner, Nature, 1963, 199, 354–356 CrossRef CAS PubMed.
- H. Gao, E. F. Yamasaki, K. K. Chan, L. L. Shen and R. M. Snapka, Cancer Res., 2000, 60, 5937–5940 CAS.
- J. C. Adkins and J. A. Balfour, Drugs Aging, 1998, 12, 225–241 CrossRef CAS PubMed.
- R. G. Ulrich, J. A. Bacon, E. P. Brass, C. T. Cramer, D. K. Petrella and E. L. Sun, Chem.-Biol. Interact., 2001, 134, 251–270 CrossRef CAS PubMed.
- C. Oncken, D. Gonzales, M. Nides, S. Rennard, E. Watsky and C. B. Billing, Varenicline Study Group, Arch. Intern. Med., 2006, 166, 1571–1577 CrossRef CAS PubMed.
- C. H. Nguyen, E. Fan, J. F. Riou, M. C. Bissery, P. Vrignaud, F. Lavelle and E. Bisagni, Anticancer Drug Des., 1995, 10, 277–297 CAS.
- S. Keinan, W. D. Paquette, J. J. Skoko, D. N. Beratan, W. Yang, S. Shinde and P. Wipf, Org. Biomol. Chem., 2008, 6, 3256–3263 RSC.
- Z. An, L. Zhao, M. Wu, J. Ni, Z. Qi, G. Yu and R. Yan, Chem. Commun., 2017, 53, 11572–11575 RSC.
- A. Burguete, E. Pontiki, D. Hadjipavlou-Litina, S. Ancizu, R. Villar, B. Solano and A. Monge, Chem. Biol. Drug Des., 2011, 77, 255–267 CrossRef CAS PubMed.
- E. Carosati, G. Sforna, M. Pippi, G. Marverti, A. Ligabue, D. Guerrieri and G. Cruciani, Bioorg. Med. Chem., 2010, 18, 7773–7785 CrossRef CAS PubMed.
- (a) J. Yuan, J. Ouyang, V. Cimrová, M. Leclerc, A. Najari and Y. Zou, J. Mater. Chem. C, 2017, 5, 1858–1879 RSC; (b) D. Gedefaw, M. Prosa, M. Bolognesi, M. Seri and M. R. Andersson, Adv. Energy Mater., 2017, 7, 1700575 CrossRef.
- S. Dailey, W. J. Feast, R. J. Peace, I. C. Sage, S. Till and E. L. Wood, J. Mater. Chem., 2001, 11, 2238–2243 RSC.
- (a) K. R. Justin Thomas, M. Velusamy, J. T. Lin, C. H. Chuen and Y. T. Tao, Chem. Mater., 2005, 17, 1860–1866 CrossRef; (b) D. O'Brien, A. Bleyer, D. D. C. Bradley and S. Meng, Synth. Met., 1996, 76, 105–108 CrossRef.
- (a) H. Wang, G. Chen, Y. Liu, L. Hu, X. Xu and S. Ji, Dyes Pigm., 2009, 83, 269–275 CrossRef CAS; (b) C. T. Chen, Y. Wei, J. S. Lin, M. V. Moturu, W. S. Chao, Y. T. Tao and C. H. Chien, J. Am. Chem. Soc., 2006, 128, 10992–10993 CrossRef CAS PubMed.
- (a) Z. Feng, Z. Gao, W. Qu, T. Yang, J. Li and L. Wang, RSC Adv., 2019, 9, 10789–10795 RSC; (b) P. Ledwon, R. Motyka, K. Ivaniuk, A. Pidluzhna, N. Martyniuk, P. Stakhira and H. Ågren, Dyes Pigm., 2020, 173, 108008 CrossRef CAS.
- (a) J. Yang, M. A. Uddin, Y. Tang, Y. Wang, Y. Wang, H. Su and X. Guo, ACS Appl. Mater. Interfaces, 2018, 10, 23235–23246 CrossRef CAS PubMed; (b) D. Zimmermann, C. Sprau, J. Schröder, V. G. Gregoriou, A. Avgeropoulos, C. L. Chochos and H. Krüger, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1457–1467 CrossRef CAS.
- (a) H. Hayashi, A. S. Touchy, Y. Kinjo, K. Kurotobi, Y. Toude, S. Ito and H. Imahori, ChemSusChem, 2013, 6, 508–517 CrossRef CAS PubMed; (b) A. Amacher, C. Yi, J. Yang, M. P. Bircher, Y. Fu, M. Cascella and S. X. Liu, Chem. Commun., 2014, 50, 6540–6542 RSC; (c) D. W. Chang, H. J. Lee, J. H. Kim, S. Y. Park, S. M. Park, L. Dai and J. B. Baek, Org. Lett., 2011, 13, 3880–3883 CrossRef CAS PubMed.
- D. O’brien, M. S. Weaver, D. G. Lidzey and D. D. C. Bradley, Appl. Phys. Lett., 1996, 69, 881–883 CrossRef.
- (a) X. Chen, P. Chen, Z. An, K. Chen and K. Okamoto, J. Power Sources, 2011, 196, 1694–1703 CrossRef CAS; (b) P. Chen, X. Chen, Z. An, K. Chen and K. Okamoto, Int. J. Hydrogen Energy, 2011, 36, 12406–12416 CrossRef CAS.
- (a) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127–2198 CrossRef CAS PubMed; (b) D. M. D'Souza and T. J. Mueller, Chem. Soc. Rev., 2007, 36, 1095–1108 RSC; (c) K. D. Dwivedi, M. S. Reddy, N. S. Kumar and L. R. Chowhan, J. Chem. Sci., 2018, 130, 129 CrossRef.
- (a) C. L. Sun and Z. J. Shi, Chem. Rev., 2014, 114, 9219–9280 CrossRef CAS PubMed; (b) T. Chatterjee, S. Bhadra and B. C. Ranu, Green Chem., 2011, 13, 1837–1842 RSC.
- (a) S. Roscales and A. G. Csákÿ, Chem. Soc. Rev., 2014, 43, 8215–8225 RSC; (b) M. S. Reddy, N. S. Kumar and L. R. Chowhan, RSC Adv., 2018, 8, 35587–35593 RSC; (c) K. D. Dwivedi, B. Borah and L. R. Chowhan, Front. Chem., 2020, 7, 944 CrossRef PubMed; (d) B. Borah, K. D. Dwivedi and L. R. Chowhan, RSC Adv., 2021, 11, 13585–13601 RSC; (e) S. R. Lakshmi, V. Singh and L. R. Chowhan, RSC Adv., 2020, 10, 13866–13871 RSC.
- (a) Y. V. D. Nageswar, K. H. V. Reddy, K. Ramesh and S. N. Murthy, Org. Prep. Proced. Int., 2013, 45, 1–27 CrossRef CAS; (b) J. A. Pereira, A. M. Pessoa, M. N. D. Cordeiro, R. Fernandes, C. Prudêncio, J. P. Noronha and M. Vieira, Eur. J. Med. Chem., 2015, 97, 664–672 CrossRef CAS PubMed; (c) C. F. daCosta, M. V. Nora deSouza, C. R. Brandao Gomes and V. Facchinetti, Curr. Microwave Chem., 2017, 4, 277–286 Search PubMed; (d) L. Biesen and T. J. Müller, Adv. Synth. Catal., 2021, 363, 980–1006 CrossRef CAS; (e) H. Khatoon and E. Abdulmalek, Molecules, 2021, 26, 1055 CrossRef CAS PubMed.
- V. K. Maikhuri, A. K. Prasad, A. Jha and S. Srivastava, New J. Chem., 2021, 45, 13214–13246 RSC.
- G. Yashwantrao and S. Saha, Org. Chem. Front., 2021, 8, 2820–2862 RSC.
- (a) G. Körner, Ber. Dtsch. Chem. Ges., 1884, 17, 572–573 CrossRef; (b) D. J. Brown, Quinoxalines, in The Chemistry of Heterocyclic Compounds, ed. E. C. Taylor and P. Wipf, John Wiley & Sons, Hoboken, NJ, 2004, pp. 1−510 Search PubMed.
- O. Hinsberg, Ber. Dtsch. Chem. Ges., 1884, 17, 318–323 CrossRef.
- (a) D. F. Saifina and V. A. Mamedov, Russ. Chem. Rev., 2010, 79, 351 CrossRef CAS; (b) V. A. Mamedov, and N. A. Zhukova, Progress in quinoxaline synthesis (Part 1), in Progress in Heterocyclic Chemistry, Elsevier, 2012, vol. 24, pp. 55–88 Search PubMed; (c) V. A. Mamedov, and N. A. Zhukova, Progress in quinoxaline synthesis (Part 2), in Progress in Heterocyclic Chemistry, Elsevier, 2013, vol. 25, pp. 1–45 Search PubMed; (d) Y. Taek Han, J. W. Jung and N. J. Kim, Curr. Org. Chem., 2017, 21, 1265–1291 CrossRef.
- S. Fathi and A. R. Sardarian, Phosphorus, Sulfur Silicon Relat. Elem., 2015, 190, 1471–1478 CrossRef CAS.
- B. Roy, S. Ghosh, P. Ghosh and B. Basu, Tetrahedron Lett., 2015, 56, 6762–6767 CrossRef CAS.
- F. Lassagne, F. Chevallier, T. Roisnel, V. Dorcet, F. Mongin and L. R. Domingo, Synthesis, 2015, 47, 2680–2689 CrossRef CAS.
- (a) Z. S. Qureshi, K. M. Deshmukh and B. M. Bhanage, Applications of ionic liquids in organic synthesis and catalysis, Clean Technol. Environ. Policy, 2014, 16, 1487–1513 CrossRef; (b) A. R. Hajipour and F. Rafiee, Org. Prep. Proced. Int., 2015, 47, 249–308 CrossRef CAS.
- S. V. Moghaddam and H. Valizadeh, J. Iran. Chem. Soc., 2016, 13, 1517–1524 CrossRef CAS.
- A. G. MulikD, R. Chandam, D. R. Patil, P. P. Patil, G. N. Mulik, S. T. Salunkhe and M. B. Deshmukh, Res. Chem. Intermed., 2015, 41, 10085–10096 CrossRef.
- S. Rezayati, M. Mehmannavaz, E. Salehi, S. Haghi, R. Hajinasiri and S. Afshari Sharif Abad, J. Sci., Islamic Repub. Iran, 2016, 27, 51–63 Search PubMed.
- M. Shamsi-Sani, F. Shirini, M. Abedini and M. Seddighi, Res. Chem. Intermed., 2016, 42, 1091–1099 CrossRef CAS.
- N. Fahimi, A. Sardarian and M. Kazemnejadi, Iran. J. Catal., 2016, 6, 161–166 Search PubMed.
- K. S. Indalkar, C. K. Khatri and G. U. Chaturbhuj, J. Chem. Sci., 2017, 129, 141–148 CrossRef CAS.
- Z. T. Bhutia, G. Prasannakumar, A. Das, M. Biswas, A. Chatterjee and M. Banerjee, ChemistrySelect, 2017, 2, 1183–1187 CrossRef CAS.
- S. A. Fazeli-Attar and B. B. F. Mirjalili, Environ. Chem. Lett., 2018, 16, 671–676 CrossRef CAS.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed.
- A. Mishra, S. Singh, M. A. Quraishi and V. Srivastava, Org. Prep. Proced. Int., 2019, 51, 345–356 CrossRef CAS.
- D. Jaiswal, J. Tiwari, S. Singh, A. K. Sharma, J. Singh and J. Singh, ChemistrySelect, 2019, 4, 8713–8718 CrossRef CAS.
- S. Bhargava, P. Soni and D. Rathore, J. Mol. Struct., 2019, 1198, 126758 CrossRef CAS.
- G. Kaur, A. Singh, N. Kaur and B. Banerjee, Synth. Commun., 2021, 51, 1121–1131 CrossRef CAS.
- R. H. Vekariya, K. D. Patel, N. P. Prajapati and H. D. Patel, Synth. Commun., 2018, 48, 1505–1533 CrossRef CAS.
- K. Kumar, S. R. Mudshinge, S. Goyal, M. Gangar and V. A. Nair, Tetrahedron Lett., 2015, 56, 1266–1271 CrossRef CAS.
- S. Alavi, M. H. Mosslemin, R. Mohebat and A. R. Massah, Res. Chem. Intermed., 2017, 43, 4549–4559 CrossRef CAS.
- K. Kiyokawa, T. Nagata and S. Minakata, Synthesis, 2018, 50, 485–498 CrossRef CAS.
- G. F. dos Santos Fernandes, E. Moreno-Viguri, M. Santivañez-Veliz, R. Paucar, C. M. Chin, S. Pérez-Silanes and J. L. dos Santos, J. Heterocycl. Chem., 2017, 54, 2380–2388 CrossRef CAS.
- C. Zhou, P. Diao, X. Li, Y. Ge and C. Guo, Chin. Chem. Lett., 2019, 30, 371–374 CrossRef CAS.
- L. A. Paquette and J. E. Hofferberth, Org. React., 2004, 62, 477–567 Search PubMed.
- C. Xie, Z. Zhang, B. Yang, G. Song, H. Gao, L. Wen and C. Ma, Tetrahedron, 2015, 71, 1831–1837 CrossRef CAS.
- Y. B. Wang, L. Shi, X. Zhang, L. R. Fu, W. Hu, W. Zhang, X. Zhu, X. Q. Hao and M. P. Song, J. Org. Chem., 2020, 86, 947–958 CrossRef PubMed.
- D. Sarma, B. Majumdar, B. Deori, S. Jain and T. K. Sarma, ACS Omega, 2021, 6, 11902–11910 CrossRef CAS PubMed.
- K. S. Vadagaonkar, H. P. Kalmode, K. Murugan and A. C. Chaskar, RSC Adv, 2015, 5, 5580–5590 RSC.
- S. D. Pardeshi, P. A. Sathe, K. S. Vadagaonkar and A. C. Chaskar, Adv. Synth. Catal., 2017, 359, 4217–4226 CrossRef CAS.
- J. Shen, X. Wang, X. Lin, Z. Yang, G. Cheng and X. Cui, Org. Lett., 2016, 18, 1378–1381 CrossRef CAS PubMed.
- D. Hazarika and P. Phukan, Tetrahedron, 2017, 73, 1374–1379 CrossRef CAS.
- J. Wu, H. Zhang, X. Ding, X. Tan, J. Chen, W. He and W. Cao, J. Org. Chem., 2018, 83, 9422–9429 CrossRef CAS PubMed.
- A. Z. A. Elassar and A. A. El-Khair, Tetrahedron, 2003, 59, 8463–8480 CrossRef CAS.
- S. Cao, S. Zhong, L. Xin, J. P. Wan and C. Wen, ChemCatChem, 2015, 7, 1478–1482 CrossRef CAS.
- X. Sun, W. Wang, Y. Li, J. Ma and S. Yu, Org. Lett., 2016, 18, 4638–4641 CrossRef CAS PubMed.
- C. C. Tran, S. I. Kawaguchi, F. Sato, A. Nomoto and A. Ogawa, J. Org. Chem., 2020, 85, 7258–7266 CrossRef CAS PubMed.
- (a) Y. X. Jiao, L. L. Wu, H. M. Zhu, J. K. Qin, C. X. Pan, D. L. Mo and G. F. Su, J. Org. Chem., 2017, 82, 4407–4414 CrossRef CAS PubMed; (b) K. D. Dwivedi, B. Kumar, M. S. Reddy, B. Borah, J. N. Babu and L. R. Chowhan, Results Chem., 2021, 3, 100201 CrossRef.
- (a) V. A. Mamedov and A. A. Kalinin, Chem. Heterocycl. Compd., 2010, 46, 641–664 CrossRef CAS; (b) A. A. Kalinin, L. N. Islamova and G. M. Fazleeva, Chem. Heterocycl. Compd., 2019, 55, 584–597 CrossRef CAS; (c) J. Guillon, E. Mouray, S. Moreau, C. Mullié, I. Forfar, V. Desplat and P. Grellier, Eur. J. Med. Chem., 2011, 46, 2310–2326 CrossRef CAS PubMed.
- (a) G. Campiani, F. Aiello, M. Fabbrini, E. Morelli, A. Ramunno, S. Armaroli and S. Caccia, J. Med. Chem., 2001, 44, 305–315 CrossRef CAS PubMed; (b) J. Guillon, I. Forfar, M. Mamani-Matsuda, V. Desplat, M. Saliège, D. Thiolat and D. Mossalayi, Bioorg. Med. Chem., 2007, 15, 194–210 CrossRef CAS PubMed; (c) J. Guillon, M. Le Borgne, C. Rimbault, S. Moreau, S. Savrimoutou, N. Pinaud and J. Jose, Eur. J. Med. Chem., 2013, 65, 205–222 CrossRef CAS PubMed; (d) P. T. Lin, D. B. Salunke, L. H. Chen and C. M. Sun, Org. Biomol. Chem., 2011, 9, 2925–2937 RSC; (e) H. Xu and L. L. Fan, Eur. J. Med. Chem., 2011, 46, 1919–1925 CrossRef CAS PubMed; (f) G. Szabó, R. Kiss, D. Páyer-Lengyel, K. Vukics, J. Szikra, A. Baki and G. M. Keserű, Bioorg. Med. Chem. Lett., 2009, 19, 3471–3475 CrossRef PubMed.
- (a) A. A. Kalinin and V. A. Mamedov, Chem. Heterocycl. Compd., 2011, 46, 1423 CrossRef CAS; (b) O. A. Brazhko, A. S. Yevlash, M. P. Zavgorodnii, M. M. Kornet, O. O. Brazhko and A. V. Lagron, Vopr. Khim. Khim. Tekhnol., 2019, 6, 6–16 Search PubMed.
- C. Xie, L. Feng, W. Li, X. Ma, X. Ma, Y. Liu and C. Ma, Org. Biomol. Chem., 2016, 14, 8529–8535 RSC.
- C. Xie, Z. Zhang, D. Li, J. Gong, X. Han, X. Liu and C. Ma, J. Org. Chem., 2017, 82, 3491–3499 CrossRef CAS PubMed.
- P. N. Allan, M. I. Ostrowska and B. Patel, Synlett, 2019, 30, 2148–2152 CrossRef CAS.
- M. Viji, M. Vishwanath, J. Sim, Y. Park, C. Jung, S. Lee and J. K. Jung, RSC Adv., 2020, 10, 37202–37208 RSC.
- L. M. Reddy, V. V. Reddy, C. S. Putta, V. Satteyyanaidu, C. K. Reddy and B. V. Subba Reddy, ChemistrySelect, 2018, 3, 9881–9884 CrossRef CAS.
- A. Preetam and M. Nath, RSC Adv., 2015, 5, 21843–21853 RSC.
- C. Wang, Y. Li, R. Guo, J. Tian, C. Tao, B. Cheng and H. Zhai, Asian J. Org. Chem., 2015, 4, 866–869 CrossRef CAS.
- M. Ramamohan, R. Sridhar, K. Raghavendrarao, N. Paradesi, K. B. Chandrasekhar and S. Jayaprakash, Synlett, 2015, 26, 1096–1100 CrossRef CAS.
- B. N. Patil, J. J. Lade, S. D. Pardeshi, P. Patil and A. C. Chaskar, ChemistrySelect, 2019, 4, 11362–11366 CrossRef CAS.
- S. D. Pardeshi, B. N. Patil, P. Patil and A. C. Chaskar, Tetrahedron Lett., 2019, 60, 151250 CrossRef.
- Q. Sun, L. Liu, Y. Yang, Z. Zha and Z. Wang, Chin. Chem. Lett., 2019, 30, 1379–1382 CrossRef CAS.
- H. Liu, F. Zhou, W. Luo, Y. Chen, C. Zhang and C. Ma, Org. Biomol. Chem., 2017, 15, 7157–7164 RSC.
- (a) Z. Zghaib, J. F. Guichou, J. Vappiani, N. Bec, K. Hadj-Kaddour, L. A. Vincent and P. A. Bonnet, Bioorg. Med. Chem., 2016, 24, 2433–2440 CrossRef CAS PubMed; (b) A. A. Kalinin, A. D. Voloshina, N. V. Kulik, V. V. Zobov and V. A. Mamedov, Eur. J. Med. Chem., 2013, 66, 345–354 CrossRef CAS PubMed; (c) V. S. Hariharakrishnan, K. Hariprasad, B. V. Rao and A. N. Singh, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 2008, 47, 1281–1283 Search PubMed; (d) H. Potschka, W. Löscher, P. Wlaź, B. Behl, H. P. Hofmann, H. J. Treiber and L. Szabo, Br. J. Pharmacol., 1998, 125, 1258–1266 CrossRef CAS PubMed; (e) G. Moarbess, J. F. Guichou, S. Paniagua-Gayraud, A. Chouchou, O. Marcadet, F. Leroy and P. A. Bonnet, Eur. J. Med. Chem., 2016, 115, 268–274 CrossRef CAS PubMed.
- A. A. Kalinin, L. N. Islamova and G. M. Fazleeva, Chem. Heterocycl. Compd., 2020, 56, 663–673 CrossRef CAS.
- Z. Zhang, C. Xie, X. Tan, G. Song, L. Wen, H. Gao and C. Ma, Org. Chem. Front., 2015, 2, 942–946 RSC.
- L. H. Chen, C. H. Kao, S. Dhole, I. J. Barve, L. C. Shen, W. S. Chung and C. M. Sun, RSC Adv., 2016, 6, 76123–76127 RSC.
- G. Joshi, M. Chauhan, R. Kumar, A. Thakur, S. Sharma, R. Singh and R. Kumar, Org. Chem. Front., 2018, 5, 3526–3533 RSC.
- Z. Rezaei, M. M. Didehvar, M. Mahdavi, H. Azizian, H. Hamedifar, E. H. Mohammed and M. Amini, Bioorg. Chem., 2019, 90, 103055 CrossRef CAS PubMed.
- S. Das, S. Maity, P. Ghosh, B. K. Paul and A. Dutta, ChemistrySelect, 2019, 4, 2656–2662 CrossRef CAS.
- I. A. Ibarra, A. Islas-Jácome and E. González-Zamora, Org. Biomol. Chem., 2018, 16, 1402–1418 RSC.
- R. Afshari and A. Shaabani, ACS Comb. Sci., 2018, 20, 499–528 CrossRef CAS PubMed.
- (a) B. Borah, K. D. Dwivedi and L. R. Chowhan, Arkivoc, 2021, 273–328 Search PubMed; (b) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 2709–2762, DOI:10.1002/ajoc.202100427; (c) B. Borah, K. D. Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021 DOI:10.1002/ajoc.202100550.
- (a) B. Borah, K. D. Dwivedi and L. R. Chowhan, Polycyclic Aromat. Compd., 2021, 1–45 Search PubMed; (b) P. Ramesh, M. S. Reddy, N. S. Kumar, B. Rathod, R. S. Prakasham and L. R. Chowan, ChemistrySelect, 2018, 3, 9096–9101 CrossRef CAS; (c) K. D. Dwivedi, M. S. Reddy and L. R. Chowhan, Tetrahedron Lett., 2021, 61, 152664 Search PubMed.
- (a) A. Váradi, T. C. Palmer, R. Notis Dardashti and S. Majumdar, Molecules, 2016, 21, 19 CrossRef PubMed; (b) S. Sadjadi, M. M. Heravi and N. Nazari, RSC Adv., 2016, 6, 53203–53272 RSC.
- M. Rouhani and A. Ramazani, J. Iran. Chem. Soc., 2018, 15, 2375–2382 CrossRef CAS.
- A. Alizadeh, H. Ghasemzadeh, R. Rezaiyehraad and H. P. Xiao, J. Sulfur Chem., 2019, 40, 614–628 CrossRef CAS.
- R. Mohebat, A. Y. E. Abadi and M. T. Maghsoodlou, Res. Chem. Intermed., 2016, 42, 6039–6048 CrossRef CAS.
- A. Yazdani-Elah-Abadi, M. T. Maghsoodlou, R. Mohebat and R. Heydari, J. Chem. Sci., 2017, 129, 691–698 CrossRef CAS.
- M. R. P. Heravi and F. Norouzy, Res. Chem. Intermed., 2017, 43, 4265–4282 CrossRef.
- B. Banerjee, Ultrason. Sonochem., 2017, 35, 15–35 CrossRef CAS PubMed.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794–806 RSC.
- A. M. Akondi, S. Mekala, M. L. Kantam, R. Trivedi, L. R. Chowhan and A. Das, New J. Chem., 2017, 41, 873–878 RSC.
- Y. Li, J. Lei, J. Xu, D. Y. Tang, Z. Z. Chen, J. Zhu and C. Xu, Chin. Chem. Lett., 2017, 28, 541–545 CrossRef CAS.
- M. S. Reddy, L. R. Chowhan, N. S. Kumar, P. Ramesh and S. B. Mukkamala, Tetrahedron Lett., 2018, 59, 1366–1371 CrossRef CAS.
- S. Mandal and A. Pramanik, J. Org. Chem., 2021, 86, 5047–5064 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |