
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ growth of CuS NPs on 3D porous cellulose macrospheres as recyclable biocatalysts for organic dye degradation
Zhouquan Suna,
Yi Zhong*a,
Hong Xud,
Bijia Wanga,
Linping Zhanga,
Xiaofeng Suia,
Xueling Feng ab and
Zhiping Mao*abc
ab and
Zhiping Mao*abc
aKey Lab of Science & Technology of Eco-Textile, Ministry of Education, College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, No. 2999 North Renmin Road, Shanghai, 201620, China. E-mail: zhpmao@dhu.edu.cn; zhongyi@dhu.edu.cn; Fax: +86-21-67792707; Tel: +86-21-67792720
bNational Engineering Research Center for Dyeing and Finishing of Textiles, Donghua University, Shanghai, 201620, China
cInnovation Center for Textile Science and Technology of Donghua University, Shanghai, 201620, China
dLu Thai Textile Co., LTD, Zibo, 255000, China
First published on 12th November 2021
Abstract
Aiming at recyclable catalyst carriers, porous cellulose macrospheres from wood pulp dissolved in an alkaline urea system were regenerated by simple injection regeneration. After solvent exchange, porous cellulose macrospheres (CMs) with a high specific surface area of 325.3 m2 g−1 were obtained by lyophilization, and CuS nanoparticles (CuS NPs) were coated on CMs by in situ growth in the liquid phase to achieve CuS-supported CM macrospheres (CuS@CM). The results indicated that the CuS@CM biocatalyst was successfully prepared with an average diameter of approximately 1.2 mm. In addition, CuS@CM was further used as a heterogeneous catalyst for the catalytic degradation of methylene blue (MB) and methyl orange (MO) model dyes during the oxidation of hydrogen peroxide (H2O2). In the presence of low doses of H2O2, the degradation rate of MB reached 94.8% within 10 min, showing high catalytic activity under neutral and alkaline conditions. After five cycles, 90.1% of the original catalytic activity was still retained, indicating that the prepared CuS@CM composite possessed excellent catalytic activity and reusability.
1. Introduction
With the rapid development of textile industries accompanied by high energy consumption and increased waste discharge, industrial wastewater containing a great deal of organic dye pollutants has an adverse effect on the environment, aquatic organisms and human health. Because of its non-biodegradability, toxicity, possible carcinogenicity and mutagenicity,1 waste discharge poses a serious threat to the aquatic ecosystem. In particular, the treatment of dye wastewater containing a large number of persistent and toxic substances with various chemical structures is a difficult and challenging task.2,3 To this end, various wastewater treatment technologies of adsorption,4 photocatalytic degradation,5 ozonization,6 electrochemistry,7 biological methods,8 and others,9,10 have been used to treat dye wastewater.11–13 However, most of these methods have a lower removal efficiency and high processing costs. Fenton-like reagents, which used transition metal oxides or sulfides as catalysts to release hydroxyl groups, present extremely high reactivity and have proved to be effective materials for converting some organic dye macromolecules into non-toxic carbon dioxide and water,14–16 and hence, they are widely used in the field of wastewater treatment. However, in practical applications, the catalyst is usually dispersed in a solution in the form of fine particles, which makes it more difficult to separate and recover the catalyst in the form of fine powder.The effective solution to this problem is to find a suitable supporting substrate for the catalyst. Cellulose, as the most abundant natural polymer material in nature,17,18 usually exists in the form of a variety of biomass such as cotton, wood pulp and flax.19–21 It is environmentally friendly, non-toxic and biodegradable as a supporting substrate material. Therefore, many prepared cellulose composites such as Pickering emulsions,22 cellulose composite membranes,23 functionalized nanocrystals,24 cellulose fibers and fabrics,25,26 hydrogels and aerogels27–29 have been reported. Cellulose gel is a three-dimensional mesh structure material, which has a high specific surface area,30 and abundant active sites that could increase the contact opportunity between contaminants and catalysts, and the pores in the structure also facilitated the mass transport of reactants and products.31 In general, spherical porous materials possess better fluidity and a higher specific surface area, making cellulose into a spherical form, and can not only increase the catalyst load, but also more easily disperse evenly in the wastewater solution during the treatment process.
It has been reported that copper sulfide (CuS), as a typical p-type semiconductor, has been widely used in various fields such as sensing,32 catalysis,33 solar cell fabrication,34 and biomedicine35 because of its excellent optical, electronic and other physical and chemical properties.36,37 In recent years, some researchers have shown that the one-dimensional or three-dimensional structure of copper sulfide is desirable for the photocatalytic activity, while all its structures are suitable for the catalytic activity.38–41 Therefore, the combination of copper sulfide and porous cellulose gel spheres was conducive to reusability and maximum utilization of the catalyst.
In this work, we used an alkaline urea system to dissolve wood pulp cellulose42,43 and form gel spheres by simple injection regeneration. After solvent exchange, porous cellulose macrospheres (CM) with a high specific surface area were obtained by lyophilization, and CuS was loaded on CM by in situ growth (CuS@CM). The prepared CuS immobilized on CM macrospheres in the presence of H2O2 showed excellent catalytic activity for the degradation of methylene blue (MB) and methyl orange (MO) due to its wide application and toxicity. In addition, CuS@CM has a wide range of pH applicability under environmental conditions, and still maintains more than 90% of the catalytic efficiency after five cycles. Therefore, the CuS@CM composite could be used as a standard candidate for recoverable Fenton reagents.
2. Materials and methods
2.1 Materials
Cellulose (wood pulp board, DP500, purity of 92.8%) was supplied by Baoding Swan New Fiber Manufacturing Co., Ltd (China). Lithium hydroxide (LiOH), sodium hydroxide (NaOH), nitric acid (HNO3), urea, tert-butanol (TBA), sodium bicarbonate solution (NaHCO3), 30% hydrogen peroxide (H2O2), and thioacetamide (CH3CSNH2) were provided by Shanghai Sinopharm Chemical Reagents Co., Ltd (China). Copper(II) nitrate trihydrate (Cu(NO3)2·3H2O) was purchased from Shanghai Macklin Biochemical Co., Ltd (China). All reagents were of analytical grade and used as received without further purification. Methylene blue (C16H18ClN3S·3H2O) and methyl orange (C14H14N3NaO3S) dyes were purchased from Shanghai Sinopharmaceutics Chemical Reagents Co., Ltd.2.2 Preparation of cellulose macrosphere (CM) and CuS-supported CM (CuS@CM) biocatalysts
The preparation process of cellulose macrospheres is as follows: 2.5 g cellulose was put into a LiOH/urea/water solution with a mass ratio of 4.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:80.5, vigorously stirred at room temperature, and uniformly dispersed. This mixture was refrigerated at −80 °C until completely frozen, thawed at room temperature and then stirred constantly. Cellulose was completely dissolved after two freeze–thaw cycles. The cellulose solution was injected into 5% sodium bicarbonate solution (NaHCO3) with constant current pump at a certain speed and stirred at 200 rpm for regeneration. It was filtered and rinsed with a large amount of deionized water until neutral pH. Then, the beads were put into tert-butanol (TBA) for solvent exchange at 120 rpm for 24 h, and after filtration and lyophilization, cellulose macrospheres with a high specific surface area were obtained.
:15:80.5, vigorously stirred at room temperature, and uniformly dispersed. This mixture was refrigerated at −80 °C until completely frozen, thawed at room temperature and then stirred constantly. Cellulose was completely dissolved after two freeze–thaw cycles. The cellulose solution was injected into 5% sodium bicarbonate solution (NaHCO3) with constant current pump at a certain speed and stirred at 200 rpm for regeneration. It was filtered and rinsed with a large amount of deionized water until neutral pH. Then, the beads were put into tert-butanol (TBA) for solvent exchange at 120 rpm for 24 h, and after filtration and lyophilization, cellulose macrospheres with a high specific surface area were obtained.
A CuS-supported cellulose macrosphere biocatalyst was prepared by in situ growth in the liquid phase.44 Briefly, 300 mg of CM was immersed in 12.5–100 mM Cu(NO3)2·3H2O solution. After stirring at 200 rpm for 1 h to reach adsorption equilibrium, the pH was further adjusted to 3 using dilute nitric acid solution. Then 25 mL constant concentration of CH3CSNH2 solution was added drop by drop, and the color of the solution gradually changed from blue to yellow. After 1 h heating in an oil bath at 90 °C, the solution turned dark green, indicating the formation of different CuS-supported CM (CuS@CM-12.5, CuS@CM-25, CuS@CM-50, and CuS@CM-100). After filtration, cleaning with deionized water, and lyophilization with tert-butanol solvent replacement, the CuS@CM catalyst was obtained. The pure copper sulfide sample was prepared by the reaction of Cu(NO3)2·3H2O with CH3CSNH2.
2.3 Characterizations
The morphology and structure of CM and CuS@CM were studied using a field emission scanning electron microscope (FESEM, Hitachi S-4800, Japan). The BET test was performed using a nitrogen adsorption desorption analyzer (TriStar II 3020M). Transmission electron microscopy (TEM, JEM-2100, Japan) was performed to observe CuS nanoparticles. The crystallographic structures of copper sulfide and supported copper sulfide were analyzed by X-ray (DX-2700BH, HAOYUAN). The content of CuS loaded on cellulose macrospheres was measured using a plasma spectrometer (Prodigy-ICP, America). Fourier transform infrared spectroscopy (FTIR, Spectrum Two, PerkinElmer, America) and microscopic confocal Raman spectroscopy (Raman, DXR2xi, Thermo Fisher Scientific, America) were performed to analyze the product ingredients. The CuS@CM macrosphere was characterized by X-ray photoelectron spectroscopy (XPS, ESCALAB 250Xi). Thermogravimetric analysis was performed in a nitrogen atmosphere at a heating rate of 10 °C min−1 to 900 °C (TG 209 F3 Tarsus, NETZSCH, Germany). The catalytic activity was tested with ultraviolet-visible spectra (UV-1800, Shimadzu, Japan).2.4 Evaluation of dye degradation by CuS@CM biocatalysts
MB and MO were commonly used as probe molecules in heterogeneous catalytic reactions and as indicators for various reactions, respectively. Therefore, CuS@CM was used to degrade these two dyes to evaluate the catalytic performance of biocatalysts. The specific catalytic process is as follows: 100 mg of different loads of CuS@CM were added to the dye solution (100 mL, 20 mg L−1). The catalytic reaction was then started by the addition of H2O2 (500 μL, 30 wt%) and oscillating at 120 rpm, 50 °C. Then, 1 mL of residual solution was taken out at a standard time to measure the dye concentration immediately using a UV-visible light spectrophotometer. After the reaction, the solution was filtered to recover the catalyst, and then rinsed with DI water, and the cyclic usability of CuS@CM was measured under the same conditions. The pseudo-first-order kinetics model was used to calculate the initial reaction rate. The degradation efficiency formula and the pseudo-first-order equation of the dye are expressed as follows:| Degradation efficiency (%) = (C0 − Ct)/C0 − ln(Ct/C0) = kt | (1) |
In addition, the effects of different reaction conditions on the degradation efficiency were studied. Catalytic experiments were carried out at different pH values (3–11), H2O2 amounts (0–1 mL), temperatures (30–60 °C) and dye concentrations (10–40 mg L−1). Other experimental conditions were the same as above.
3. Result and discussions
3.1 Formation mechanism of CuS particles on porous cellulose macrospheres
The synthesis schematic diagram of cellulose macrospheres and CuS-supported CM catalysts is shown in Scheme 1. In this work, CuS was grown in situ on porous CM with a large specific surface area of 325.3 m2 g−1 in an aqueous solution at 90 °C. Compared with the method of preparing copper sulfide in a high-temperature autoclave,45 our method is simpler and faster. It is reported that the specific mechanism of CuS nanostructure formation is still being studied, which is quite complex.32,46 In our experiment, we inferred from the following reaction equation (eqn (2)–(4)). The CuNO3 aqueous solution was acidic due to the hydrolysis of Cu2+. In the reaction, thioacetamide could be complexed with Cu2+ and hydrolyzed in an acidic solution to produce H2S, acting as a precipitator to deposit CuS particles on CM. Therefore, a dilute HNO3 solution was used to adjust the reaction system to the optimal pH value, so that CuS could grow rapidly on CM. The entire formation process of CuS particles can be expressed by the following equation:| CH3CSNH2 + H2O → CH3CONH2 + H2S | (2) |
| H2S + Cu2+ → CuS + 2H+ | (3) |
| CH3CSNH2 + Cu2+ + H2O → CuS + CH3CONH2 + 2H+ | (4) |
 | ||
| Scheme 1 Schematic diagram of the preparation process of 3D porous CM and CuS-supported CM composite biocatalysts. | ||
As the porous structure of cellulose microsphere is conducive to the effective adsorption of Cu2+, CuS particles could be formed in situ on the surface and channels of the three-dimensional cellulose microsphere.
3.2 Morphological and chemical composition analysis of CuS-supported CM biocatalysts
Fig. 1 shows the morphology of CM and a series of CuS@CM catalysts. As illustrated in Fig. 1A and B, CM presented a regular spherical shape with a diameter of about 1.2 mm and a porous network structure on the surface, which was beneficial to the loading of catalyst. Fig. 1C–F show the morphology of CuS-supported CM at different initial Cu2+ concentrations, which demonstrate that CuS nanoparticles successfully aggregate and grow on the surface and channels of CM. The TEM images (Fig. 1G and H) reveal that CuS nanoparticles have similar sizes and structures to CuS NPs grown on CM, with a diameter of about 20 nm, indicating that this in situ growth method does not damage the original structure and properties of CuS. Meanwhile, the elemental mapping images (Fig. 2A–D) showed that sulfur (S) and copper (Cu) were uniformly distributed in CuS@CM-50 biocatalysts, and EDS analysis revealed the chemical composition of copper and sulfur. The atomic percentage of Cu and S is 32.77 and 30.36 with an atomic ratio of Cu:S = 1:1. Fig. 2F further shows the increase in the amount of copper sulfide by increasing the concentration of copper salt from 12.5 mM to 100 mM. However, with the increase in copper source, the amount of CuS gradually reached equilibrium or even slightly decreased, and the maximum load was 135.2 mg g−1, which may be due to the excessive aggregation of copper sulfide nanoparticles leading to some separation. The above-mentioned results indicate that CuS nanoparticles are uniformly and densely anchored on the cellulose macrospheres with three-dimensional porous structures, which not only make the dye more easily adsorbed to contact CuS, but also provide more contact sites for catalysis.
 | ||
| Fig. 1 Apparent SEM images of (A and B) cellulose macrospheres, (C) CuS@CM-12.5, (D) CuS@CM-25, (E) CuS@CM-50 and (F) CuS@CM-100. (G and H)TEM images of CuS nanoparticles. | ||
 | ||
| Fig. 2 (A–D) Corresponding elemental mapping image and (E) EDS spectrum of CuS@CM-50. (F) Amount of CuS in CuS@CM composites as a function of CuNO3 concentration and changes of specific surface area of different samples. | ||
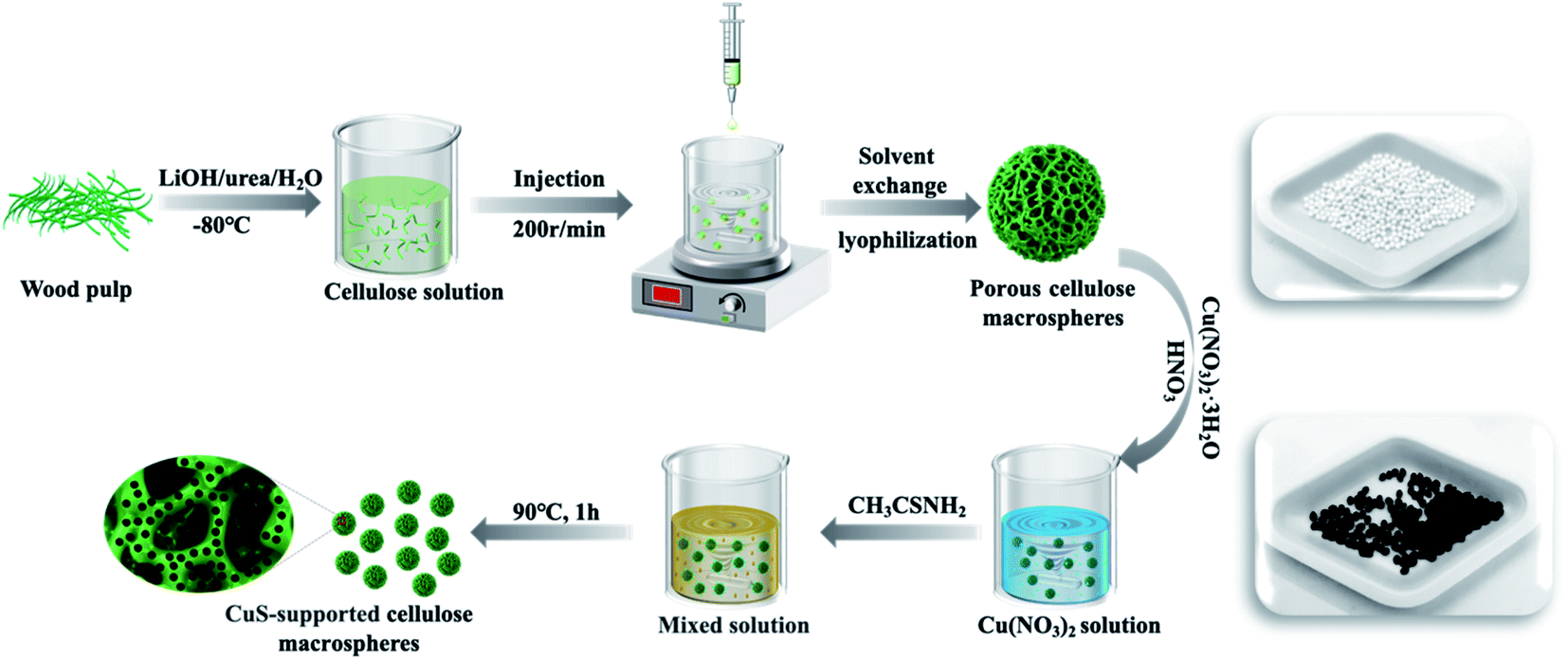
To confirm the preparation of crystalline copper sulfide on the macrosphere surface of cellulose, XRD analysis was performed, as shown in Fig. 3A. It can be seen from the XRD pattern that all diffraction peaks of pure CuS particles and CuS@CM catalyst can be assigned to cobalt-phase copper sulfide, corresponding to the standard data (PDF card no. 00-001-1281). The crystal peaks at 29.04°, 31.96°, 33.22°, 47.86°, and 59.34° were attributed to the planar structures of (102), (103), (006), (110), and (116) respectively.44 The sharp peak at 20.7° and the low peak at 12.1° belonged to the characteristic diffraction peaks of cellulose II, corresponding to the crystal planes of (110) and (200), respectively.47 This indicated that CuS has good crystallinity and stability on CM.
 | ||
| Fig. 3 (A) XRD patterns and (B) TGA curve of CM, CuS, and CuS@CM-50. | ||
TGA measurement was used to study the thermal stability of CuS, CM, and CuS@CM in a N2 atmosphere. As shown in Fig. 3B, the TGA curve of the CuS sample presented several obvious steps of mass loss. At a temperature of 200 °C, the thermal weight loss (8.9%) was mainly attributed to the loss of residual solvent and sulfur source in the sample.48 This indicated that the sample was extremely unstable at this temperature, and CuS decomposed into various crystalline CuxS. The weight loss (10.2%) from 330 °C to 450 °C was classified as CuS decomposition into Cu2S and phase transition of sulfur elements.49 For CM and CuS@CM samples, there was an initial peak at 50–60 °C, which was the weightlessness (5.6%) peak of water adsorption.50 The main decomposition temperature of CM was approximately 350–360 °C, which was mainly the decomposition and oxidative degradation of cellulose (80.7%).51 Obviously, CuS@CM integrated the two above-mentioned decomposition steps and decomposed gradually in the range of 200–400 °C, which was sufficient to prove that the CuS-supported CM biocatalyst was successfully prepared.
FTIR and Raman spectra were used to further determine the binding of CuS on cellulose macrospheres, and the results are shown in Fig. 4. As shown in Fig. 4A, the FTIR spectrum of CM showed characteristic peaks at 890 and 1430 cm−1, and the absorption peak was wider at 3440 cm−1, which was considered to be the typical energy band peak of regenerated cellulose.47,52,53 In the CM sample deposited by CuS, the Cu–S tensile vibration band appears at the peak of 620 cm−1, and the peak at 3440 cm−1 moved to the high wave digit, indicating the attraction between CuS and –OH groups on cellulose.54,55 As shown in Fig. 4B, Raman spectra also showed characteristic peaks of cellulose and CuS, with 345–390 cm−1, 978–1178 cm−1, and 1280–1411 cm−1 respectively belonging to the β-D-glucose ring, glycosidic bond (C–O–C), and subunit methyl (–CH2) absorption bands.56–58 The sharp peak at 471 cm−1 belonged to the CuS Raman characteristic absorption peak.59 The above-mentioned results indicated that CuS was stably deposited on CM after complexing with CH3CSNH2 in the liquid phase.
 | ||
| Fig. 4 (A) FTIR and (B) Raman spectra of the as-prepared CM, CuS and CuS@CM-50. | ||
To further investigate the chemical structure of CuS@CM, X-ray photoelectron spectroscopy was performed to analyze the sample surface. Fig. 5A presents the wide-scan XPS spectra of the prepared CuS@CM macrospheres. It can be seen that C, O, Cu, and S elements are significantly detected, indicating their presence in the sample. The Auger line of Cu (Cu LMM) appears at 569.0 eV, which is the typical binding energy value for CuS, indicating that the Cu element is present in the form of bivalent states.32 The peaks of Cu 2p (Fig. 5B) and S 2p (Fig. 5C) were further identified by high-resolution XPS spectra. In Fig. 5B, the two main peaks at 932.4 and 952.3 eV were attributed to Cu 2p3/2 and Cu 2p1/2, respectively.54 Fig. 5C shows that the binding energies of S 2p3/2 and S 2p1/2 were 161.7 and 163.3, respectively, 168.1 eV corresponding to the S2− state in CuS.60 All measurements confirmed that CuS and not Cu2S or other substances were bound to cellulose macrospheres.
 | ||
| Fig. 5 XPS spectra of CuS@CM microsphere: (A) full spectrum, (B) Cu 2p spectrum, and (C) S 2p spectrum. | ||
3.3 Catalytic activity of the prepared CuS@CM composite macrospheres
A series of prepared CuS@CM (12.5, 25, 50, and 100 mM) composite macrospheres were used to degrade MB and MO in H2O2. During the reaction process, CuS initiated hydrogen peroxide to release hydroxyl radicals of high reaction, which could convert organic dyes into non-toxic small molecules.61,62 The mechanism of catalysis can be expressed by the following equations (eqn (5)–(8)):| Cu2+ + H2O2 → H+ + CuOOH+ | (5) |
| CuOOH+ → HOO˙ + Cu+ | (6) |
| Cu+ + H2O2 → Cu2+ + ˙OH + OH− | (7) |
| MB/MO + HO˙ → CO2 + H2O | (8) |
The evolution of UV-vis spectra with respect to time is shown in Fig. 6A and B. The strong absorption peaks located at 660 nm and 609 nm belonged to the maximum absorption of the monomer and dimer of MB in aqueous solutions, and the absorption peak at 463 nm belonged to the characteristic absorption peak of MO. It is obvious that the original peaks of MB and MO decreased gradually within 90 min, and no new peaks can be observed after degradation reaction. The results indicated that CuS@CM composite spheres can successfully degrade MB and MO in H2O2.
 | ||
| Fig. 6 Changes in the UV-vis absorbance spectra of (A) MB and (B) MO aqueous solutions in the presence of CuS@CM-50 composites and H2O2. The degradation rates of (C) MB and (D) MO in different catalytic systems for 60 min, described by the decrease in the intensity of peak absorption of MB and MO solutions: (a) only CM; (b) only H2O2; (c) CuS@CM-12.5 + H2O2; (d) CuS@CM-25 + H2O2; (e) CuS@CM-50 + H2O2; and (f) CuS@CM-100 + H2O2. (E and F) First-order kinetic plot of ln(Ct/C0) vs. time (0–10 min) degradation of MB and MO by different biocatalyst samples. | ||
Fig. 6C and D show the influence of different catalytic systems on the catalytic effects of MB and MO. All of the catalytic reactions were performed under the set experimental conditions. When CuS@CM alone was added, the removal rates of MB and MO were 3.57% (Fig. 6C, curve a) and 3.67% (Fig. 6D, curve a) within 1 h, which was due to the effect of porous macrospheres on dye adsorption, while only H2O2 was added, the degradation rates of MB and MO were 5.65% (Fig. 6C, curve b) and 3.67% (Fig. 6D, curve b), indicating that the higher temperature promoted the generation of free radicals by H2O2 leading to the oxidative degradation of dyes. However, in the presence of CuS@CM macrospheres and H2O2, the degradation of MB and MO in the prepared four samples within 1 h reached more than 95% (Fig. 6C, curve c–f; Fig. 6D, curve c–f), which fully indicates that the as-prepared catalysts has excellent catalytic efficiency. In addition, the initial reaction rate constants of the four catalysts were calculated using the pseudo-first-order equation, and the obtained fitting curve conformed to this equation (Fig. 6E and F). It can be seen that the CuS@CM-50 composite material has the fastest reactive rate k (MB: 0.28729 min−1; MO: 0.101 min−1), and the results were because this sample has the largest loading capacity and a relatively high specific surface area, which made the dye molecules more receptive to contact with CuS, thus improving the reaction probability.
3.4 Effects of pH, temperature, H2O2 and dye concentration on catalytic efficiency
The CuS@CM-50 biocatalyst with the best degradation effect was used for the reaction condition influence determination, with MB as the test dye. As shown in Fig. 7A, degradation rates within 20 min were 75.4%, 82.8%, 95.6%, 96.6% and 96.7%, corresponding to pH values of 3, 5, 7, 9, and 11, respectively. Obviously, the catalytic effect was better than that of acid under alkaline conditions, and the degradation efficiency was maintained highly at pH ≥ 7, this is because the solution in this pH range has an isoelectric point higher than that of copper sulfide, the cationic MB dye has better affinity for the surface catalyst, and H2O2 is dominant under acidic conditions, while HO2− or O22− is more under alkaline conditions. The results were consistent with the test results of Wang et al.,54 indicating that this catalyst has a wider range of pH applicability. | ||
| Fig. 7 Effect of (A) initial pH, (B) temperature and (C) H2O2 dosage on the catalytic degradation of MB using CuS@CM-50 biocatalyst and the degradation efficiency of (D) different concentrations of MB solution. | ||
Fig. 7B shows the catalytic efficiency of CuS@CM composites at different temperatures. The results illustrated that the degradation efficiency increased significantly with the increase in reaction temperature. When the reaction temperature was higher than 45 °C, the degradation rate of MB reached more than 95% within 10 min, while when the reaction temperature was 25 °C or 35 °C, only 62.6% and 84.5% of MB were degraded within 20 min. The results indicate that higher temperatures are conducive to rapid dye removal, which is consistent with the report by Zhan et al.63
The influence of H2O2 concentration on the catalytic efficiency was also investigated, because it is directly related to the number of free radicals generated in the reaction, and has an significant influence on the reaction rate, especially the initial reaction rate. As can be seen from Fig. 7C, degradation efficiency gradually increased with the dosage of H2O2 from 65 μL to 500 μL. When the dosage was further increased to 1 mL, the catalytic efficiency did not significantly increase, indicating that the reaction rate reached the maximum when 500 μL of H2O2 was added, and the hydroxyl free radical in the dye solution had reached saturation. On the contrary, the dosage was further increased, and the reaction rate would decrease, which may be due to its scavenging effect on the hydroxyl radical caused by low reaction.64 Therefore, in order to maintain a high level of reaction rate, a dosage of 500 μL of H2O2 was selected in this experiment.
At the same time, different concentrations of MB were degraded under the above-mentioned optimal conditions. As shown in Fig. 7D, the degradation rates of dyes within 20 min were 96.8%, 95.6%, 92.6% and 90.3%, which were attributed to MB concentrations of 10, 20, 30 and 40 g L−1 respectively. For a series of dye concentrations, the degradation rates were all over 90% in a short time. The results indicated that at a given amount of catalyst, it also had excellent catalytic activity for dyes with high initial concentrations.
3.5 Reusability of CuS@CM biocatalysts
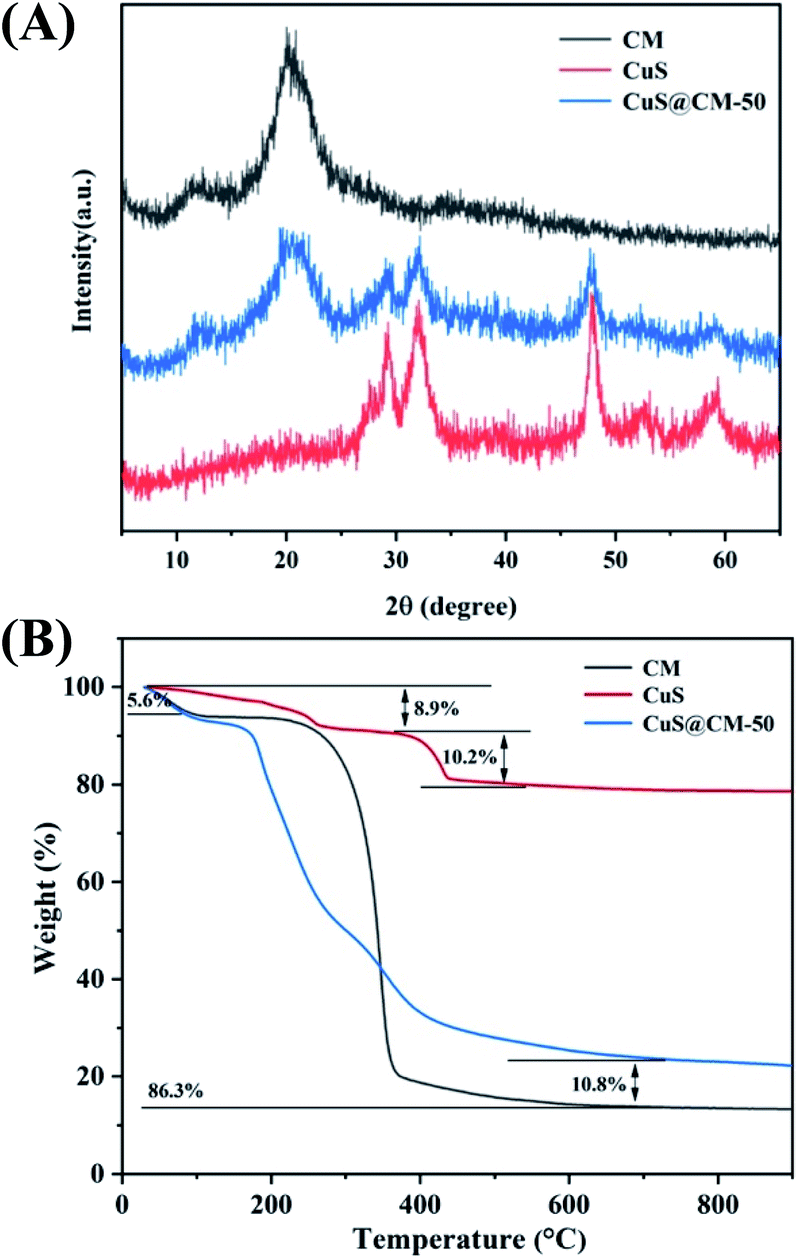
Catalyst reusability and stability are critical in industrial applications because it is an important way to save costs. In order to confirm the reusability of the synthetic CuS@CM, five cycles were carried out in the process of MB degradation under the same catalytic conditions. As illustrated in Fig. 8A, the initial degradation rate of MB was 95.6%. Although the catalytic activity decreased to a certain extent during use, the degradation degree of CuS@CM was still 90.1% after five cycles, indicating that this material is a potential candidate for recoverable Fenton-like reagents. | ||
| Fig. 8 (A) Catalytic cycle of the prepared CuS@CM-50 biocatalyst for the MB degradation under optimal conditions for 20 min. (B) XRD patterns of the CuS@CM-50 composite after consecutive recycling for five times. SEM images (C) before and (D) after five cycles of degradation. | ||
Furthermore, the morphology and structural stability of the recovered CuS@CM were characterized by SEM and XRD respectively. The SEM image after cycling (Fig. 8D) showed that the porosity of cellulose was significantly lower than that of cellulose before catalytic degradation (Fig. 8C), which may be due to the partial shedding or collapse of cellulose during the degradation process, resulting in a decrease in the catalytic activity, and the XRD spectra (Fig. 8B) also proved that no new diffraction peaks were generated after five cycles compared with the fresh catalyst, both of which showed CuS characteristic peaks at 29.04°, 31.96° and 47.86°, indicating that CuS supported on cellulose macrospheres remained stable after catalysis. However, the intensity of characteristic peaks decreases after cycling, which may be due to the lower crystallinity of CuS caused by a higher catalytic temperature. It is worth noting that CuS itself did not suffer any significant damage and showed excellent cyclic catalytic stability. In addition, the results of this study were compared with those of previous studies (Table 1), and the activity of peroxide oxidation and photocatalytic oxidation of organic dyes by copper sulfide is summarized. Briefly, the CuS-supported CM composite macrosphere prepared in this work had higher catalytic activity in the presence of less H2O2 and was a kind of green and promising material with excellent catalytic activity, stability and reusability.
| Materials | Synthesis method | Dye conc. (mg L−1) | Means | Degradation (%) | Time (min) | Ref. |
|---|---|---|---|---|---|---|
| CuS/cellulose aerogel | Ion exchange | 20 of MB | 3 mL H2O2 | 94.1 | 60 | 60 |
| CuS/PAN | Hydrothermal approach | 10 of MB | 1 mL H2O2 | 93.6 | 45 | 65 |
| CuS–CdS | Hydrothermal approach | 10 of MB | 2 mL H2O2 and visible light | 94.7 | 10 | 66 |
| CuS/rGO | Solvothermal | 15 of MB | UV-vis. light | 80 | 140 | 67 |
| CNT/CuS | Co-precipitation method | 5 of RhB | Sun light | 120 | 92.4 | 68 |
| GO–FA–CuS | Hydrothermal approach | 10 of MB | NIR | 94.3 | 20 | 69 |
| CuS/cellulose macrospheres | Liquid phase deposition | 20 of MB | 0.5 mL H2O2 | 94.8 | 10 | This work |
4. Conclusion
In summary, a 3D porous cellulose macrosphere with high specific surface area has been successfully prepared and CuS was stably immobilized on the porous cellulose macrosphere with high capacity. The prepared CuS@CM exhibited excellent catalytic activity in the presence of a small amount of H2O2 and a wide range of pH applicability (5–11) for the degradation of MB and MO dyes. After several cycles of catalysis, more than 90% of the initial activity still remained and the loaded CuS nanoparticles did not suffer any serious damage, showing good recycling stability. The results indicated that CuS@CM is promising to solve the recycling problem of Fenton reagents and has great application potential in the removal of dyes from wastewater. In this study, only laboratory-simulated wastewater was treated. Future research should use more complex industrial wastewater to further develop practical water treatment technologies.Conflicts of interest
Authors declare that they have no conflict of interest.Acknowledgements
This work was supported by Identification, emission scenario construction and substitution technology of toxic and hazardous substances in printing and dyeing industry (2018YFC1801502) and Program of Shanghai Academic/Technology Research Leader (Grant Number 19XD1434300).References
- M. Visa, Appl. Surf. Sci., 2012, 263, 753–762 CrossRef CAS
.
- A. Rodríguez, G. Ovejero, J. L. Sotelo, M. Mestanza and J. García, Ind. Eng. Chem. Res., 2010, 49, 498–505 CrossRef
- K. Pakshirajan and S. Singh, Ind. Eng. Chem. Res., 2010, 49, 7484–7487 CrossRef CAS
- U.-J. Kim, D. Kim, J. You, J. W. Choi, S. Kimura and M. Wada, Cellulose, 2018, 25, 2615–2628 CrossRef CAS
- H. E. Emam and H. B. Ahmed, Int. J. Biol. Macromol., 2019, 138, 450–461 CrossRef CAS PubMed
- G. Ciardelli and N. Ranieri, Water Res., 2001, 35, 567–572 CrossRef CAS PubMed
- M. F. Elahmadi, N. Bensalah and A. Gadri, J. Hazard. Mater., 2009, 168, 1163–1169 CrossRef CAS PubMed
- A. Baban, A. Yediler, N. Ciliz and A. Kettrup, Chemosphere, 2004, 57, 731–738 CrossRef CAS PubMed
- A. Khan, S. J. Shah, K. Mehmood, Awais, N. Ali and H. Khan, J. Mater. Sci.: Mater. Electron., 2018, 30, 406–414 CrossRef
- M. S. Mia, P. Yao, X. Zhu, X. Lei, T. Xing and G. Chen, RSC Adv., 2021, 11, 8290–8305 RSC
- J. Lunagariya, A. Dhar and R. L. Vekariya, RSC Adv., 2017, 7, 5412–5420 RSC
- A. Dhar, N. S. Kumar, M. Asif and R. L. Vekariya, New J. Chem., 2018, 42, 6990–6996 RSC
- A. Dhar, N. Siva Kumar, M. Khimani, A. S. Al-Fatesh, A. A. Ibrahim, A. H. Fakeeha, H. Patel and R. L. Vekariya, RSC Adv., 2020, 10, 15282–15292 RSC
- G. Liu, Q. Deng, H. Wang, D. H. L. Ng, M. Kong, W. Cai and G. Wang, J. Mater. Chem., 2012, 22, 9704–9713 RSC
- J. H. Ramirez, C. A. Costa, L. M. Madeira, G. Mata, M. A. Vicente, M. L. Rojas-Cervantes, A. J. López-Peinado and R. M. Martín-Aranda, Appl. Catal., B, 2007, 71, 44–56 CrossRef CAS
- H. R. Pouretedal, A. Norozi, M. H. Keshavarz and A. Semnani, J. Hazard. Mater., 2009, 162, 674–681 CrossRef CAS PubMed
- A. D. French, Cellulose, 2017, 24, 4605–4609 CrossRef CAS
- H. Shaghaleh, X. Xu and S. Wang, RSC Adv., 2018, 8, 825–842 RSC
- N.-M. Park, S. Choi, J. E. Oh and D. Y. Hwang, Carbohydr. Polym., 2019, 223, 115114 CrossRef PubMed
- A. Farooq, M. K. Patoary, M. Zhang, H. Mussana, M. Li, M. A. Naeem, M. Mushtaq, A. Farooq and L. Liu, Int. J. Biol. Macromol., 2020, 154, 1050–1073 CrossRef CAS PubMed
- J. L. Slavin, P. M. Brauer and J. A. Marlett, J. Nutr., 1981, 111, 287–297 CrossRef CAS PubMed
- H. Dong, Q. Ding, Y. Jiang, X. Li and W. Han, Carbohydr. Polym., 2021, 265, 118101 CrossRef CAS PubMed
- D. Georgouvelas, H. N. Abdelhamid, J. Li, U. Edlund and A. P. Mathew, Carbohydr. Polym., 2021, 264, 118044 CrossRef CAS PubMed
- N. Li, W. Lu, J. Yu, Y. Xiao, S. Liu, L. Gan and J. Huang, Mater. Sci. Eng., C, 2018, 91, 179–189 CrossRef CAS PubMed
- A. Batool and S. Valiyaveettil, J. Hazard. Mater., 2021, 413, 125301 CrossRef CAS PubMed
- P. Zhou, L. Zhang, X. Sui, Y. Zhong, B. Wang, Z. Chen, X. Feng, H. Xu and Z. Mao, Cellulose, 2020, 27, 3517–3530 CrossRef CAS
- R. M. Barajas-Ledesma, L. Hossain, V. N. L. Wong, A. F. Patti and G. Garnier, J. Colloid Interface Sci., 2021, 599, 140–148 CrossRef CAS PubMed
- X. He, T. Chen, T. Jiang, C. Wang, Y. Luan, P. Liu and Z. Liu, Carbohydr. Polym., 2021, 260, 117790 CrossRef CAS PubMed
- D. Li, Y. Wang, F. Long, L. Gan and J. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 1549–1557 CrossRef CAS PubMed
- Y. Zhao, Y. Zhang, A. Liu, Z. Wei and S. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 4006–4014 CrossRef CAS PubMed
- W. Lyu, J. Li, L. Zheng, H. Liu, J. Chen, W. Zhang and Y. Liao, Chem. Eng. J., 2021, 414, 128931 CrossRef
- L. Qian, J. Mao, X. Tian, H. Yuan and D. Xiao, Sens. Actuators, B, 2013, 176, 952–959 CrossRef CAS
- G. Nie, Z. Li, X. Lu, J. Lei, C. Zhang and C. Wang, Appl. Surf. Sci., 2013, 284, 595–600 CrossRef CAS
- L. Zheng, F. Teng, X. Ye, H. Zheng and X. Fang, Adv. Energy Mater., 2020, 10, 1902355 CrossRef CAS
- S. Goel, F. Chen and W. Cai, Small, 2014, 10, 631–645 CrossRef CAS PubMed
- W. He, H. Jia, X. Li, Y. Lei, J. Li, H. Zhao, L. Mi, L. Zhang and Z. Zheng, Nanoscale, 2012, 4, 3501–3506 RSC
- T. Zhu, B. Xia, L. Zhou and X. Wen Lou, J. Mater. Chem., 2012, 22, 7851–7855 RSC
- A. Manzi, T. Simon, C. Sonnleitner, M. Döblinger, R. Wyrwich, O. Stern, J. K. Stolarczyk and J. Feldmann, J. Am. Chem. Soc., 2015, 137, 14007–14010 CrossRef CAS PubMed
- J. Kundu, S. Khilari and D. Pradhan, ACS Appl. Mater. Interfaces, 2017, 9, 9669–9680 CrossRef CAS PubMed
- X.-j. Yang, X.-m. Xu, J. Xu and Y.-f. Han, J. Am. Chem. Soc., 2013, 135, 16058–16061 CrossRef CAS PubMed
- C. Zhou, J. Luo, Q. Chen, Y. Jiang, X. Dong and F. Cui, Chem. Commun., 2015, 51, 10847–10849 RSC
- J. Cai and L. Zhang, Macromol. Biosci., 2005, 5, 539–548 CrossRef CAS PubMed
- J. Yang, J. Duan, L. Zhang, B. Lindman, H. Edlund and M. Norgren, Cellulose, 2016, 23, 3105–3115 CrossRef CAS
- J. Huang, J. Zhou, J. Zhuang, H. Gao, D. Huang, L. Wang, W. Wu, Q. Li, D. P. Yang and M. Y. Han, ACS Appl. Mater. Interfaces, 2017, 9, 36606–36614 CrossRef CAS PubMed
- V. K. Gupta, D. Pathania, S. Agarwal and P. Singh, J. Hazard. Mater., 2012, 243, 179–186 CrossRef CAS PubMed
- Q. Y. Lu, F. Gao and D. Y. Zhao, Nano Lett., 2002, 2, 725–728 CrossRef CAS
- X. Luo and L. Zhang, Biomacromolecules, 2010, 11, 2896–2903 CrossRef CAS PubMed
- J. Madarász, M. Krunks, L. Niinistö and G. Pokol, J. Therm. Anal. Calorim., 2015, 120, 189–199 CrossRef
- K. R. Nemade and S. A. Waghuley, Mater. Sci. Semicond. Process., 2015, 39, 781–785 CrossRef CAS
- A. M. A. Nada and M. L. Hassan, Polym. Degrad. Stab., 2000, 67, 111–115 CrossRef CAS
- L. Zhou, J. He, J. Zhang, Z. He, Y. Hu, C. Zhang and H. He, J. Phys. Chem. C, 2011, 115, 16873–16878 CrossRef CAS
- M. L. Nelson and R. T. O'Connor, J. Appl. Polym. Sci., 1964, 8, 1311–1324 CrossRef CAS
- P. Langan, Y. Nishiyama and H. Chanzy, J. Am. Chem. Soc., 1999, 121, 9940–9946 CrossRef CAS
- J. Qian, K. Wang, Q. Guan, H. Li, H. Xu, Q. Liu, W. Liu and B. Qiu, Appl. Surf. Sci., 2014, 288, 633–640 CrossRef CAS
- J. Guo, H. Tian and J. He, Chem. Eng. J., 2021, 408, 127336 CrossRef CAS
- R. H. Atalla and B. E. Dimick, Carbohydr. Res., 1975, 39, C1–C3 CrossRef CAS
- K. Schenzel, H. Almlöf and U. Germgård, Cellulose, 2009, 16, 407–415 CrossRef CAS
- A. Jahn, M. W. Schroder, M. Futing, K. Schenzel and W. Diepenbrock, Spectrochim. Acta, Part A, 2002, 58, 2271–2279 CrossRef CAS
- Y. Zou, L. Jiang, T. Zhai, T. You, X. Jing, R. Liu, F. Li, W. Zhou and S. Jin, J. Alloys Compd., 2021, 865, 158919 CrossRef CAS
- R. M. Y. Saeed, Z. Bano, J. Sun, F. Wang, N. Ullah and Q. Wang, J. Appl. Polym. Sci., 2019, 136, 47404 CrossRef
- A. Y. Satoh, J. E. Trosko and S. J. Masten, Environ. Sci. Technol., 2007, 41, 2881–2887 CrossRef CAS PubMed
- Z. K. Yang, L. X. Song, Y. Teng and J. Xia, J. Mater. Chem. A, 2014, 2, 20004–20009 RSC
- Y. Zhan, H. Li and Y. Chen, J. Hazard. Mater., 2010, 180, 481–485 CrossRef CAS PubMed
- N. N. Fathima, R. Aravindhan, J. R. Rao and B. U. Nair, Chemosphere, 2008, 70, 1146–1151 CrossRef CAS PubMed
- Y. Lu, Y. Wang, S. Cui, W. Chen and L. Mi, RSC Adv., 2018, 8, 40589–40594 RSC
- M. Mahanthappa, N. Kottam and S. Yellappa, Appl. Surf. Sci., 2019, 475, 828–838 CrossRef CAS
- X.-S. Hu, Y. Shen, Y.-T. Zhang and J.-J. Nie, J. Phys. Chem. Solids, 2017, 103, 201–208 CrossRef CAS
- Y. Wang, F. Jiang, J. Chen, X. Sun, T. Xian and H. Yang, Nanomaterials, 2020, 10, 178 CrossRef CAS PubMed
- G. M. Neelgund and A. Oki, Appl. Surf. Sci., 2021, 566, 150648 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2021 |