
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking thiacalix[4]arene into pieces – a novel synthetic approach to higher calixarenes bearing mixed (–S–, –CH2–) bridges†
Lukáš Kaiser a,
Tomáš Landovskýa,
Karolína Salvadorib,
Václav Eignerc,
Hana Dvořákovád and
Pavel Lhoták*a
a,
Tomáš Landovskýa,
Karolína Salvadorib,
Václav Eignerc,
Hana Dvořákovád and
Pavel Lhoták*a
aDepartment of Organic Chemistry, University of Chemistry and Technology, Prague (UCTP), Technická 5, 166 28 Prague 6, Czech Republic. E-mail: lhotakp@vscht.cz
bDepartment of Physical Chemistry, UCTP, 166 28 Prague 6, Czech Republic
cDepartment of Solid State Chemistry, UCTP, 166 28 Prague 6, Czech Republic
dLaboratory of NMR Spectroscopy, UCTP, 166 28 Prague 6, Czech Republic
First published on 17th November 2021
Abstract
A novel approach to calix[5–7]arenes possessing mixed (S and CH2) bridges within the skeleton is based on the reaction of thiacalix[4]arene monosulfoxide with BuLi leading to a linear phenolic tetramer in essentially quantitative yield. This key intermediate is then cyclized with suitable building blocks to give macrocyclic calixarene analogues. Compared to the traditional stepwise construction of similar systems, this procedure based on thiacalixarene cleavage represents a scalable, robust, and straightforward synthesis and enables the preparation of larger calixarenes on a gram scale. As shown by 1H NMR and UV-vis titration experiments, the mixed-bridge calix[7]arene is able to recognize fullerenes C60 and C70, thus showing possible applications of such systems. The structures of the mixed bridge systems were confirmed by single crystal X-ray analysis, and the behavior of novel macrocyclic skeletons in solution was studied using dynamic NMR techniques.
Introduction
Calix[n]arenes1 are a well-established family of macrocyclic compounds due to their preorganization and easy derivatization, and are frequently used as molecular scaffolds in the design and synthesis of more complex supramolecular systems, including various receptors. The introduction of sulfur instead of methylene bridges has opened2 many new possibilities, as the chemistry of so-called thiacalixarenes offers many new features compared to “classical” calixarenes.As recently documented,3 the presence of heteroatoms brings about different complexation properties, novel types of chemical transformations unknown for pristine macrocycles, and different conformational preferences leading to 3D structures/conformations and/or substitution patterns which are otherwise hardly accessible for classical calixarenes. All these features make thiacalixarene an attractive platform for applications in supramolecular chemistry.
In this context, it seems interesting to study macrocycles formed by “interbreeding” the two parent systems – calixarene and thiacalixarene. Structures having both –S– and –CH2– bridges could, on the one hand, retain some of the characteristics of the parent systems, on the other hand, exhibit new behavior compared to the originals, offering thus new applications.
While the chemistry of calixarenes and thiacalixarenes is already well-established, mixed bridge systems remain almost untouched in the literature, mainly because of their hard accessibility. For the time being, the preparation of calix[4]arene analogues bearing one, two, or three sulfur bridges using the stepwise construction of linear tetramers that were finally cyclized (Scheme 1) was reported.4
 | ||
| Scheme 1 Various approaches to mixed (S/CH2) bridge systems. | ||
Alternative approach is based on the cyclization of bisphenolic building blocks with formaldehyde, leading to a mixture of tetrameric, hexameric, or octameric condensation products.5 Recently, our group succeeded with simple and scalable fragment condensation synthesis6 of 2,14-dithiacalix[4]arene possessing the alternating –S– and –CH2– bridges, which enabled the study of basic properties of this analogue.7
Unfortunately, the above-mentioned procedures rely on the electrophilic reaction of phenols with SCl2, which is the key reagent in all these approaches. While just a few years ago it was a common commercially available chemical sold by, e.g.: Aldrich company, recently, this compound became unavailable due to significant safety concerns regarding its possible use in the preparation of chemical warfare agents (sulfur mustard = yperite). Hence, as a direct consequence of the worldwide counter terrorism measures, the above approaches currently cannot be used because of the total unavailability of the starting compound.
In this contribution, we report an entirely novel approach overcoming the above-mentioned difficulties, and moreover, paving the path to so far inaccessible bigger calixarene systems having mixed bridges (S and CH2) within the basic macrocyclic skeleton.
Results and discussion
The whole synthetic procedure starts with pristine thiacalix[4]arene 1 (Scheme 2), which is easily accessible in 100 g scale using the condensation of p-tert-butylphenol and sulfur under basic conditions.2 Subsequent alkylation with methyl iodide8 to the corresponding methoxy derivative 2 and selective monooxidation (NaBO3) leads to monosulfoxide9 3 in good yield after column chromatography on silica gel. | ||
| Scheme 2 Synthesis of bigger calixarenes bearing mixed bridges (S and CH2) starting from thiacalix[4]arene. | ||
The key step is based on the recently discovered unexpected reactivity of thiacalixarene monosulfoxides with organolithium compounds.10 It was shown that irrespective of the conformation of the starting sulfoxide the macrocycle can be cleaved via a ligand exchange mechanism yielding linear thiacalixarene analogues which would be otherwise hardly obtainable. Thus, the reaction of compound 3 with an excess of n-BuLi (10 equiv.) in THF at −78 °C and subsequent quenching with MeOH provided a linear tetramer 4 (Scheme 2) in 90% yield and dibutyl sulfoxide as a byproduct. Methoxy derivative 4 was subjected to demethylation using BBr3, thus leading to tetraphenol 5 in excellent yield (94%). The use of Me3SiI as the dealkylation agent did not result into complete dealkylation.
In fact, tetramer 5 represents a key intermediate, which can be used for further synthesis of the mixed-bridge systems. So far, it has only been prepared via time-consuming stepwise procedure (as shown in Scheme 1) with very low overall yield,11 which hindered the preparation of compound 5 on a gram scale. In contrast, the procedure shown in Scheme 2 allows the preparation of 5 on a large scale, and, except for sulfoxide formation step, without any chromatographic separation.
The condensation reactions of 5 with suitable building blocks were carried out in chloroform where all reactants exhibited good solubility. Thus, the reaction of 5 with 2,6-bis(hydroxymethyl)-p-tert-butylphenol 6 (ref. 12) with PTSA under high-dilution conditions led to calix[5]arene derivative 8 in 47% yield (Scheme 2). Similarly, condensation of 5 with bisphenol 7 (ref. 7) provided calix[6]arene derivative13 9 in 32% yield.
The structures of products 8 and 9 were confirmed by HRMS ESI+ analysis, which excluded the formation of higher macrocycles (e.g.: 2 + 2 products). Thus, macrocycle 8 showed the most intense signals at m/z = 887.3808 and 903.3548, which is in good agreement with the calculated values 887.3805 and 903.3538 for the [M + Na]+ and [M + K]+ ions. Similar agreement was found also for 9 (found: 1067.4417 vs. calc.: 1067.4417) for [M + Na]+ species.
The 1H NMR spectrum of 8 (CDCl3, 298 K) reflects the expected Cs symmetry of the compound showing three singlets of OH groups A, C, and B (Scheme 2) at 8.31, 8.25, and 8.16 ppm with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 intensity ratio, respectively (Fig. 1). The shape of the signal of the methylene bridges – singlet at 3.85 ppm – indicates a fast conformational motion of the molecule at room temperature (time averaged signal).
:1:2 intensity ratio, respectively (Fig. 1). The shape of the signal of the methylene bridges – singlet at 3.85 ppm – indicates a fast conformational motion of the molecule at room temperature (time averaged signal).
 | ||
| Fig. 1 The partial 1H NMR spectra of 8 (phenolic OH and CH2 area) at various temperatures (CD2Cl2, 500 MHz) reflecting the cone–cone interconversion and the flip-flop motion of the circular HB array on the lower rim of calixarene. | ||
As shown by the dynamic NMR study (298–153 K), cooling CD2Cl2 solution of 8 (Fig. 1) resulted in the extensive broadening of the CH2 singlet, leading finally to the disappearance at around 225 K (coalescence temperature). Further cooling resulted in the appearance of two new broad signals at 3.60 and 4.10 ppm (203 K) resembling the axial and equatorial signals for the cone conformer of calix[5]arene derivatives.14 This phenomenon could be explained as a fast cone – inverted cone interconversion15 of the two identical cone conformations held by hydrogen bond array on the lower rim. The corresponding activation free energy (ΔG*) was calculated using Eyring equations for the rate constant k:
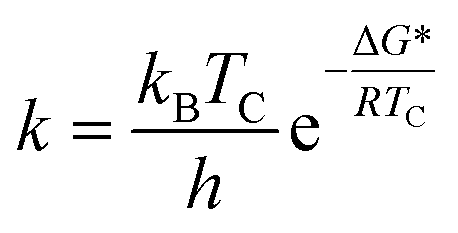
Remarkably, further lowering the temperature down to 153 K led to the appearance of five different main signals of the OH groups (7.59, 8.62, 9.00, 9.54, and 10.05 ppm). This observation can be ascribed to the flip-flop motion of the circular hydrogen bond array at the lower rim of 8. The orientation of HB array (clockwise vs. anti-clockwise) makes the system chiral, and originally equivalent signals of the OH protons (3 signals under fast exchange conditions) become inequivalent under slow exchange conditions (Fig. 1). The presence of other small peaks in the OH area of the spectrum suggests that there are probably some other minor conformations besides the cone in the equilibrium.
The single crystal X-ray diffraction analysis revealed that compound 8 crystallized in a triclinic crystal system, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Fig. 2) as a complex with EtOH and H2O. The macrocycle adopted the cone conformation held by two circular hydrogen bonds between phenolic OH groups (O–H⋯O distances are 2.035 and 2.039 Å) completed by several HBs between OH, EtOH and H2O (from 1.863 Å for EtO–H⋯OH2 interaction to 2.308 Å for H–O–H⋯OH bonding). Moreover, the overall supramolecular motif is complemented by the S⋯O (3.304 Å) and S⋯H–O (2.854 Å) close contacts (Fig. 2).
(Fig. 2) as a complex with EtOH and H2O. The macrocycle adopted the cone conformation held by two circular hydrogen bonds between phenolic OH groups (O–H⋯O distances are 2.035 and 2.039 Å) completed by several HBs between OH, EtOH and H2O (from 1.863 Å for EtO–H⋯OH2 interaction to 2.308 Å for H–O–H⋯OH bonding). Moreover, the overall supramolecular motif is complemented by the S⋯O (3.304 Å) and S⋯H–O (2.854 Å) close contacts (Fig. 2).
 | ||
| Fig. 2 Single crystal X-ray structures of compound 8·EtOH·H2O: (a) top-view showing the array of HBs, (b) side-view (interacting atoms shown as balls for better clarity). | ||
Compound 9 has crystallized in triclinic system in space group P as a complex with DMSO (see ESI Fig. 57†). Only half of the calix[6]arene molecule is present in the asymmetric unit, resulting in complete disorder of methylene and sulfur bridges. The solvent is disordered over two positions, with both positions held by two hydrogen bonds between OH groups and oxygen atom of DMSO with distances 1.936 Å and 2.085 Å for main positions and 2.037 Å and 2.041 Å. The remaining hydroxyl group forms intramolecular hydrogen bond with one of the solvent binding hydroxyl groups with distance of 2.075 Å.
To prepare bigger calixarenes while showing a general use of our synthetic approach, tetramer 5 was reacted with trisphenols 10 (ref. 16) and 11 (ref. 17) to provide the corresponding calix[7]arenes 12 and 13 in 16 and 31% yields, respectively (Scheme 2).
It is well known that higher calixarenes can form complexes with fullerenes based on a mutual size and shape complementarity (concave vs. convex).18 Derivative 13 was selected for the complexation study using 1H NMR titration experiments with C60 fullerene in toluene-d8. The addition of C60 led to a reasonable change of the NMR spectrum, particularly in the aromatic range, shifting the signals towards higher field (CIS ≈ 0.1 ppm) as shown in ESI (Fig. 52†). To avoid the problems associated with overlapping of the aromatic signals the CH2 bridges were selected for building the binding isotherm. Surprisingly, the shape of the titration curve indicated that the stoichiometry of the complex formed did not correspond to a simple 1:1 model. There are two plateaus indicating the more complex behavior which was confirmed also by Job plot analysis (Fig. 3) reflecting the 2:1 stoichiometry (C60:13).
 | ||
| Fig. 3 The complexation induced chemical shifts of the CH2 signals in 1H NMR spectra of 13 upon addition of C60. Upper inset: the corresponding titration curve (toluene-d8, 298 K, 400 MHz); lower inset: the Job plot of the same system. | ||
To shed more light into the stoichiometry, we have carried out the UV-vis titration in toluene to achieve a higher C60/13 ratio. The titration was carried out in toluene with a constant concentration of fullerene. To avoid the influence of self-absorption of 13 (λmax = 290 nm) the region of wavelengths >400 nm was chosen to build the binding isotherms. Fitting procedure using program Bindfit19 confirmed the 2:1 stoichiometry with an overall complexation constant β = 4900 (K1 × K2). Interestingly, the same conditions used for the complexation of C70 resulted in the expected 1:1 stoichiometry with the corresponding association constant K = 3100.
We also tried to extend this methodology to even larger calix[8]arenes derivatives. Tetramer 5, therefore, was hydroxymethylated by standard procedure4 (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/KOH) to provide disubstituted derivative 14 in 59% yield (Scheme 3). The condensation of building blocks 5 and 14 under high dilution conditions, however, did not lead to expected product 16, but rather, homooxacali[4]arene 17 was isolated as the only product (95% yield) of self-condensation20 of 14. The condensation in the presence of C60, to use the possible template effect of the fullerene ball, had no success as well since only 14 was isolated again as a single product. To overcome this issue, we employed mono(hydroxymethyl) compound 15 (ref. 4) (a byproduct in the preparation of 14) as it cannot provide the homooxa product. The attempted self-dimerization of 15 resulted in the exclusive formation of calix[4]arene derivative 18 (90% yield) representing the self-(mono)condensation product.
O/KOH) to provide disubstituted derivative 14 in 59% yield (Scheme 3). The condensation of building blocks 5 and 14 under high dilution conditions, however, did not lead to expected product 16, but rather, homooxacali[4]arene 17 was isolated as the only product (95% yield) of self-condensation20 of 14. The condensation in the presence of C60, to use the possible template effect of the fullerene ball, had no success as well since only 14 was isolated again as a single product. To overcome this issue, we employed mono(hydroxymethyl) compound 15 (ref. 4) (a byproduct in the preparation of 14) as it cannot provide the homooxa product. The attempted self-dimerization of 15 resulted in the exclusive formation of calix[4]arene derivative 18 (90% yield) representing the self-(mono)condensation product.
 | ||
| Scheme 3 Attempted synthesis of calix[8]arene derivative. | ||
Conclusion
In conclusion, a novel approach to calix[5–7]arenes having mixed (S and CH2) bridges within the skeleton is based on the reaction of thiacalix[4]arene monosulfoxide with BuLi leading to a linear phenolic tetramer in an essentially quantitative yield. This key intermediate is then cyclized with suitable building blocks to give macrocyclic calixarene analogues. Compared to the traditional stepwise construction of similar systems, this procedure based on thiacalixarene cleavage represents a scalable, robust, and straightforward synthesis enabling the preparation of larger calixarenes in a gram scale. As shown by titration experiments the mixed-bridge calix[7]arene 13 is able to recognize fullerenes C60 and C70 in solution, with remarkable differences in the stoichiometry of the corresponding complexes, thus indicating possible applications of such systems.Experimental
General experimental procedures
All chemicals were purchased from commercial sources, and used without further purification. Chloroform, tetrahydrofuran and dichloromethane used for the reactions were dried with CaH2 or MgSO4 and stored over molecular sieves. Melting points were measured on a Heiztisch Mikroskop-Polytherm A (Wagner & Munz, Germany). The IR spectra were measured on a FT-IR spectrometer Nicolet 6700 combined with an ATR cell GladiATR (PIKE). NMR spectra were recorded on Agilent 400-MR DDR2 (1H, 400 MHz; 13C, 100 MHz), Bruker Avance DRX 500 (1H, 500 MHz; 13C, 125 MHz), and Bruker 600 Avance III (1H, 600.1 MHz; 13C, 150.9 MHz) spectrometers at 298 K. Structural assignments were made with additional information from gCOSY, gHMQC, and gHMBC experiments. The mass analyses were performed using the ESI technique on a FT-MS (LTQ Orbitrap Velos) spectrometer. Column chromatography was performed on silica gel 60 with particle sizes 0.063–0.200 mm (Merck). Radial thin layer chromatography was carried out using Chromatotron on glass disks covered by silica gel 60 PF254 containing CaSO4 (Merck). Purity of the substances and courses of the reactions were monitored by TLC using TLC aluminium sheets with Silica gel 60 F254 (Merck) and analysed at 254 nm.Compound 2 was prepared according to the described procedure.8
:1 v/v) as an eluent provided monosulfoxide 3 as a white powder (3.95 g, 49%). 1H-NMR (CDCl3, 300 MHz, 298 K) δ 7.63 (bs, 2H, Ar-H), 7.56 (d, J = 2.3 Hz, 2H, Ar-H), 7.43 (bs, 4H, Ar-H), 3.78 (s, 6H, Ar-O-CH3), 3.46 (s, 6H, Ar-O-CH3), 1.23 (s, 36H, Ar-C(CH3)3) ppm. The data are in agreement with ref. 9.:C6H12 = 10:1 v/v). Calix[6]arene 9 was isolated as a pinkish powder (48 mg, 32%) m.p. > 350 °C (decomp). 1H-NMR (CD2Cl2, 600 MHz, 298 K) δ 9.75 (s, 2H, Ar-OH), 9.57 (s, 2H, Ar-OH), 9.50 (s, 2H, Ar-OH), 7.71–7.66 (m, 4H, Ar-H), 7.61–7.56 (m, 4H, Ar-H), 7.35 (d, J = 2.3 Hz, 2H, Ar-H), 7.30 (d, J = 2.3 Hz, 2H, Ar-H), 4.00 (bs, 4H, Ar-CH2-Ar), 1.29 (s, 18H, Ar-C(CH3)3), 1.27 (s, 36H, Ar-C(CH3)3) ppm. 13C-NMR (CD2Cl2, 125 MHz, 298 K) δ 155.2, 151.4, 150.9, 144.8, 144.7, 144.7, 136.0, 135.5, 132.4, 132.3, 132.0, 129.6, 129.5, 126.9, 126.8, 121.4, 120.7, 120.4, 120.3, 34.1, 34.0, 32.8, 31.1, 31.0, 30.9, 29.7, 29.7 ppm. IR (ATM) ν 3236, 2959, 2868, 1736, 1685, 1578 cm−1. HRMS (ESI+) calcd for C62H76O6S4 1067.4417 [M + Na]+, 1083.4156 [M + K]+, found m/z 1067.4417 [M + Na]+, 1083.4145 [M + K]+.:MeOH mixture (99:1 v/v) as an eluent. The first fraction provided compound 14 as a thick pinkish glue-like matter (324 mg, 59%).1H NMR (CDCl3, 400 MHz, 298 K) δ 7.40–7.36 (m, 4H, Ar-H), 7.33 (d, J = 2.4 Hz, 2H, Ar-H), 7.15 (d, J = 2.4 Hz, 2H, Ar-H), 4.78 (s, 4H, Ar-CH2-OH), 1.22 (s, 18H, Ar-C(CH3)3), 1.17 (s, 18H, Ar-C(CH3)3) ppm.
Compound 15 was isolated from the second fraction (103 mg, 24%) as a yellowish glassy solid.
1H NMR (CDCl3, 400 MHz, 298 K) δ 7.51 (d, J = 2.4 Hz, 1H, Ar-H), 7.47 (d, J = 2.4 Hz, 1H, Ar-H), 7.45 (d, J = 2.4 Hz, 1H, Ar-H), 7.38 (d, J = 2.3 Hz, 1H, Ar-H), 7.33 (d, J = 2.4 Hz, 1H, Ar-H), 7.27 (dd, J = 2.4 Hz, 7.5 Hz, 1H, Ar-H), 7.20 (d, J = 2.4 Hz, 1H, Ar-H), 7.11 (d, J = 2.4 Hz, 1H, Ar-H), 6.84 (d, J = 8.5 Hz, 1H, Ar-H), 4.89 (s, 2H, Ar-CH2-OH), 1.25 (s, 9H, Ar-C(CH3)3), 1.22 (s, 9H, Ar-C(CH3)3), 1.18 (s, 9H, Ar-C(CH3)3), 1.16 (s, 9H, Ar-C(CH3)3) ppm.
The data are in agreement with ref. 4.
Procedure A. A mixture of bis(hydroxymethyl) tetramer 14 (0.146 mmol, 110 mg) and tetramer 5 (0.146 mmol, 101.2 mg) dissolved in chloroform (25 mL) was added via syringe pump (2.5 mL h−1) into a flask containing TsOH monohydrate (2 equiv., 55.7 mg) in refluxing chloroform (100 mL). The next day, solvent was evaporated and the crude reaction mixture was separated using radial chromatography with CH2Cl2 as an eluent. Homooxacalixarene 17 was isolated as a white solid (102 mg, 95% based on 14) together with unreacted tetramer 5.
The addition of fullerene C60 (1 equiv.) into the reaction mixture as possible template did not provide any required calix[8]arene.
1H NMR (CDCl3, 400 MHz, 298 K) δ 9.10 (s, 2H, Ar-OH), 8.46 (s, 2H, Ar-OH), 7.70 (d, J = 2.5 Hz, 2H, Ar-H), 7.66 (d, J = 2.5 Hz, 2H, Ar-H), 7.60 (d, J = 2.5 Hz, 2H, Ar-H), 7.10 (d, J = 2.5 Hz, 2H, Ar-H), 4.62 (s, 4H, Ar-CH2-O-CH2-Ar), 1.26 (s, 18H, Ar-C(CH3)3), 1.22 (s, 18H, Ar-C(CH3)3) ppm. The data are in agreement with ref. 20.
Procedure B. Monohydroxymethyl tetramer 15 (0.08 mmol, 58 mg) was dissolved in 10 mL of chloroform and added via syringe pump (1 mL h−1) into a flask containing TsOH monohydrate (2 equiv., 30.4 mg) in 100 mL of CHCl3 under reflux. The next day, solvent was evaporated and the residue was separated using radial chromatography with CH2Cl2 as an eluent. Calix[4]arene 18 was isolated as a white solid (51 mg, 90% yield).
1H NMR (CDCl3, 400 MHz, 298 K) δ 9.81 (s, 2H, Ar-OH), 9.71 (s, 2H, Ar-OH), 7.64–7.60 (m, 4H, Ar-H), 7.50 (d, J = 2.4 Hz, 2H, Ar-H), 7.24 (d, J = 2.4 Hz, 2H, Ar-H), 4.24 (bs, 1H, Ar-CH2-Ar), 3.61 (bs, 1H, Ar-CH2-Ar) 1.22 (s, 18H, Ar-C(CH3)3), 1.23 (s, 18H, Ar-C(CH3)3) ppm. The data are in agreement with ref. 4.
The addition of fullerene C60 (1 equiv.) into the reaction mixture as possible template did not provide any required calix[8]arene.
X-ray crystallography
, a = 13.2330 (3) Å, b = 14.4941 (4) Å, c = 14.6750 (4) Å, α = 71.9424 (11)°, β = 82.1907 (11)°, γ = 89.1864 (12)°, Z = 2, V = 2650.05 (12) Å3, Dc = 1.165 g.cm−3, µ(Cu-Kα) = 1.66 mm−1, crystal dimensions of 0.21 × 0.11 × 0.09 mm. Data were collected at 200 (2) K on D8 Venture Photon CMOS diffractometer with Incoatec microfocus sealed tube Cu-Kα radiation. The structure was solved by charge flipping methods21 and anisotropically refined by full matrix least squares on F squared using the CRYSTALS22 to final value R = 0.055 and wR = 0.153 using 9633 independent reflections (Θmax = 68.2°), 668 parameters and 94 restrains. The hydrogen atoms attached to carbon atoms were placed in calculated positions, refined with weak restraints and then refined with a riding constrains. The hydrogen atoms attached to oxygen atoms were refined with retrained geometry. The disordered functional groups positions were found in difference electron density maps and refined with restrained geometry. The occupancy was constrained to full for each functional group. MCE23 was used for visualization of electron density maps. Diamond 3.0 (ref. 24) was used for molecular graphics. The structure was deposited into Cambridge Structural Database under number CCDC 2110419., a = 11.1603 (3) Å, b = 11.9646 (3) Å, c = 13.1785 (3) Å, α = 81.0129 (10)°, β = 70.9839 (9)°, γ = 80.0849 (10)°, Z = 1, V = 1629.33 (7) Å3, Dc = 1.225 g cm−3, µ(Cu-Kα) = 2.35 mm−1, crystal dimensions of 0.18 × 0.15 × 0.12 mm. Data were collected at 200 (2) K on a D8 Venture Photon CMOS diffractometer with Incoatec microfocus sealed tube Cu-Kα radiation. The structure was solved by charge flipping methods21 and anisotropically refined by full matrix least squares on F squared using the CRYSTALS22 to final value R = 0.033 and wR = 0.082 using 5978 independent reflections (Θmax = 68.4°), 440 parameters and 62 restrains. The hydrogen atoms attached to carbon atoms were placed in calculated positions, refined with weak restraints and then refined with a riding constrains. The hydrogen atoms attached to oxygen atoms were refined with retrained geometry. The disordered bridging atoms were refined with sum of sulfur atom occupancies restrained to 2 and the occupancy of each position constrained to 1. The disordered solvent positions were located in difference electron density maps and refined with restrained geometry. The solvent occupancy was refined with the sum constrained to 1. MCE23 was used for visualization of electron density maps. Diamond 3.0 (ref. 24) was used for molecular graphics. The structure was deposited into Cambridge Structural Database under number CCDC 2110418.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Czech Science Foundation (Grant 19-08273S).Notes and references
- For selected books on calixarenes and their applications, see: (a) P. Neri, J. L. Sessler and M. X. Wang, Calixarenes and Beyond, Springer, Cham, Switzerland, 2016 CrossRef; (b) C. D. Gutsche, Calixarenes: An Introduction, Royal Society of Chemistry, Cambridge, UK, 2nd edn, 2008 Search PubMed; (c) J. Vicens, J. Harrowfield and L. Backlouti, Calixarenes in the Nanoworld, Springer, Dordrecht, The Netherlands, 2007 CrossRef; (d) Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Calixarenes 2001, Kluwer, Dordrecht, The Netherlands, 2001 Search PubMed; (e) L. Mandolini and R. Ungaro, Calixarenes in Action, Imperial College Press, London, 2000 CrossRef.
- H. Kumagai, M. Hasegawa, S. Miyanari, Y. Sugawa, Y. Sato, T. Hori, S. Ueda, H. Kamiyama and S. Miyano, Tetrahedron Lett., 1997, 38, 3971–3972 CrossRef CAS.
- For recent reviews on thiacalixarenes see: (a) R. Kumar, Y. O. Lee, V. Bhalla, M. Kumar and J. S. Kim, Chem. Soc. Rev., 2014, 43, 4824–4870 RSC; (b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed; (c) P. Lhotak, Eur. J. Org. Chem., 2004, 1675–1692 CrossRef CAS.
- T. Sone, Y. Ohba, K. Moriya, H. Kumada and K. Ito, Tetrahedron, 1997, 53, 10689–10698 CrossRef CAS.
- N. Kon, N. Iki, Y. Yamane, S. Shirasaki and S. Miyano, Tetrahedron Lett., 2004, 45(1), 207–211 CrossRef CAS.
- M. Hucko, H. Dvorakova, V. Eigner and P. Lhotak, Chem. Commun., 2015, 51(32), 7051–7053 RSC.
- (a) D. Kortus, J. Miksatko, O. Kundrat, M. Babor, V. Eigner, H. Dvorakova and P. Lhotak, J. Org. Chem., 2019, 84, 11572–11580 CrossRef CAS PubMed; (b) D. Kortus, O. Kundrat, M. Tlusty, J. Cejka, H. Dvorakova and P. Lhotak, New J. Chem., 2020, 44, 14496–14504 RSC.
- P. Lhotak, M. Himl, I. Stibor, J. Sykora, H. Dvorakova and H. Petrickova, Tetrahedron, 2003, 59, 7581–7585 CrossRef CAS.
- The formation of 3 was described in 26% yield: N. Morohashi, N. Iki, T. Onodera, C. Kabuto and S. Miyano, Tetrahedron Lett., 2000, 41, 5093–5097 CrossRef CAS.
- J. Miksatko, V. Eigner and P. Lhotak, RSC Adv., 2017, 7, 53407–53414 RSC.
- Y. Ohba, K. Moriya and T. Sone, Bull. Chem. Soc. Jpn., 1991, 64, 576–582 CrossRef CAS.
- S. Yu, Y. Wang, Y. Ma, L. Wang, J. Zhu and S. Liu, Inorg. Chim. Acta, 2017, 468, 159–170 CrossRef CAS.
- N. Morohashi, T. Ishiwata, K. Ito and Y. Ohba, Tetrahedron Lett., 2004, 45, 799–802 CrossRef CAS.
- C. D. Gutsche and L. J. Bauer, J. Am. Chem. Soc., 1985, 107, 6052–6059 CrossRef CAS.
- S. E. Biali, V. Böhmer, I. Columbus, G. Ferguson, C. Grüttner, F. Grynszpan, E. F. Paulus and I. Thondorf, J. Chem. Soc., Perkin Trans. 2, 1998, 2261–2269 RSC.
- D. Kortus, K. Krizova, H. Dvorakova, V. Eigner and P. Lhotak, Tetrahedron Lett., 2021, 69, 152924 CrossRef CAS.
- K. Ito, Y. Yamamori, Y. Ohba and T. Sone, Synth. Commun., 2000, 30, 1167–1177 CrossRef CAS.
- (a) P. E. Georghiou, Calixarenes and Fullerenes, in Calixarenes and Beyond, ed. P. Neri, J. L. Sessler and M. X. Wang, Springer Int. Publishing Switzerland, 2016, pp. 879–919 Search PubMed; (b) J. L. Delgado and J. F. Nierengarten, Fullerenes and Calixarenes, in Calixarenes in the Nanoworld, ed. J. Vicens, J. Harrowfield and L. Backlouti, Springer, Dordrecht, The Netherlands, 2007, pp. 173–196 Search PubMed; (c) P. Lhoták and O. Kundrát, Fullerene Receptors Based on Calixarene Derivatives in Artificial Receptors for Chemical Sensors, ed. V. Mirsky and A. Yatsimirsky, Wiley-VCH Verlag, Weinheim, Germany, 2011, pp. 249–272 Search PubMed.
- The binding constants were calculated using the Bindfit application freely available at: http://supramolecular.org.
- H. Teraura, K. Ito, N. Morohashi and Y. Ohba, Heterocycl. Commun., 2003, 9, 443–448 CAS.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- J. Rohlicek and M. Husak, J. Appl. Crystallogr., 2007, 40, 600 CrossRef CAS.
- K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 1999 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, full characterization of compounds 5–8 and 12–16, X-ray data for structures 6 and 7, and the complexation studies of C60 and C70. CCDC 2110418 and 2110419. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra07464d |
| This journal is © The Royal Society of Chemistry 2021 |