
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation and crystalline transformation of functionalized poly(1-butene) containing PFPU and mPEG side chain†
Chuang Lia,
Zefeng Cuia,
Min Yang a,
Haifeng Shi*b and
Binyuan Liu*a
a,
Haifeng Shi*b and
Binyuan Liu*a
aHebei Key Laboratory of Functional Polymer Materials, School of Chemical Engineering and Science, Hebei University of Technology, Tianjin 300130, China. E-mail: byliu@hebut.edu.cn
bState Key of Separation Membranes and Membrane Process, School of Materials Science and Engineering, Tiangong University, Tianjin 300387, China. E-mail: haifeng.shi@gmail.com
First published on 22nd November 2021
Abstract
A series of 1-butene/pentafluorophenylundec-1-ene ester random copolymers were synthesized under the Ziegler–Natta catalyst system. The content of the pentafluorophenyl (PFP) group in the copolymer can reach up to 0.59 mol%. The DSC test found that the PFP groups attached to the PB main chain retard the crystalline transformation of Form II to Form I. Nucleophilic aromatic substitutions of pentafluorophenyl ester occurred with biocompatible poly(ethylene glycol) methyl ether (mPEG) introduced into the side chain of PB under very mild conditions. The results show that not only is the crystallization rate of mPEG functionalized PB increased, but also Tm, Tc, χc and the crystalline phase transition rate of Form II to Form I are also enhanced. Among them, mPEG with a Mn of 500 has the best promoting effect. On the other hand, the hydrophilicity of mPEG-functionalized PB is improved, and it is proportional to the chain length of mPEG. However, the experimental results show that the regularity of the PB structure is the determinant of the rate of crystallization and phase transition.
Introduction
Incorporating polar functional groups into polyolefin backbones by copolymerization of olefins with a polar comonomer is a powerful approach to improve the physicochemical properties of polyolefins and broaden their applications.1–4 However, it is an interesting and challenging research topic with classical Ziegler–Natta catalyst-mediated coordination copolymerization due to the limitations of catalyst poisoning and different comonomer reactivity ratios.5,6 One strategy is the utilization of comonomers bearing a long spacer between the vinyl group and the functional group which was capped by masking reagents to prohibit the polar group coordinating to the metal center.7Isotactic poly(1-butene) (iPB) is a polymorphic polyolefin that displays three major crystal forms (Form I, Form I′, and Form II), depending on the crystallization conditions.8,9 Generally, iPB crystallizes into Form II from bulk melt under quiescent conditions, which is a metastable state with 11/3 helix geometry and a tetragonal unit cell.10 The melt grown Form II spontaneously transforms to thermodynamically stable Form I with 3/1 helix conformations at room temperature in about seven days.9,11,12 Such polymorphic transformation involves remarkable variations of mechanical properties, including higher hardness, stiffness, and strength. Also the thermal properties are affected by the crystal transformation because Form I crystals have a higher melting temperature and melting enthalpy than Form II.9,11,13 Therefore, it is of particular interest to investigate phase transformation of PB from viewpoints both of academic study and of practical applications. Previous works showed that it is a feasible method to control the crystallinity and phase transformation of PB by copolymerization 1-butene with comonomers. For instance, Ma and Li successfully prepared iPB with various 4-methyl-1-pentene (4M1P) concentrations by the dimethyl-pyridylamidohafnium/organoboron catalyst. They found that the phase transition from Form II to Form I of iPB containing ≥3.40 mol% of 4M1P units was completely inhibited.14 While they found that the methylene-1,3-cyclopentane with five-membered ring introduced into PB backbone through the copolymerization with 1,5-hexadiene accelerate the phase transition rate of PB.15 Chen et al. found that the phase transition rate of II–I was greatly enhanced for the PB with a number fraction of ethylene monomers of 4.3% or higher contents.16 Furthermore, our group successfully introduced the natural antioxidant, biomass eugenol (EG), into PB by the Ziegler–Natta catalyst-initiated the copolymerization of 1-butene with EG, in which EG moieties endow the resulted PB a high thermal-oxidative stability, while suppress the crystallization rate and the Form II–Form I crystal transformation.17
Incorporation of poly(ethylene glycol) (PEG) into polymer chain often render the resultant block-/brush-copolymer attractive performances and morphological structure.18–21 PEG is a nonionic hydrophilic polymer that exhibits no immunogenicity, antigenicity, or toxicity and excellent anti-fouling properties.22 The covalent conjugation of PEG (PEGylation) has emerged as a valuable tool to overcome many of the deficiencies, particularly of protein- and peptide-based drugs.22 More related to this work, Ma et al. synthesized the PEG-grafted PB copolymer by metallocene-catalyzed copolymerization of 1-butene and 4-[4-(trimethylsilylalkynyl)-phenyl]-1-butylene and then incorporated the PEG by Cu-mediated azide–alkyne cycloaddition.23 They found that the molecular weight of PEG play positive roles in enhancing the Tc and χc of PB and in accelerating the transformation rate of Form II to From I as the PEG ranging from 750 to 4000.23 However, the modification effect of PEG with lower molecular weight on PB has not been explored in the report. Also, pentafluorophenyl ester is a kind of polar functional group with high reactivity, which makes it easy as intermediate functional groups for the further post-modification of polymers.24–26 One advantage of method lies in the transformation could be occurred under mild and metal-free conditions.
Inspired by the above-mentioned findings, we, herein, report the synthesis of functionalized PB containing pentafluorophenyl activated ester group by copolymerization of 1-butene with biomass undecylenic acid-derived pentafluoro-phenylundec-1-ene ester (PFPU) under heterogeneous MgCl2-supported TiCl4 Ziegler–Natta catalyst. Then poly(ethylene glycol) methyl ether (mPEG) is introduced into PB by nucleophilic aromatic substitution reaction with a high degree. The preparation procedure is outlined in Scheme 1. And focused on exploring the effect of PFP and mPEG with different molecular weights on the crystalline properties and II–I phase transition for PB copolymer.
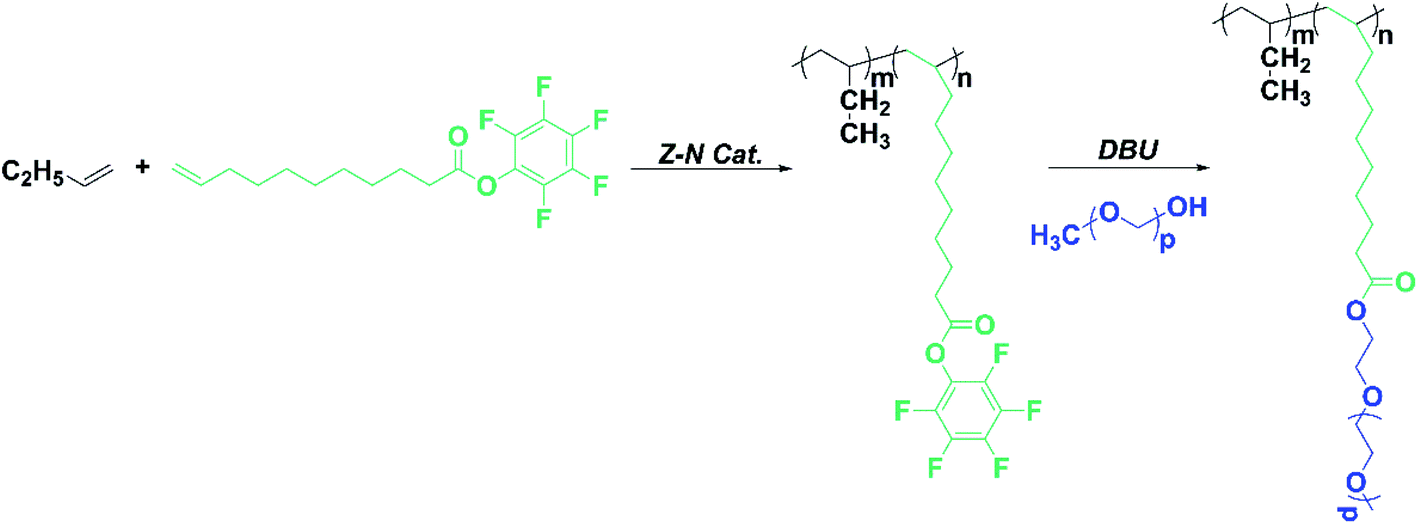 | ||
| Scheme 1 Synthesis of the copolymer of PFPU with 1-butene and trans-esterification of copolymer with mPEG. | ||
Experimental section
Materials
All involving moisture or air sensitive compound were carried out under a dry argon atmosphere. n-Hexane were first dried with 4 Å molecular sieves for 48 h, and then distilled with Na and benzophenone under argon before use. Polymer grade 1-butene (99.9%) and hydrogen (99.9%) were purchased from Saimeite gas Co., Ltd. (Tianjin, China). Cyclohexylmethyldimethoxysilane (CHMMS, 0.025 mol L−1 in n-hexane) was dried with 4 Å molecular sieves and was stored under argon atmosphere. Triethylaluminum (AlEt3) (TEA, 1.0 mol L−1 in n-hexane) was purchased from Yanfeng Science and Technology Co., Ltd. (Beijing, China) and used as received. The MgCl2-supported TiCl4 catalyst (Ti% = 2.3%) was prepared according to the patent.27 10-Undecenoic acid (95.0%) was purchased from Maikaxi Chemical Co., Ltd. (Chengdu, China) and used as received. Pentafluorophenol (99.9%) was purchased from Jiangsu Aikang Biomedical R&D Co., Ltd. (Nanjing, China) and used as received. Poly(ethylene glycol) methyl ether (mPEG) (99.9%) was purchased from Rhawn reagent (Tianjin, China). 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) (99%) was purchased from Shanghai Haiqu Chemical Co., Ltd. (Shanghai, China) and used as received. All the other chemicals were purchased from Fuchen Chemical (Tianjin, China) and used as received. PFPU was synthesized according to the previous literature and has been analyzed by 1H NMR spectroscopy.28Copolymerization of 1-butene with PFPU
A 100 mL of n-hexane was transferred into the 250 mL three-necked flask, which was purged with argon and 1-butene three times, respectively. 1-Butene, TEA and PFPU were introduced into the flask with stirring for 10 min. The required amounts of Ziegler–Natta catalyst suspension, CHMMS and H2 were added into the flask. The reaction temperature and pressure were maintained constant at 35 °C and 0.1 MPa for 2 h. The polymerization was quenched with acidified ethanol. Copolymer samples were washed several times with ethanol until there were no PFPU in ethanol filtrate. Finally, the samples were dried under vacuum at 60 °C until the weight was constant.Conversion of pentafluorophenyl ester functional group
The PB copolymer containing 0.36 mol% PFP (PB-0.36 mol%) (0.4 g) were dissolved in 10 mL CCl4 in a reaction vial. And then, 10 equiv. of mPEG (Mn = 350, 500, 750, or 2000) and DBU (10 equiv.), DMF (2.0 mL) were added. After the reaction was carried out at 30 °C for 6 h, the mixtures were precipitated with ethanol and washed several times with ethanol until there were no reactive monomer in ethanol filtrate. Then the samples were dried under vacuum at 60 °C for 48 h, and marked as PB-mPEG350, PB-mPEG500, PB-mPEG750, and PB-mPEG2000, respectively.Characterization
1H NMR spectra was performed on a Bruker AVANCE-400 spectrometer with the volume ratio of CCl4 to CDCl3 was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Chemical shifts are given in ppm relative to tetramethylsilane (TMS). IR spectrum of the copolymers were measured on a Vector 22 spectrometer (Bruker Optics, Karlsruhe, Germany) within the region of 4000–400 cm−1 by applying CCl4 solution of PB on KBr discs. PB molecular weight (Mw) and molecular weight distribution (Đ) measured by gel permeation chromatography (GPC) on a PL-GPC 220 instrument, which has a refractive index detector and calibrated using polystyrene standards. The columns were kept at 150 °C ± 0.05 °C and 1,2,4-trichlorobenzene was used as the eluents at a flow rate of 1.0 mL min−1. Samples were placed between two pieces of glass and placed under a Axioskop 40 hot stage polarizing microscope (POM, Karl Zeiss, Germany). The temperature was raised to 200 °C at a rate of 30 °C min−1, and the thermal history was eliminated for 5 min, then the temperature was lowered to 75 °C at a rate of 30 °C min−1, and the spherulites were isothermal crystallized at 75 °C to observe the growth process.
:1. Chemical shifts are given in ppm relative to tetramethylsilane (TMS). IR spectrum of the copolymers were measured on a Vector 22 spectrometer (Bruker Optics, Karlsruhe, Germany) within the region of 4000–400 cm−1 by applying CCl4 solution of PB on KBr discs. PB molecular weight (Mw) and molecular weight distribution (Đ) measured by gel permeation chromatography (GPC) on a PL-GPC 220 instrument, which has a refractive index detector and calibrated using polystyrene standards. The columns were kept at 150 °C ± 0.05 °C and 1,2,4-trichlorobenzene was used as the eluents at a flow rate of 1.0 mL min−1. Samples were placed between two pieces of glass and placed under a Axioskop 40 hot stage polarizing microscope (POM, Karl Zeiss, Germany). The temperature was raised to 200 °C at a rate of 30 °C min−1, and the thermal history was eliminated for 5 min, then the temperature was lowered to 75 °C at a rate of 30 °C min−1, and the spherulites were isothermal crystallized at 75 °C to observe the growth process.
Thermal properties of PB samples were characterized by differential scanning calorimeter (DSC, Diamond, PE Co.) under nitrogen atmosphere. PB samples were heating from 20 °C to 180 °C at the rate of 10 °C min−1 and held for 3 min to eliminate the thermal history, then cold from 180 °C to 20 °C at the rate of 10 °C min−1 held for 3 min to obtained Tc. Finally, heating from 20 to 180 °C at the rate of 10 °C min−1 to measure Tm. The crystallinity is calculated by following equation:
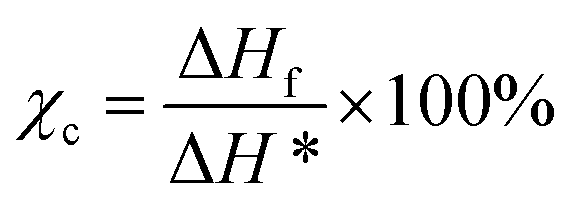 | (1) |
The phase transformation was measured by differential scanning calorimeter in nitrogen atmosphere. All samples were annealed at 180 °C for 10 min to eliminate the thermal history, and the phase transformation was carried out at 25 °C before testing. The melting enthalpies of Form I and Form II can be obtained by integrating the areas of the two melting peaks. The content of Form I was calculated according to the following equation:
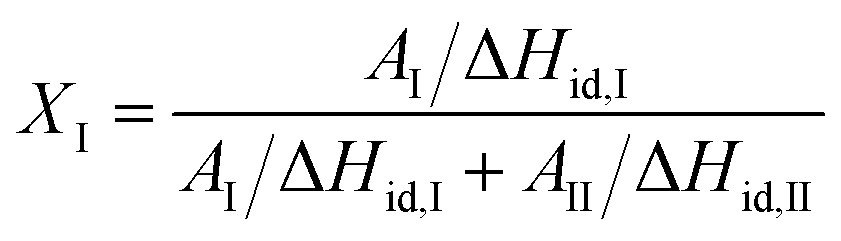 | (2) |
The static contact angles were measured on a video-based optical contact angle instrument (OCA 15 EC, Data Physics Instruments GmbH, Germany). The neat and mPEG-functionalized PB were pressed into a film at 150 °C and 10 MPa for 3 min, 6.0 μL of distilled water droplet was placed on the film and recorded. Each species was tested three times at different regions and taken average value. The relative error on the calculated parameters was in the range of 1.7–3.4%.
Results and discussion
Previous studies have found that polar monomer containing multiple methylene groups between the functional group and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond favors the copolymerization of α-olefins with polar monomers.5,29,31,32 In this regard, biomass-derived 10-undecylenic acid (10-UA) is an idea candidate of comonomer owing to the presence of the above-stated feature. Meanwhile, the active pentafluorophenyl ring (PFP) could afford the bulky hindrance which also benefit hindering ester group to poisoning active center by competing coordination with CC double bond. Therefore, PFPU has been designed and synthesized by the treatment of 10-UA with pentafluorophenol with DCC in ethyl acetate28 in the about 73% isolated yield (Scheme 1). The targeted PFPU was evidenced by the 1H NMR spectroscopy (Fig. 1b) and IR spectroscopy (Fig. 2a).
C double bond favors the copolymerization of α-olefins with polar monomers.5,29,31,32 In this regard, biomass-derived 10-undecylenic acid (10-UA) is an idea candidate of comonomer owing to the presence of the above-stated feature. Meanwhile, the active pentafluorophenyl ring (PFP) could afford the bulky hindrance which also benefit hindering ester group to poisoning active center by competing coordination with CC double bond. Therefore, PFPU has been designed and synthesized by the treatment of 10-UA with pentafluorophenol with DCC in ethyl acetate28 in the about 73% isolated yield (Scheme 1). The targeted PFPU was evidenced by the 1H NMR spectroscopy (Fig. 1b) and IR spectroscopy (Fig. 2a).
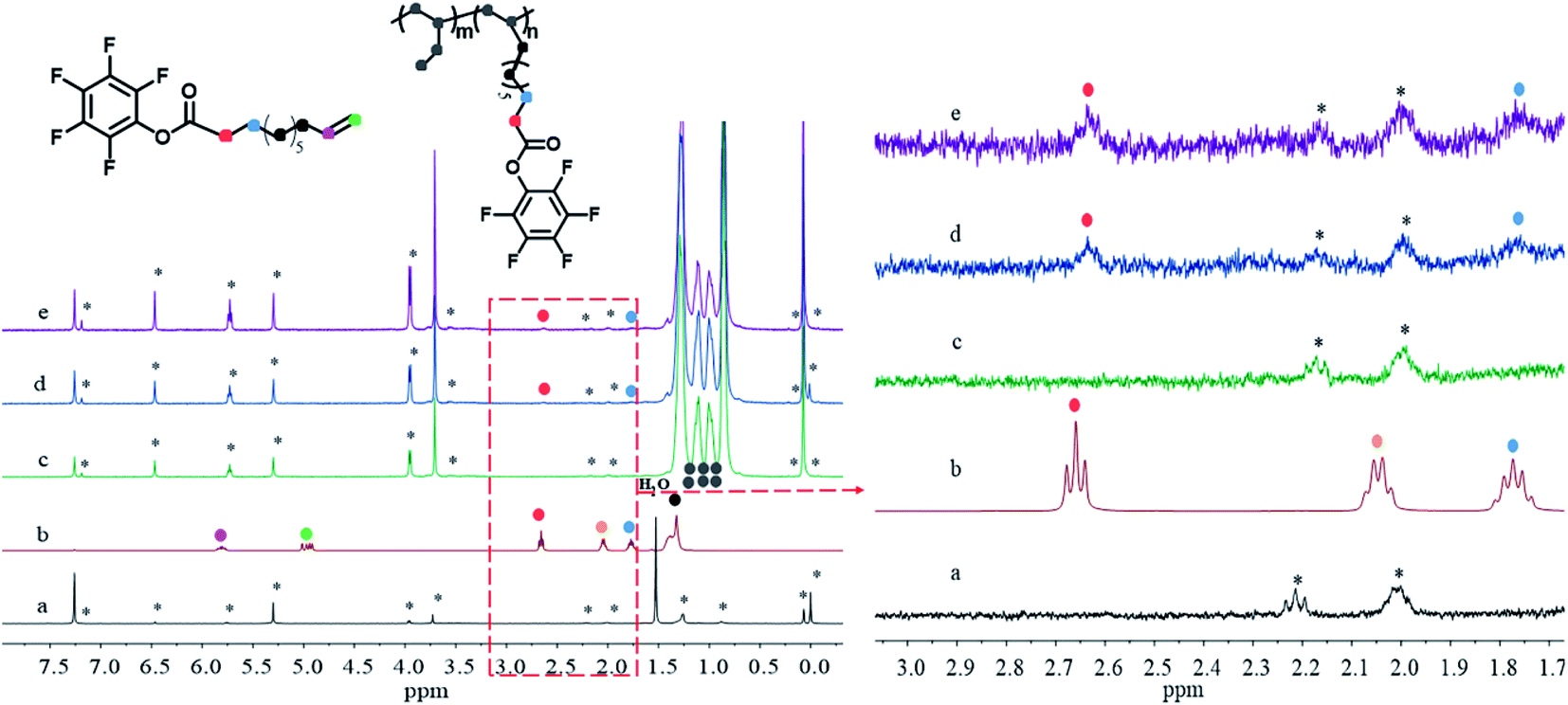 | ||
| Fig. 1 1H NMR spectra of CCl4, PFPU and PB with different PFP incorporation rate. (a) CCl4, (b) PFPU, (c) PB-0, (d) PB-0.36 mol%, (e) PB-0.59 mol%, the asterisk from CCl4. | ||
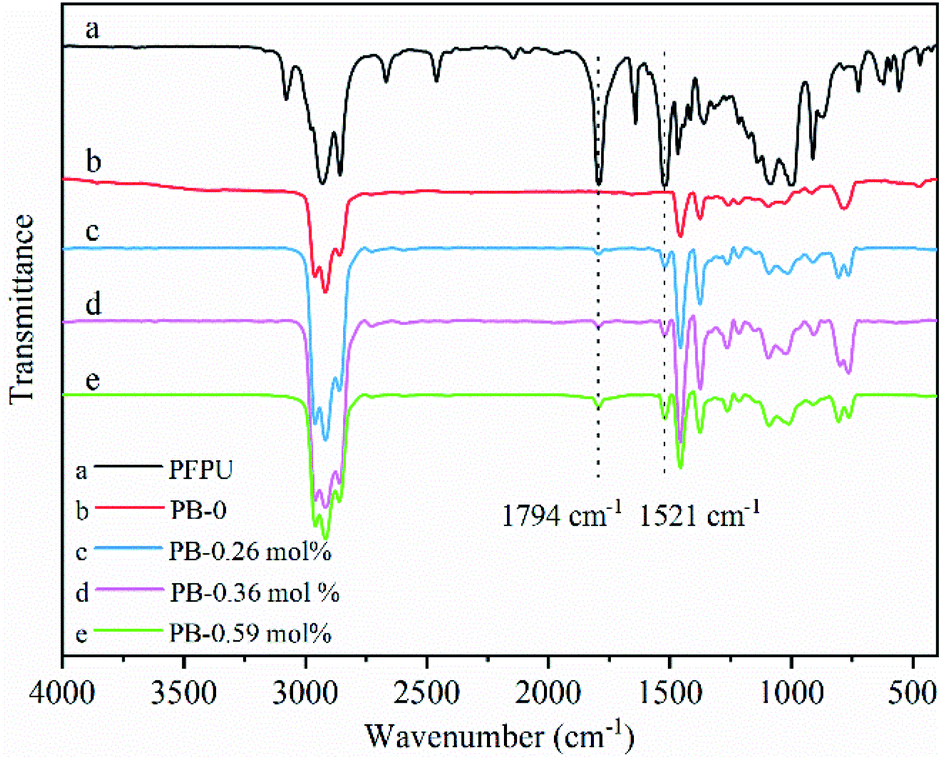 | ||
| Fig. 2 IR spectra of PB-0 and PB copolymers with different PFP contents. | ||
Table 1 summarizes the results of copolymerizations of 1-butene with PFPU mediated by MgCl2-supported TiCl4 Ziegler–Natta catalyst (the details can be found in ESI†). As indicted by Table 1, the content of PFP in resultant PB copolymers could be tuned in the slight range. Generally, the higher PFP loading is beneficial for incorporating much more PFP, while deteriorating the reaction efficiency and isotacticity of resulted PB copolymer varying from 95.3% to 77.6%. The maximum PFP content up to 0.59 mol% was obtained in this work. PFP-functionalized end groups were confirmed by the presence of characteristic proton signal corresponded to the methylene group adjacent to ester group (PFPCOO–CH2–) (at around 2.63 ppm) in the copolymers. Other evidences for the successful copolymerization of 10-UA and 1-butene resulted from the disappearance of proton signals (at 5.80 ppm and 4.94 ppm) of CH2CH2 of 10-UA monomer (Fig. 1b versus Fig. 1d and e). Moreover, we observe the characteristic absorption peak of CO bond and the stretching vibration absorption peak of CH2CH2 in the aromatic ring skeleton at 1794 cm−1 and 1521 cm−1 in PFPU/PB copolymers, respectively.33 These results further support the successful incorporation of PFP into PB backbone chain. Furthermore, the morphology of the samples gradually became fluffy with the incorporated PFP units increases, as shown in Fig. S1.†
| Sample | PFP contentb (mol%) | I.Ic (%) | Mwd (×104) | Đd |
|---|---|---|---|---|
| a Polymerization conditions: Cat = 10 mg, P = 0.1 MPa, TEA/PFPU = 2, Al/Ti = 300, Al/Si = 30, H2 = 3 mL, n-hexane = 25 mL, 35 °C, 2 h.b Molar content of incorporated PFP determined by1H NMR analyses.c Determined by the percentage of insoluble fraction after 48 h of extraction in boiling ether.d Determined by GPC. | ||||
| PB-0 | 0 | 95.3 | 37.6 | 8.3 |
| PB-0.03 mol% | 0.03 | 92.0 | 34.4 | 6.7 |
| PB-0.26 mol% | 0.26 | 81.6 | 46.7 | 5.0 |
| PB-0.36 mol% | 0.36 | 79.1 | 37.6 | 3.1 |
| PB-0.59 mol% | 0.59 | 77.6 | 45.6 | 5.6 |
With well-defined PFP-functionalized PB copolymers in hand, their reactivity toward oxygen-based nucleophiles, mPEGs with different molecular weight, was assessed, which aimed at further understanding the effect of side-chain length on the properties of PB copolymer. Herein, PB precursor containing 0.36 mol% PFP units was selected as the model substrate by nucleophilic substitution of PFP-activated ester at 30 °C under DBU base. A twofold excess of mPEGs with respect to active ester groups was used. In all cases, selective substitution of the ester cleavage was found with observed evidence of ester absorption peak at 1794 cm−1 in PB-PFPU red-shifted to 1742 cm−1 in PB-mPEG due to the change of its chemical environment,33 and the absence of characteristic peak of the CH2CH2 double bond in benzene ring at 1520 cm−1 (see Fig. S2†). Further supports arose from the observation of methylene proton signal peak in the mPEG repeat unit at 3.60 ppm in 1H-NMR spectroscopy see Fig. S3(c–f).†
Conversion of pentafluorophenyl ester functional group
The effect of PFP content on the crystallization behavior of PB was measured by DSC. By observing the DSC results (Fig. S4†), it can be found that with the increase of incorporated PFP, both the melting peak and crystallization peak of PB crystal Form II shifted to the low-temperature region, and the melting enthalpy in the heating curve gradually decreases. The dependence of Tm, Tc, and χc on the content of PFP in Fig. 3 indicated that crystallization capacity (crystal size and perfection) is deteriorated with increasing PFPU comonomer, likely due to lower stereoregularity of resulted PB chain and the steric hindrance of the side chain. The former makes it difficult for the stack of the PB chains to form crystals, thus decreasing the crystallization performance and lessening the Tm and χc.34,35 The latter could hinder the rearrangement of PB segments during the cooling process, resulting in lower Tc.36,37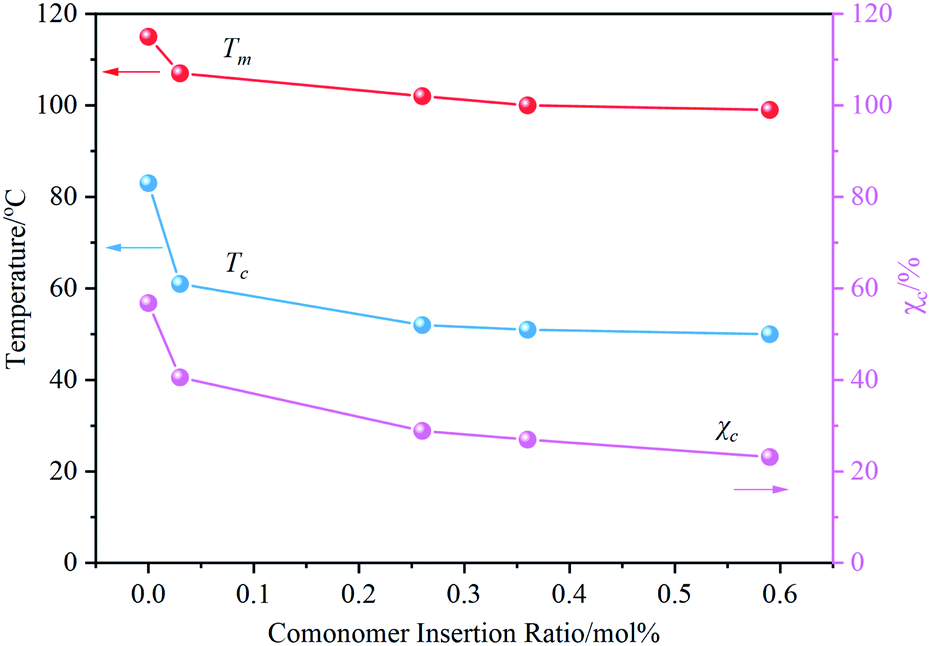 | ||
| Fig. 3 Influence of PFP insertion ratio on the melting and crystallization properties of PB. | ||
Interestingly, we found that the PFP group was substituted by mPEG has a pronounced effect on the crystallization performance of PB containing 0.36 mol% PFP, and Tc and χc of samples show a trend of increasing first and then decreasing with the molecular weight of mPEG increasing from 350, 500, 750, to 2000. Among them, PB-mPEG500 exhibits the most excellent crystallization performance, which has the highest Tm, Tc, and χc. For the convenience of comparison, we summarized the Tm, Tc, and χc of the samples in Fig. 4. The lower Tg of the mPEG segments adjacent to the backbone possibly contributes to good movement of molecular segments, thereby improving the crystallization performance of PB.19,23 On the other hand, with the increase of molecular weight of mPEG, the longer length of the chain resulting in the sterically hindered nature, which limits the movement of molecular segments and slows the growth of crystal nucleus, inhibiting PB crystal formation.14,23,36–39 The coaction lead to the findings of that PB with suitable grafted mPEG length benefit for the formation of crystal with lower defect. However, it should be pointed out that the Tm, Tc, and χc of the mPEG-PB samples are all lower than those of PB with high I.I, suggesting that the tacticity of the structure of PB plays the predominant role in affecting the crystallization performance compared with the effect of mPEG.
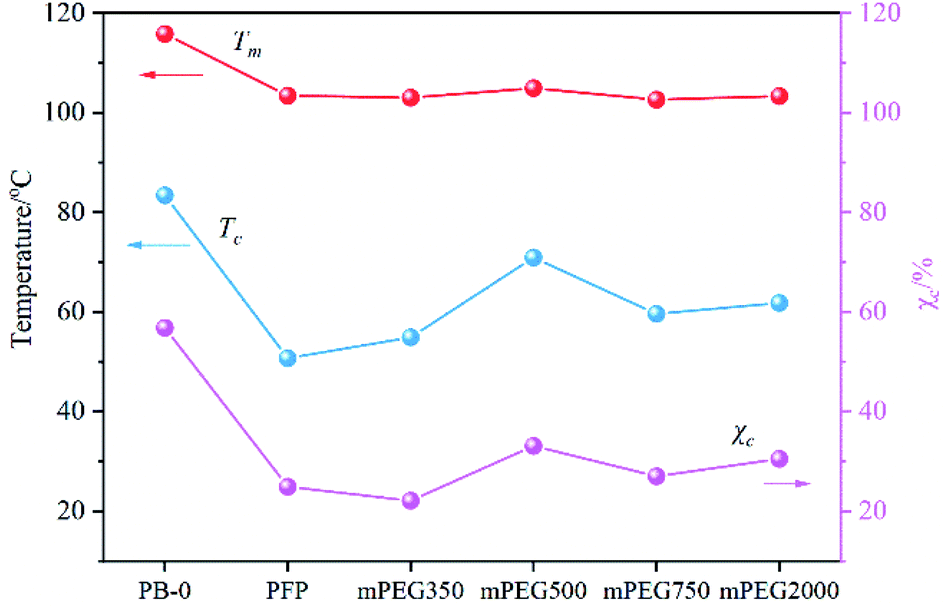 | ||
| Fig. 4 Influence of different molecular weight mPEGs on the melting and crystallization properties of PB. | ||
Fig. 5 shows the POM images for PB containing PFP and mPEG isothermally crystallized at 75 °C and various time. The results in Fig. 5 show that the content evolutions of spherulitic site and finer grainy morphology as a function of time. Note that there are obvious differences between the crystalline morphology, the crystallization rate and PB containing different functional substituents, where mPEG-functionalized PB shows more nucleus and faster growth rate of spherulite, and while smaller spherulitic size with clear indistinct boundary than that of PB with PFP group. Among mPEGs investigated, the sample modified by mPEG500 shows the smallest spherulite size and the fastest growth rate, indicating that mPEG500 is a more effective for crystalline than other mPEGs, being consistent with the findings in DSC.
 | ||
| Fig. 5 The POM images of isothermally crystallized. (a–c) PB-0, (d–f) PB-0.36 mol%, (g–i) PB-mPEG350, (j–l) PB-mPEG500, (m–o) PB-mPEG750, (p–r) PB-mPEG2000. | ||
Crystal transformation of PFP- and/or mPEG-functionalized PB
Crystal transformation is one of the important factors affecting the properties of PB, therefore, we studied the effect of PFP content and mPEG on II–I transformation of PB through DSC. As shown in Fig. 6, the PB with higher content of PFP groups inhibit the transformation of Form II to Form I. However, mPEG instead of PFP accelerate the II–I phase transition indicated by the comparison of PB. The rate of phase transition is function of length of mPEG. Among them, PB containing mPEG with Mn = 500 has the highest content of Form I after 144 h of annealing, which is 64.6%. As the Mn of grafted mPEG exceeds 500, such as Mn = 750 or 2000, the phase transition rate was reduced, but the rate is still faster than that of parent copolymer PB with 0.36 mol% PFP units. The possibility results from the steric effect of the mPEG grafted, which retards the necessary helical and positional adjustment of the crystalline stems within the crystals.14,23,38,39 However, it can be seen from Fig. 6 that the crystal transformation rate of PB is still the fastest, which shows that the regularity of the structure of PB is the main factor affecting the crystal transformation of PB.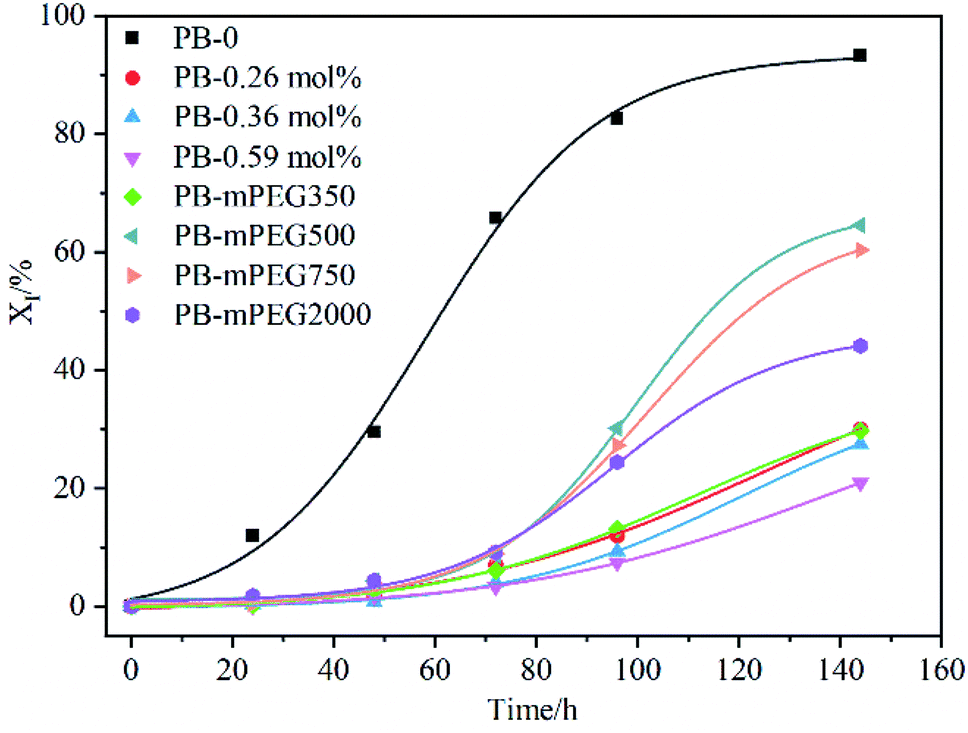 | ||
| Fig. 6 Time-variying curves of the Form I contents in different samples crystallized isothermally at 25 °C. | ||
To further verify this speculation, the mPEG2000 was blended with high isotacticity PB (Mw = 14.6 × 104 g mol−1 Đ = 4.1 I.I = 98.5%) by solution method. Three blend samples with weight content of mPEG2000 of 0.3%, 0.5% and 1.0% were obtained by dried at 60 °C under vacation to the constant. The samples were undergone annealing at 180 °C for 10 min and then isothermally crystallized at 25 °C. The evolution of DSC melting curves of blending samples shown in Fig. S7† also present two endothermic peaks varying with aging time, and the area of endothermic peaks at high temperature is enlarged, while ΔH assigned to Form II is diminished, indicating the occurrence of phase transition from Form II to Form I. For the clarity to understand the effect of mPEG2000 on the II–I phase transition, the dependency of content of Form I in different samples on the aging time are shown in Fig. 7. Compared with neat iPB, an acceleration of II–I phase transition is observed for the mPEG2000/iPB blend, and the promoting effect is positively correlated to loadings of mPEG2000.
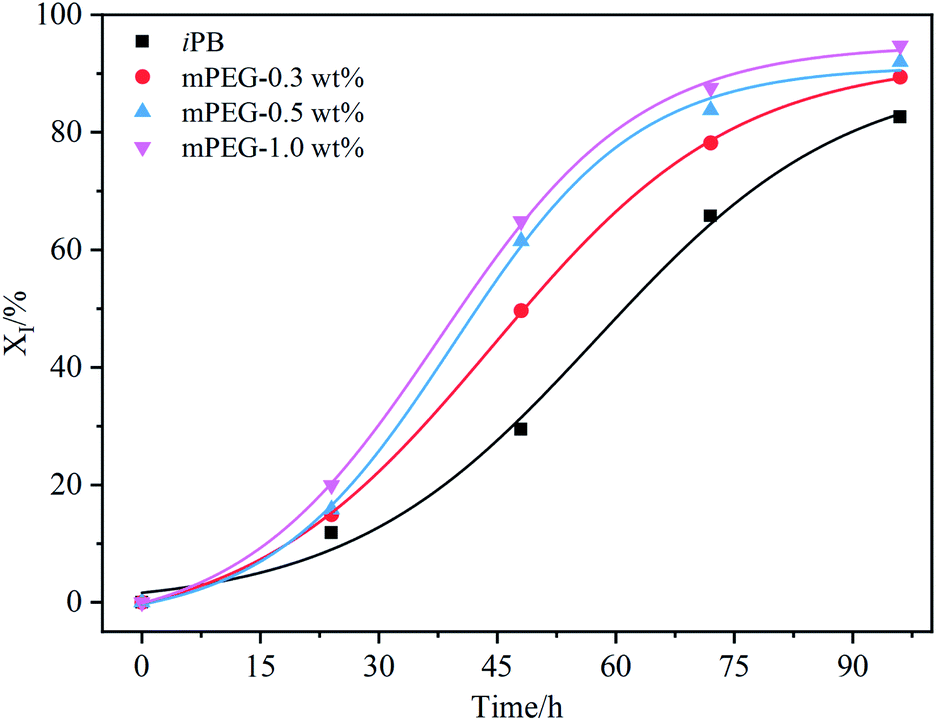 | ||
| Fig. 7 Time-varying curves of the Form I content in different mPEG2000/iPB blend samples. | ||
Contact angle study
Considering that the iPB is a promising candidate for the pipe and food packing, the relative surface hydrophilicity for the PB without or with different mPEG chain length were estimated by measuring the contact angles (θ) for water. As shown in Fig. 8, the water droplet at the film surface of PB containing 0.36 mol% PFP units shows the highest contact angle (θ = 107.9°). As expectedly, as the PFP group instead by hydrophilic mPEG, a decrease in θ for water on the PB film was observed, and such decrement (from 100.5° for PB-mPEG350, 94.1° for PB-mPEG500, 91.8° for PB-mPEG750, 83.8° for PB-mPEG2000) in water contact angle is parallel to the chain length of mPEG, indicating a higher improvement in surface hydrophilicity for PB with pendent longer mPEG chain. The chemical heterogeneity and enhancement of polar property of polymer surface by the presence of water-soluble mPEG moieties attached to hydrophobic PB backbone could be accounted, in part, for the increase in hydrophilicity. A model for heterogeneous surfaces which is analogous to that for rough surfaces was proposed by Johnson and Dettre.40 | ||
| Fig. 8 Water contact angle of different samples. (a) PB-0.36 mol%, (b) PB-mPEG350, (c) PB-mPEG500, (d) PB-mPEG750, (e) PB-mPEG2000. | ||
Conclusions
In this work, a series of 1-butene/pentafluorophenylundec-1-ene ester random copolymers with a narrow range of counit content from 0 to 0.59 mol% were synthesized to study the effects of the molecular factor of incorporated counits on the phase transition from kinetically favored Form II to the thermodynamically stable Form I, of which the PFP groups attached to PB main chain not only significantly restricts the kinetics of II–I phase transition but also lessens the ultimate Form I fraction in the transition plateau. Conveniently, the biocompatible mPEG segment was incorporated into PB side chain through the nucleophilic aromatic substitutions with the pentafluorophenyl ester under very mild basic conditions. DSC results showed that the improvement of Tm, Tc, and χc of the mPEG-functionalized PB depend on the employed mPEG molecular weight, and the PB modified by mPEG500 has the highest Tm, Tc, and χc. Moreover, the hydrophilicity of mPEG-functionalized PB is also the function as the mPEG molecular weight, being proportional to chain length of mPEG. In contrast to the inhibited role in the II–I phase transition, the incorporated mPEG segment plays an accelerated role in the phase transition evolution from Form II to Form I. However, in all polymer samples investigated, high isotactic PB homopolymer shows the best crystallization performance and the fastest phase transformation rate, indicating that the regularity of PB structure is the rate-determining factor for the crystallization and phase transformation.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 51873055 and 52173004) and Natural Science Foundation of Hebei Province (No. B2018202112).References
- J. Dong and Y. Hu. Coord, Chem. Rev., 2006, 250, 47–65 CAS.
- G. Zhang, C. Nam, L. Petersson, J. Jämbeck, H. Hillborg and T. C. M. Chung, Macromolecules, 2018, 51, 1927–1936 CrossRef CAS.
- C. Zou and C. Chen, Angew. Chem., Int. Ed., 2020, 59, 395–402 CrossRef CAS PubMed.
- H. Mu, G. Zhou, X. Hu and Z. Jian, Coord. Chem. Rev., 2021, 435, 213802 CrossRef CAS.
- H. Huang, L. Zhang, H. Li and Y. Hu, Chin. J. Catal., 2010, 31, 1077–1082 CAS.
- E. Chiellini, G. Cantoni and R. Solaro, J. Appl. Polym. Sci., 1996, 61, 285–291 CrossRef CAS.
- Z. Chen, J. Li, W. Tao, X. Sun, X. Yang and Y. Tang, Macromolecules, 2013, 46, 2870–2875 CrossRef CAS.
- I. D. Rubin, J. Polym. Sci., Part B: Polym. Lett., 1964, 2, 747–749 CrossRef CAS.
- G. Alfonso, F. Azzurri and M. Castellano, J. Therm. Anal. Calorim., 2001, 66, 197–207 CrossRef CAS.
- S. Kopp, J. Wittmann and B. Lotz, Polymer, 1994, 35, 908–915 CrossRef CAS.
- Y. Fujiwara, Polym. Bull., 1985, 13, 253–258 CrossRef CAS.
- M. Maruyama, Y. Sakamoto, K. Nozaki, T. Yamamoto, H. Kajioka, A. Toda and K. Yamada, Polymer, 2010, 51, 5532–5538 CrossRef CAS.
- M. L. Di Lorenzo, M. C. Righetti and B. Wunderlich, Macromolecules, 2009, 42, 9312–9320 CrossRef CAS.
- L. Zheng, L. Liu, C. Shao, W. Wang, B. Wang, L. Pan, Y. Li and Z. Ma, Macromolecules, 2019, 52, 1188–1199 CrossRef.
- L. He, B. Wang, F. Yang, Y. Li and M. Ma, Macromolecules, 2016, 49, 6578–6589 CrossRef CAS.
- C. Liu, Z. Zhang, S. Huang and Q. Chen, Polymer, 2018, 149, 146–153 CrossRef CAS.
- X. Cui, G. Gu, C. Li, N. Liu, Y. Gong and B. Liu, Polymer, 2020, 202, 122739 CrossRef CAS.
- Y. Zheng, M. L. Bruening and G. Baker, Macromolecules, 2007, 40, 8212–8219 CrossRef CAS.
- S. Xiang, D. Zhou, L. Feng, X. Bian, G. Li, X. Chen and T. Wang, Chin. J. Polym. Sci., 2018, 37, 258–267 CrossRef.
- D. Neugebauer, M. Theis, T. Pakula, G. Wegner and K. Matyjaszewski, Macromolecules, 2006, 39, 584–593 CrossRef CAS.
- K. Inomata, Y. Sasaki and T. Nose, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1904–1912 CrossRef CAS.
- B. Obermeier, F. Wurm, C. Mangold and H. Frey, Angew. Chem., Int. Ed., 2011, 50, 7988–7997 CrossRef CAS PubMed.
- C. An, Y. Li, Y. Lou, D. Song, B. Wang, L. Pan, Z. Ma and Y. Li, Polymers, 2019, 11, 837 CrossRef PubMed.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- G. Delaittre and L. Barner, Polym. Chem., 2018, 9, 2679–2684 RSC.
- M. B. Houck, T. J. Fuhrer, C. R. Phelps, L. C. Brown and S. T. Iacono, Macromolecules, 2021, 54, 5586–5594 CrossRef CAS.
- S. Xiao, G. Xie, H. Wang, L. Zhou and D. He, CN Patent, 1110281A, 18 October, 1995 Search PubMed.
- F. Martin, US Pat., 9657042B2, Montpellier, 23 May, 2017 Search PubMed.
- C. An, Y. Lou, Y. Li, B. Wang, L. Pan, Z. Ma and Y. Li, Macromolecules, 2019, 52, 4634–4645 CrossRef CAS.
- M. L. Di Lorenzo, R. Androsch and M. C. Righetti, Eur. Polym. J., 2015, 67, 264–273 CrossRef CAS.
- W. Wang, L. Hou, S. Luo, G. Zheng and H. Wang, Macromol. Chem. Phys., 2013, 214, 2245–2249 CAS.
- M. D. Purgett and O. Vogl, J. Appl. Polym. Sci., 1989, 27, 2051–2063 CAS.
- A. Das and P. Theato, Macromolecules, 2015, 48, 8695–8707 CrossRef CAS.
- J. Arranz-Andrés, B. Peña, R. Benavente, E. Pérez and M. L. Cerrada, Eur. Polym. J., 2007, 43, 2357–2370 CrossRef.
- T. Konishi, K. Nishida, T. Kanaya and K. Kaji, Macromolecules, 2005, 38, 8749–8754 CrossRef CAS.
- C. De Rosa, F. Auriemma, O. Ruiz de Ballesteros, L. Resconi and I. Camurati, Macromolecules, 2007, 40, 6600–6616 CrossRef CAS.
- B. Poon, M. Rogunova, A. Hiltner, E. Baer, S. Chum, A. Galeski and E. Piorkowska, Macromolecules, 2005, 38, 1232–1243 CrossRef CAS.
- A. T. Jones, Polymer, 1966, 7, 23–59 CrossRef CAS.
- O. Tarallo, O. Ruiz de Ballesteros, A. Bellissimo, M. Scoti, A. Malafronte, F. Auriemma and C. De Rosa, Polymer, 2018, 158, 231–242 CrossRef CAS.
- R. E. Johnson Jr and R. H. Dettre, J. Phys. Chem., 1964, 68, 1744–1750 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07698a |
| This journal is © The Royal Society of Chemistry 2021 |