
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSequence-specific recognition of a coding segment of human DACH1 gene via short pyrimidine/purine oligonucleotides†
Shoaib Khana,
Anju Singhb,
Nishu Naina,
Srishty Gulatia and
Shrikant Kukreti *a
*a
aNucleic Acid Research Laboratory, Department of Chemistry, University of Delhi, Delhi-110007, India. E-mail: skukreti@chemistry.du.ac.in
bDepartment of Chemistry, Ramjas College, University of Delhi, Delhi-110007, India
First published on 16th December 2021
Abstract
With growing in vivo evidence of the roles of triplexes in biological processes, oligonucleotide-directed targeting of double-helical DNA for selective modulation of gene functions has become imperative in their therapeutic aspects. This study comprises a comparative investigation of 17-mer Py- and Pu-TFO for the formation of an intermolecular triplex with a 27-bp genomic homopurine–homopyrimidine track present in the transcriptional element of the human DACH1 gene. The biochemical and biophysical studies have revealed that triplex formation takes place only with Py-TFO and not with its Pu-counterpart. Non-denaturating gel electrophoresis indicated the formation of an intermolecular triplex in Py-motif with an increasing amount of Py-TFO, whereas no such interaction was observed for the Pu-counterpart. UV-thermal melting (Tm), circular dichroism (CD) and thermal difference spectra (TDS) studies confirmed the pyrimidine motif triplex formation, which was observed to be significantly pH-dependent and stable at acidic pH (5.2) in the presence of 100 mM Na+ ions. Contrarily, Pu-TFO was not found to bind to the target predominantly, owing to its self-association properties. Further studies have revealed that the GA-rich Pu-TFO adopts a homoduplex structure leading to a limit in its availability for triplex formation. These results may add to our understanding of sequence-specific gene targeting and give insight into designing more specific TFOs depending on genomic targets.
Introduction
Enrichment of eukaryotic genomes in tracts of polypurine–polypyrimidine repeats confirmed its biological relevance. These repeats in genomic sequences make them capable of adopting multiple non-canonical structures depending on environmental conditions.1 In vivo existence of these structures undoubtedly guided therapeutics to develop more effective drugs. Beyond the existence of various supramolecular assemblies adopted by nucleic acids, the interest has continuously been growing in triple helical nucleic acids. Triple helical DNA structures have provided a brand new perspective to DNA-based gene regulation. The biological importance of triple-helical structures is associated with their functional involvement in genomic instability, gene silencing, and mutagenesis.2 The triplex formation has evolved as a novel targeting technology that brought the possibility of modulating gene functions such as gene expression, recombination, and gene repair, by forming sequence-specific triple helical structures. Triple helical structures are formed by the interaction of the third base with the exposed groups of purine of the Watson–Crick base pair through Hoogsteen-bonding or reverse Hoogsteen-binding.3 Triplexes are mainly classified as intramolecular and intermolecular triplexes. Intramolecular triplexes, or H-DNA, involve folding of a strand of the same duplex in polypyrimidine–polypurine mirror repeat sequences. On the other hand, intermolecular triplexes are formed when free triplex-forming oligonucleotides (TFOs) bind the target duplex through major groove. The recognition of triplex-forming oligonucleotides is sequence-specific and extended sequences can also be recognized with triplex-forming oligonucleotides in contrast to most recognizing agents such as polyamides, thus they have drawn much attention for their potential use in therapeutics.4 A schematic representation of base pairing involved in a triplex is depicted in Fig. 1.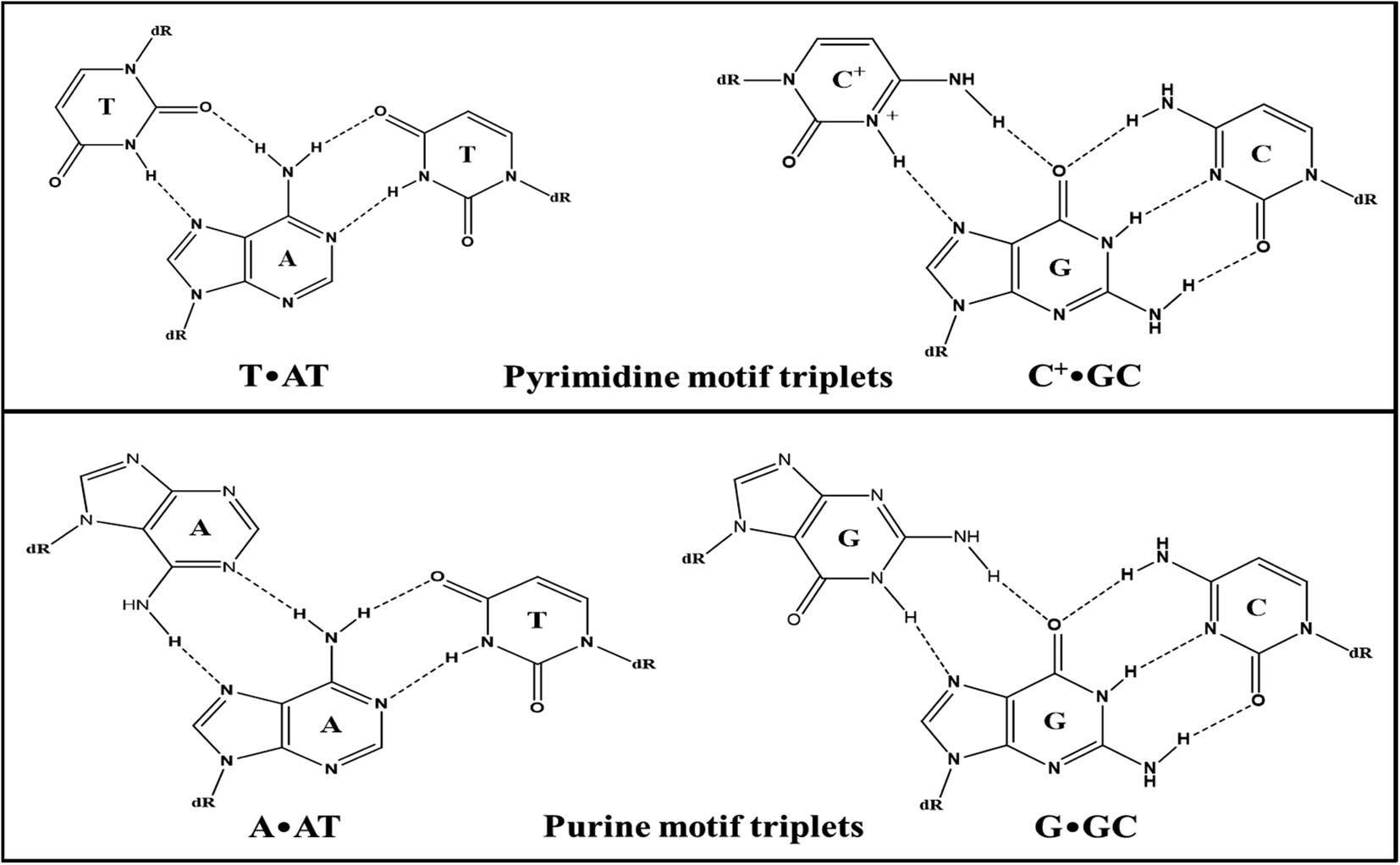 | ||
| Fig. 1 Possible base triplet schemes of Watson–Crick and Hoogsteen hydrogen bonding in a triplex. | ||
Triplex forming oligonucleotides can align either parallel or anti-parallel relative to the exposed purine strand of the target duplex giving rise to a parallel or antiparallel motif respectively.5 Parallel triplexes typically involve binding of pyrimidine-rich TFOs to the purine-rich strand via Hoogsteen-hydrogen bonds forming T·AT and C+·GC base triplets where thymine and protonated cytosine bind to AT and GC base pairs, respectively through Hoogsteen-bonding. The stability of these triplexes requires protonation of cytosine at N3 or substitution of cytosine with 5-methylcytosine6 while anti-parallel triplexes involve purine-rich sequences as TFOs binding through reverse Hoogsteen-bonds to form A·AT and G·GC base triplets. Moreover, binding of the G and T containing TFOs is independent of the orientation i.e., it can either form parallel or anti-parallel triplexes generating T·AT and G·GC base triplets.7 Though both types of triplexes are very significant for gene-targeting applications, they have restricted applications because of low stability and slow kinetics under physiological conditions. The biggest disadvantage with pyrimidine motif triplexes is that the triplet C+·GC requires cytosine protonation which makes them unavailable for canonical Watson–Crick pairing in physiological conditions. Nevertheless, in parallel triplexes, the triplets (C+·GC and T·AT) are more isomorphic than the triplets (T·AT & G·GC and A·AT & G·GC) in anti-parallel triplexes which reduce backbone distortion in the third strand of parallel triplexes making them structurally more stable than antiparallel triplexes.8 However, their stability depends on various other factors such as the position of triplets, length of the strands, and solution conditions.9 The triple-helical assembly is associated with increased charge repulsion. Thus triplexes are stabilized by the presence of multivalent cations particularly divalent cation (Mg2+).10,11 However, the poor stability of some of these structures might limit their use under physiological conditions. Specific ligands can interact/intercalate into DNA triple helices and stabilize them.12–15 Several efforts have been made to design and modify the third strand i.e., TFOs to improve the stability of the triple-helical structures under physiological conditions, but the formation of triplexes with natural oligonucleotides is still not well established.16–19
In the present study, we attempted to investigate the potential of a 27-bp homopurine–homopyrimidine genomic sequence for intermolecular triplex formation with pyrimidine (Py) and purine (Pu) TFOs in various solution conditions. The 27-mer purine-rich sequence studied here was identified to be present in the transcriptional region of the gene DACH1 on chromosome 13q21–q22. It encodes for dachshund family transcription factor 1. A chromatin-associated transcription factor encoding gene DACH1 is found to be widely expressed in various tissues and involved in important biological processes such as gene regulation, coactivator binding, and cell fate determination process.20–23 Selective silencing of DACH1 via triplex formation in a particular model cell type or tissue can aid in elucidating the tissue-specific physiological function of DACH1. Such an approach will enhance the understanding of disease pathophysiology resulting from loss of DACH1 expression. We have previously reported the possibility of the formation of an intramolecular (Pu-motif) triplex in the absence of Mg2+.24 Herein, we report that while the presence of Mg2+ is not mandatory for the formation of intermolecular parallel (Py-motif) triplexes, contrary to the requirement of Mg2+, it could not facilitate/induce the formation of antiparallel (Pu-motif) triplex structures. The tendency of purine-rich TFOs to form stable self-associate structures such as G-quadruplexes and GA-homoduplexes restricted the formation of purine motif triplexes, owing to their unavailability to bind to the duplex targets. This study might give more insight about designing of TFOs to genomic duplex targets, as physiological solution conditions.
Materials and methods
All DNA oligonucleotides used in this study were purchased from Helix Biosciences, Delhi, India. Oligonucleotides received in lyophilized powder form, were synthesized and HPLC purified on a micromolar scale and then stored at −20 °C conditions without further purification. The conventional nearest-neighbour approximation was applied to drive the extinction coefficient (ε) values, summarized in Table 1, for all the oligonucleotides used in the present study.25,26 These values were further used to determine the concentration of oligonucleotides spectrophotometrically by measuring the absorbance at 260 nm. The oligonucleotides in lyophilized powder form were dissolved directly in Milli-Q water to make the stock solutions. All analytical grade chemicals were used in mimicking the cellular physiological conditions.| S. no. | Oligonucleotide sequences | ε (M−1 cm−1) |
|---|---|---|
| 1 | 5′-GGAGGGGAAGGGGAAAAAAGGGGGGAG-3′ (SH27) | 297, 900 |
| 2 | 5′-CTCCCCCCTTTTTTCCCCTTCCCCTCC-3′ (SH27c) | 203, 600 |
| 3 | 5′-CCTTCCCCTTTTTTCCC-3′ (SH17Py) | 129, 800 |
| 4 | 5′-GGGAAAAAAGGGGAAGG-3′ (SH17Pu) | 190, 700 |
| 5 | 5′-CTTGAGCTCAAG-3′ (PAL12) | 113, 700 |
| 6 | 5′-CTTGAGCTTGAGCTCAAGCTCAAG-3′ (PAL24) | 226, 100 |
| 7 | 5′-GACTGACTTAAGCGCATAGCTAGCTAGCTCGATAGCTGA-3′ (M35) | 342, 600 |
| 8 | 5′-TCAGCTATCGAGCTAGCTAGCTATGCGCTTAAGTCAGTC-3′ (M35c) | 333, 100 |
Non-denaturating gel electrophoresis
The used DNA oligonucleotides were first checked for their purity by running gel electrophoresis in denaturating conditions on 20% polyacrylamide gel (PAGE) in 7 M urea. All oligomers exhibited a single band with appropriate size markers representing their purity. Non-denaturating gel electrophoresis was performed on 15% PAGE under native physiological conditions. To prepare the samples, oligonucleotides (desired concentration) were heated, in 20 mM sodium cacodylate buffer containing 100 mM Na+ (with 15 mM Mg2+ in case of Pu-TFO) and 0.1 mM EDTA, to a high temperature of ∼95 °C for 5 min. All the samples were allowed to cool slowly to room temp followed by overnight incubation at ∼4 °C. 15% PAGE prepared to contain 20 mM sodium cacodylate buffer with 100 mM Na+ (with 15 mM Mg2+ in case of Pu-TFO) and 0.1 mM EDTA was pre-equilibrated at ∼4 °C for few hours. Prior to loading the samples onto the gel, loading dye orange G was added to samples for tracking purposes. 15% gel was then loaded with samples and run in a cold environment (∼4 °C) with the help of a running buffer of 1× TBE containing the same amount of Na+ (and Mg2+ in case of Pu-TFO) and EDTA at a constant voltage of 65 V. Gel stained with stains-all solutions, was examined and imaged under trans-white light in Gel Dock by AlphaImager™ 2200 (Alpha Infotech Corp.).UV-thermal denaturation
UV-Vis thermal melting absorption spectra were recorded on a UV-1650PC Shimadzu spectrophotometer (Kyoto, Kyoto, Japan) equipped with a thermoprogrammer (TMSPC-8(E)-200). The thermal profiles were recorded at 260 nm and 295 nm where absorbance was measured as a function of temperature. The samples of desired concentrations were scanned from 20 °C to 95 °C using the quartz cuvettes of 1 cm optical path length and 1 mL volume capacity with a ramp rate of 0.5 °C min−1. The thermal melting temperature (Tm) values were derived from the first derivative curves of the melting profiles. The accuracy of the instrument was ±1 °C.Circular dichroism spectroscopy
CD spectra were recorded on a D-camphor sulfonic acid calibrated JASCO-815 spectropolarimeter equipped with a compatible computer. In order to eliminate any contamination present, the instrument was air cleaned by dry N2 purging. The buffer conditions were first scanned for a baseline at room temperature. In a stoppered quartz cuvette of path length 1 cm and volume 1 mL, the samples were scanned over a wavelength range of 200 to 320 nm. Three accumulative scans of the spectrum were collected and averaged for each sample at a scan rate of 100 nm min−1. The spectra were plotted in units of millidegrees with respect to wavelength (nm).Thermal differential spectra
UV-absorbance thermal difference spectra (TDS) were carried out for triplex formation on a UV-1800PC Shimadzu UV-visible spectrophotometer (equipped with a Peltier thermo-programmer TMSPC-8(E)-200) using a 10 mm path length quartz cuvette. The absorbance spectra were recorded at scan rate of 100 nm min−1 from 200–320 nm above and below melting temperature (Tm) of triplex formed. TDS was determined by normalising the arithmatic difference between the UV-absorbance at high temperature (first transition, triplex dissociation) and at low temperature (structured form). Accordingly, the graph was plotted in terms of normalised ΔAbs vs. wavelength (nm) as TDS.Result and discussion
Non-denaturating gel electrophoresis
Non-denaturating gel electrophoresis has always been a very sensitive and informative tool to determine the strandedness/molecularity and structural status of the oligonucleotides based on their differential mobility, under the influence of counter cations. In order to check the molecularity status of each sequence (SH27, SH27c, SH17Py, and SH17Pu), a 15% non-denaturating gel electrophoresis was carried out in 20 mM sodium cacodylate buffer containing 100 mM Na+ and 0.1 mM EDTA at physiological pH 7.4, is presented in Fig. 2. The molecularity of the structure formed was compared with control size markers PAL12, PAL24, M35, and M35 + M35c (duplex). Herein, PAL12 (lower band in lane 1) and PAL24 (upper band in lane 1) are 12-mer and 24-mer palindromic sequences respectively, that migrate as perfect duplexes, whereas M35 and M35c are the 35-mer single strand random markers complementary to each other. SH27, a 27-mer purine-rich sequence (lane 2), migrated slower than the PAL24 duplex indicating its dimeric status which can be assigned to a bimolecular species whereas 27-mer pyrimidine sequence, SH27c migrated almost equivalent to PAL12 duplex representing its unstructured single-strand status at physiological pH 7.4. On the other hand, SH17Py and SH17Pu both 17-mer sequences moved slower than PAL12 duplex but faster than M35 indicating the formation of dimeric species involving two strands in structure formation. | ||
| Fig. 2 15% native PAGE mobility pattern of oligonucleotide sequences: lane 1 → PAL12 + PAL24, lane 2 → SH27, lane 3 → SH17Pu, lane 4 → SH17Py, lane 5 → SH27c, lane 6 → M35 and lane 7 → M35 + M35c containing 20 mM sodium cacodylate buffer with 100 mM Na+ in 0.1 mM EDTA. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TFO = 1:0.25 → 1) incubated with the target duplex in 20 mM sodium cacodylate buffer (pH 5.7 and 7.4 respectively) containing 100 mM Na+ and 0.1 mM EDTA. The mobility of the structure formed was compared with control size markers PAL12, PAL24, and M35 + M35c (duplex). At pH 5.7, 17-mer Py-TFO (lane 2), migrated faster than the PAL12 duplex and exhibited a dispersed single band representing its unstructured single-stranded status. The duplex of SH27 and SH27c (lane 3) migrated slower than the duplex of PAL24, giving an intense single band. It is known that under slightly acidic conditions, cytosine of pyrimidine sequence get protonated at N3 position and thus Py-TFO bind to target duplex in a parallel fashion. Another band appears (upper band, lane 4) with the addition of TFO (SH17Py), which migrated retarded than the target duplex band (lower band, lane 4). The intensity of this band got improved with increasing concentration of Py-TFO (lane 4 → 8), in contrast to the gradually decreasing intensity of the duplex band and which disappeared finally after being converted to higher molecular weight triples structure. The upper band was assigned to the formation of structural species involving three strands, most possibly an intermolecular triplex. Interestingly, no such band was observed at pH 7.4, indicating no triplex formation. The additional dispersed band observed (lane 4 → 8), moving equivalent to SH17Py alone, is due to the leftover (residual) Py-TFO strand.
:TFO = 1:0.25 → 1) incubated with the target duplex in 20 mM sodium cacodylate buffer (pH 5.7 and 7.4 respectively) containing 100 mM Na+ and 0.1 mM EDTA. The mobility of the structure formed was compared with control size markers PAL12, PAL24, and M35 + M35c (duplex). At pH 5.7, 17-mer Py-TFO (lane 2), migrated faster than the PAL12 duplex and exhibited a dispersed single band representing its unstructured single-stranded status. The duplex of SH27 and SH27c (lane 3) migrated slower than the duplex of PAL24, giving an intense single band. It is known that under slightly acidic conditions, cytosine of pyrimidine sequence get protonated at N3 position and thus Py-TFO bind to target duplex in a parallel fashion. Another band appears (upper band, lane 4) with the addition of TFO (SH17Py), which migrated retarded than the target duplex band (lower band, lane 4). The intensity of this band got improved with increasing concentration of Py-TFO (lane 4 → 8), in contrast to the gradually decreasing intensity of the duplex band and which disappeared finally after being converted to higher molecular weight triples structure. The upper band was assigned to the formation of structural species involving three strands, most possibly an intermolecular triplex. Interestingly, no such band was observed at pH 7.4, indicating no triplex formation. The additional dispersed band observed (lane 4 → 8), moving equivalent to SH17Py alone, is due to the leftover (residual) Py-TFO strand.
 | ||
| Fig. 3 15% native electropherogram of target duplex with Py-TFO in 20 mM sodium cacodylate buffer with 100 mM Na+ and 0.1 mM EDTA: lane 1: PAL12 + PAL24, lane 2: SH17Py, lane 3: SH27 + SH27c (duplex), lane 4 → 8: duplex + SH17Py (1:0.25 → 2) and lane 9: M35 + M35c at (a) pH 5.7, (b) pH 7.4. | ||
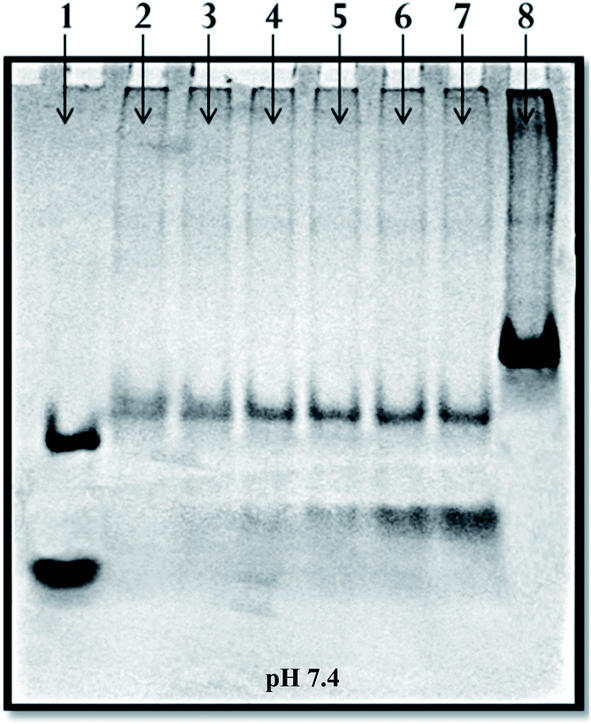 | ||
| Fig. 4 15% native electropherogram of target duplex with Pu-TFO in 20 mM sodium cacodylate buffer with 100 mM Na+, 15 mM Mg2+ and 0.1 mM EDTA at pH 7.4. Lane 1: PAL12 + PAL24, lane 2: SH27 + SH27c (duplex), lane 3 → 7: duplex + SH17Py (1:0.25 → 2) and lane 8: M35 + M35c. | ||
UV-thermal denaturation studies
Thermal denaturation study is employed to monitor the thermal stability as well as for determining the thermodynamics of the secondary structure of nucleic acids. UV-Vis thermal denaturation study monitors the increase in absorbance when a folded structure of DNA or RNA unfolds with a rise in temperature. A simple numerical value for this transition called temperature of melting (Tm) can be derived. Thermal melting was carried out to confirm the triplex formation by the target duplex with Py-TFO and further to check the thermal stability of the triplex formed. Fig. 5a displayed the absorbance versus temperature profile of the target duplex with Py-TFO sequence compared with that of duplex alone in 20 mM sodium cacodylate buffer (pH 5.7) containing 0.1 mM EDTA in the presence of 100 mM Na+. The melting profile of the target duplex-Py TFO complex was observed to be biphasic, in contrast to that of the monophasic curve of the free duplex. The biphasic nature of the curve can be assigned to the two-step dissociation of triplex, first the lower temperature transition, as dissociation of TFO strand that hybridizes with the target duplex and then dissociation of target duplex at higher temperature transition. Since, the pyrimidine rich TFO sequence contains about 50% cytosines, the formation of triplex predominantly depends on the cytosine protonation of Py-TFO. The stability of the triplex was further investigated at varied pH values. The Tm values derived from the first derivative curves, dA/dT vs. temp., (represented in insets) are presented in Table 2. The stability of the Py-motif triplex decreases significantly with an increase in solution pH, showing a Tm value of 40 °C at pH 5.2. The biphasic status disappears, leaving only the monophasic curve, as pH increases to physiological pH (7.4), representing almost no triplex formation at neutral pH (Fig. 5b). | ||
| Fig. 5 Thermal melting profile of target duplex with Py-TFO at (a) pH 5.7 (red for duplex with TFO and black for duplex alone) and (b) varied pH, in 20 mM sodium cacodylate buffer with 100 mM Na+ and 0.1 mM EDTA. | ||
| pH | Tm values (°C) |
|---|---|
| 5.2 | 40 |
| 5.7 | 33 |
| 6.2 | 30 |
| 7.4 | Not detected |
On the other hand, the thermal melting profile for the target duplex with Pu-TFO was observed to be monophasic indicating no triplex formation (Fig. 6a). The absence of triplex formation may be linked to the unavailability of Pu-TFO for triplex formation due to the self-association of G-rich purine-rich TFO. Meeting our expectations, the thermal melting of Pu-TFO alone exhibited an upright monophasic curve at 295 nm. The upright nature of the curve ruled out the possibility of quadruplex formation since the quadruplexes exhibit inverted melting curves,27 which rather indicates the possibility of formation of a homoduplex structure due to the presence of GA bases (Fig. 6b). The first derivative plot (dA/dT vs. temp.) shows that homoduplex structure is quiet stable.
 | ||
| Fig. 6 Thermal melting profiles of (a) target duplex with Pu-TFO (red for duplex with TFO and black for duplex alone) and (b) Pu-TFO alone (red curve represents dA/dT vs. temp.) in 100 mM Na+, 15 mM Mg2+ and 0.1 mM EDTA in sodium cacodylate buffer (pH 7.4). | ||
Circular dichroism study
Circular dichroism spectroscopy is a sensitive technique, employed to map the secondary structure conformation of nucleic acids. The interpretation of CD spectra is mainly empirical, based on the characteristic spectra exhibited by the reported DNA secondary structures. In order to confirm the formation of triplex suggested by thermal melting and gel electrophoresis, circular dichroism was carried out. The CD spectra for the target duplex with Py-TFO and duplex alone in 20 mM sodium cacodylate buffer (pH 5.7) containing 100 mM Na+ in 0.1 mM EDTA, is shown in Fig. 7a and b. CD spectrum for duplex alone exhibited a broad positive peak at 260–280 nm region and a negative peak of moderate-intensity around 240–245 nm assigned to Watson–Crick base pairing in the duplex. Furthermore, the CD spectrum for the target duplex with Py-TFO exhibited a strong peak at 275–280 and a negative peak around 240–245 nm assigned to Watson–Crick of the duplex. An additional negative peak at 210–220 nm was observed and assigned to Hoogsteen base pairing of Py-TFO with purine part of the target duplex in the intermolecular triplex.28 These results are in good correlation with gel and thermal melting studies confirming the intermolecular triplex formation of the 27-mer triplex forming sequence with Py-TFO. In order to check the stability of triplex formed, circular dichroism at varied pH was also carried out. The structures having Watson–Crick base pairing is manifested with a positive peak at around 275–280 nm and negative at 240–245 nm showed no significant change. However, the triplex signature peak at negative 212 nm gets stronger with decreasing pH. The higher ellipticity is associated with more triplex formation. The results confirmed the formation of a triplex at lower pH and are in accordance with that shown in thermal melting studies. | ||
| Fig. 7 CD spectra of target duplex with Py-TFO at (a) pH 5.7 and (b) varied pH, in 20 mM sodium cacodylate buffer with 100 mM Na+ and 0.1 mM EDTA. | ||
CD titration of target duplex with Pu-TFO is shown in Fig. 8a. The spectra exhibited an increase in the peak around 260–265 nm and 240–245 nm with the addition of Pu-TFO. However, no signature for triplex was observed supporting no triplex formation by Pu-TFO. Furthermore, CD spectra for Pu-TFO alone exhibited a signatory combination of a strong positive peak around 220 nm with a positive peak at 265 nm and negative peak at 245 nm confirming the possibility of homoduplex formation of 17-mer purine sequence29 as indicated by thermal melting (Fig. 8b).
 | ||
| Fig. 8 CD spectra of (a) target duplex with successive Pu-TFO addition and (b) Pu-TFO alone in 100 mM Na+, 15 mM Mg2+ and 0.1 mM EDTA in sodium cacodylate buffer (pH 7.4). | ||
Thermal differential spectra
UV-absorbance thermal difference spectra (TDS) is another complimentary and convenient approach to characterize nucleic acid structures. TDS is obtained from the arithmatic difference between the UV-absorbance of nucleic acids at higher temperature (unfolded form) and low temperature (folded form). This difference in absorbance when normalized and plotted provides a very unique signatory curve for each nucleic acid structural conformation. Mergny et al. have already reported thermal difference spectra for a variety of nucleic acid structures.30 TDS study for target duplex with Py-TFO in 20 mM sodium cacodylate buffer (pH 5.7) containing 100 mM Na+ in 0.1 mM EDTA was carried out to confirm/correlate the results obtained by the above mentioned techniques. The UV-absorbance spectra were recorded at 20 °C and 60 °C (as Tm for triplex was found to be 33 °C, already mentioned in UV-thermal denaturation study). Thermal difference spectrum (Fig. 9) exhibited a signatory combination of two maxima peaks at ∼240 nm and ∼270 nm along with one minimum peak at ∼295 nm compatible to the reported Py-motif triplex structures.30 This result validates the proposed findings for the formation of triplex of the target duplex with Py-TFO. | ||
| Fig. 9 Thermal differential spectra of target duplex with Py-TFO in 20 mM sodium cacodylate buffer (pH 5.7) with 100 mM Na+ and 0.1 mM EDTA. | ||
Proposed model for target duplex with TFOs
The results of the native gel electrophoresis, thermal melting, and circular dichroism studies were found to be in good correlation. Fig. 10 depicts the schematic representation of the intermolecular treatment of respective TFOs with target duplex. Py-TFO was reported to form an intermolecular triplex with nine C+·GC and eight T·AT triads. The extensive presence of the C+·GC triad reflects its pH dependence with greater stability at pH 5.2. In addition, the isomorphic nature of C+·GC and T·AT triads also helps in reducing backbone distortion thus stabilizing the triplex.8 | ||
| Fig. 10 Schematic representation of the proposed model of intermolecular triplex and homoduplex of used DNA sequences. | ||
In contrast, Pu-TFO has been found to form a stable self-associated homoduplex structure leaving the target duplex uninterrupted. The homoduplex comprises ten G·A and four G·G mismatch base pairs which provide higher stability to it even in the presence of three AA base-pair bulges. The proposed homoduplex is antiparallel in nature. The canonical Watson–Crick G·C involves three hydrogen bonds whereas G·A and G·G base pairs form two hydrogen bonds. Fig. 11 demonstrates the bonding pattern of mis-match base pairs. The stability of mis-match base pairs is well documented in literature which follows the order G·G > G·T > G·A respectively.31–36 It is also well established that Pu·Py mis-matches are more stable than Py·Py mis-match pairs.37 The G·GC and A·AT triads, owing to its non-isomorphic nature, reduce the possibility of triplex formation. However, the formation of G-quadruplex structure by SH27 cannot be ruled out in both cases because of the presence of consecutive guanine tracts in the sequence.
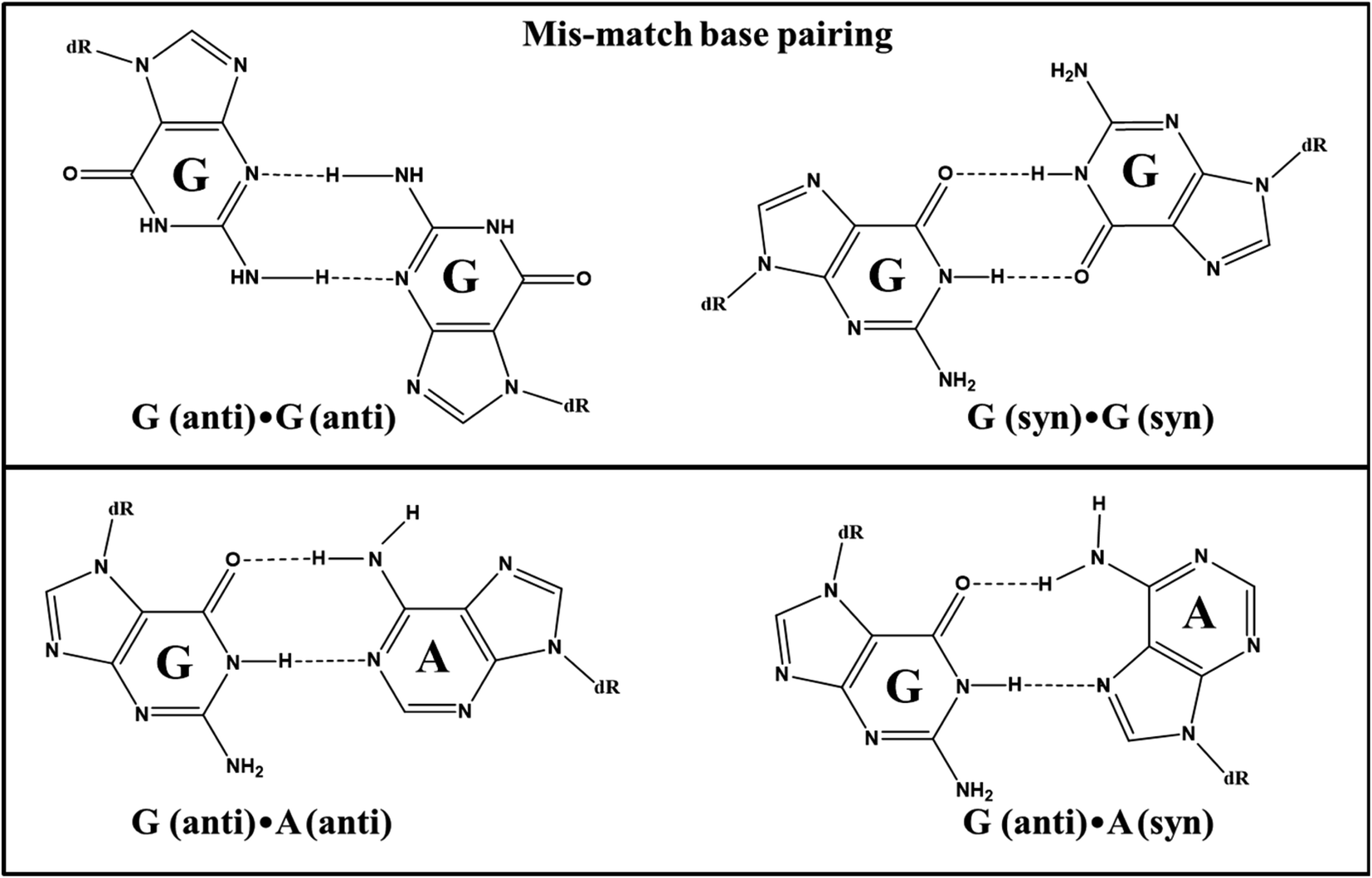 | ||
| Fig. 11 Possible G·G and G·A mis-match base pairing in homoduplex formed by 17Pu sequence. | ||
Biological significance
The polymorphic nature of DNA (ability to adopt multiple structures at specific genomic sites) affects various cellular processes reflecting the involvement of these structures as a target in the treatment of various diseases.38 The earlier therapeutic research relied on developing selective drugs that target only the pathological molecules responsible for the disease. New approaches focus more on targeting genes and blocking the synthesis of pathological proteins either by inhibiting the translation of mRNA (antisense therapy) or transcription of specific genes (antigene therapy).39 This prospect of oligonucleotide-based gene regulation has extensive implications in therapeutics, diagnostics, and molecular biology. One of the major advantages of DNA-based gene regulation (at the transcriptional site) is its efficiency unlike RNA or protein-based gene regulation where a large number of candidates need to be targeted.40 With the advancements in the sequencing of the human genome, we now have a better understanding of the involvement of genes in various important cellular processes that can provide us with more specific disease-related targeting of genes.41In triplex strategy, triplex forming oligonucleotides (TFOs) are designed to target double-helical genomic DNA in order to modulate functions of specific genes by forming a local triple-helical complex. In vitro applications of gene regulation by oligonucleotide-directed approach have been a success in the inhibition of genes.42–44 An excellent summary comprising of specific-site recognition of duplex and thus inhibition of gene expression using a variety of TFOs was published by the Vasquez group.45 The data accumulated suggest that the mammalian genome is evidently rich with potential TFO binding sites in the promoter and coding regions.45,46
The polypurine–polypyrimidine duplex regions are potential targets for TFOs leading to the formation of intermolecular triple helical structures and can be exploited for gene regulation. However, there are limitations with the triplexes to prevent its practical uses which comprise accessibility and unavailability of optimal binding sites, triplex stability, and dependency on solution conditions, TFO's delivery into cells, and finally its competence with DNA metabolic processes. Several chemical approaches have been used to modify TFOs in order to overcome these limitations such as base modification, sugar modification, and backbone modification.47
Over the years, it has been established that RNA molecule interacts with genomic DNA to form hybrid triple helix structures that play functional genomic roles in performing biological catalytic and regulatory activities.48 DNA–RNA triple helix has very recently suggested a potential auto-regulatory mechanism in vivo, by participating in feedback regulation of genes in cis.49 Thus, the focus is now on how these structures are functionally recognized and utilized in biological systems. Though triplex-based gene regulation mainly focused on transcription inhibition of genes however its use in direct gene inactivation through DNA damage has shown significant potential whether in recombination, mutagenesis, and DNA repair.50–52
The genomic 27-bp Pu·Py duplex target studied here is located in the transcriptional region of the DACH1 gene. This gene encodes for a chromatin-associated protein, DACH1 that with other transcription factors like c-Jun, estrogen receptor alpha, androgen receptor, plays a role in such important cellular activities as gene regulation and cell fate determination. Expression of this gene is associated with various diseases which include cancer, nephropathy, and diabetes, etc.53–56 Belonging to the Sno/Ski family of co-repressors,57 DACH1 protein was originally found to regulate eye development in Drosophila.58 Genetic association studies in human subjects and expression studies in DACH1 knockout mouse models and human tissues have revealed its role in tissue differentiation and organ development.59 Reduced DACH1 levels have been observed to be associated with poor prognosis and lower survival rate in breast cancer patients, tumor invasiveness in prostate and uterine cancer, etc. indicating its role as a tumor suppressor.57 Moreover, decreased expression of DACH1 was also seen in human renal biopsy samples from nephropathic patients, which suggested that DACH1 is involved in affecting disease severity of glomerular diseases.60 Additionally, a homozygous missense mutation in DACH1 in a patient with renal hypodysplasia (RHD) supports its role in RHD development.55 Being a crucial gene in the regulation of cell differentiation and cell fate determination, DACH1 needs to be explored in-depth to uncover its diverse functions in normal physiology. For this, the gene silencing approach to creating DACH1 knockdown in vitro/in vivo models can be efficient and promising.61–63 Selective silencing of DACH1 in a particular model cell type or tissue can aid in elucidating the tissue-specific physiological function of DACH1. Second, such an approach will enhance the understanding of disease pathophysiology resulting from loss of DACH1 expression. Third, it will help to unveil essential signaling pathways and other proteins interacting with DACH1. Hence, the studies related to the silencing of DACH1 may also provide a potential target protein, whose activation could compensate for the loss of DACH1 function, with further applications in the development of novel therapeutics.
Furthermore, Multiple Sequence Alignment (MSA) shows that the target 27-mer sequence is fully conserved in humans and other species like a chimpanzee and rhesus monkey. Further, using AliBaba (version 2.1) several transcription factor binding sites were revealed in the proximity of the studied target sequence. The presence of various transcription factors (i.e. Sp1, TBP, PU.1, etc.) found overlapping (ESI Fig. 2 and 3†), in the vicinity of the sequence, further adds to the importance of this gene segment. The proposed formation of triplex may interfere in the recognition of this sequence by the transcription factors, thus may silence the gene in a particular model cell type.
Present study can provide the potential of designed Py-, Pu-TFO sequences in controlled experimental conditions, but due to limitation, is still far from their implementation in vivo. The major limitations to this approach include TFO binding under physiologic conditions. Py-TFOs forming triplexes are pH sensitive as they require a protonated cytosine (C+·GC) for Hoogsteen hydrogen bond formation. Though Pu-TFOs bind well to the target in a pH-independent fashion, physiologic K+ concentrations can inhibit the binding in particularly G-rich TFOs. Self-association of G-rich oligomers (TFOs) may result in formation of homoduplexes or G-quadruplexes or other secondary structures.
Thus, TFO delivery and uptake into cells, lack of optimal target site binding affinity and specificity due to intracellular salt concentrations and pH, and chromatin structure which may present a barrier to target site accessibility.64 Moreover, in an intracellular environment, charge repulsion between the three polyanionic (duplex and TFO) strands and accessibility of binding sites in a chromosomal context can create obstacles to stable intermolecular triplex formation. To add to that TFO inside the cell, can also behave differently from its intended purpose of binding to target duplex i.e., TFOs exhibit aptameric properties where they bind transcription factors, such that they are not available to bind their duplex consensus sequences for transcription activation.65 The relevance of this study may not seem direct to gene regulation, but the results of the biophysical study presented here may help in understanding the behavior of sequences and cations in intermolecular triplex formation.
Conclusion
To sum up, the results obtained based on biochemical and biophysical studies for the structural status of 27-mer-purine rich sequence SH27 of DACH1 gene as target duplex for triplex formation by 17-mer Pu- and Py-TFOs are concluded here. The gel essay could demonstrate the formation of the triplex structure at DACH1 gene target sequence, only in Py-motif (using Py TFO), not in Pu-motif (using Pu TFO). Further, the triplex formation was also confirmed by UV melting studies, as the addition of 17Py-TFO displayed a biphasic melting profile of the TFO–duplex complex, indicating two-step dissociation phenomena. UV-thermal denaturation carried at varied pH also confirmed the triplex formation at acidic pH. No triplex formation was observed when targeted by Pu-TFO. A monophasic melting profile obtained for 17Pu-TFO alone, supported the self-associated status of this oligonucleotide, in solution. Since the nature of the curve is sigmoidal, unlike the well-reported inverted melting profile obtained generally for G-quadruplexes at 295 nm, the possibility of 17Pu-TFO to exist as a self-associated G-quadruplex structure is ruled out. Therefore, it was hypothesized that due to the presence of consecutive GA-tracts 17Pu-TFO tends to self-associate facilitating the formation of homoduplex structure, restricting it to be available for the triplex formation. The stability of this homoduplex structure is attributed to the presence of G·G and G·A mis-match pairs between two strands. Furthermore, CD studies revealed the secondary structure of the target duplex with both Py-, Pu-TFOs and confirmed the Py-motif triplex formation while no triplex signatures were attained for Pu-motif. Occurrence of the thermal difference spectra is also in accordance to this conclusion. CD studies are in good correlation with gel studies supporting pH dependency of the triplex formation. The present study proposes that a 27-nt Pu·Py DNA segment of the DACH1 gene adopts an intermolecular triplex with 17Py-TFO. In contrast, purine-rich (17Pu-TFO) was not able to target the said duplex, this failure of Pu-TFO not able to approach the target, can be attributed to the predominance of self-association of guanines at physiological conditions. It is also noteworthy that even the presence of Mg2+ could not facilitate/induce the formation of intermolecular Pu-motif triplex. However, an earlier report from the author's laboratory has demonstrated that Mg2+ is not mandatory for the intramolecular/unimolecular Pu-motif (antiparallel) triplex formation, which is not the case here. The substantial role of mis-matches is also shown in the formation of unusual structures i.e. homoduplex via non-Watson–Crick base pairing. Though this hypothesis is far from the in vivo picture, extensive studies are required to understand the complete status in detail. Undoubtedly, in vitro structural analysis will provide approximate information about the structural status of the studied oligonucleotides in physiological solution conditions. The formation and role of triplex structure in biological processes are still skeptical, we propose that this study may add more understanding about the triplex formation and pivotal role played by sticky guanines in obstruction of triplex formation. These studies bolster the formation and equilibrium of more unusual structures in a physiological condition which open new avenues for designing more specific TFOs, drugs, and therapeutics.Conflicts of interest
The authors declare that the publication of this research article does not contain any conflict of interest.Acknowledgements
The authors are thankful to the DU-DST/PURSE Grant (CD/2016/1761) from the University of Delhi, Delhi, and Shoaib also acknowledges the UGC for the JRF & SRF support.References
- M. Kaushik, S. Kaushik, K. Roy, A. Singh, S. Mahendru, M. Kumar and S. Kukreti, Biochem. Biophys. Rep., 2016, 5, 388–395 Search PubMed.
- F. A. Buske, J. S. Mattick and T. L. Bailey, RNA Biol., 2011, 8, 427–439 CrossRef CAS PubMed.
- K. R. Fox, Curr. Med. Chem., 2000, 7, 17–37 CrossRef CAS PubMed.
- J. J. Bissler, Front. Biosci., 2007, 12, 4536–4546 CrossRef CAS PubMed.
- V. N. Soyfer and V. N. Potaman, Triple-helical nucleic acids, Springer Science & Business Media, New York, 1996 Search PubMed.
- J. L. Asensio, A. N. Lane, J. Dhesi, S. Bergqvist and T. Brown, J. Mol. Biol., 1998, 275, 811–822 CrossRef CAS PubMed.
- S. P. Chandler and K. R. Fox, Biochemistry, 1996, 35, 15038–15048 CrossRef CAS PubMed.
- G. Goldsmith, T. Rathinavelan and N. Yathindra, PLoS One, 2016, 11, e0152102 CrossRef PubMed.
- C. N. Kunkler, J. P. Hulewicz, S. C. Hickman, M. C. Wang, P. J. McCown and J. A. Brown, Nucleic Acids Res., 2019, 47, 7213–7222 CrossRef CAS PubMed.
- N. Sugimoto, P. Wu, H. Hara and Y. Kawamoto, Biochemistry, 2001, 40, 9396–9405 CrossRef CAS PubMed.
- B. I. Kankia, Nucleic Acids Res., 2003, 31, 5101–5107 CrossRef CAS PubMed.
- S. Kukreti, J. S. Sun, T. Garestier, C. Helene, D. Loakes, D. M. Brown and E. Bisagni, Nucleic Acids Res., 1998, 26, 2179–2183 CrossRef CAS PubMed.
- C. Escudé, C. H. Nguyen, S. Kukreti, Y. Janin, J. S. Sun, E. Bisagni and C. Hélène, Proc. Natl. Acad. Sci., 1998, 95, 3591–3596 CrossRef PubMed.
- M. D. Keppler, P. L. James, S. Neidle, T. Brown and K. R. Fox, Eur. J. Biochem., 2003, 270, 4982–4992 CrossRef CAS PubMed.
- L. Strekowski, M. Hojjat, E. Wolinska, A. N. Parker, E. Paliakov, T. Gorecki and W. D. Wilson, Bioorg. Med. Chem. Lett., 2005, 15, 1097–1100 CrossRef CAS PubMed.
- H. Okamura, Y. Taniguchi and S. Sasaki, Org. Biomol. Chem., 2013, 11, 3918–3924 RSC.
- H. Okamura, Y. Taniguchi and S. Sasaki, Angew. Chem., Int. Ed., 2016, 128, 12633–12637 CrossRef.
- Y. Taniguchi and S. Sasaki, Development of Triplex Forming Oligonucleotide Including Artificial Nucleoside Analogues for the Antigene Strategy, in Synthesis of Therapeutic Oligonucleotides, ed. S. Obika and M. Sekine, Springer, Singapore, 2018, pp. 253–269 Search PubMed.
- Y. Taniguchi, M. Miyazaki, N. Matsueda, L. Wang, H. Okamura and S. Sasaki, Chem. Pharm. Bull., 2018, 66, 624–631 CrossRef CAS PubMed.
- K. Wu, S. Katiyar, A. Witkiewicz, A. Li, P. McCue, L. N. Song and R. G. Pestell, Cancer Res., 2009, 69, 3347–3355 CrossRef CAS PubMed.
- V. M. Popov, J. Zhou, L. A. Shirley, J. Quong, W. S. Yeow, J. A. Wright and R. Kumar, Cancer Res., 2009, 69, 5752–5760 CrossRef CAS PubMed.
- J. Zhou, Y. Liu, W. Zhang, V. M. Popov, M. Wang, N. Pattabiraman and C. Wang, J. Biol. Chem., 2010, 285, 40342–40350 CrossRef CAS PubMed.
- K. Wu, K. Chen, C. Wang, X. Jiao, L. Wang, J. Zhou and M. P. Lisanti, Cancer Res., 2014, 74, 829–839 CrossRef CAS PubMed.
- S. Kaushik, M. Kaushik, F. Svinarchuk, C. Malvy, S. Fermandjian and S. Kukreti, Biochemistry, 2011, 50, 4132–4142 CrossRef CAS PubMed.
- T. Xia, J. SantaLucia Jr, M. E. Burkard, R. Kierzek, S. J. Schroeder, X. Jiao and D. H. Turner, Biochemistry, 1998, 37, 14719–14735 CrossRef CAS PubMed.
- F. Tanaka, A. Kameda, M. Yamamoto and A. Ohuchi, Biochemistry, 2004, 43, 7143–7150 CrossRef CAS PubMed.
- J. L. Mergny, A. T. Phan and L. Lacroix, FEBS Lett., 1998, 435, 74–78 CrossRef CAS PubMed.
- J. Kypr, I. Kejnovská, D. Renčiuk and M. Vorlíčková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- H. Porumb, M. Monnot and S. Fermandjian, Electrophoresis, 2002, 23, 1013–1020 CrossRef CAS PubMed.
- J. L. Mergny, J. Li, L. Lacroix, S. Amrane and J. B. Chaires, Nucleic Acids Res., 2005, 33, e138 CrossRef PubMed.
- Y. Li, G. Zon and W. D. Wilson, Biochemistry, 1991, 30, 7566–7572 CrossRef CAS PubMed.
- S. H. Ke and R. M. Wartell, Nucleic Acids Res., 1993, 21, 5137–5143 CrossRef PubMed.
- E. M. Evertsz, K. Rippe and M. T. Jovin, Nucleic Acids Res., 1994, 22, 3293–3303 CrossRef CAS PubMed.
- N. G. Dolinnaya, A. Ulku and J. R. Fresco, Nucleic Acids Res., 1997, 25, 1100–1107 CrossRef CAS PubMed.
- O. I. Kaplan, B. Berber, N. Hekim and O. Doluca, Nucleic Acids Res., 2016, 44, 9083–9095 CAS.
- A. Singh and S. Kukreti, J. Biomol. Struct. Dyn., 2018, 36, 2773–2786 CrossRef CAS PubMed.
- C. R. Geyer, T. R. Battersby and S. A. Benner, Structure, 2003, 11, 1485–1498 CrossRef CAS PubMed.
- R. Thys, DNA Secondary Structure in Human Disease and Gene Regulation, PhD Thesis, Wake Forest University, North Carolina, 2015.
- M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS PubMed.
- D. A. Rusling and K. R. Fox, Methods, 2014, 67, 123–133 CrossRef CAS PubMed.
- O. Humbert, L. Davis and N. Maizels, Crit. Rev. Biochem. Mol. Biol., 2012, 47, 264–281 CrossRef CAS PubMed.
- M. M. Seidman and P. M. Glazer, J. Clin. Invest., 2003, 112, 487–494 CrossRef CAS PubMed.
- A. Jain, M. Magistri, S. Napoli, G. M. Carbone and C. V. Catapano, Biochimie, 2010, 92, 317–320 CrossRef CAS PubMed.
- K. Seio, K. Yamaguchi, A. Yamazaki, T. Kanamori and Y. Masaki, Nucleosides, Nucleotides Nucleic Acids, 2020, 39, 892–904 CrossRef CAS PubMed.
- Q. Wu, S. S. Gaddis, M. C. MacLeod, E. F. Walborg, H. D. Thames, J. DiGiovanni and K. M. Vasquez, Mol. Carcinog., 2007, 46, 15–23 CrossRef CAS PubMed.
- S. S. Gaddis, Q. Wu, H. D. Thames, J. Digiovanni, E. F. Walborg, M. C. MacLeod and K. M. Vasquez, Oligonucleotides, 2006, 16, 196–201 CrossRef CAS PubMed.
- K. R. Fox, T. Brown and D. A. Rusling, DNA Recognition by Parallel Triplex Formation, RSC, 2018, pp. 1–32 Search PubMed.
- A. Bacolla, G. Wang and K. M. Vasquez, PLoS Genet., 2015, 11, e1005696 CrossRef PubMed.
- Z. Zhou, K. E. Giles and G. Felsenfeld, Proc. Natl. Acad. Sci., 2019, 116, 6130–6139 CrossRef CAS PubMed.
- K. M. Vasquez, J. Christensen, L. Li, R. A. Finch and P. M. Glazer, Proc. Natl. Acad. Sci., 2002, 99, 5848–5853 CrossRef CAS PubMed.
- Q. Wu, L. A. Christensen, R. J. Legerski and K. M. Vasquez, EMBO Rep., 2005, 6, 551–557 CrossRef CAS PubMed.
- A. Mukherjee and K. M. Vasquez, Biochimie, 2011, 93, 1197–1208 CrossRef CAS PubMed.
- K. Wu, S. Katiyar, A. Li, M. Liu, X. Ju, V. M. Popov and R. G. Pestell, Proc. Natl. Acad. Sci., 2008, 105, 6924–6929 CrossRef CAS PubMed.
- A. Watanabe, H. Ogiwara, S. Ehata, A. Mukasa, S. Ishikawa, D. Maeda and M. Fukayama, Proc. Natl. Acad. Sci., 2011, 108, 12384–12389 CrossRef CAS PubMed.
- R. Schild, T. Knüppel, M. Konrad, C. Bergmann, A. Trautmann, M. J. Kemper and D. E. Müller-Wiefel, Nephrol., Dial., Transplant., 2013, 28, 227–232 CrossRef CAS PubMed.
- A. H. Chang, B. C. Raftrey, G. D'Amato, V. N. Surya, A. Poduri, H. I. Chen and K. Red-Horse, Genes Dev., 2017, 31, 1308–1324 CrossRef CAS PubMed.
- V. M. Popov, K. Wu, J. Zhou, M. J. Powell, G. Mardon, C. Wang and R. G. Pestell, Trends Endocrinol. Metab., 2010, 21, 41–49 CrossRef CAS PubMed.
- T. A. Heanue, R. J. Davis, D. H. Rowitch, A. Kispert, A. P. McMahon, G. Mardon and C. J. Tabin, Mech. Dev., 2002, 111, 75–87 CrossRef CAS PubMed.
- X. Zheng, Q. Liu, M. Yi, S. Qin and K. Wu, OncoTargets Ther., 2018, 11, 6479–6487 CrossRef CAS PubMed.
- Q. Q. Liu, Y. Q. Zhou, H. Q. Liu, W. H. Qiu, H. Liu, T. Y. Hu and K. M. Wu, Oncotarget, 2016, 7, 86547–86560 CrossRef PubMed.
- J. Harborth, S. M. Elbashir, K. Bechert, T. Tuschl and K. Weber, J. Cell Sci., 2001, 114, 4557–4565 CrossRef CAS PubMed.
- S. D. Buckingham, B. Esmaeili, M. Wood and D. B. Sattelle, Hum. Mol. Genet., 2004, 13, R275–R288 CrossRef CAS PubMed.
- T. Miwa, R. Minoda, Y. Ishikawa, T. Kajii, Y. Orita and T. Ohyama, Biol. Open, 2019, 8, bio043612 CrossRef CAS PubMed.
- A. Jain, G. Wang and K. M. Vasquez, Biochimie, 2008, 90, 1117–1130 CrossRef CAS PubMed.
- M. Borgatti, I. Lampronti, A. Romanelli, C. Pedone, M. Saviano, N. Bianchi and R. Gambari, J. Biol. Chem., 2003, 278, 7500–7509 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06604h |
| This journal is © The Royal Society of Chemistry 2021 |