
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic laboratory approach to produce Mg-rich carbonates at low temperature†
Zulfa Ali Al Disi *a,
Nabil Zouaria,
Essam Attiac,
Mazen Al-Asalib,
Hamad Al Saad Al-Kuwarib,
Fadhil Sadoonib,
Maria Dittrichbd and
Tomaso R. R. Bontognalibef
*a,
Nabil Zouaria,
Essam Attiac,
Mazen Al-Asalib,
Hamad Al Saad Al-Kuwarib,
Fadhil Sadoonib,
Maria Dittrichbd and
Tomaso R. R. Bontognalibef
aEnvironmental Sciences Program, Department of Biological & Environmental Sciences, College of Arts Sciences, Qatar University, P. O. Box 2713, Doha, Qatar. E-mail: zaldisi@qu.edu.qa
bEnvironmental Science Centre, Qatar University, P. O. Box 2713, Doha, Qatar
cCentral Laboratory Unit, Qatar University, P. O. Box 2713, Doha, Qatar
dDepartment of Physical and Environmental Sciences, University of Toronto Scarborough, 1265 Military Trail, Toronto, M1C 1A4, Canada
eSpace Exploration Institute (SPACE-X), 68 Faubourg de l'Hopital, 2000 Neuchatel, Switzerland
fDepartment of Environmental Sciences, University of Basel, Klingelbergstrasse 27, 4056 Basel, Switzerland
First published on 18th November 2021
Abstract
Dolomite is a common Mg-rich carbonate in the geological record, but the mechanism of its formation remains unclear. At low temperature, the incorporation of magnesium ions into the carbonate minerals necessary to form dolomite is kinetically inhibited. Over the decades, several factors that possibly allow for overcoming this kinetic barrier have been proposed, and their effectiveness debated. Here, we present the results of a large number of laboratory precipitation experiments that have been designed to identify and compare the factors that promote the formation of Mg-rich carbonates. Under the tested conditions, the most interesting observations include: (1) from solutions that mimic evaporitic seawater, the maximum mol% of Mg incorporated in high Mg calcite is 35, (2) carbonates with a mol% of Mg above 40 were obtained exclusively in the presence of organic molecules, (3) no correlation was observed between the charge of the organic molecules and the incorporation of Mg, (4) the mode (i.e., slow vs. fast mixing) used to add carbonate to the solution obtaining supersaturation has a significant impact on the forming mineral phase (aragonite vs. nesquehonite vs. high Mg calcite) and its Mg content. These findings allow for a more informed evaluation of the existing models for dolomite formation, which are based on the study of natural environments and ancient sedimentary sequences.
1 Introduction
Dolomite – MgCa(CO3)2 – is a common carbonate mineral in ancient sedimentary sequences. Despite the fact that seawater is supersaturated with respect to dolomite, its formation as a primary or early diagenetic precipitate is very rare in modern environments.1,2 Laboratory experiments corroborate this observation showing that at temperatures lower than 60–80 °C, the incorporation of Mg into anhydrous carbonate minerals is kinetically inhibited.3–6 Because the thick strata of dolomite present in ancient sedimentary sequences are practically impossible to explain as a result of a high temperature metamorphic/replacement process (e.g., through a replacement process whereby Mg is transported by diffusion),7,8 factors that under some circumstances allow for overcoming such a kinetic barrier leading to the formation of dolomite as a primary or early diagenetic mineral must exist. The identification of such factors is at the center of a debate that is often referred to as “the dolomite problem”.2,4,9,10During the last two centuries, many models for dolomite formation have been proposed, without providing an unanimously accepted solution to the problem. In the last decades, the hypothesis that gained more momentum than others posits that microbes may play a mediating role in the mineralization process (e.g. ref. 4 and 11–17). Extracellular polymeric substances (EPS) that microorganisms secrete from their cells to obtain ecological advantages – including both large polymers and low molecular weight organic matter18,19 – have been shown to be of a particular importance for the process. Besides providing nucleation sites, such organic molecules may promote the dehydration of Mg, which is a recognized kinetic barrier for the formation of Mg-rich carbonates.2,5,15,20–22 It is known that EPS are secreted by microorganisms into their microenvironment as response to ecological stress.18,23 EPS are usually produced in huge amounts as a protective strategy to overcome extreme environmental variations (e.g., evaporation and subsequent desiccation and increase in salinity).24
Sabkhas – the Arabic term for flat saline mud areas – are modern dolomite-forming environments where such stresses are exerted on microbial populations.25 Sabkhas occur in regions characterized by extreme climatic and environmental conditions, such as high temperature, salinity, light intensity, etc.26,27 Since long, sabkhas have been studied as modern analogues for interpreting some ancient dolomite-rich sedimentary sequences, and several models for dolomite formation are based on the study of these depositional settings.28 More recent studies indicate that not only evaporation, but also microbial processes may play a key role in these evaporitic environments.2,29,30 It has been suggested that the strong evaporation causing consequently high salinity and increased supersaturation, combined with microbial EPS produced under extreme ecological stress, may be linked to the formation of dolomite at ambient temperature. Supersaturated pore-waters, as well as high alkalinity and suitable redox conditions sustained by microbial respiration in buried microbial mats,2 may create the chemical conditions required for low temperature dolomite formation. Recent works have focused on the community composition of microbial mats (with top layers comprised mostly of cyanobacteria and anoxygenic phototrophs) that promote the formation of dolomite in a sabkha environment, revealing that anoxygenic phototrophic microbes are particularly important for the mineralization process.10,27 Specifically, a cyclic shift in microbial community between cyanobacteria and anoxygenic phototrophs results in the production of EPS with an increased concentration of carboxylic functional groups, which in turn favors dolomite formation.
Considering the almost ubiquitous presence of microbes and EPS in modern environments where dolomite is not forming, it can be deduced that only EPS with a specific composition play a catalytic role (e.g. ref. 15 and 27), or that EPS need to co-occur with other factors. The identification of co-occurring factors essential for dolomite formation cannot be easily inferred by reviewing and comparing the results of published studies, which are often conducted using different methodologies. For this reason, we have conducted laboratory precipitation experiments with the goal of systematically comparing which minerals form under various conditions, using different methods to reach supersaturation. To make the results of the experiment as relevant as possible for studies of natural environments, we performed experiments with artificial solutions that mimic the pore waters composition of the Dohat Faishakh sabkha in Qatar28,30 (see Map in ESI†). This sabkha has been the focus of previous studies, which led to the isolation of microbes capable of precipitating Mg-rich carbonates in the laboratory14 and the characterization of their EPS.15 Because the method used in laboratory experiments to reach supersaturation with respect to carbonate minerals may affect the kinetic of the reaction resulting in the formation of different phases31 we have used and compared three methods: ammonium free-drift (AFD), slow mixing with sodium bicarbonate method (BSM) and fast mixing with sodium bicarbonate method (BFM). To simulate EPS, we performed experiments adding different organic molecules that occur in natural EPS and provide nucleation surfaces and charged functional groups.32 We selected molecules characterized by different charges (i.e., negative, positive, and neutral). This approach allowed us to test the hypothesis5,33–35 that negatively charged carboxyl groups are essential to promote Mg dehydration and consequent incorporation in Mg-rich carbonates.
2 Material and methods
2.1 Precipitation solutions
An artificial solution was prepared based on the results of ICP-mass analysis of some natural pore waters of a sabkha sediment (i.e., the Dohat Faishakh sabkha) in which dolomite is currently forming28,30 (see also Fig. S1 and Table S1 in ESI†). The composition was (mM): MgCl2 379, CaCl2 25, NaCl 4053, KCl 67, NaSO4 100, SrCl2 0.3 and LiCl 0.1. For each experimental setup, different types of amino acid (w/v 0.1%) and/or xanthan (w/v 0.1%) were added. All solutions were prepared using Milli-Q water and sterilized by filtration through a 0.2 μm pore size membranes. Parallel control experiments were performed in the absence of organic molecules. As an additional control, experiments were also conducted with natural pore water from the Dohat Faishakh sabkha (labelled SW4), which was sterilized by filtration. The measured concentrations of the major ions of SW4 were similar to those of the artificial solution. Visual checks were regularly performed to exclude contamination and microbial growth in the vials. All experiments were carried out-at least-in duplicates. The saturation indices of the artificial solution at various pH were calculated as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Q/K using SpecE8-The Geochemist's Workbench (GWB2021)-Community Edition.
Q/K using SpecE8-The Geochemist's Workbench (GWB2021)-Community Edition.
2.2 Organic compounds
Xanthan gum (Xan) is an extracellular polysaccharide produced by Xanthomonas campestris bacterium. Xanthan is composed of repeating units of mannose, glucose, glucuronic acid and pyruvic acid.36 Xanthan forms homogeneous aqueous dispersions and increases viscosity.37,38 With respect to artificially synthesized organic compounds, xanthan produced in different facilities or in different times may show slight compositional variabilities. Moreover, not only xanthan composition but also its use at different concentrations, which in turn affect viscosity of the precipitation environment, may impact crystal morphology and mineralogy.37,38Six amino acids providing different functional groups were selected and used in this study. L-Glutamine (Gln) (C5H10N2O3) is polar with a side chain containing two amino groups. L-Glutamic acid (Glu) (C5H9NO4) and L-aspartic acid (Asp) (C4H7NO4) are acidic, polar, containing two carboxyl groups. Phenylalanine (Phe) (C9H11NO2) and alanine (Ala) (C3H7NO2) are non-polar. Arginine (Arg) (C6H14N4O2) is a basic amino acid (see Table S2 in ESI†). Stock solutions of amino acids were prepared and sterilized by filtration through a hydrophilic polyethersulfone (PES) filter membrane with a 0.22 micron pore size to remove biological contaminants, including bacteria. The xanthan stock solutions were sterilized and thus homogenized by autoclaving at 121 °C for 20 min.
The approach of using “single” amino acids with respect to xanthan or natural biofilms allows for evaluating the influence of specific functional groups for mineral precipitation. On the other hand, it fails to mimic the complexity of natural biofilms, in which a large variety of organic molecules with various functional groups co-occur at the mineral nucleation site.
2.3 Mineral precipitation experiments
SEM images were obtained using Nova Nano Scanning Electron Microscope equipped with Bruker EDS Detector with 5 nm resolution and a magnification of 200000×. The EDS analyses were performed following the “ASTM standard method E1508-12a”.40
To determine their mineralogical composition, the recovered precipitates in form of dried powder were subjected to a discrete X-ray analysis. The bulk mineralogical composition was determined using a PANalytical-multipurpose Empyrean X-ray diffractometer. The analysis of XRD spectra was performed using the MATCH! Software, Version 3.11.2.188, CRYSTAL IMPACT, Kreuzherrenstr. 102, 53227 Bonn, Germany. The Mg mol% of carbonate minerals were calculated according to the position of the (d104) peak in the XRD pattern using the formula of Goldsmith et al., (1961).41 No quartz was added as internal standard to the bulk precipitate. Nevertheless, most of the samples contained minor amounts of halite, which forms when the bulk mineral phases are separated from the artificial solution and dried. The halite (200) was therefore used to correct for minor shifts in the position of the d(104) reflection of our synthetic Mg calcite.
For practical reasons (e.g., use of Petri dishes vs. flasks and flasks connected to a gas flushing system), the volume of the precipitation solutions, the incubation-time, and the incubation temperature, varied depending on the used method. For details, see the paragraph above, the “Results and discussion” section, and Table 1. Maintaining sterility avoiding microbial contamination was more difficult with the AFD method with respect to the BSM and BFM methods. For this reason, the incubation times of the experiments conducted with the AFD method were kept shorter (i.e., 2 weeks to 1 month vs. 1 to 3 months). However, one duplicate set of the experiments performed with the AFD method was transferred to centrifuge tubes and kept for a longer incubation period (i.e., more than 3 months). No changes in the bulk mineral phases were observed, suggesting that the difference in incubation time does not represent a major bias for the comparison of the results obtained with the 3 different methods.
| No. | Exp. | Ammonia free-drift (AFD) at 25 °C and 1 month incubation | Slow mixing conditions (BSM) at 30 °C and 1–3 months incubation | Fast mixing conditions (BFM) at 30 °C and 1–3 months incubation | |||||
|---|---|---|---|---|---|---|---|---|---|
| Ending pH | Ending precip. | Ending pH | Ending precip. | Ending pH | Yield of ending precipitates | Mol% Mg | |||
| 1 | Cont.1 | 9.85 ± 0.32 | Nesquehonite/minor amounts of aragonite & Mg-calcite | 8.16 ± 0.21 | Aragonite/miner amounts of calcite & huntite/hydromagnesite | 8.10 ± 0.13 | Aragonite (63%) | HMC (37%) | 33.7 ± 0.9 |
| 2 | Cont.2 (SW4) | 9.67 ± 0.47 | 8.19 ± 0.11 | 8.01 ± 0.25 | Aragonite (76%) | HMC (33%) | 34.5 ± 1.2 | ||
| 3 | Xan | 9.73 ± 0.54 | 8.28 ± 0.07 | 7.88 ± 0.16 | Aragonite (15%) | HMC (85%) | 37.3 ± 2.3 | ||
| 4 | Glu | 9.48 ± 0.21 | 8.01 ± 0.01 | 7.80 ± 0.1 | Aragonite (87%) | HMC (13%) | 37.5 ± 1.2 | ||
| 5 | Glu–Xan | 9.50 ± 0.11 | 8.21 ± 0.13 | 7.94 ± 0.1 | Aragonite (96%) | HMC (4%) | 33.0 ± 2.3 | ||
| 6 | Gln | 9.88 ± 0.34 | 8.31 ± 0.07 | 8.40 ± 0.08 | Aragonite (100%) | — | — | ||
| 7 | Gln–Xan | 9.72 ± 0.17 | 8.40 ± 0.08 | 8.23 ± 0.11 | Aragonite (7%) | HMC (93%) | 38.9 ± 1.1 | ||
| 8 | ASP | 9.41 ± 0.04 | 8.27 ± 0.08 | 7.84 ± 0.11 | Aragonite (87%) | HMC (13%) | 36.2 ± 1.3 | ||
| 9 | ASP–Xan | 9.39 ± 0.26 | 8.20 ± 0.13 | 7.72 ± 0.19 | Aragonite (94%) | HMC (6%) | 31.28 ± 0.1 | ||
| 10 | Phe | 9.68 ± 0.09 | 8.33 ± 0.04 | 8.26 ± 0.24 | Aragonite (18%) | HMC (82%) | 42.19 ± 1.9 | ||
| 11 | Phe–Xan | 9.54 ± 0.52 | 8.37 ± 0.05 | 8.23 ± 0.17 | Aragonite (6%) | HMC (94%) | 40.2 ± 2.7 | ||
| 12 | Ala | 9.85 ± 0.37 | 8.33 ± 0.06 | 8.24 ± 0.02 | Aragonite (100%) | — | — | ||
| 13 | Ala–Xan | 9.82 ± 0.14 | 8.26 ± 0.07 | 7.99 ± 0.21 | Aragonite (10%) | HMC (90%) | 35.65 ± 0.3 | ||
| 14 | Arg | 9.98 ± 0.12 | 8.41 ± 0.08 | 8.25 ± 0.03 | Aragonite (17%) | HMC (83%) | 38.42 ± 0.8 | ||
| 15 | Arg–Xan | 9.95 ± 0.08 | 8.26 ± 0.06 | 8.22 ± 0.04 | Aragonite (19%) | HMC (81%) | 41.68 ± 0.5 | ||
3 Results and discussion
3.1 pH evolution at different mineral formation conditions and corresponding saturation indices
The pH was monitored in all experimental conditions using a bench pH meter (i.e., pHenomenal IS 2100L). For the precipitation experiments performed using ADFM, the pH was adjusted to 7.00 at the beginning of the precipitation experiments. The pH evolved to 9.30 within 15 min and remained almost at same level until the end of the incubation period. For the BSM and BFM precipitation experiments, the reactant NaHCO3 was added to the precipitation solutions once the pH reached 7.00 by N2 bubbling. The pH increased to a range between 8.00 and 8.41 at the end of BSM incubation period. With the BFM method, final pH ranged from 7.72 to 8.25. The increase in solution's pH observed at the end of the experiments conducted with both the BFM and BSM methods (i.e., slow and fast mixing of bicarbonate) is due to the bubbling of N2, which results in degassing of CO2. The significant increase in pH observed at the end of the experiments performed with the ammonium free-drift method (AFD) is due to the decomposition of (NH4)2CO3. The NH3 gas rapidly dissolves in solution leading to an increase in pH.The calculated saturation indices show that the solutions were saturated with respect to dolomite, aragonite and calcite at pH 7. Upon the increase of pH to 8, the solutions become additionally saturated with respect to monohydrocalcite and hydromagnesite. At pH of 9 and above, the saturation index of nesquehonite is reached (see Table S3 in ESI†). Information on main ions activity and cations to anions ratios are reported in Tables S4 and S5.†
3.2 SEM/EDS and XRD analysis
The SEM/EDS analysis of the minerals obtained at the different conditions, revealed high variations in the morphology and the elemental composition of the recovered minerals. In the experiments performed using AFD, the majority (>85%) of crystals formed in the presence/absence of amino acids and/or xanthan were columnar shaped magnesium carbonates (identified with XRD analysis as nesquehonite – (Mg(HCO3)(OH)·2(H2O))) besides minor amounts of spherical-shaped Mg–calcium carbonate crystals with variable Mg content (Fig. 1).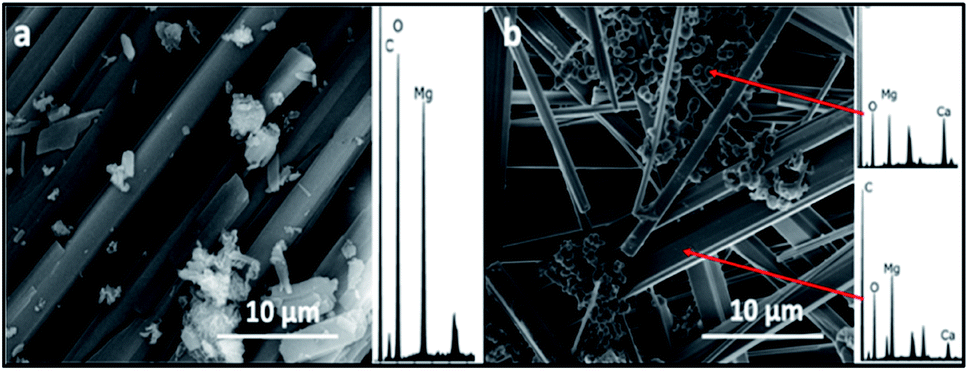 | ||
| Fig. 1 Representative SEM/EDS images and spectra of crystals recovered from precipitation experiments performed using ammonium free drift method, (a) control showing nesquehonite crystals, (b) spherical Mg calcite and columnar nesquehonite crystals formed in the presence of phenylalanine. | ||
XRD analysis of the minerals formed using the AFD method (Table 1 and Fig. 2) revealed that nesquehonite is the main mineral phase together with minor (<10%) amounts of calcite and Mg-calcite.
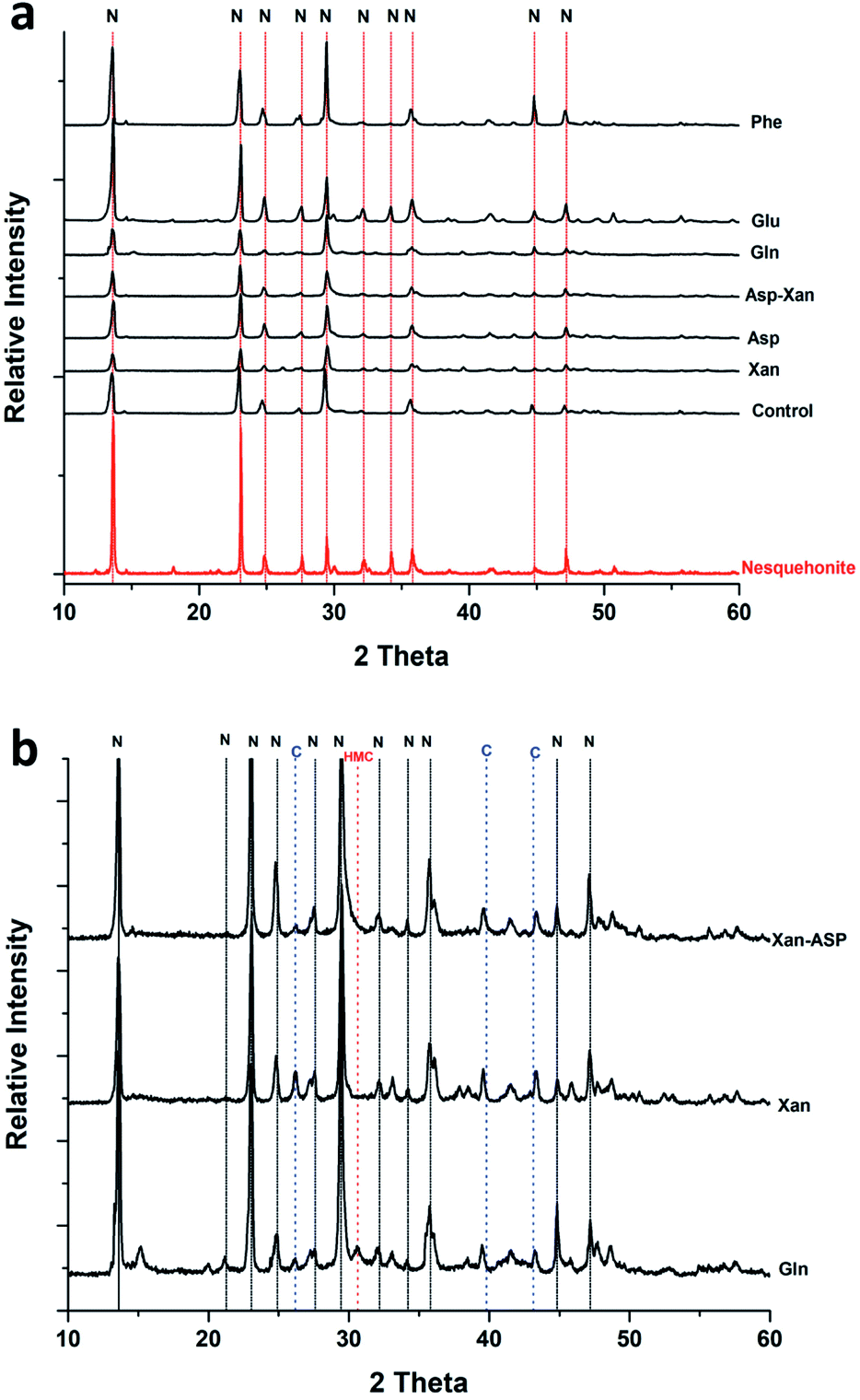 | ||
| Fig. 2 (a) Representative XRD patterns of minerals obtained from the precipitation experiments performed using the ammonium free-drift method. (b) Close up of X-ray diffraction patterns showing minor phases. N: nesquehonite, C: calcite, HMC: high magnesium calcite. | ||
With the BSM method, the minerals recovered after 24 h were identified by SEM/EDS as calcium carbonate and Mg–calcium aggregates with minor amounts of columnar nesquehonite crystals. However, the SEM/EDS of the minerals recovered after longer incubation periods (1–3 months or more) showed growing semi-rounded shaped calcium carbonate crystals, but with lower amounts of incorporated magnesium. Moreover, the nesquehonite morphology transformed into a rosette aggregate (huntite/hydromagnesite), as illustrated in (Fig. 3).
 | ||
| Fig. 3 Representative SEM/EDS images and spectra of crystals recovered from the precipitation experiments performed using the BSM method, (a) crystals formed in a control experiment without organic molecules in 24 h, (b) crystals formed in a control experiment without organic molecules in 2 months, (c) crystals formed in the presence of aspartic acid in one month, (d) crystals formed in the presence of aspartic acid in 3 months, (e) crystals formed in the presence of xanthan in one month, (f) crystals formed in the presence of glutamine and xanthan in 3 months. | ||
The XRD analysis of the minerals that formed in the precipitation experiments performed with the BSM method confirmed that aragonite is the main forming-phase in the presence of xanthan, of all the individually tested amino acids, and with the combination of xanthan and glutamine. Instead, monohydrocalcite/calcite were formed in the presence of glutamic acid or aspartic acid combined with xanthan (Table 1 and Fig. 4). This observation is consistent with the results of a recent study on microbially induced formation of monohydrocalcite, showing that glutamic and aspartic acids are preferentially adsorbed and mixed in the monohydrocalcite crystal lattice, and may have a role for its formation.42 In our experiments, the metastable monohydrocalcite phase transformed to aragonite after longer incubation periods (3 months and above) (ESI Fig. S2†). Such a transformation has been reported in previous studies, suggesting that monohydrocalcite that nucleates during simultaneous dissolution of Mg-bearing, amorphous calcium carbonate may be an important intermediate for aragonite and calcite that form at high Mg/Ca ratios.43,44 The minor amounts of nesquehonite transformed into huntite/hydromagnesite (Table 1, Fig. 4 and ESI Fig. S2†)
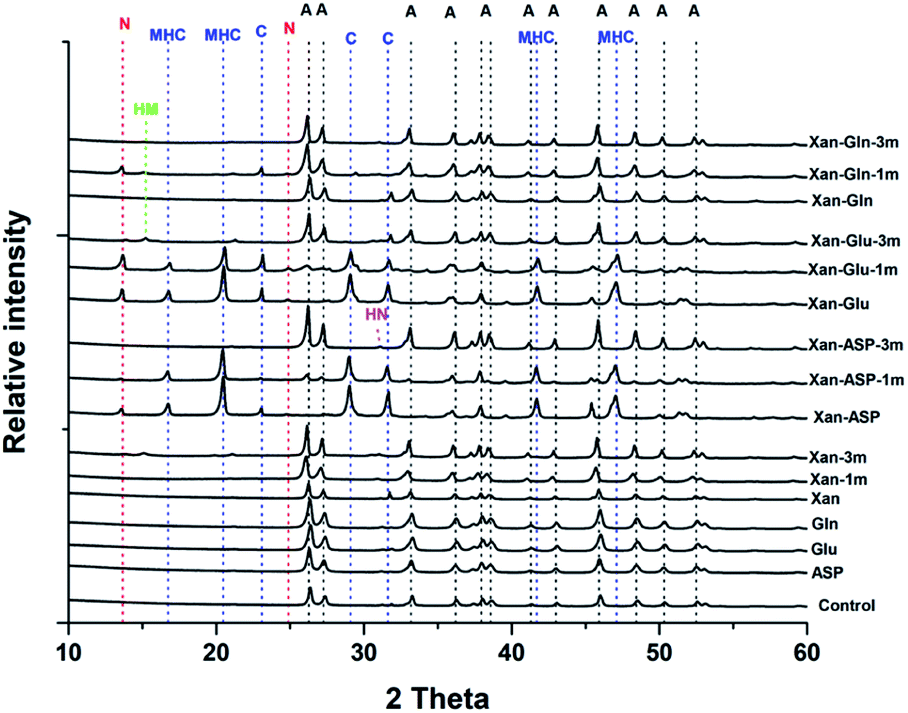 | ||
| Fig. 4 Representative XRD patterns of minerals recovered from the precipitation experiments performed with the BSM method. A: aragonite, MHC: monohydrocalcite, C: calcite, N: nesquehonite, HN: huntite, HM: hydromagnesite. | ||
The majority (>90%) of crystals formed with the BFM method in the control (i.e., without organic molecules) or using amino acids were oval shaped with an average length of almost 1 μm. Mg-calcite crystals with various mol% Mg were observed in the precipitates that formed in the presence of organic molecules (Fig. 5). Crystals formed in the presence of xanthan have two different morphologies: oval-shaped (Fig. 5B and F) with higher content of Mg and aggregates forming semi-spherical shapes with lower Mg content. No major changes were observed in the morphology and composition of the minerals recovered after longer incubation periods (of 1–3 months).
 | ||
| Fig. 5 SEM/EDS of crystals recovered from precipitation experiments performed with the BFM method. (a) Control without organic molecules (b) experiment with xanthan, (c) with phenylalanine, (d) with phenylalanine and xanthan (e) with glutamine and xanthan, and (f) with alanine and xanthan. | ||
Interestingly, with the BFM method high magnesium calcites (HMC) phases with variable mol% of Mg were obtained (Table 1 and Fig. 6). In the control samples, aragonite and HMC with a mol% up to 35 were formed. Aragonite was the mineral formed in presence of glutamine and alanine in absence of xanthan. In presence of xanthan, aragonite and HMC were formed with or without amino acids. The amounts of aragonite and HMC in each mixture were semi-quantitatively assessed using MATCH software. The highest amounts of HMC (81–94%) were obtained in the presence of phenylaniline, phenylaniline–xanthan, glutamine–xanthan, alanine–xanthan, arginine, and arginine–xanthan. The highest mol% Mg (38.9 ± 1.1 to 42.19 ± 1.9) was obtained in the presence of phenylaniline, phenylaniline–xanthan, glutamine–xanthan, and arginine.
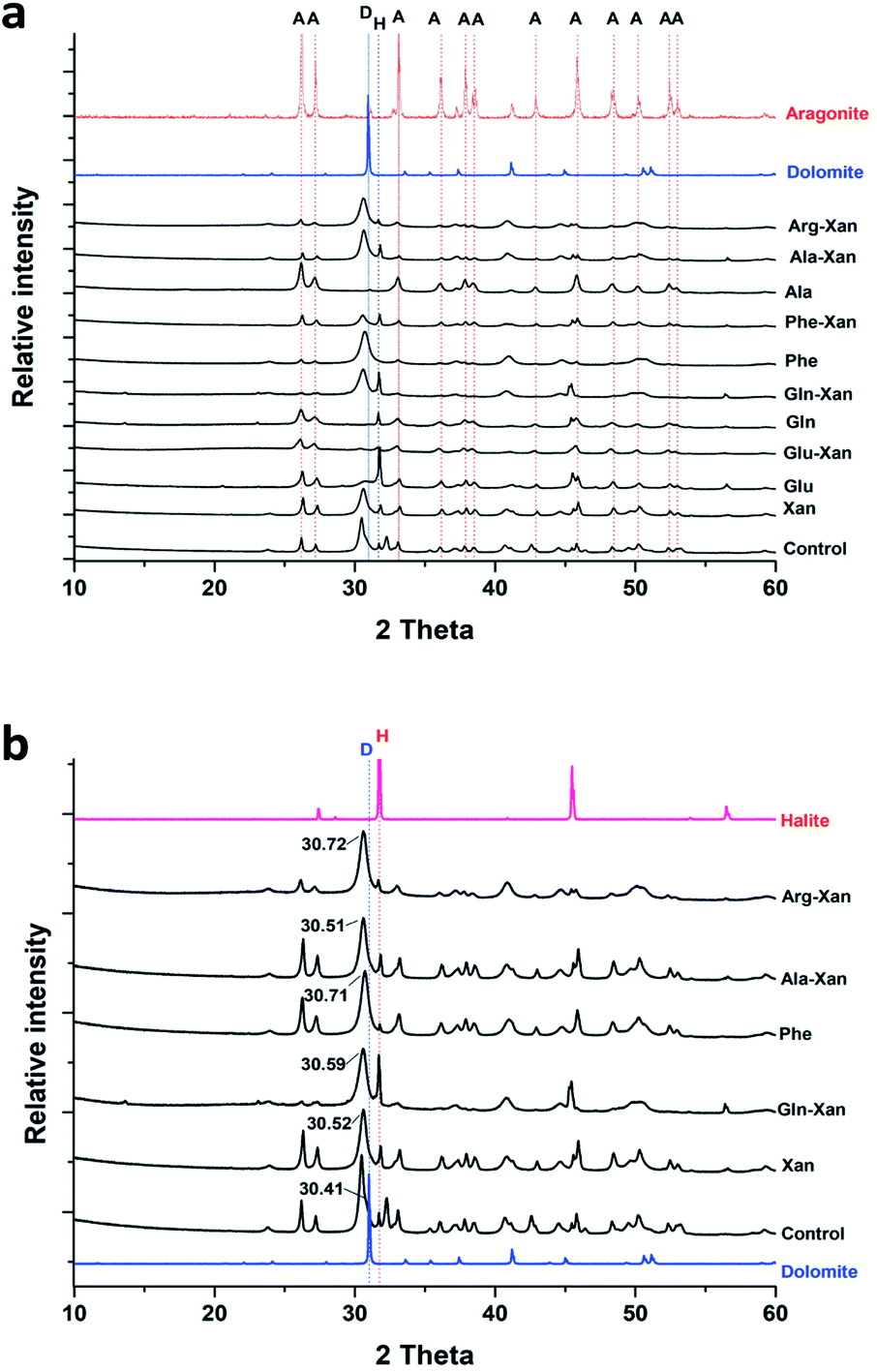 | ||
| Fig. 6 (a) Representative XRD patterns of minerals recovered from the precipitation experiments performed with the BFM method, (b) XRD patterns of the recovered HMC minerals showing the 2-theta values of the main peaks. A: aragonite, D: dolomite, H: halite. | ||
The results of the control precipitation-experiments performed with natural pore waters (i.e., Cont. 2–SW4) are broadly similar to those performed using the artificial solution (see Table 1).
Anhydrous Mg-carbonates with a mol% Mg higher than 40 – a phase that is considered as a possible precursor to ordered dolomite, referred in the literature to as protodolomite, non-ordered dolomite, Ca-dolomite, or very high Mg calcite2,45 – has been detected only in some of the experiments conducted with the bicarbonate fast mixing method (BFM) and in the presence of organic molecules (Table 1). Most of the tested experimental conditions only led to the formation of aragonite, calcite, and hydrous Mg-rich phases (i.e., nesquehonite and hydromagnesite). These results are consistent with previous works, indicating that (1) incorporation of Mg into anhydrous carbonate minerals is kinetically inhibited at low temperature (e.g. ref. 46), (2) that aragonite and not calcite is forming from solutions with a high Mg/Ca ratio (e.g. ref. 47, 48 and 54) (3) that rapid rise in pH promotes the formation of nesquehonite,49 and (4) that the presence of organic molecules – produced by microbes in most natural environments – promote the incorporation of Mg into the carbonate mineral (e.g. ref. 5, 10 and 50). It is worth noting that our experiments were conducted in the absence of any mineral seed, which are also known to influence the nucleation of Mg-rich carbonates.51–53 In particular, a recent study has shown that the presence of seeds comprised of negatively charged clay minerals promote the incorporation of Mg into the carbonate mineral.34 Besides confirming the results of the abovementioned previous studies, our experiments provided novel insight on the factors influencing the incorporation of Mg into carbonate minerals at low temperature, which we discuss in more detail below.
Having conditions that promote the dehydration of Mg shells has often been described as essential in many microbial models for dolomite formation (e.g. ref. 2). Functional groups present in EPS have been often proposed as promoting dehydration due to their negatively charged functional groups.33 In the range of pH at which the BSM and BFM precipitation experiments were performed (i.e., 7 to 8.5), the carboxyl groups of all the tested amino acids were deprotonated, and their amine groups were protonated (Table S2†). The total charge of the acidic and basic amino acids was determined by the charge of their side chain (see Table S2†). The results of our experiments do not point to any significant correlation between specific groups of amino acids and the incorporation of Mg into the carbonate mineral. For example, the presence of Glu and Asp – that under our experimental conditions had a negative charge – resulted in the formation of HMC with a mol% Mg that is not higher than that of the HMC formed in the presence of positively charged (i.e., Arg) or neutrally charged (i.e., Ala, Phe, Gln) amino acids (Table 1). In the case of the experiments conducted with the AFD method, pH values up to 9.9 were reached. Therefore, also the amine groups (expect that of arginine) were deprotonated (Table S2†). However, only minor amounts of HMC were detected. It could be the case that under the conditions mimicked by our experiments, the organic matrices tested exhibited higher affinity for Ca2+ than for Mg2+ ions. Additional experiments considering also binding site concentrations and competitive interactions between Mg2+ and Ca2+ for the available ligands, and at higher pH ranges, would help in concluding further on this matter.
Xu et al., (2013)48-conducted precipitation experiments at low temperature in the absence of water and showed Mg hydration is probably not the only kinetic barrier preventing the formation of Mg carbonates. They proposed a more intrinsic inhibitory effect that may be due to lattice restraints on the spatial distribution of CO3 groups in the MgCO3 crystals. Without being able to provide a molecular scale model elucidating the role of organic molecules for the nucleation process, we concur with the hypothesis of Xu et al.,48 and suggest that kinetic barriers other than dehydration, which are more easily overcome in the presence of organic molecules50 but independently from their charge, probably exist.
Another interesting outcome of our experiments is the demonstration that the mode of carbonate supply (i.e., not only the total inorganic carbon present in solution, but also the time at which supersaturation is reached), has a significant impact on the forming mineral phase (i.e., aragonite vs. hydromagnesite vs. nesquehonite vs. high Mg calcite) and its Mg content. Indeed, at equal Mg2+:Ca2+ ratios, the formation of high Mg calcite is favored by a rapid addition and mixing of carbonate ions (i.e., experiments conducted with the BFM method). The concept that high alkalinity deriving from both physicochemical (e.g., evaporation) and biological processes (e.g., microbial metabolisms) may favor dolomite formation is not new (e.g. ref. 2, 55 and 56). However, despite several modern dolomite-forming environments are characterized by an alkalinity that surpasses that of average seawater (e.g. ref. 11, 27 and 50) there is evidence indicating that alkalinity is not the ultimate factor controlling whether Mg-carbonates vs. other carbonate phases will form at low temperature (e.g. ref. 57 and 58). Nevertheless, it is important to note that the debate about the importance of high alkalinity for the formation of Mg-rich carbonates mostly focuses on the concentration in solution of carbonate ions in solution and not on the mode and velocity/rate they are brought to the site of mineral nucleation and growth. Based on the results of this study, we propose that the mode of carbonate supply may play an important and, as yet, limitedly investigated role for the formation of Mg-rich carbonates.
It is possible that the formation of Mg-rich carbonate at low temperature does not follow the “classical nucleation theory”, but rather begins with the precipitation of an amorphous phase.59–61 The existence of a hypothetical precursor phase that is Mg-rich, amorphous, and forms exclusively under high saturation conditions is consistent with our observations. Our results show that precipitation of HMC is favored in the experiments whereby bicarbonate was mixed quickly with the solution (i.e., BFM method). If the mixing is instead slow (e.g., BSM method), the formation of other minerals such as aragonite reduces the saturation, which may in turn prevent the formation of the required precursor phase. Therefore, although the artificial solution is supersaturated with respect to dolomite already at pH 7 (but dolomite is not forming due to kinetic barriers3), it is possible that its hypothetical amorphous precursor requires a higher saturation. Such conditions should be reached fast enough to outcompete the formation of other phases. Among the methods tested in this study, only the BFM method would produce the mass-transfer rate necessary for the formation of the precursor phase. Future investigations including a continuous monitoring of the solid phases and high-resolution surface characterization of the precipitates may provide key insight to evaluate the hypothesis described above.
The influence of the precipitation method on the resulting mineral phase has been studied for calcium carbonate,31 showing that kinetics factors are important for determining polymorphism.62 In contrast, less is known on whether the dynamic of carbonate supply control the incorporation of Mg into carbonate minerals at low temperature. In 2012, Wang et al.,60 presented a conceptual model to explain the relationship between Mg2+:Ca2+ ratio, carbonate supply and CaCO3 mineralization. They suggested that the combination “high Mg2+:Ca2+ ratio” and “high saturation” favors the rapid formation of amorphous Mg-carbonates that eventually transforms into high Mg calcites. Although our results emphasize the importance of the “mode of carbonate supply” rather than the “carbonate ions concentration”, the outcome of our precipitation experiments is broadly consistent with their model. Indeed, from solutions characterized by an elevated Mg2+:Ca2+ ratio, very high magnesium calcite (VHMC) formed exclusively with the BFM method (i.e., with a fast supply of bicarbonates), whereas mostly aragonite formed by adding bicarbonate slowly (i.e., BSM method). Nevertheless, also with a fast bicarbonate supply, it has not been possible to produce carbonate phases with a mol% Mg higher than 35. VHMC with a mol% Mg exceeding 40 was obtained exclusively in the presence of organic molecules. Because several factors other than Mg/Ca ratio are known to favor the incorporation of Mg in carbonate minerals (e.g., temperature, presence of cell walls, extracellular polymeric substances, clay minerals, presence of some trace metals…) (e.g. ref. 15, 33, 34, 51, 63 and 64) it would be virtually impossible to produce a model that correctly predict mineral formation in different natural environments just as a function of velocity/mode of carbonate supply and Mg/Ca ratios.
Considering the results of our laboratory experiments, we hypothesize that also in natural sedimentary systems different carbonate minerals may form depending on the mode of carbonate supply. No values measured directly in the field are to our knowledge available for evaluating whether a carbonate supply analogue, for example, to that of the BMF method used in our experiments can be produced by a natural process. Nevertheless, we speculate that progressive evaporation of seawater may correspond to a carbonate supply that is not ideal for formation of Mg-rich carbonates, even in the presence of organic molecules. Instead, microbial metabolic reactions that releases at high-rate carbonate ions in pore-waters or within a microbial mat may result in a relatively faster carbonate supply, which is more similar to that obtained with the BFM method. For instance, the mode of carbonate supply could play an important role in freshwater–seawater mixing-zone, which have since long been studied to formulate models for low temperature dolomite formation.2,65,66 In mixing zones, not only there is a rapid mixing of solutions characterized by different chemical compositions, but conditions are particularly favorable for sustaining microbial metabolisms (e.g., manganese-, iron-, and sulfate-reduction coupled to organic matter oxidation) that generates high rate of alkalinity in pore waters rich in organic molecules.67 Similarly, in supratidal organic-rich pore waters of modern sabkhas30,50 dolomite vs. aragonite formation may be determined not only by the presence of organic molecules (i.e., living and degrading microbial cells and extracellular polymeric substances)22 but also by cyclic mixing of seawater brought by the tide with more evaporated seawater present in the sediment pores, in which the presence of hotspots of alkalinity production associated to microbial respiration of organic matter. Future studies measuring alkalinity production-rate and daily fluctuations in sabkhas, and other modern dolomite-forming environments will be essential to test the abovementioned hypothesis.
Regarding the morphologies of the precipitates, spherical shaped Mg–calcium carbonate crystals were observed in the ADFM precipitation experiments. Crystals with semi-spherical shapes were observed in the BSM experiments, while oval shaped crystals were obtained in the experiments performed at BFM method. These observations suggest that factors other than mol% Mg incorporated into the carbonate mineral are at play in determining crystal morphology (e.g. ref. 20). Thus, crystal morphology can hardly predict the composition of the carbonate nor the mechanisms through which it originally formed.
Each experiment was conducted at least in duplicate. In some cases, 3 or 4 parallel experiments were conducted. The error reported next to the pH and mol% Mg values refers to the differences among duplicates/parallel experiments.
4 Conclusions
The results of our laboratory precipitation experiments showed that, at equal physicochemical conditions, a fast supply of carbonate ions promote the incorporation of Mg into carbonate minerals at low temperatures. In an artificial solution that simulates evaporitic seawater (i.e., sabkha water), Mg-carbonates with a mol% Mg of about 35 are formed exclusively when bicarbonate is mixed fast to the solution. If the solution also contains organic molecules, the mol% of Mg in the carbonate surpasses 40, forming a phase that is a possible precursor to ordered dolomite. These observations provide a possible explanation for why Mg-rich mineral phases rarely form in modern natural environments, despite the fact the microbes and extracellular polymeric substances are almost ubiquitous in all low temperature sedimentary systems: the mode of carbonate supply may be equally important as the presence of organic molecules. No correlation between the charge of the organic molecules and the mol% Mg of the carbonate minerals has been identified in our experiments. However, more tests covering a larger pH range are necessary to draw general conclusions on the importance of charged functional groups in natural environments. In future studies aimed at finding a definitive solution to the long-standing enigma that surrounds the incorporation of Mg into carbonate minerals at low temperature—leading to dolomite formation, it will be important to focus not only on biological factors, saturation indexes, and the total alkalinity of the system, but also on the dynamics controlling how carbonate ions are supplied to the site of mineral formation.Author contributions
ZA: conceptualized & designed the research, performed the experiments, analyzed the data, and wrote the manuscript. TB, NZ, HK, and FS: conceptualized & designed the research, analyzed the data, provided resources, equipment and infrastructure, and reviewed & edited the manuscript. MD: helped with conceptualization and formal analysis of the data, ES and MA helped with the experimental setup and data analysis.Conflicts of interest
The authors declare that this research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. There are no conflicts to declare.Acknowledgements
The authors acknowledge all the staff of the Central Laboratories Unit (QUCLU) of the college of Arts and Sciences at Qatar University for providing support in performing SEM/EDS analysis. Also, we would like to thank Dr Peter Kazak and Mr Abdullah Alashraf from the Center for Advanced Materials (CAM-QU) for their great help with the XRD analysis. This work was made possible by the grants NPRP10-0214-170462 and NPRP13S-0207-200291 from the Qatar National Research Fund (a member of the Qatar Foundation). The statements made herein are solely the responsibility of the authors.Notes and references
- H. D. Holland and H. Zimmermann, The Dolomite Problem Revisited, Int. Geol. Rev., 2000, 42(6), 481–490, DOI:10.1080/00206810009465093.
- D. A. Petrash, O. M. Bialik, T. R. Bontognali, C. Vasconcelos, J. A. Roberts, J. A. McKenzie and K. O. Konhauser, Microbially catalyzed dolomite formation: from near-surface to burial, Earth-Sci. Rev., 2017, 171, 558–582, DOI:10.1016/j.earscirev.2017.06.015.
- L. S. Land, Failure to Precipitate Dolomite at 25 °C from Dilute Solution Despite 1000-Fold Oversaturation after 32 Years, Aquat. Geochem., 1998, 4, 361–368, DOI:10.1023/A:1009688315854.
- R. S. Arvidson and F. T. Mackenzie, The dolomite problem; control of precipitation kinetics by temperature and saturation state, Am. J. Sci., 1999, 1, 257–288, DOI:10.2475/ajs.299.4.257.
- J. A. Roberts, P. A. Kenward, D. A. Fowle, R. H. Goldstein, L. A. González and D. S. Moore, Surface chemistry allows for abiotic precipitation of dolomite at low temperature, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(36), 14540–14545, DOI:10.1073/pnas.1305403110.
- X. Wang, I.-M. Chou, W. Hu, S. Yuan, H. Liua, Y. Wan and X. Wang, Kinetic inhibition of dolomite precipitation: insights from Raman spectroscopy of Mg2+–SO42− ion pairing in MgSO4/MgCl2/NaCl solutions at temperatures of 25 to 200 °C, Chem. Geol., 2016, 435, 10–21, DOI:10.1016/j.chemgeo.2016.04.020.
- F. M. V. Tuyl, The Origin of Dolomite, Lowa Geological Survey Annual Report, 1914, vol. 25, pp. 251–422, DOI:10.17077/2160-5270.1180.
- M. Al-Awadi, W. j. Clark, W. R. Moore, M. Herron, T. Zhang, W. Zhao, N. Hurley, D. Kho, B. Montaron and F. Sadooni, Dolomite: Perspectives on a Perplexing Mineral, Oilfield Rev., 2009, 32–45 CAS.
- F. Sadooni, F. Howari and A. El-Saiy, Microbial dolomites from carbonate-evaporite sediments of the coastal sabkha of Abu Dhabi and theri exploration inplication, J. Pet. Geol., 2010, 33(4), 289–298, DOI:10.1111/j.1747-5457.2010.00480.x.
- T. R. Bontognali, Anoxygenic phototrophs and the forgotten art of making dolomite, Geology, 2019, 47(6), 591–592, DOI:10.1130/focus062019.1.
- C. Vasconcelos and J. A. McKenzie, Microbial mediation of modern dolomite precipitation and diagenesis under anoxic conditions (Lagoa Vermelha, Rio de Janeiro, Brazil), J. Sediment. Res., 1997, 67(3), 378–390, DOI:10.1306/D4268577-2B26-11D7-8648000102C1865D.
- M. Sanchez-Roman, C. Vasconcelos, R. Warthmann, M. Rivadeneyra and J. A. Mckenzie, Microbial dolomite precipitation under aerobic conditions: results from Brejo do Espinho Lagoon (Brazil) and culture experiments, Sedimentolgy, 2009, 41, 167–178, DOI:10.1002/9781444312065.ch11.
- T. R. R. Bontognali, J. A. McKenzie, R. J. Warthmann and C. Vasconcelos, Microbially influenced formation of Mg-calcite and Ca-dolomite in the presence of exopolymeric substances produced by sulphate-reducing bacteria, Terra Nova, 2014, 26, 72–77, DOI:10.1111/ter.12072.
- Z. A. Al Disi, S. Jaoua, T. R. R. Bontognali, E. S. M. Attia, H. A. A. S. Al-Kuwari and N. Zouari, Evidence of a Role for Aerobic Bacteria in High Magnesium Carbonate Formation in the Evaporitic Environment of Dohat Faishakh Sabkha in Qatar, Front. Environ. Sci., 2017, 5, 1, DOI:10.3389/fenvs.2017.00001.
- Z. A. Al Disi, N. Zouari, M. Dittrich, S. Jaoua, H. A. S. l-Kuwari and T. R. R. Bontognali, Characterization of the extracellular polymeric substances (EPS) of Virgibacillus strains capable of mediating the formation of high Mg-calcite and protodolomite, Mar. Chem., 2019, 216, 103693, DOI:10.1016/j.marchem.2019.103693.
- Z. A. Al Disi, T. R. R. Bontognali, S. Jaoua, E. Attia, H. A. S. Al-Kuwari and N. Zouari, Influence of temperature, salinity and Mg2+:Ca2+ ratio on microbially-mediated formation of Mg-rich carbonates by Virgibacillus strains isolated from a sabkha environment, Sci. Rep., 2019, 9, 9633, DOI:10.1038/s41598-019-56144-0.
- D. T. Wright, The role of sulphate-reducing bacteria and cyanobacteria in dolomite formation in distal ephemeral lakes of the Coorong region, South Australia, Sediment. Geol., 1999, 126(1–4), 147–157 CrossRef CAS.
- A. W. Decho and T. Gutierrez, Microbial Extracellular Polymeric Substances (EPSs) in Ocean Systems, Front. Microbiol., 2017, 8, 922, DOI:10.3389/fmicb.2017.00922.
- M. Obst, J. Dynes, J. Lawrence, G. Swerhone, K. Benzerara, C. Karunakaran, K. Kaznatcheev, T. Tyliszczak and A. Hitchcock, Precipitation of amorphous CaCO3 (aragonite-like) by cyanobacteria: a STXM study of the influence of EPS on the nucleation process, Geochim. Cosmochim. Acta, 2009, 73(14), 4180–4198, DOI:10.1016/j.gca.2009.04.013.
- O. Braissant, G. Cailleau, C. Dupraz and E. P. Verrecchia, Bacterially Induced Mineralization of Calcium Carbonate in Terrestrial Environments: The Role of Exopolysaccharides and Amino Acids, J. Sediment. Res., 2003, 73(3), 485–490, DOI:10.1306/111302730485.
- S. Krause, V. Liebetrau, S. Gorb, T. Treude, J. A. McKenz and T. Treude, Microbial nucleation of Mg-rich dolomite in exopolymeric substances under anoxic modern seawater salinity: new insight into an old enigma, Geology, 2012, 40(v), 587–590, DOI:10.1130/G32923.1.
- M. M. Esther, C. Óscar and M. Sánchez-Román, Microbial Mg-rich Carbonates in an Extreme Alkaline Lake (Las Eras, Central Spain), Front. Microbiol., 2019, 10, 148, DOI:10.3389/fmicb.2019.00148.
- O. Y. A. Costa, J. M. Raaijmakers and E. E. Kuramae, Microbial Extracellular Polymeric Substances: Ecological Function and Impact on Soil Aggregation, Front. Microbiol., 2018, 9, 14 CrossRef PubMed.
- W. Yin, Y. Wang, L. Liu and J. He, Biofilms: The Microbial “Protective Clothing” in Extreme Environments, Int. J. Mol. Sci., 2019, 20(14), 3423 CrossRef CAS PubMed.
- Z. A. DiLoreto, T. R. R. Bontognali, Z. A. A. Disi, H. A. S. Al-Kuwari, K. H. Williford, C. J. Strohmenger, F. Sadooni, C. Palermo, J. M. Rivers, J. A. McKenzie, M. Tuite and M. Dittrich, Microbial community composition and dolomite formation in the hypersaline microbial mats of the Khor Al-Adaid sabkhas, Qatar, Extremophiles, 2019, 2019, 1–18, DOI:10.1007/s00792-018-01074-4.
- A. Alsharhan and C. Kendallb, Holocene coastal carbonates and evaporites of the southern Arabian Gulf and their ancient analogues, Earth-Sci. Rev., 2003, 61(3–4), 91–243, DOI:10.1016/S0012-8252(02)00110-1.
- Z. A. Diloreto, S. Garg, T. R. R. Bontognali and M. Dittrich, Modern dolomite formation caused by seasonal cycling of oxygenic phototrophs and anoxygenic phototrophs in a hypersaline sabkha, Sci. Rep., 2021, 11, 4170, DOI:10.1038/s41598-021-83676-1.
- L. V. Illing, A. J. Wells and J. C. M. Taylor, Penecontemporary dolomite in the Persian Gulf, Society for Sedimentary Geology, Special Publications (SPEM), 1965, vol. 13, pp. 89–111, DOI:10.2110/pec.65.07.0089.
- T. R. R. Bontognali, C. Vasconcelos, R. J. Warthmann, R. Lundberg and J. A. McKenzie, Dolomite-mediating bacterium isolated from the sabkha of Abu Dhabi (UAE), Terra Nova, 2012, 24(3), 248–254, DOI:10.1111/j.1365-3121.2012.01065.x.
- M. Brauchli, J. A. McKenzie, C. J. Strohmenger, F. Sadooni, C. Vasconcelos and T. R. R. Bontognali, The importance of microbial mats for dolomite formation in the Dohat Faishakh sabkha, Qatar, Carbonates Evaporites, 2016, 31, 339–345, DOI:10.1007/s13146-015-0275-0.
- W. House, The kinetics of calcite precipitation and related processes, in Fifty-second annual report for the year ended 31st March 1984, Ambleside, Freshwater Biological Association, 1984 Search PubMed.
- J. Wingender, T. R. Neu and H.-C. Flemming, Microbial Extracellular Polymeric Substances, Characterization, Structure and Function, Springer, Berlin, Heidelberg, 1999 Search PubMed.
- P. A. Kenward, D. A. Fowle, R. H. Goldstein, M. Ueshima, L. A. Gonzalez and J. A. Roberts, Ordered low-temperature dolomite mediated by carboxyl-group density of microbial cell walls, AAPG Bull., 2013, 97(11), 2113–2125, DOI:10.1306/05171312168.
- D. Liu, Y. Xu, D. Papineau, N. Yu, Q. Fan, X. Qiu and H. Wang, Experimental evidence for abiotic formation of low-temperature proto-dolomite facilitated by clay minerals, Geochim. Cosmochim. Acta, 2019, 274, 83–95, DOI:10.1016/j.gca.2018.12.036.
- D. Liu, Y. Xu, Q. Yu, N. Yu, X. Qiu, H. Wang and D. Papineau, Catalytic effect of microbially-derived carboxylic acids on the precipitation of Mg-calcite and disordered dolomite: implications for sedimentary dolomite formation, J. Asian Earth Sci., 2020, 193, 104301, DOI:10.1016/j.jseaes.2020.104301.
- F. Katzen, D. U. Ferreiro, C. G. Oddo, M. V. Ielmini, A. Becker, A. Pühler and L. Ielpi, Xanthomonas campestris pv. campestris gum Mutants: Effects on Xanthan Biosynthesis and Plant Virulence, J. Bacteriol., 1998, 180(7), 1607–1617 CrossRef CAS PubMed.
- R. Moorhouse, M. D. Walkinshaw and S. Arnott, Xanthan Gum—Molecular Conformation and Interactions, in Extracellular Microbial Polysaccharides, American Chemical Society, 1977, pp. 7–90 Search PubMed.
- L. Zhong, M. Oostrom, M. J. Truex, V. R. Vermeul and J. E. Szecsody, Rheological behavior of xanthan gum solution related to shear thinning fluid delivery for subsurface remediation, J. Hazard. Mater., 2013, 244–245, 160–170 CrossRef CAS PubMed.
- B. Lian, Q. Hu, J. Chen, J. Ji and H. H. Teng, Carbonate biomineralization induced by soil bacterium Bacillus megaterium, Geochim. Cosmochim. Acta, 2006, 70(22), 5522–5535 CrossRef CAS.
- I. ASTM, Standard Guide for Quantitative Analysis by Energy-Dispersive Spectroscopy, 2019 Search PubMed.
- J. R. Goldsmith, D. L. Graf and H. C. Heard, Lattice constants of the calcium-magnesium carbonates, Am. Mineral., 1961, 46, 451–457 Search PubMed.
- Y. Zhao, Z. Han, H. Yan, H. Zhao, M. E. Tucker, X. Gao, N. Guo, R. Meng and D. C. Owusu, Selective Adsorption of Amino Acids in Crystals of Monohydrocalcite Induced by the Facultative Anaerobic Enterobacter ludwigii SYB1, Front. Microbiol., 2021, 12, 696557, DOI:10.3389/fmicb.2021.696557.
- R. Nishiyama, T. Munemoto and K. Fukushi, Formation condition of monohydrocalcite from CaCl2–MgCl2–Na2CO3 solutions, Geochim. Cosmochim. Acta, 2013, 1, 217–231 CrossRef.
- J. Rodriguez-Blanco, S. B. P. Shaw, T. Roncal-Herrero and L. Benning, The role of Mg in the crystallization of monohydrocalcite, Geochim. Cosmochim. Acta, 2014, 127, 204–220 CrossRef CAS.
- J. M. Gregg, D. L. Bish, S. E. Kaczmarek and H. G. Machel, Mineralogy, nucleation and growth of dolomite in the laboratory and sedimentary environment: a review, Sedimentology, 2015, 62, 1749–1769 CrossRef CAS.
- I. M. Power, P. A. Kenward, G. M. Dipple and M. Raudsepp, Room Temperature Magnesite Precipitation, Cryst. Growth Des., 2017, 17(11), 5652–5659, DOI:10.1021/acs.cgd.7b00311.
- W. Sun, S. Jayaraman, W. Chen, K. A. Persson and G. Ceder, Nucleation of metastable aragonite CaCO3 in seawater, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(11), 3199–3204, DOI:10.1073/pnas.1423898112.
- J. Xu, C. Yan, F. Zhang, H. Konishi, H. Xu and H. H. Teng, Testing the cation-hydration effect on the crystallization of Ca–Mg–CO3 systems, Proc. Natl. Acad. Sci. U. S. A., 2013, 6, DOI:10.1073/pnas.1307612110.
- Z. Zhang, Y. Zheng, Y. Ni, Z. Liu, J. Chen and X. Liang, Temperature- and pH-Dependent Morphology and FT-IR Analysis of Magnesium Carbonate Hydrates, J. Phys. Chem. B, 2006, 110(26), 2969–12973, DOI:10.1021/jp061261j.
- T. R. R. Bontognali, C. g. Vasconcelos, R. J. Warthmann, S. M. Bernasconi, C. Dupraz, C. j. Strohmenger and J. A. Mckenzie, Dolomite formation within microbial mats in the coastal sabkha of Abu Dhabi (United Arab Emirates), Sedimentology, 2010, 57, 824–844, DOI:10.1111/j.1365-3091.2009.01121.x.
- F. Zhang, H. Xu, H. Konishi, J. M. Kemp, E. E. Roden and Z. Shen, Dissolved sulfide-catalyzed precipitation of disordered dolomite: implications for the formation mechanism of sedimentary dolomite, Geochim. Cosmochim. Acta, 2012, 97, 148–165, DOI:10.1016/j.gca.2012.09.008.
- D. L. Graf and J. R. Goldsmith, Some Hydrothermal Syntheses of Dolomite and Protodolomite, J. Geol., 1956, 64(2), 173–186 CrossRef CAS.
- F. Zhang, H. Xu, H. Konishi, E. Shelobolina and E. Roden, Polysaccharide-catalyzed nucleation and growth of disordered dolomite: a potential precursor of sedimentary dolomite, Am. Mineral., 2012, 97(4), 556–567, DOI:10.2138/am.2012.3979.
- Y. Xu and N. A. J. M. Sommerdijk, Aragonite formation in confinements: a step toward understanding polymorph control, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(34), 8469–8471, DOI:10.1073/pnas.1811696115.
- J. Warren, Dolomite: occurrence, evolution and economically important associations, Earth-Sci. Rev., 2000, 52, 1–81, DOI:10.1016/S0012-8252(00)00022-2.
- C. Dupraz, R. P. Reid, O. Braissant, A. W. Decho, R. S. Norman and P. T. Visscher, Processes of carbonate precipitation in modern microbial mats, Earth-Sci. Rev., 2009, 96(3), 141–162, DOI:10.1016/j.earscirev.2008.10.005.
- K. Stanienda-Pilecki, Magnesium calcite in Muschelkalk limestones of the Polish part of the Germanic Basin, Carbonates Evaporites, 2018, 33, 801–821, DOI:10.1007/s13146-018-0437-y.
- J. McCormack, T. R. R. Bontognali, A. Immenhauser and O. Kwiecien, Controls on Cyclic Formation of Quaternary Early Diagenetic Dolomite, Geophys. Res. Lett., 2018, 45(8), 3625–3634, DOI:10.1002/2018GL077344.
- W. Jin, S. Jiang, H. Pan and R. Tang, Amorphous Phase Mediated Crystallization: Fundamentals of Biomineralization, Minerals, 2018, 8(1), 48, DOI:10.3390/cryst8010048.
- D. Wang, L. M. Hamm, A. J. Giuffre, T. Echigo, J. D. Rimstidt, J. J. D. Yoreo, J. Grotzinger and P. M. Dove, Revisiting geochemical controls on patterns of carbonate deposition through the lens of multiple pathways to mineralization, Faraday Discuss., 2012, 159, 371–386, 10.1039/C2FD20077E.
- J. J. De Yoreo, P. U. P. A. Gilbert, N. A. J. M. Sommerdijk, R. L. Penn, S. Whitelam, D. Joester, H. Zhang, J. D. Rimer, A. Navrotsky, J. F. Banfield, A. F. Wallace, F. M. Michel, F. C. Meldrum, H. Cölfen and P. M. Dove, Crystallization by particle attachment in synthetic, biogenic, and geologic environments, Science, 2015, 349(6247), 1–9, DOI:10.1126/science.aaa6760.
- V. Choudens-Sanchez and L. Gonzalez, Calcite and Aragonite Precipitation Under Controlled Instantaneous Supersaturation: Elucidating the Role of CaCO3 Saturation State and Mg/Ca Ratio on Calcium Carbonate Polymorphism, J. Sediment. Res., 2009, 79(9), 363–376, DOI:10.2110/jsr.2009.043.
- D. A. L. Petrash, S. V. González-Arismendi, G. Gordon, R. A. Méndez, J. A. Gingras, M. K. Konhauser and K. O. Konhauser, Can Mn-S redox cycling drive sedimentary dolomite formation? A hypothesis, Chem. Geol., 2015, 404, 27–40 CrossRef CAS.
- M. Daye, J. Higgins and T. Bosak, Formation of ordered dolomite in anaerobic photosynthetic biofilms, Geology, 2019, 6, 509–512 CrossRef.
- S. J. Burns and V. J. Rossinsky, Late Pleistocene mixing zone dolomitization, southeastern Barbados, West Indies, Sedimentology, 1989, 36, 1139–1142, DOI:10.1111/j.1365-3091.1989.tb01547.x.
- S. Mazzullo, Organogenic Dolomitization in Peritidal to Deep-Sea Sediments, J. Sediment. Res., 2000, 7(1), 10, DOI:10.1306/2DC408F9-0E47-11D7-8643000102C1865D.
- D. A. Petrash, O. M. Bialik, P. T. Staudigel, K. O. Konhauser and D. A. Budd, Biogeochemical reappraisal of the freshwater–seawater mixing-zone diagenetic model, Sedimentology, 2021, 68(5), 1797–1830, DOI:10.1111/sed.12849.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06206a |
| This journal is © The Royal Society of Chemistry 2021 |