
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign and synthesis of water-soluble grifolin prodrugs for DNA methyltransferase 1 (DNMT1) down-regulation†
Liguo Wang‡
 a,
Yue Wu‡a,
Zhenzhen Li‡bcde,
Tianlong Lana,
Xu Zhaobcde,
Wenxing Lva,
Feng Shibcde,
Xiangjian Luo*bcde,
Yu Rao*a and
Ya Cao*bcde
a,
Yue Wu‡a,
Zhenzhen Li‡bcde,
Tianlong Lana,
Xu Zhaobcde,
Wenxing Lva,
Feng Shibcde,
Xiangjian Luo*bcde,
Yu Rao*a and
Ya Cao*bcde
aKey Laboratory of Protein Sciences, School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China. E-mail: yrao@tsinghua.edu.cn
bKey Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Central South University, Changsha 410078, China. E-mail: ycao98@vip.sina.com
cCancer Research Institute, School of Basic Medicine, Central South University, Changsha, 410078, China
dMolecular Imaging Research Center of Central South University, Changsha, China
eNational Joint Engineering Research Center for Genetic Diagnostics of Infectious Diseases and Cancer, Changsha 410078, China. E-mail: luocsu@hotmail.com; ycao98@vip.sina.com
First published on 3rd December 2021
Abstract
DNA methylation and gene silencing play indispensable roles in the epigenetic landscape and gene expression. DNA methyltransferase 1 (DNMT1), a member of the DNMT family, which catalyzes the addition of methyl groups on DNA has been identified to have a close relationship with tumorigenesis. But DNMT1 inhibitors are rare except for the highly toxic nucleoside derivates. Grifolin is a unique natural product which down-regulates DNMT1 and has low toxicity. However, the poor solubility and stability of grifolin limit its application. Herein, we synthesized PEG5-Grifolin as a water-miscible prodrug of grifolin. The half-life of PEG5-Grifolin at 25 °C was considerably extended, revealing excellent stability. Meanwhile, PEG5-Grifolin suppressed tumor growth of by downregulating DNMT1 and reactivating the expression of several tumor suppressor genes in vivo. PEG5-Grifolin might be a promising demethylation agent for DNMT1 associated diseases and benefit much against various types of DNMT1 associated cancer.
1. Introduction
DNA methylation and gene silencing reprogram the epigenetic landscape and tremendously contribute to cancer etiology.1 CpG island methylator phenotype (CIMP) is defined as the high activity of global and non-random CpG island methylation. CIMP of tumor suppressor genes (TSGs) is one of the most characteristic abnormalities in cancers.2 Methylation and inactivation of TSGs, which are involved in the cell cycle (e.g., p16), apoptosis (e.g., death-associated protein kinase (DAPK1)), detoxification (GSTP1), migration (e.g., E-cadherin (E-cad)) and in the response to growth factors (e.g., phosphatase and tensin homolog (PTEN)) have essential roles in tumorigenesis and malignant development of cancer.1,3 Especially, the CpG island promoter methylation of HOPX,4 TIPE3 (ref. 5) and RAB37 (ref. 6) genes has been defined as the marker of metastatic and radio-resistant nasopharyngeal carcinoma. DNA hypermethylation has become a biomarker of the screening, prognosis and therapy surveillance of multiple types of cancers.DNA methyltransferases (DNMTs) belong to a class of enzymes involved in the regulation of DNA methylation and gene expression.7 Transcriptional repression of chromatin is highly impacted by DNMTs overexpression. Thus, DNMTs play important roles in neoplasia, growth and metabolism.8 Overexpression of DNMTs has been well-documented to inactivate TSGs in many solid and hematological cancers.8 Three DNMT members have been identified in mammals: DNMT1, DNMT3a and DNMT3b.1 DNMT1 is responsible for the most potent methylation of CpG sites in neoplasia among these DNMTs subtypes.9 The expressions of TSGs, such as p16, DAPK1, CDH1 and PTEN were suppressed by overactivation of DNMT1.1,3 The use of reagents which induces DNMT1 loss-of-function and reverses methylation-induced inactivation of TSGs has become a promising approach for cancer therapy. Although DNMT1 is vital to neoplasia and other diseases, the nucleoside derivatives (e.g., 5-azacytidine (5-AD)) appear to be the only reported epidrug targeting DNMTs, whereas the high toxicity and instability of nucleoside derivatives cause serious side effects and toxicities.
Natural products provide a unique source for the discovery of innovative leading compounds and drugs targeting cancer and other diseases.10–13 Several natural products including polyphenolic compounds, e.g., epigallocatechin-3-gallate (EGCG) and curcumin, and flavonoids, e.g., genistein and quercetin, have shown demethylating effects by targeting DNMTs.14 Grifolin and its derivates are natural secondary metabolites with small molecular weights extracted from mushrooms Albatrellus confluens and Boletus pseudocalopus.15 Since 2005, we and others have demonstrated that grifolin could down-regulate DNMT1 and consequently exert the anti-growth, anti-invasion, and anti-metastasis effects in multiple types of tumor cells, especially nasopharyngeal carcinoma (NPC) cells.16–19 Thus, grifolin might be a promising demethylation agent for the treatment of NPCs and other DNMT1 associated diseases. However, poor solubility and stability limited its application. As a way out, corn oil was used as the solvent to facilitate the absorption and administration, however, serious side effects such as fat accumulation and fatty liver were observed in mice model.20
Herein, we developed a series of prodrugs of grifolin to improve its water-solubility and stability (Fig. 1). Poly ethylene glycol (PEG), which has good water solubility and low toxicity, is widely used in many solubilizers and prodrugs.21 The PEG modified prodrug, PEG5-Grifolin is miscible with phosphate buffer saline pH = 7.4 (PBS) at room temperature. Meanwhile, we assessed the in vivo bioactivity of PEG5-Grifolin. It efficiently hampered tumor growth without significant toxicity in an NPC xenograft mice model. PEG5-Grifolin treatment down-regulates DNMT1 and up-regulates the expression of TSGs. Moreover, PEG5-Grifolin induced a more potent demethylating effect at the promoter region of TSGs than 5-AD did, revealing it could be a potential demethylation agent for DNMT1-related diseases.
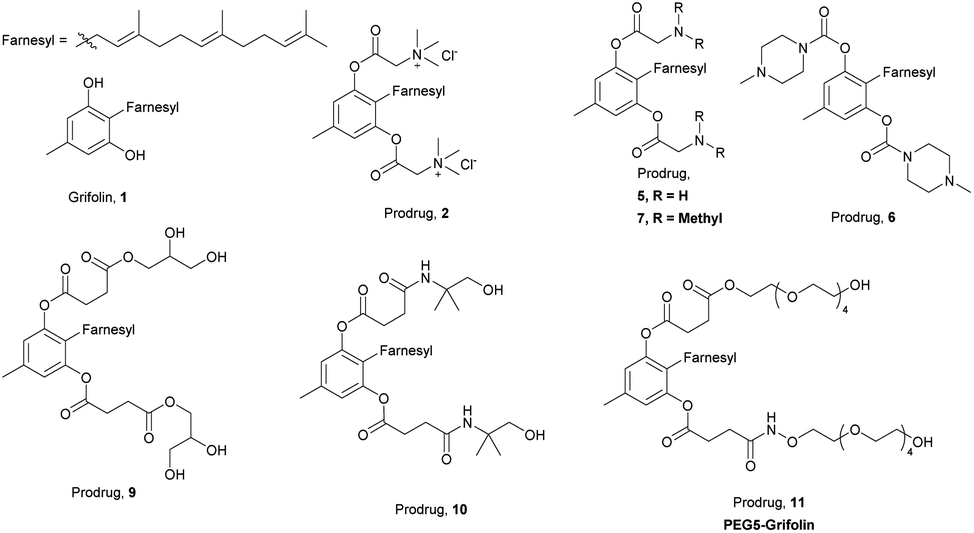 | ||
| Fig. 1 Chemical structures of grifolin and prodrugs. | ||
2. Results and discussion
We analysed the structure of grifolin and proposed several potential modification strategies. Grifolin is a phenol derivative with two hydroxyl groups, a methyl substituent and a farnesyl substituent. The phenol ring is very electron-rich and can be readily oxidized into quinones. Meanwhile, a pericyclic reaction possibly happens between farnesyl function group and the ortho-phenolic hydroxyl group under acid or base conditions.22,23 Thus, the phenolic hydroxyl groups are the key factors for stability.As shown in Scheme 1, we designed and synthesized the following compounds based on the principle that these solubilizing groups should be efficiently released in vivo. In order to optimize reaction conditions, we tested the stability of grifolin under acid and base. Even acetic acid or pyridine could lead to severe, complex and unidentifiable degradation of the original grifolin. However, the degradation products seemed to differ from each other. Inert atmosphere could only help to stabilize grifolin under pyridine and was non effective under acetic acid. With these stability data in hand, relatively more mild reaction conditions with inert gas protection were adopted. First, we tried to introduce betaine (compound 2) on the hydroxyl groups. However, the desired molecular weight of compound 2 could only be detected by mass spectrometer at the initial of the reaction. We speculated that the installation of two large hinder groups into the farnesyl ortho position might cause a large bond tension to the overall structure, which was unstable. Meanwhile, quaternary amine compounds might have significant toxic effects. Thus, we did not further investigate such type of prodrugs. Next, hydrophilic alkali prodrugs were tried. Boc-glycine and Fmoc-glycine was also successfully introduced to grifolin through esterification (intermediate 3, 4). However, they failed to give compound 5, the deprotection product. Not only that, the compounds 6 and 7, containing tertiary amine, totally decomposed after condensation reaction. The alkalinity of amine might be too strong for the prodrugs of grifolin to maintain stability.
 | ||
| Scheme 1 Design and synthetic route towards prodrugs of grifolin. Reagents and conditions: (a), grifolin, betaine chloride, pyridine, THF, 0 °C – >r. t., soon decomposed; (b), for compound 3: grifolin, Boc-glycine, EDCI, DMAP, DCM, r. t., 5 h, 74%; for compound 4: grifolin, Fmoc-glycine, EDCI, DMAP, DCM, r. t., 5 h, 81%; (c), compound 3 or 4, 4 M HCl in dioxane or piperidine in DMF, −10 °C – >r. t., decomposed; (d), for compound 6: grifolin, phosgene, 1-methylpiperazine, DCM, 0 °C – >r. t., 6 h, 24%; for compound 7: grifolin, N,N-dimethylglycine, EDCI, DMAP, DCM, r. t., 5 h decomposed; (e), succinic anhydride, pyridine, DMAP, DCM, 12 h, 51%; (f), glycerol, pentaglycol or 2-amino-2-methylpropan-1-ol, EDCI, DMAP, THF, r. t., corresponding solvent, 43–69%. | ||
Because of the instability of ionic-type solubilizing groups with large steric hindrance and alkaline solubilizing groups, neutral and nonionic hydrophilic groups were adopted in the next design and synthesis. To avoid the steric hindrance, grifolin reacted with succinic anhydride to generate a diester derivate (intermediate 8) under inert gas protection. Succinic anhydride also generates carboxylic acid terminals as reactive cohesive ends. Using these two small volume succinic acids as linkers, this modification strategy reduced the steric hindrance of the benzene ring and farnesyl. Then, we tried to install glycerol and alkamine on the cohesive ends by condensation reactions to produce prodrugs 9 and 10. PEG is a kind of linear, water-soluble and biocompatible molecule with low toxicity, immunogenicity and teratogenicity, which has been widely used in drug formula and prodrugs.21 Thus, we also tried to introduce PEG chains to the cohesive ends of intermediate 8 to form prodrug 11 (PEG5-Grifolin).
Next, we tested the solubility of 1, 9, 10 and PEG5-Grifolin by HPLC. The solubility of each compound might be affected significantly by different pH, because of the protonated form and formation of hydrogen bonds, especially for prodrug 10 which contained two amide bonds. Herein, PBS was used as the solvent to simulate the mild storage and administration condition. As shown in Table 1, compared with grifolin, all prodrugs in PBS have improved solubilities. Much to our delight, PEG5-Grifolin, containing two 5-PEG chains, was miscible with PBS at 25 °C. Like most glycol prodrug and polyoxyethylene ethers nonionic surfactants, PEG5-Grifolin separated out when warming up. This was possibly due to the broken hydrogen bonds between PEG5-Grifolin molecules and water molecules when the temperature rises. However, this feature would not affect the administration of PEG5-Grifolin in most situations.
| Compound | Solubility (μg ml−1) | Foldsc |
|---|---|---|
| a The solubility of grifolin is lower than the minimum concentration of standard linear calibration curve (0.32 μg ml−1). 0.2 μg ml−1 is calculated by the linear regression equation.b PEG5-Grifolin is miscible with water under room temperature.c Folds represented the multiple of prodrugs compared to the compound 1, grifolin. | ||
| Grifolin, 1 | <0.32a | — |
| 9 | 5.20 | 26 folds |
| 10 | 0.44 | 2.2 folds |
| PEG5-grifolin, 11 | Miscibleb | >1000 folds |
After obtaining the water-miscible PEG5-Grifolin, we tested its stability. PEG5-Grifolin was dissolved in PBS to prepare a 40 mM solution, followed by splitting into small aliquots. The concentration of PEG5-Grifolin in each aliquot which was placed at room temperature for different times was assessed by HPLC. Much to our delight, even at room temperature, the half-life period of the PEG5-Grifolin in PBS was expected to be about 87 days (Fig. S1A,† ESI). Meanwhile, no significant degradation was observed when PEG5-Grifolin in PBS was stored at −20 °C. Thus, the lower storage temperature is still more preferred to keep the stability. Additionally, we evaluated the stability profile of PEG5-Grifolin in mouse plasma model. PEG5-Grifolin was dissolved in fresh mice plasma, followed by HPLC analysis (Fig. S1B,† ESI). During the 6 h treatment, no significant degradation was observed. These data suggested PEG5-Grifolin is stable in normal solvent, storage conditions as well as plasma and could be an easily used agent.
Then, we evaluated the anti-proliferative activity of PEG5-Grifolin and 5-AD on representative non-cancerous cell lines (Beas2b cells and 293T cells) and NPC cell line (C666-1 cells) (Fig. 2 and Table S1,† ESI). The inhibitory curve (Fig. 2) and IC50 (Table S1,† ESI) proved that PEG5-Grifolin induced more potent growth inhibition on tumor cell line (C666-1) than on non-cancerous cell lines (293T and Beas2b). Meanwhile, NPC cell line C666-1 was not so sensitive to 5-AD treatment, while 5-AD still potently inhibited the growth of non-cancerous cell lines (293T and Beas2b). Consequently, the data revealed the improved inhibitory selectivity on non-cancerous cells and NPC cells and demonstrated the advantages of PEG5-Grifolin over 5-AD.
 | ||
| Fig. 2 The effect of PEG5-Grifolin and 5-AD on growth inhibition of tumor cells (C666-1) and non-cancerous cells (293T and Beas2b). (A) PEG5-Grifolin (B) 5-AD. (A and B) The growth inhibition assessment was conducted by CCK-8 method. The cells were seeded into 96-wells plates. Small molecules were diluted and added in the plates at indicated concentrations. The cells were treated for 3 days. The data are individual replicate values and shown as means ± s. d. (n = 3 for biologically independent samples per group). | ||
The anticancer activity of PEG5-Grifolin was measured in NPC xenograft mice model bearing C666-1 cells to further evaluate drug efficacy. 5-AD, the current DNMT1 inhibitor, was used as the positive control. As shown in Fig. S2† ESI, treatment with PEG5-Grifolin at a dose of 90 mg kg−1 resulted in a significant inhibition of tumor load compared to the vehicle group. However, 5-AD treatment had no evident tumor suppression at a dose of 1 mg kg−1 while showed a more severe toxic side-effect. During the experiment, two mice died in the 5-AD treatment group, while the toxicity of PEG5-Grifolin was not observed. The toxicity of 5-AD was also reported by previous literatures.24 Thus, the dosage of 5-AD was highly restricted. All the data indicated that PEG5-Grifolin might be a more effective and safer agent than 5-AD.
In order to clarify the underlying mechanism of anti-cancer effect, the expressions of DNMT1 and several TSGs in tumor tissue were identified by immunohistochemistry (IHC) after the mice were sacrificed (Fig. 3A). DAPK1, PTEN, CDH1 and p16 were chosen as representative TSGs, as DNMT1 contributes to their hypermethylation in promoter region and low expression. Consistent with the anti-cancer effects, the expression of DNMT1 was significantly suppressed by PEG5-Grifolin and 5-AD. Notably, PEG5-Grifolin treatment induced much more potent DNMT1 down-regulation than 5-AD did. Meanwhile, the expressions of DAPK1, PTEN, E-cadherin and p16 were substantially up-regulated compared with the control. These results indicate that the PEG5-Grifolin might act as a demethylation agent to inhibit tumor growth in vivo through reactivating a panel of TSGs expression.
 | ||
| Fig. 3 PEG5-Grifolin down-regulates DNMT1 to reactivate TSG expressions in vivo. Nude mice bearing C666-1 cells were randomly separated into 3 groups (n = 8) and treated with PBS (vehicle), PEG5-Grifolin (90 mg kg−1) or 5-azacytidine (1 mg kg−1) every other day for 17 days. (A) Images of tumor sections in each group stained with indicated antibodies. Antibody staining is in brown and nuclear counter staining is in blue. (B) Methylation of promoter region of DAPK1 and p16. Tumor tissue was extracted from the xenograft mice and analysed by BSP method. | ||
To further confirm that up-regulated expressions of TSGs are correlated with their promoter methylation status, bisulfite sequencing PCR (BSP) were performed to analyze the methylation at the promoter region of the representative DAPK1 and p16 genes. Genomic DNA was extracted from the tumor tissue of xenograft mice. Compared with the vehicle control, PEG5-Grifolin treatment resulted in 17% and 11% demethylation in DAPK1 and p16 promoter region, respectively. However, little demethylation effect has been observed with 5-AD treatment (Fig. 3B). The data revealed that PEG5-Grifolin suppressed tumor growth by DNMT1 down-regulation and consequently demethylation of TSGs. PEG5-Grifolin as a potent DNMT1 modulator could be a promising supplement to existing DNMT1 inhibitors.
The preliminary in vivo xenograft experiment demonstrated that PEG5-Grifolin exerted potent anti-tumor activity with low toxicity. However, single administration of 5-AD had no obvious effect on NPC tumor growth, which accorded with previous studies.25 The expressions of four representative TSGs, including DAPK1, PTEN, CDH1 and p16, were substantially increased after PEG5-Grifolin treatment. BSP assay further proved PEG5-Grifolin induced much more potent DNA demethylation of TSGs than 5-AD did. In general, compared with 5-AD, PEG5-Grifolin treatment exerts more effective demethylation and anti-tumor activity in NPC tumors. This reflects the importance of our research to develop such a modulator with high efficacy and low toxicity to expand the drug categories for DNMT1 down-regulation.
Up-regulation of DNMTs is a universal epigenetic alteration in tumors, which links CIMP, chromatin modification and the subsequent gene silencing, suggesting that DNMTs might serve as valuable targets for designing specific antitumor agents to reverse CIMP.8 The methylation status of p16 and DAPK1 genes in bodily fluids have been used for early detection of head and neck squamous cell carcinomas (HNSCCs).26 Urinary p16 and CDH1 methylation have been used for the detection of prostate cancer.27 p16 methylation detection has also been applied for the pathological typing of EBV-positive gastric cancer.28 CIMP of DAPK1, PTEN, CDH1 and p16 genes could represent a typical epigenetic change in cancers. Moreover, DNMT1 does not specifically regulate the four representative genes, but induces broad methylation changes of TSGs. Hence, this new-developed DNMT1 modulator might benefit much for various types of CIMP-associated cancer.
Up to date, the nucleoside analogues DNMT inhibitors, including Vidaza® (5-azacytidine), Dacogen® (decitabine or 5-aza-2′-deoxycytidine) and 5-fluoro-2′-deoxycytidine have been applied for various diseases. In addition to hematological neoplasms, DNMT inhibitors are also applied for solid tumors, such as head and neck, lung and breast cancers and metastatic melanoma. In most cases, DNMT inhibitors are used in combination with other anti-cancer drugs such as Oxaliplatin,29 Temozolomide (Temodar)1 and histone deacetylase inhibitor Vorinostat (Zolinza).30 Most recently, in a phase II study, CC-486 (oral azacitidine) did not show sufficient clinical activity as a monotherapy in locally advanced or metastatic NPC.31 The common feature of all these inhibitors is to induce DNMT loss-of-function. This further supports our study that PEG5-Grifolin exerts much better demethylation activity than 5-AD in an NPC model.31 PEG5-Grifolin, in combination with other conventional antitumor drugs, might have profound influence on clinic treatment of CIMP-associated cancers.
3. Conclusion
In this work, we developed a series of prodrugs of grifolin to improve the solubility and stability of grifolin. Among them, a representative compound, PEG5-Grifolin down-regulated DNMT1 to demethylate the promoter region of TSGs, resulting in the activation of the TSGs expression, thereby inhibiting the proliferation of NPC cells in vivo. Collectively, our finding suggests that PEG5-Grifolin can overcome the weaknesses of grifolin and 5-AD, and may serve as a potential demethylation agent for DNMT1 down-regulation.4. Experimental section
Materials and methods
All commercial materials (Adamas-beta, Bidepharmatech, Energy Chemical, etc.) were used without further purification. All solvents were analytical grade. The grifolin was prepared by the procedure reported by Justin T. Mohr in 2016.32 The 1H-NMR and 13C-NMR spectra were recorded on a Bruker AVANCEIII 400 MHz spectrometer in CDCl3 using solvent peak as a standard. All 13C-NMR spectra were recorded with complete proton decoupling. Normal-resolution mass spectral analyses were performed with Waters AQUITY UPLC-MS/MS. High-resolution mass spectral analyses were performed with Waters Xevo G2 QTof. Analytical TLC was performed on Yantai Chemical Industry Research Institute silica gel 60 F254 plates and flash column chromatography was performed on Qingdao Haiyang Chemical Co. Ltd silica gel 60 (200–300 mesh). 5-AD (Decitabine) was purchased from MedChemExpress. The reagents for genomic DNA extraction were purchased from QIAGEN.Compounds synthesis and characterization
The general procedures for compounds synthesis and NMR spectra could be found in ESI.†Antibody
The antibody for detecting P16 was from Proteintech (Illinois, NA, USA). The antibodies against PTEN and E-cadherin were purchased from Cell Signalling Technology (Danvers, MA, USA). The anti-DAPK1 was obtained Sigma-Aldrich (St. Louis, MO, USA). The antibody against DNMT1 was from Santa Cruz Biotechnology (Santa Cruz, CA, USA).Method of solubility test
The solubility test was conducted by HPLC method. First, a linear regression curve was established. Samples were weighted out in gradient and dissolved in acetonitrile. The concentration of each sample was calculated by the weight and the volume of acetonitrile. The peak area of each sample was measured by HPLC. Based on the concentrations and peak area, the linear regression curve was established.Then, excess amount of sample was dissolved in PBS buffer (pH = 7.4). The solution was shaken vigorously, followed by sonicated for 15 min at 25 °C. Then the solution was centrifuged under 6000g for 3 min. Then, the supernatant solution was passed through ultrafiltration membrane (0.45 μM, HY-RMF05). Next, the filtrate was analysed by HPLC. The peak area was substituted into the linear regression curve to get the concentration.
Method of stability test
PEG5-Grifolin was dissolved in PBS buffer (pH = 7.4) to get the 40 mM storage solution. This storage solution was separated in several small fraction packages in EP tubes (1.5 ml, ThermoFisher). These tubes were kept at −78 °C. Then, these EP tubes could warm to room temperature after indicated time. At every point of time, three tubes were taken out to room temperature. The EP tubes were marked with the take-out time. Finally, the new thawed storage solution and the solution in room temperature was analysed by HPLC. The area of product peak S0 in new thawed storage solution was set to 1. The other PE tubes were analysed by HPLC. S represent the area of the samples in room temperature. The figure was drawn with S/S0 as y axis and time as x axis. S/S0 determines the percentage of remaining percent of PEG5-Grifolin. The data was analysed by GraphPad Prism 7, exponential model.For the in vitro stability experiment in plasma, PEG5-Grifolin and internal standard substrate sulfanilamide was dissolved in new collected mice plasma. Then, the solutions were placed in 37 °C. After indicated time, the solutions were frozen with liquid nitrogen and stored at −80 °C until the HPLC analysis. For HPLC analysis, the plasma was injected into HPLC via a 20 μL needle and the peak area of sulfanilamide (A0) and PEG5-Grifolin (A1) were collected at each time point. The ratio A1/A0 represented the relative concentration of PEG5-Grifolin. The A1/A0 at the first time point was set to 100%.
Viability assessment experiment
The viability assessment experiment was performed referred to reported method.33 5000 cells in 50 μL corresponding culture medium were seeded into 96-wells plate. After two hours, 50 μL full culture medium containing double indicated concentration drug was supplement into each well. Then, the plates were incubated in cell incubator for 3 days, followed by adding 10 μL CCK-8 into each well. After 1–4 h treatment, the OD value of each well of 96-well plates was detected at 490 nm by SpectraMax Plus Microplate Reader. The data were analysed by GraphPad Prism 7 nonlinear regression curve fit.Cell line and cell culture
Thanks to the Department of Anatomy and Cellular Pathology, The Chinese University of Hong Kong for constructing and donating C666-1 cell. 293T and Beas2b cells were from National Collection of Authenticated Cell Cultures of China. The human nasopharyngeal carcinoma cell line C666-1 was grown in RPMI-1640 media (Gibco) supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS), 1% w/v glutamine and 1% w/v antibiotics and cultured at 37 °C in a humidified incubator containing 5% CO2. 293T and Beas2b cells were grown in DMEM media (Gibco) supplemented with 10% v/v heat-inactivated fetal bovine serum (FBS), 1% w/v glutamine and 1% w/v antibiotics and cultured at 37 °C in a humidified incubator containing 5% CO2.Tumor xenograft study
To study the inhibitory effect of PEG5-Grifolin on NPC in vivo, a 6 weeks xenograft experiment using C666-1 cells was performed by subcutaneously injecting 4 × 106 cells into 5 weeks-old female BALB/c nu/nu mice. After the tumor grew to about 80–100 mm3 (tumor volume was calculated according to the formula (V = length × width2/2)), the mice were randomly divided into three groups (n = 8, each group): untreated, intraperitoneally injected with PBS (vehicle), PEG5-Grifolin (90 mg kg−1) or 5-AD (1 mg kg−1) every other day. At the end of experiments, the mice were euthanized by CO2 inhalation and the tumors were stripped and weighed. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Xiangya hospital of Central South University, and approved by the Animal Ethics Committee of Xiangya hospital of Central South University.Immunohistochemical analysis
The tumor tissue sections were deparaffinized in environmentally friendly dewaxing agent (Solarbio, China) and rehydrated with an ethanol-aqueous solution of decreasing concentrations. For antigen retrieval, tissue sections were incubated in 10 mM sodium citrate buffer (pH = 6.0) for 20 min in a microwave oven. The endogenous peroxidase activity was removed by incubating with 3% hydrogen peroxide for 10 min and was blocked in normal donkey serum for 30 min. The primary antibodies (anti-DNMT1, anti-DAPK, anti-PTEN, anti-E-cadherin) were applied at 4 °C overnight. Chromogen was developed using DAB (Zsgbbio, China) and counterstained with hematoxylin staining kit. Immuno-histochemical staining of these sections was evaluated based on all of the available tumor cells or epithelial cells meeting the typical morphological criteria by 3 pathologists using the qualitative scale that is described in the literature.DNA bisulfite sequencing PCR
Genomic DNA was extracted using a Tissue DNA kit (QIAGEN, Inc., Hilden, Germany) and treated with bisulfate according to the EZ DNA Methylation-Gold™ Kit instruction manual (Zymo Research, USA). The promoter sequence was amplified from the isolated DNA using touchdown PCR, extracted from an agarose gel and loaded into the Puc18-T vector (Sangon Biotech Co., Ltd, Shanghai, China) for TA cloning and sequencing by Shanghai Sangon Biotech Company. The PCR conditions were as follows: 95 °C for 5 min; 35 cycles of 30 s at 94 °C, 30 s at 55 °C, and 50 s at 72 °C; and a final extension at 72 °C for 8 min. The BSP primer sequences for CpG region of DAPK1 were as follows: forward, 5′-TTTTTTAAAAAGTAAATAGGTGAGGT-3′ and reverse, 5′-CACCTCCAAAATTCAAATAATTC-3′. The BSP primer sequences for CpG region of p16 were as follows: forward, 5′-TTTGTAGTTAAGGGGGTAGGAGT-3′ and reverse, 5′-CTTTCCTACCTAATCTTCTAAAAAAC-3′. In detail, 10 individual clones from each group were selected and the number of detected methylated CpG sites was divided by the total 10 clones to evaluate the methylation percentage of each CpG site. The total methylation status of the core CpG region within the promoter from each group was calculated by averaging the methylation rate of each CpG site.Genomic DNA extraction
The tumor tissues were cut into small pieces (<25 mg) and placed in 1.5 ml centrifuged tubes. 20 μl proteinase K and 180 μl ATL buffer were added, followed by violent vortexing. The sample was incubated under 56 °C for 2 h. Then, 200 μl AL buffer was added to the tubes and mixed thoroughly by vortexing. After incubated under 70 °C for 10 min, 200 μl ethanol was added, followed by vortexing for 15 s and centrifuging. Then, the samples were transferred to QIAamp mini spin columns placed in a 2 ml collection tube. The samples were centrifuged at 6000g for 1 min. Discard flow-through and collection tube. Place the QIAamp mini spin column in a 2 ml tube. Add 500 μl AW1 and centrifuge at 6000g for 1 min. Discard flow-through and collection tube. Place the QIAamp mini spin column in a new 2 ml tube. Add 500 μl AW2 and centrifuge at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 3 min. Discard flow-through and collection tube. Place the QIAamp mini spin column in a 1.5 ml tube. Add 200 μl AE buffer and incubate for 1 min. Centrifuge at 6000g for 1 min to provide DNA samples for BSP analysis.
000g for 3 min. Discard flow-through and collection tube. Place the QIAamp mini spin column in a 1.5 ml tube. Add 200 μl AE buffer and incubate for 1 min. Centrifuge at 6000g for 1 min to provide DNA samples for BSP analysis.
Author contributions
L. W, Y. W and Z. L contributed equally to this work. X. L, Y. R and Y. C designed the experiments. L. W, X. L, Y. R and Y. C wrote the manuscript. T. L and W. L help organized the manuscript. L. W, Y. W, T. L and W. L designed and synthesized the molecules. X. Z and Z. L conducted the xenograft experiment and IHC experiment. X. Z and Z. L extracted DNA and conducted BSP experiment. F. S provided support for some experiments. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by grants from the Natural Science Foundation of China (81874172, 81874195), Open Funding of Key Laboratory of Carcinogenesis and Invasion of Chinese Ministry of Education (201701), Natural Science Foundation of Hunan Province (2020JJ4769) and the fellowship of China Postdoctoral Science Foundation (2021M701953).Notes and references
- Y. Watanabe and M. Maekawa, in Adv. Clin. Chem., ed. G. S. Makowski, Elsevier, 2010, vol. 52, pp. 145–167 Search PubMed.
- M. Toyota and J.-P. J. Issa, Semin. Cancer Biol., 1999, 9, 349–357 CrossRef CAS PubMed.
- R. Hino, H. Uozaki, N. Murakami, T. Ushiku, A. Shinozaki, S. Ishikawa, T. Morikawa, T. Nakaya, T. Sakatani, K. Takada and M. Fukayama, Cancer Res., 2009, 69, 2766–2774 CrossRef CAS PubMed.
- X. Ren, X. Yang, B. Cheng, X. Chen, T. Zhang, Q. He, B. Li, Y. Li, X. Tang, X. Wen, Q. Zhong, T. Kang, M. Zeng, N. Liu and J. Ma, Nat. Commun., 2017, 8, 14053 CrossRef CAS PubMed.
- X. Y. Ren, X. Wen, Y. Q. Li, J. Zhang, Q. M. He, X. J. Yang, X. R. Tang, Y. Q. Wang, P. P. Zhang, X. Z. Chen, B. Cheng, J. Ma and N. Liu, J. Exp. Clin. Cancer Res., 2018, 37, 227 CrossRef PubMed.
- Y. Li, X. Yang, X. Du, Y. Lei, Q. He, X. Hong, X. Tang, X. Wen, P. Zhang, Y. Sun, J. Zhang, Y. Wang, J. Ma and N. Liu, Clin. Cancer Res., 2018, 24, 6495–6508 CrossRef CAS PubMed.
- A. Shehzad, R. Shahzad and Y. S. Lee, Enzymes, 2014, 36, 149–174 CAS.
- W. Zhang and J. Xu, Biomark Res, 2017, 5, 1 CrossRef PubMed.
- B. Yang, in Cell and Tissue Based Molecular Pathology, ed. R. R. Tubbs and M. H. Stoler, Churchill Livingstone, Philadelphia, 2009, pp. 84–90, DOI:10.1016/b978-044306901-7.50014-6.
- C. Cheng, Z. Li, X. Zhao, C. Liao, J. Quan, A. M. Bode, Y. Cao and X. Luo, Eur. J. Pharmacol., 2020, 870, 172922 CrossRef CAS PubMed.
- C. Liao, M. Li, X. Li, N. Li, X. Zhao, X. Wang, Y. Song, J. Quan, C. Cheng, J. Liu, A. M. Bode, Y. Cao and X. Luo, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2020, 1865, 158540 CrossRef CAS PubMed.
- X. Luo, N. Li, X. Zhao, C. Liao, R. Ye, C. Cheng, Z. Xu, J. Quan, J. Liu and Y. Cao, J. Exp. Clin. Cancer Res., 2019, 38, 300 CrossRef PubMed.
- X. Zhao, J. Quan, Y. Tan, Y. Liu, C. Liao, Z. Li, W. Liao, J. Liu, Y. Cao and X. Luo, Am. J. Cancer Res., 2021, 11, 729–745 CAS.
- M. Fang, D. Chen and C. S. Yang, J. Nutr., 2007, 137, 223S–228S CrossRef CAS PubMed.
- D. Q. Luo, H. J. Shao, H. J. Zhu and J. K. Liu, Z. Naturforsch., C: J. Biosci., 2005, 60, 50–56 CrossRef CAS PubMed.
- M. Ye, J. K. Liu, Z. X. Lu, Y. Zhao, S. F. Liu, L. L. Li, M. Tan, X. X. Weng, W. Li and Y. Cao, FEBS Lett., 2005, 579, 3437–3443 CrossRef CAS PubMed.
- M. Ye, X. Luo, L. Li, Y. Shi, M. Tan, X. Weng, W. Li, J. Liu and Y. Cao, Cancer Lett., 2007, 258, 199–207 CrossRef CAS PubMed.
- X. J. Luo, L. L. Li, Q. P. Deng, X. F. Yu, L. F. Yang, F. J. Luo, L. B. Xiao, X. Y. Chen, M. Ye, J. K. Liu and Y. Cao, Eur. J. Cancer, 2011, 47, 316–325 CrossRef CAS PubMed.
- X. J. Luo, W. Li, L. F. Yang, X. F. Yu, L. B. Xiao, M. Tang, X. Dong, Q. P. Deng, A. M. Bode, J. K. Liu and Y. Cao, Eur. J. Pharmacol., 2011, 670, 427–434 CrossRef CAS PubMed.
- X. Luo, L. Yang, L. Xiao, X. Xia, X. Dong, J. Zhong, Y. Liu, N. Li, L. Chen, H. Li, W. Li, W. Liu, X. Yu, H. Chen, M. Tang, X. Weng, W. Yi, A. Bode, Z. Dong, J. Liu and Y. Cao, Oncotarget, 2015, 6, 42704–42716 CrossRef PubMed.
- S. S. Banerjee, N. Aher, R. Patil and J. Khandare, J. Drug Delivery, 2012, 2012, 103973 Search PubMed.
- M. Okada, K. Saito, C. P. Wong, C. Li, D. Wang, M. Iijima, F. Taura, F. Kurosaki, T. Awakawa and I. Abe, Org. Lett., 2017, 19, 3183–3186 CrossRef CAS PubMed.
- Y. Asakawa, T. Hashimoto, D. Ngoc Quang and M. Nukada, Heterocycles, 2005, 65 CrossRef.
- M. M. D. Hossain, A. Takashima, H. Nakayama and K. Doi, Exp. Toxicol. Pathol., 1997, 49, 201–206 CrossRef CAS PubMed.
- W. Jiang, Y. Q. Li, N. Liu, Y. Sun, Q. M. He, N. Jiang, Y. F. Xu, L. Chen and J. Ma, PLoS One, 2014, 9, e93273 CrossRef PubMed.
- D. A. Ovchinnikov, M. A. Cooper, P. Pandit, W. B. Coman, J. J. Cooper-White, P. Keith, E. J. Wolvetang, P. D. Slowey and C. Punyadeera, Transl. Oncol., 2012, 5, 321–326 CrossRef PubMed.
- J. Y. Park, Cancer Control, 2010, 17, 245–255 CrossRef PubMed.
- Q. N. Vo, J. Geradts, M. L. Gulley, D. A. Boudreau, J. C. Bravo and B. G. Schneider, J. Clin. Pathol., 2002, 55, 669–675 CrossRef CAS PubMed.
- M. Hosokawa, S. Tanaka, K. Ueda, S. Iwakawa and K. I. Ogawara, Biochem. Biophys. Res. Commun., 2019, 509, 249–254 CrossRef CAS PubMed.
- F. Tang, E. Choy, C. Tu, F. Hornicek and Z. Duan, Cancer Treat. Rev., 2017, 59, 33–45 CrossRef CAS PubMed.
- R. Mesia, P. Bossi, A. R. Hansen, C. Y. Hsieh, L. F. Licitra, E. H. Tan, P. Chen, J. Miller, L. L. Siu and R. I. Haddad, Eur. J. Cancer, 2019, 123, 138–145 CrossRef CAS PubMed.
- G. A. Grabovyi and J. T. Mohr, Org. Lett., 2016, 18, 5010–5013 CrossRef CAS PubMed.
- L. Wang, X. Shao, T. Zhong, Y. Wu, A. Xu, X. Sun, H. Gao, Y. Liu, T. Lan, Y. Tong, X. Tao, W. Du, W. Wang, Y. Chen, T. Li, X. Meng, H. Deng, B. Yang, Q. He, M. Ying and Y. Rao, Nat. Chem. Biol., 2021, 17, 567–575 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06648j |
| ‡ These authors made equal contributions to this work. |
| This journal is © The Royal Society of Chemistry 2021 |