
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiofunctional hollow γ-MnO2 microspheres by a one-pot collagen-templated biomineralization route and their applications in lithium batteries†
Huixia Hea,
Caihong Fua,
Yongling Anb,
Jinkui Feng b and
Jianxi Xiao*a
b and
Jianxi Xiao*a
aState Key Laboratory of Applied Organic Chemistry, Key Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: xiaojx@lzu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan 250100, China
First published on 17th November 2021
Abstract
γ-MnO2 nanomaterials play an essential role in the development of advanced electrochemical energy storage and conversion devices with versatile industrial applications. Herein, novel dandelion-like hollow microspheres of γ-MnO2 mesocrystals have been fabricated for the first time by a one-pot biomineralization route. Recombinant collagen with unique rod-like structure has been demonstrated as a robust template to tune the morphologies of γ-MnO2 mesocrystals, and a very low concentration of collagen can alter the nanostructures of γ-MnO2 from nanorods to microspheres. The as-prepared γ-MnO2 mesocrystals formed well-ordered hollow microspheres composed of delicate nanoneedle-like units. Among all the reported γ-MnO2 with various nanostructures, the γ-MnO2 microspheres showed the most prowess to maintain high discharge capacities after 100+ cycles. The superior electrochemical performance of γ-MnO2 likely results from its unique hierarchical micro-nano structure. Notably, the γ-MnO2 mesocrystals display high biocompatibility and cellular activity. Collagen plays a key dual role in mediating the morphology as well as endowing the biofunction of the γ-MnO2 mesocrystals. This environmentally friendly biomineralization approach using rod-like collagen as the template, provides unprecedented opportunity for the production of novel nanostructured metal oxides with superior biocompatibility and electrochemical performance, which have great potential in advanced implantable and wearable health-care electronic devices.
1. Introduction
Electrochemical energy storage and conversion systems such as batteries, fuel cells and supercapacitors are critical enabling technologies for renewable energy, and have wide applications in various industry sectors such as smart electrical grids, electric vehicles and wearable devices.1–3 The development of active electrode materials plays an essential role in the construction of advanced energy storage and conversion devices.4,5 Among the large number of possible electrode materials, manganese dioxides have received special attention due to their fabulous features such as high natural abundance, low cost as well as unique structures and morphologies. Manganese dioxides have very broad applications such as ion-sieves, catalysts and particularly as electrode materials in Zn/MnO2 and Li/MnO2 batteries.6–12MnO2 can form different kinds of polymorphs including α-, β-, γ- and δ- types, while each type possesses distinctive electrochemical features.13,14 Compared with other MnO2 structures, γ-MnO2 shows slower reduction in the electrical potential during the discharge process. γ-MnO2 is therefore the most commonly used Mn-based oxides as the cathode materials in primary batteries.6,14 With the advancement of nanoscience and nanotechnology, nanostructured materials have attracted increasing interests in the field of electrochemical energy storage and conversion. Metal oxide nanomaterials with small crystalline particle size have been reported to display excellent electrochemical activities, since their large specific surface areas result in the improved contact between the active materials and the electrolyte, leading to fast proton diffusion, low internal resistance and high utilization efficiency.15,16
Extensive efforts have been performed to create γ-MnO2 nanomaterials with well-defined morphologies. Electrodeposition and hydrothermal techniques are two mostly used methods to synthesize a variety of MnO2 nanostructures such as nanowires, nanoflakes, nanorods and nanotubes.13,17–22 PEG (polyethylene glycol) has been used as a polymer precursor to produce γ-MnO2 nanowires/nanotubes, which displayed good charge performance under low and heavy loads.6 These one-dimensional γ-MnO2 nanomaterials have been shown to reduce the electronic transport and ionic diffusion distance along their 1D direction, resulting in better rate performance.23,24 However, these nanomaterials may be liable to undesirable side reactions, leading to low volumetric energy density. Therefore, mesocrystals with hierarchical micro-nano structures have been recently developed to create improved electrode materials, which can maintain the advantages of micromaterials and nanomaterials.25–29
Biomineralization is a widespread synthetic route by various living organisms to produce functional inorganic crystals with exquisite hierarchical micro-nano structures.30 Biomolecules such as proteins play a critical role as the template to modulate the proper assembly of inorganic nanomaterials to form bones, teeth, shells and fish scales in nature. Collagen is the most abundant protein in animals, and it has a distinct triple helical structure resulted from its repetitive (Gly–X–Y)n amino acid sequences.31 The rod-like structure and self-assembly properties makes collagen robust building blocks, and collagen has been selected as the main biotemplate to synthesize inorganic mesocrystals in human body.32
Advanced implantable and wearable devices such as hearing aids, cardiac peacemakers and neuro-stimulators are playing increasingly important role in improving the quality of our life. The development of high-performance biocompatible electrode nanomaterials is critical for those personalized health-care electronics. Herein we have for the first time reported the fabrication of γ-MnO2 mesocrystals with hierarchical hollow microspherical structures. Recombinant collagen with uniform size and rod-like shape has been demonstrated as an excellent biotemplate to mediate the morphologies of γ-MnO2 mesocrystals. The unique hierarchical hollow micro-nano structures endow collagen with exceptional electrochemical performance. Furthermore, collagen plays as a functional agent to empower γ-MnO2 mesocrystals with high biocompatibility and bioactivity. The one-pot, green synthetic route using collagen as the key biomineralization template greatly contribute to the development of advanced inorganic mesocrystals with attractive biofunction and electrochemical performance, which may have promising applications in health-care energy systems.
2. Experimental section
2.1 Preparation of recombinant collagen
Recombinant collagen with high purity was obtained following previously reported protocols.33 Briefly, 50 mL overnight culture was transferred into 1000 mL LB medium containing ampicillin (50 mg mL−1), and was grown at 37 °C until optical density (OD) at 600 nm reaching 0.8. Protein expression was induced by 1 mM isopropyl beta-D-thiogalactopyranoside (IPTG) at 25 °C. Cells were collected after overnight incubation and re-suspended in 50 mL binding buffer (20 mM PB, 500 mM NaCl, 20 mM imidazole, pH 7.4). Cells were disrupted by sonication, and cell debris was removed. The supernatants were loaded into a Ni-NTA-Sepharose column, and eluted using the binding buffer supplemented with 500 mM imidazole. The collected protein was immediately dialyzed against 50 mM glycine buffer, pH 8.6. Recombinant collagen was harvested by trypsin digestion of the purified protein as previously described. The identity and purity of recombinant collagen was confirmed by SDS-PAGE. The pure recombinant collagen was lyophilized and stored at −20 °C for future use.2.2 Synthesis of γ-MnO2 mesocrystals
5 mM anhydrous manganese sulfate was added to 45 mL solution of 0.11 wt% collagen. After stirring for 30 minutes, 5 mL solution of 1.25 mol L−1 ammonium persulfate was slowly added. The mixture was vigorously stirred to form colourless transparent solution, and was then transferred to a 100 mL Teflonlined stainless steel autoclave. The autoclave was heated to 80 °C for 12 h and then cooled to room temperature. The final black precipitate was harvested by centrifugation, washed three times with distilled water, and dried in air at 60 °C. In order to investigate the effect of collagen on the morphology of MnO2 nanoparticles, the concentration of collagen was varied from 0 to 0.5 wt%, while [Mn(II)] was set as 0.1 mol L−1. Meanwhile, collagen concentration was set as 0.1 wt% and the concentration of [Mn(II)] was varied from 0.01 to 0.2 mol L−1 in order to investigate the role of Mn(II) in the formation of MnO2 nanostructures.2.3 Characterization of γ-MnO2 mesocrystals
FESEM (field emission scanning electron microscopy) measurements were performed on a Hitachi S-4800 field mission scanning electron microscope (Hitachi limited, Japan) with an operating voltage of 5.0 kV. TEM (transmission electron microscopy), HRTEM (high-resolution transmission electron microscopy), SAED (selected area electron diffraction), and electron diffraction (EDX) measurements were carried out using a JEM-2100 transmission electron microscope (JEOL, Japan) at 200 kV. X-ray diffraction (XRD) patterns were recorded on a Rigaku D/ma-2500 diffractometer with Cu Kα radiation (40 kV, 200 mA) at a scanning rate of 0.02° s−1 in the 2θ range from 20 to 80°. X-ray photoelectron spectroscopy (XPS) measurements were carried out on a Kratos Axis UltraDLD X-ray photoelectron spectrometer (England) with a monochrome X-ray source using Al Kα (1486.6 eV) radiation. The binding energies measured by XPS were corrected by referencing the C 1s line to 284.5 eV. FT-IR (Fourier transform infrared spectroscopy) spectra were obtained using a Nicolet NEXUS 670 infrared spectrometer. Thermogravimetric analysis (TGA) was performed on a TGA/NETZSCH STA449F3 instrument under a nitrogen atmosphere, employing a heating rate of 10 °C min−1 from 25 °C to 800 °C.2.4 Electrochemical properties of γ-MnO2 nanostructures
MnO2 nanomaterials prepared with 0.1 wt% collagen and 0.1 mol L−1 [Mn(II)] were used for further evaluation of electrochemical properties. 60 wt% of active material (γ-MnO2), 20 wt% super P carbon, and 20 wt% binder polyacrylic acid (PAA) were mixed to form a uniform slurry material. The slurry material was coated on a copper foil as a current collector and then dried at 100 °C under vacuum conditions for 12 h to form the working electrodes. 1 M lithium hexafluorophosphate (LiPF6) was prepared in the cosolvents of ethylene carbonate/diethyl carbonate (EC/DEC, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as the liquid electrolyte. The Celgard 2400 was used as the separator. To measure the electrochemical performance, the 2016-coin cells were assembled using the γ-MnO2 as the working electrodes, while the Li foil was used as the reference electrode and counter electrode. All the cells were installed in a glovebox with water, and the oxygen content was lower than 1 ppm. The galvanostatic discharge/charge cycles were measured on the NEWARE battery tester between 0.01 and 3 V (vs. Li+/Li) at different current densities.
:1, v/v) as the liquid electrolyte. The Celgard 2400 was used as the separator. To measure the electrochemical performance, the 2016-coin cells were assembled using the γ-MnO2 as the working electrodes, while the Li foil was used as the reference electrode and counter electrode. All the cells were installed in a glovebox with water, and the oxygen content was lower than 1 ppm. The galvanostatic discharge/charge cycles were measured on the NEWARE battery tester between 0.01 and 3 V (vs. Li+/Li) at different current densities.
2.5 Cytotoxicity of γ-MnO2
Cell counting kit-8 (CCK-8) was used to evaluate the cytotoxicity of γ-MnO2 nanomaterials using HFF-1 cells. HFF-1 cells were incubated in DMEM culture medium supplemented with 15% bovine serum solution in a humidified atmosphere of 5% CO2 at 37 °C. 100 μL of HFF-1 cell suspension was added in a 96-well cell-culture plate at a density of 5 × 103 cells per well, and incubated for 24 h to allow attachment. 100 μL microsphere-like γ-MnO2 nanoparticle suspension with four different final concentrations (0.1, 1, 10, and 50 μg mL−1) was then added into the wells. 100 μL DMEM medium was added in other wells as control groups. After the incubation of 24 h, 10 μL CCK-8 solution (2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfobenzene)-2H-tetrazole monosodium salt) was added into each well. The 96-well plate was incubated for 2 h (5% CO2 at 37 °C). The optical density at 450 nm was measured by Tecan Infinite F200/M200 Multifunction Microplate Reader (Tecan, Männedorf, Switzerland). Cell viability was calculated as the mean absorption value of four measurements of each condition divided by the mean absorption value of the DMEM control group.2.6 Cell adhesion assay
Nunclon Delta TC Microwell plates were coated with one thin layer of microsphere-like γ-MnO2. The plates coated with heat-denatured BSA were used as control groups. The plates were washed with PBS three times. Then 100 μL HFF-1 cell suspension (1 × 106 cells per mL) in serum-free DMEM medium was added, and incubated for 12h at 37 °C. Unattached cells was removed by washing with PBS buffer (10 mM). The amount of the attached cells was determined by a total deoxyribonucleic acid (DNA) quantification assay (Hoechst 33258, Solarbio). The cells were lysed by three freeze–thaw cycles in ultrapure water. Hoechst 33258 with a final concentration of 5 μg mL−1 was added to the cell lysates, and the mixtures were incubated in the dark for 1 hour. The fluorescence intensity was measured on a microplate reader (Tecan Infinite M200) at an excitation wavelength of 360 nm and an emission wavelength of 465 nm. The measurements were performed in triplicates.2.7 Immunofluorescence staining
Fluorescent confocal dishes (nontreated) were coated with one thin layer of microsphere-like γ-MnO2. The coverslips were washed 3 times with 10 mM, pH 7.4 PBS buffer. Then the HFF-1 cells were added on the coverslips at a density of 400 cells per mm2, and incubated at 37 °C for 24 h. The attached cells were fixed in cold 4% paraformaldehyde for 10 min, and permeabilized with 0.1% Triton X-100 for 5 min. 1% BSA in PBS buffer (10 mM, pH 7.2–7.4) was employed as a blocking agent for 0.5 h at room temperature. The actin cytoskeleton and cell nucleus were stained by incubating the cells with phalloidin-tetramethylrhodamine isothiocyanate for 1 hour and DAPI (1 mL, 5 μg mL−1) for 10 min at 37 °C. The images were recorded on Leica (Leica Microsystems Inc., Wetzlar, Germany) fluorescence microscope.3. Results and discussion
3.1 Synthesis and characterization of MnO2 mesocrystals
Recombinant collagen was produced following previously reported procedures, and was investigated as biotemplates to control the synthesis of manganese dioxide nanoparticles. Briefly, the mixture of collagen, manganese sulfate and ammonium persulfate was prepared and incubated at 80 °C for 12 h to obtain manganese dioxide mesocrystals. The final product was harvested, and characterized by X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) techniques. The XRD results indicated that the observed diffraction peaks were totally the same as the standard diffraction peaks of the orthogonal phase of γ-MnO2 (JCPDS no. 14-0644) (Fig. S1†). The XPS spectrum indicated the as-prepared mesocrystals consisted of Mn 3s, C 1s, N 1s, O 1s and Mn 2p, while the C and N elements confirmed the presence of collagen (Fig. S2a†). The high-resolution XPS spectrum of Mn 2p showed two strong peaks of 654.8 eV and 643.2 eV, which corresponded to Mn 2p1/2 and Mn 2p3/2, respectively, confirming the presence of MnO2 (Fig. S2b†). The XRD and XPS results demonstrated that pure γ-MnO2 was obtained via this collagen-templated biomineralization process.Field-emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) techniques were employed to characterize the morphology and detailed structure of the γ-MnO2 particles. The SEM images showed uniform microspheres with diameters of approximately 4 μm (Fig. 1a and b). The magnified SEM image indicated that the dandelion-like microsphere consisted of nanoneedle-like units. The TEM images indicated that the dandelion-like γ-MnO2 mesocrystals were assembled via an oriented growth with primary nanoparticles (Fig. 1c and d). The related selected area electron diffraction (SAED) graph showed a single crystal spot pattern with slightly elongated spots, demonstrating that the assembly of the primary γ-MnO2 nanoparticles was highly oriented (Fig. 1d).
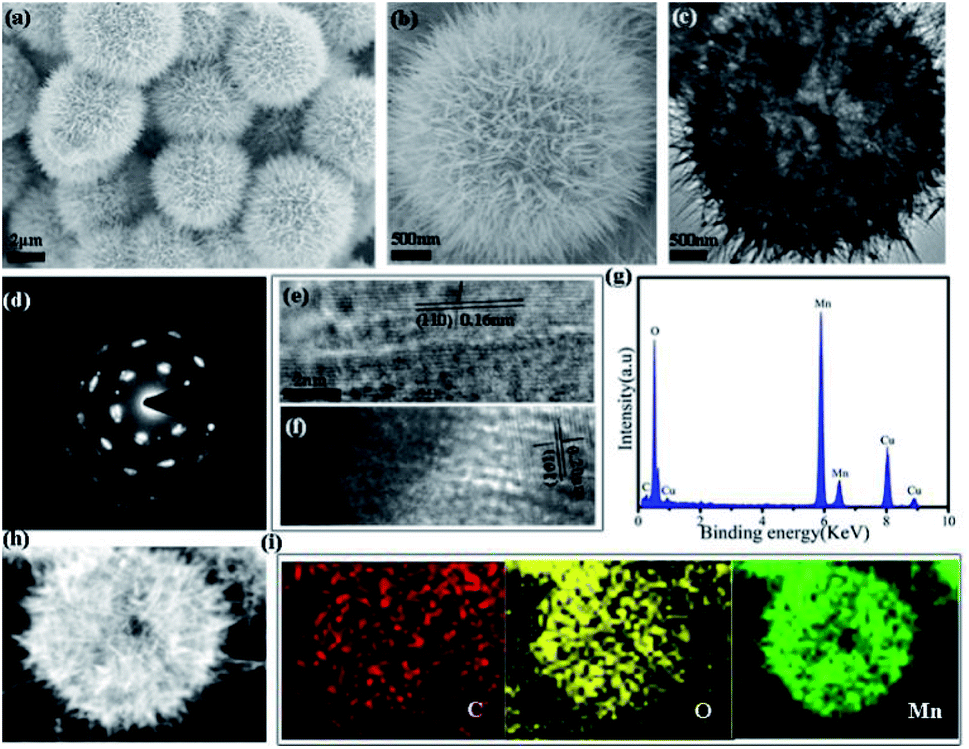 | ||
| Fig. 1 Characterization of the morphology of the γ-MnO2 mesocrystals obtained after 12 h via collagen-templated biomineralization ([collagen] = 0.1 wt%, [Mn(II)] = 0.1 mol L−1): (a and b) FESEM images; (c) TEM image; (d) the SAED pattern; (e and f) HRTEM images; (g) EDX spectrum; (h) HAADF-STEM images; (i) EDS mapping images of C, O, and Mn for the γ-MnO2 in (h). | ||
The high-resolution TEM image (HRTEM) showed the characteristic (104) lattice spacing (0.16 nm) and (110) lattice spacing (0.20 nm) of γ-MnO2, again confirming the well-ordered alignment of the nanoparticles (Fig. 1e and f). The electron diffraction (EDX) image showed the presence of the C, O and Mn elements of collagen and MnO2 in the mesocrystals (Fig. 1g). The high-angle annular dark-field scanning TEM (HAADF-STEM) image suggested that the primary MnO2 nanoparticles gradually assemble to form porous mesocrystals with hierarchical nanostructures (Fig. 1h). Elemental mapping by energy dispersive X-ray spectroscopy (EDS) results further indicated that the elements of C, O and Mn were evenly distributed in the mesocrystals (Fig. 1i).
The specific surface area and pore size distribution of the as-prepared γ-MnO2 mesocrystals were measured by the BET (Brunauer–Emmett–Teller) and BJH (Barrett–Joyner–Halenda) methods (Fig. S3†). The specific surface area was determined as ∼94.99 m2 g−1, while the average pore diameter was estimated as ∼20 nm, confirming the porous nature of the γ-MnO2 mesocrystals (Fig. S3†). FT-IR spectra of the mesocrystals prepared using different concentrations of collagen all showed the vibration peaks corresponding to the bonds of O–H, N–H, C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, again supporting the inclusion of collagen in the γ-MnO2 mesocrystals (Fig. S4†). Thermogravimetric analysis (TGA) was performed to further evaluate the percentage of collagen in the γ-MnO2 mesocrystals, and two stages of mass reduction were observed (Fig. S5†). The first stage resulted from the removal of water at the temperatures below 200 °C, while the second stage corresponded to the breakdown of collagen between 200–600 °C (Fig. S5†). It demonstrated that a significant portion of recombinant collagen was packed in the γ-MnO2 mesocrystals during the biomineralization process. To conclude, γ-MnO2 mesocrystals with exquisite nanostructures were obtained by the mild biomineralization approach using collagen as the biotemplate.
O, again supporting the inclusion of collagen in the γ-MnO2 mesocrystals (Fig. S4†). Thermogravimetric analysis (TGA) was performed to further evaluate the percentage of collagen in the γ-MnO2 mesocrystals, and two stages of mass reduction were observed (Fig. S5†). The first stage resulted from the removal of water at the temperatures below 200 °C, while the second stage corresponded to the breakdown of collagen between 200–600 °C (Fig. S5†). It demonstrated that a significant portion of recombinant collagen was packed in the γ-MnO2 mesocrystals during the biomineralization process. To conclude, γ-MnO2 mesocrystals with exquisite nanostructures were obtained by the mild biomineralization approach using collagen as the biotemplate.
3.2 Time-dependent growth of the dandelion-like γ-MnO2 mesocrystals
FESEM and XRD techniques were applied to monitor the time-dependent growth of γ-MnO2 mesocrystals in order to examine its underlying assembly mechanism (Fig. 2 and S6†). The SEM images indicated that nanoparticles were observed within 10 min of reaction, and well-defined dandelion-like microspheres became the dominant products after 45 min of incubation (Fig. 2a–d). When the incubation time got longer, the dandelion-like microspheres gradually became larger, and they reached a steady state with the diameters of approximately 4 μm after 90 min of cultivation (Fig. 2e–i). Meanwhile, the mesocrystals collected at different incubation time from 10 min to 90 min all showed similar XRD patterns, indicating that the nanoparticles were consistently orthogonal γ-MnO2 (Fig. S6†). | ||
| Fig. 2 FESEM images of the γ-MnO2 mesocrystals obtained via collagen-templated biomineralization ([collagen] = 0.1 wt%, [Mn(II)] = 0.1 mol L−1) by different incubation time: (a) 10 min; (b) 15 min; (c) 30 min; (d) 45 min; (e) 60 min; (f) 90 min; (g) 120 min; (h) 2.5 h; (i) 4 h. | ||
Notably, cracked dandelion-like microspheres were also observed in some SEM images of the γ-MnO2 mesocrystals prepared after 120 min of incubation (Fig. 3). The inner side of these incomplete microspheres were clearly visualized, indicating that the microspheres were completely hollow. In addition, the membranes of the microspheres were composed of nanoneedles of approximately 500 nm in length. Furthermore, the observation of a series of holes of different sizes in the microspheres gave a vivid picture of the formation of the microspheres, suggesting that the nanoneedles of similar sizes gradually assembled in a highly oriented way along the surface of the microspheres, and finally formed a complete membrane of the hollow microspheres (Fig. 3).
 | ||
| Fig. 3 Observation of some incomplete microspheres in the FESEM images of the γ-MnO2 mesocrystals obtained after 2 h via collagen-templated biomineralization ([collagen] = 0.1 wt%, Mn(II) = 0.1 mol L−1). | ||
3.3 The effect of collagen concentration on the MnO2 mesocrystals
The role of collagen in the biomineralized synthesis of MnO2 mesocrystals was further examined by varying the concentrations of collagen (Fig. 4). Mn(II) was kept at a constant concentration of 0.1 mol L−1. In the absence of collagen, rod-like nanoparticles were formed (Fig. 4a). The addition of very low concentration of collagen (0.02 wt%) led to the formation of uniform γ-MnO2 microspheres (Fig. 4b). The sharp geometric transition from nanorods to microspheres demonstrated that collagen provided a very robust template to control the morphologies of MnO2 mesocrystals. When the collagen concentration was further increased to 0.05 wt% or 0.2 wt%, well-ordered dandelion-like microspheres were consistently obtained (Fig. 4). These results have demonstrated that collagen played a key role in modulating the morphologies of MnO2 mesocrystals, and the collagen-templated biomineralization steadily tended to create microsphere-like nanostructures under a broad range of reaction conditions. | ||
| Fig. 4 FESEM and TEM images of the γ-MnO2 mesocrystals obtained after 12 h with a constant Mn(II) concentration of 0.1 mol L−1 and various collagen concentrations: (a) 0 wt%; (b) 0.02 wt%; (c) 0.05 wt%; (d) 0.2 wt%. | ||
3.4 The effect of Mn(II) concentration on the MnO2 mesocrystals
The role of Mn(II) in the biomineralized synthesis of MnO2 mesocrystals was also evaluated (Fig. S7†). The concentration of collagen was kept constant as 0.1 wt%, while the concentration of Mn(II) was varied. When the Mn(II) concentration was 0.05 mol L−1, nanoflowers with diameters of approximately 1.5 μm were formed. When the Mn(II) concentration was increased to 0.2 mol L−1, uniform nanorods of lengths of ∼2 μm became predominant (Fig. S7†). These results indicated that Mn(II) also played a critical role in the morphology of biomineralized γ-MnO2 mesocrystals.3.5 Electrochemical properties of dandelion-like γ-MnO2 mesocrystals
The electrochemical properties of the as-prepared γ-MnO2 mesocrystals were determined to evaluate its potential applications in batteries. The cycling performance of γ-MnO2 particles prepared using different concentrations of collagen was compared (Fig. 5). The nanorod-like γ-MnO2 mesocrystals prepared without collagen showed a high initial discharge capacity of 2318.1 mA h g−1 on the first cycle, and immediately decreased to 886.4 mA h g−1 on the second cycle (Fig. 5a). The discharge capacity reached the minimum value of 280 mA h g−1 after 40 cycles. It was consistent with previous reports that the discharge capacity of MnO2 nanomaterials showed rapid decrease during the cycling processes.20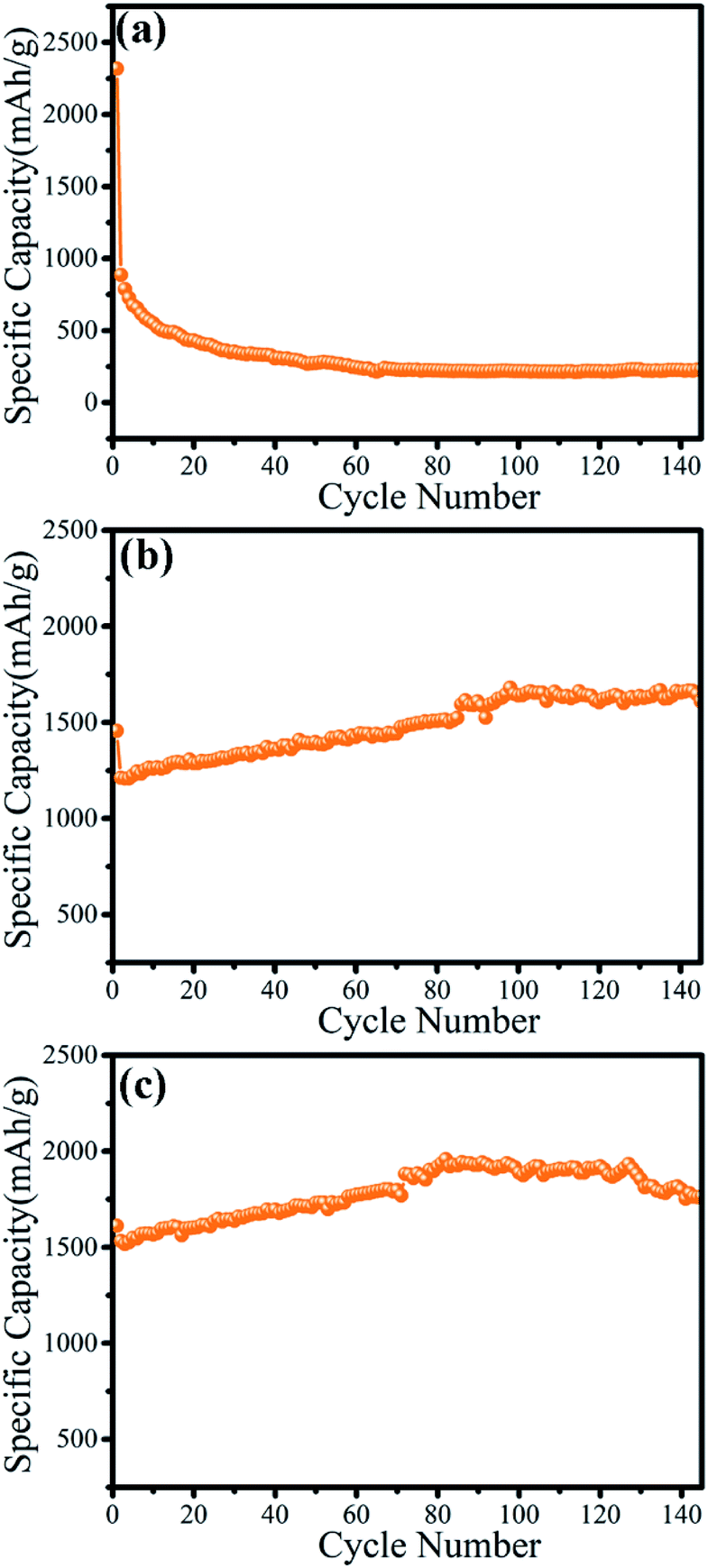 | ||
| Fig. 5 The cycling performance of γ-MnO2 mesocrystals prepared by different concentrations of collagen: (a) 0; (b) 0.05 wt%; (c) 0.1 wt%. | ||
Notably, the microsphere-like γ-MnO2 mesocrystals prepared with 0.05 wt% collagen showed a very unexpected trend (Fig. 5b). Despite a slight decrease at the first cycle, the discharge capacity started to increase from the 1212.0 mA h g−1 (second cycle) to 1640.9 mA h g−1 (100th cycle). Surprisingly, the microsphere-like γ-MnO2 mesocrystals maintained the high discharge capacity after 160 cycles (1597.5 mA h g−1). Furthermore, the microsphere-like γ-MnO2 mesocrystals prepared with 0.1 wt% collagen displayed an even better cycling performance. Its discharge capacity kept increasing from the second cycle (1532.6 mA h g−1) to the 80th cycle (1919.9 mA h g−1), and maintained the high discharge capacity after 140 cycles (1800 mA h g−1). The even better performance of the microsphere-like γ-MnO2 mesocrystals prepared with 0.1 wt% collagen than those obtained using 0.05 wt% collagen may contribute to its more uniformly distributed architecture. These results demonstrated that the morphology determined the cycling performance of γ-MnO2 nanomaterials.
The electrochemical performance of the microsphere-like γ-MnO2 mesocrystals prepared with 0.1 wt% collagen was further examined (Fig. 6). The first and second discharge/charge profiles of the γ-MnO2 were recorded in the voltage range of 0.01–3.00 V (vs. Li+/Li) at 0.2C (Fig. 6a). The first discharge capacity and charge capacity were 1614.7 mA h g−1 and 1537.6 mA h g at a rate of 0.2C, respectively. The irreversible capacity loss during the first discharge cycle may be attributed to the formation of solid electrolyte interfaces (SEI).34,35 The specific capacity of γ-MnO2 electrodes at different current densities of 0.2C, 0.5C, 1C and 2C were 1539.7 mA h g−1, 1516.4 mA h g−1, 1415.1 mA h g−1 and 1098.3 mA h g−1, respectively, while the corresponding retention rates were 95.2%, 97.6%, 94.3% and 86.1% (Fig. 6b and c). All the results have consistently demonstrated that the cathode materials for lithium ion batteries using the microsphere-like γ-MnO2 mesocrystals have displayed highly stable electrochemical performance. Compared with the γ-MnO2 mesocrystals with other reported morphologies such as nanocubes, the γ-MnO2 microspheres showed the most prowess to maintain high discharge capacities after 100+ cycles.36
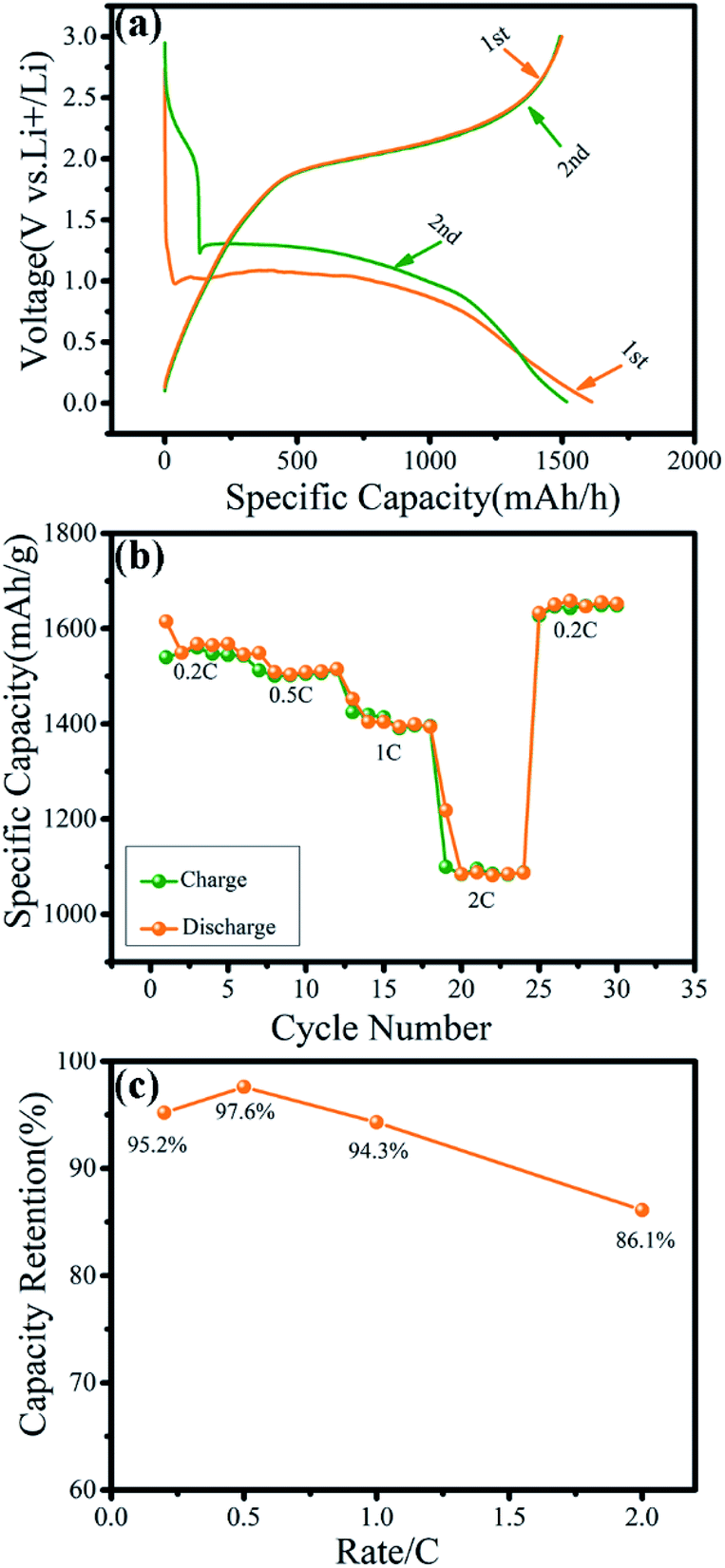 | ||
| Fig. 6 Electrochemical properties of the γ-MnO2 electrodes. (a) The charge/discharge profiles of γ-MnO2 electrode at 0.2C between 0.01 to 3.0 V; (b) rate capacities of γ-MnO2 at different current densities from 0.2C to 2.0C; (c) capacity retention of γ-MnO2 at different rates. | ||
The superior electrochemical performance of the dandelion-like γ-MnO2 nanomaterials may be attributed to their unique morphology. The dandelion-like hollow microspheres of γ-MnO2 have orderly packed ultrathin nanoneedles, leading to a much shorter diffusion distance of lithium ions and more efficient contact with the electrolyte. A short lithium ion diffusion path has been proved to promote surface electrochemical reactions and thus increase the capacity during the charge–discharge process.37–41 Furthermore, the exquisite geometry of γ-MnO2 mesocrystals could facilitate better accommodation of the strain energy associated with lithium insertion-removal, resulting in suitable electrode–electrolyte contact area and reduced polarization of the electrode, which would accelerate the discharge capacity.42,43 Most importantly, the highly orderly organization of γ-MnO2 microspheres may resist the volume expansion and morphological changes, which have been reported to cause particle pulverization and loss of capacity.44,45
3.6 Biocompatibility and bioactivity of microsphere-like γ-MnO2
The biocompatibility and bioactivity of the as-prepared microsphere-like γ-MnO2 nanomaterials was further examined (Fig. 7). In vitro cytotoxicity of microsphere-like γ-MnO2 nanomaterials was determined using HFF-1 cells by the CCK-8 assay (Fig. 7a). The microsphere-like γ-MnO2 exhibited similarly high cell viability at four different concentrations (0.1, 1, 10, and 50 μg mL−1), demonstrating the high biocompatibility of the synthesized γ-MnO2 nanomaterials, which likely resulted from the inclusion of biocompatible collagen in the γ-MnO2 nanomaterials (Fig. 7a).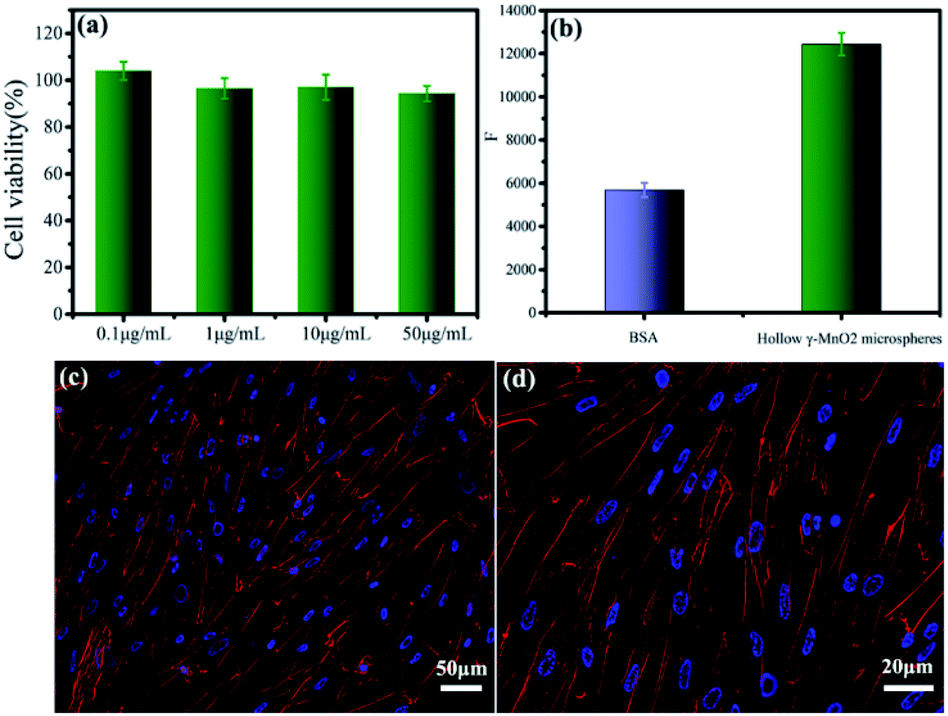 | ||
| Fig. 7 The biocompatibility and bioactivity of the microsphere-like γ-MnO2. In vitro cytotoxicity of microsphere-like γ-MnO2 nanomaterials with different concentrations was evaluated by examining the viability of HFF-1 cells (a). The adhesion ability of HFF-1 cells to microsphere-like γ-MnO2 nanomaterials versus the control BSA (b). Adhesion and spreading of HFF-1 cells on the surface of microsphere-like γ-MnO2 (c and d). Cells were fixed and stained for actin stress fibers (red) and nuclei (blue). | ||
The bioactivity of microsphere-like γ-MnO2 nanomaterials was evaluated using HFF-1 cells by cell adhesion assay (Fig. 7b). HFF-1 cells were cultured on the plate wells coated with microsphere-like γ-MnO2 and heat-denatured bovine serum albumin (BSA), respectively. The fluorescence intensity of the wells coated with microsphere-like γ-MnO2 was remarkably higher than those coated with the control BSA, indicating that a significant amount of HFF-1 cells attached to the as-prepared microsphere-like γ-MnO2 nanomaterials (Fig. 7b). It showed that collagen can functionalize γ-MnO2 nanomaterials with nice cell adhesion activity.
Cell adhesion and spreading properties were investigated by confocal fluorescence microscopy (Fig. 7c and d). Actin stress fibers and nuclei in HFF-1 cells were stained with phalloidin-tetramethylrhodamine isothiocyanate and DAPI, respectively. The HFF-1 cells on the γ-MnO2-coated substrates displayed well-developed actin cytoskeletal structure (Fig. 7c and d). The extensive cell adhesion and spreading indicated that collagen endowed γ-MnO2 nanomaterials with cellular function. These results demonstrated that collagen not only provides a superior template to tune the morphology of MnO2, but also plays as a functional agent to enable the nanomaterials with fabulous bioactivity, which could be essential characteristics for implantable electrical devices.
3.7 Possible mechanism of the collagen-templated biomineralized synthesis of γ-MnO2
Collagen has been demonstrated as a determinant role in the biomineralization route to modulate the nanostructures of γ-MnO2 mesocrystals (Scheme 1). Firstly, recombinant collagen has a high content of charged and polar amino acids, whose interaction with the Mn(II) ions results in the formation of a complex of collagen and Mn(II). Secondly, the addition of (NH4)2S2O8 drives the formation of manganese dioxide due to the redox reaction between Mn2+ and S2O82−: MnSO4 (aq) + (NH4)2S2O8 (aq) + 2H2O (l) → MnO2 (s) + (NH4)2SO4 (aq) + 2H2SO4 (aq). Thirdly, the free MnO2 in the reaction solution gets crystallized, leading to the formation of primary nanoparticles; while collagen covers the surface of the primary MnO2 nanoparticles, stabilizing them in the aqueous solution. Finally, these primary nanoparticles further assemble into larger nanoparticles and finally form dandelion-like hollow microspheres. Collagen provides a unique triple helical template to assist the assembly in a highly oriented way.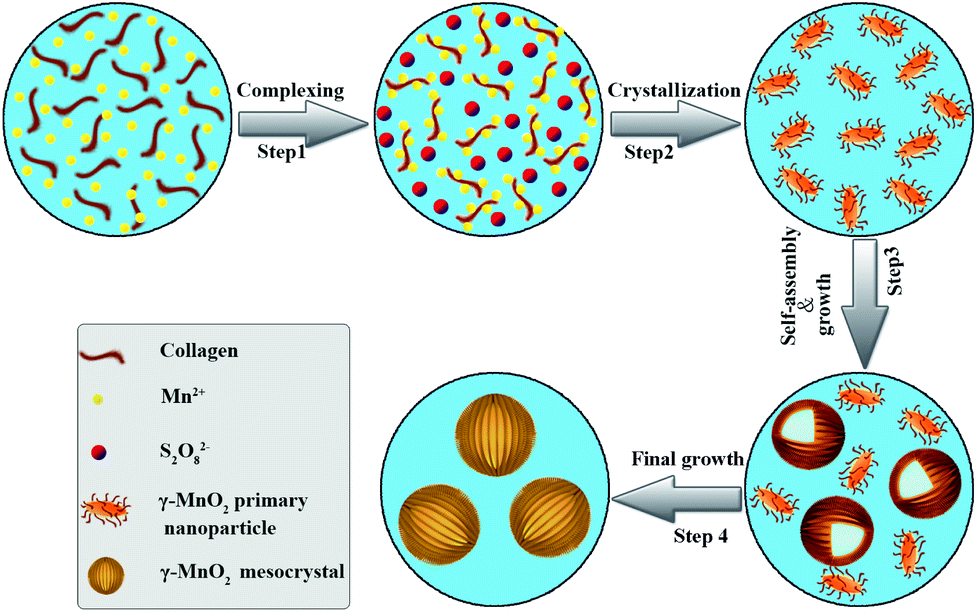 | ||
| Scheme 1 Schematic illustration of the essential role of collagen in the modulation of hollow dandelion-like microspheres of γ-MnO2 mesocrystals. | ||
4. Conclusions
The controlled synthesis of γ-MnO2 nanostructures plays a critical role in the construction of active materials for electrodes. Herein, γ-MnO2 mesocrystals with novel dandelion-like hollow microspheres have been created through a one-pot biomineralization approach using recombinant collagen as the template. The XRD and XPS results indicated that pure γ-MnO2 was harvested via this collagen-templated biomineralization process, while the SEM and TEM studies demonstrated that the as-prepared γ-MnO2 mesocrystals formed well-defined hollow microspheres composed of delicate nanoneedle-like units. The FT-IR and TGA characterization confirmed the inclusion of recombinant collagen in the γ-MnO2 mesocrystals. Collagen provides a robust template for tuning the nanostructures of γ-MnO2, and a very low concentration of collagen can shift the morphology of γ-MnO2 from nanorods to microspheres. A possible mechanism has been proposed for the biomineralized synthesis of γ-MnO2 that the special amino acid content and triple helical structure endows collagen as the unique template to mediate the nanostructures of γ-MnO2 mesocrystals.Notably, the as-prepared hollow microsphere-like γ-MnO2 mesocrystals displayed superior electrochemical performance. Compared with the γ-MnO2 mesocrystals with other nanostructures, the hollow γ-MnO2 microspheres showed the most prowess to maintain high discharge capacities after 100+ cycles. It indicated the morphology played a dominant role in the cycling performance of γ-MnO2 nanomaterials. The environmentally amiable collagen-mediated biomineralization strategy can consistently produce γ-MnO2 mesocrystals with delicate microsphere-like morphologies, which shows great potential for the fabrication of active cathode materials of lithium ion batteries. Furthermore, the microsphere-like γ-MnO2 mesocrystals possessed superior biocompatibility and cellular activity. Collagen has been demonstrated not only as a unique biotemplate to modulate the morphologies of γ-MnO2 mesocrystals, but also as a functional agent to enable their biocompatibility and bioactivity. This novel collagen-assisted green synthetic route provides promising opportunities for the creation of high-performance biofunctional γ-MnO2 mesocrystals and other metal oxide nanomaterials, which have great potential in implantable health-care devices.
Conflicts of interest
The authors declare no competing conflict of interests.Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (Grant No. 22074057, 21775059), the National Natural Science Foundation of Gansu Province (Grant No. 20YF3FA025, 18YF1NA004), and Lanzhou Talent Innovation and Entrepreneurship Project (Grant No. 2019-RC-43).References
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- Q. F. Zhang, E. Uchaker, S. L. Candelaria and G. Z. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- M. S. Islam and C. A. J. Fisher, Chem. Soc. Rev., 2014, 43, 185–204 RSC.
- F. Y. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- F. Y. Cheng, J. Chen, X. L. Gou and P. W. Shen, Adv. Mater., 2005, 17, 2753–2756 CrossRef CAS.
- S. W. Lee, J. Kim, S. Chen, P. T. Hammond and Y. Shao-Horn, ACS Nano, 2010, 4, 3889–3896 CrossRef CAS PubMed.
- J. G. Yang, X. P. Han, X. L. Zhang, F. Y. Cheng and J. Chen, Nano Res., 2013, 6, 679–687 CrossRef CAS.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- A. Mauger, M. Armand, C. M. Julien and K. Zaghib, J. Power Sources, 2017, 353, 333–342 CrossRef CAS.
- R. Chitrakar, H. Kanoh, Y. S. Kim, Y. Miyai and K. Ooi, J. Solid State Chem., 2001, 160, 69–76 CrossRef CAS.
- J. F. Xiao, R. T. Gao, B. Niu and Z. M. Xu, J. Hazard. Mater., 2021, 407, 124704–124712 CrossRef CAS PubMed.
- X. Wang and Y. D. Li, J. Am. Chem. Soc., 2002, 124, 2880–2881 CrossRef CAS PubMed.
- S. L. Chou, F. Y. Cheng and J. Chen, J. Power Sources, 2006, 162, 727–734 CrossRef CAS.
- S. Liu, S. H. Sun and X. Z. You, Nanoscale, 2014, 6, 2037–2045 RSC.
- M. H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- M. Ghaemi, L. Khosravi-Fard and J. Neshati, J. Power Sources, 2005, 141, 340–350 CrossRef CAS.
- K. Katakura, S. Nishimura and Z. Ogumi, J. Power Sources, 2005, 146, 217–221 CrossRef CAS.
- C. C. Hu and T. W. Tsou, Electrochim. Acta, 2002, 47, 3523–3532 CrossRef CAS.
- F. Y. Cheng, J. Z. Zhao, W. Song, C. S. Li, H. Ma, J. Chen and P. W. Shen, Inorg. Chem., 2006, 45, 2038–2044 CrossRef CAS PubMed.
- X. Wang and Y. D. Li, Chem. Commun., 2002, 764–765, 10.1039/b111723h.
- V. Subramanian, H. W. Zhu, R. Vajtai, P. M. Ajayan and B. Q. Wei, J. Phys. Chem. B, 2005, 109, 20207–20214 CrossRef CAS PubMed.
- S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Roziere, Energy Environ. Sci., 2011, 4, 4761–4785 RSC.
- S. Xin, Y. G. Guo and L. J. Wan, Acc. Chem. Res., 2012, 45, 1759–1769 CrossRef CAS PubMed.
- J. Xiao, J. M. Zheng, X. L. Li, Y. Y. Shao and J. G. Zhang, Nanotechnology, 2013, 24, 424004–424011 CrossRef PubMed.
- Y. Lu, R. Gao, J. M. Song, W. F. Li and G. Hu, J. Nanosci. Nanotechnol., 2020, 20, 2239–2246 CrossRef CAS PubMed.
- X. W. Lou, L. A. Archer and Z. C. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- S. D. Sun, Q. Yang, S. H. Liang and Z. M. Yang, CrystEngComm, 2017, 19, 6225–6251 RSC.
- X. J. Wang, J. Feng, Y. C. Bai, Q. Zhang and Y. D. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- E. Ruiz-Hitzky, M. Darder, P. Aranda and K. Ariga, Adv. Mater., 2010, 22, 323–336 CrossRef CAS PubMed.
- J. A. M. Ramshaw, J. A. Werkmeister and G. J. Dumsday, Bioengineered, 2014, 5, 227–233 CrossRef PubMed.
- O. A. Tertuliano and J. R. Greer, Nat. Mater., 2016, 15, 1195–1202 CrossRef CAS PubMed.
- Y. Y. Peng, A. Yoshizumi, S. J. Danon, V. Glattauer, O. Prokopenko, O. Mirochnitchenko, Z. X. Yu, M. Inouye, J. A. Werkmeister, B. Brodsky and J. A. M. Ramshaw, Biomaterials, 2010, 31, 2755–2761 CrossRef CAS PubMed.
- M. S. Wu and P. C. J. Chiang, Electrochem. Commun., 2006, 8, 383–388 CrossRef CAS.
- L. Wang, Y. L. Zheng, X. H. Wang, S. H. Chen, F. G. Xu, L. Zuo, J. L. Wu, L. L. Sun, Z. Li, H. Q. Hou and Y. H. Songt, ACS Appl. Mater. Interfaces, 2014, 6, 7117–7125 CrossRef CAS PubMed.
- J. Z. Zhao, Z. L. Tao, J. Liang and J. Chen, Cryst. Growth Des., 2008, 8, 2799–2805 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- L. N. Cong, H. M. Xie and J. H. Li, Adv. Energy Mater., 2017, 7, 1601906–1601942 CrossRef.
- H. K. Liu, Mater. Res. Bull., 2013, 48, 4968–4973 CrossRef CAS.
- H. K. Liu, G. X. Wang, Z. P. Guo, J. Z. Wang and K. Konstantinov, J. Nanosci. Nanotechnol., 2006, 6, 1–15 CrossRef CAS PubMed.
- Y. M. Sun, N. A. Liu and Y. Cui, Nat. Energy, 2016, 1, 16071–16083 CrossRef CAS.
- S. P. Ma, X. Q. Zhang, S. M. Li, Y. X. Cui, Y. L. Cui, Y. Zhao and Y. H. Cui, Ionics, 2020, 26, 2165–2176 CrossRef CAS.
- H. Ma, S. Y. Zhang, W. Q. Ji, Z. L. Tao and J. Chen, J. Am. Chem. Soc., 2008, 130, 5361–5367 CrossRef CAS PubMed.
- Y. S. Hu, Y. G. Guo, W. Sigle, S. Hore, P. Balaya and J. Maier, Nat. Mater., 2006, 5, 713–717 CrossRef CAS PubMed.
- J. Jamnik, R. Dominko, B. Erjavec, M. Remskar, A. Pintar and M. Gaberscek, Adv. Mater., 2009, 21, 2715–2719 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S7. See DOI: 10.1039/d1ra06899g |
| This journal is © The Royal Society of Chemistry 2021 |