
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStability and structural properties of vacancy-ordered and -disordered ZrCx†
Theresa Davey *,
Ken Suzuki,
Hideo Miura and
Ying Chen
*,
Ken Suzuki,
Hideo Miura and
Ying Chen
School of Engineering, Tohoku University, Sendai, 980-8579, Japan. E-mail: theresa@tohoku.ac.jp
First published on 4th October 2021
Abstract
Vacancy-ordered superstructural phases of zirconium carbide have been intermittently observed at low temperatures for over 50 years. However, little is known about these ordered phases as they have proven to be challenging to fabricate experimentally, although theoretical predictions suggest that they should be significantly more stable than the more-observed vacancy-disordered solid solution ZrCx (x ≤ 1) phase at low temperatures. The stability and structural properties of the vacancy-ordered and vacancy-disordered phases are investigated using first-principles calculations. The stability of the ordered superstructural phases is related to the driving force from the relative instability of certain vacancy configurations, which are preferred or avoided in ordered structures. The trend of the vacancy ordering and the underlying mechanisms of the relative instability are explained in terms of the geometry of the crystal structures and the electronic charge distribution and atomic bonding features.
1. Introduction
The carbon–zirconium system has been widely investigated for over 150 years,1 but there are still many unanswered questions about the intermediate intermetallic phase, ZrCx where x ≤ 1, which is widely used within the nuclear and aerospace industries because of its extremely high melting point (∼3700 K) and strength at high temperatures.2 Zirconium monocarbide has a rocksalt (NaCl B1) structure with zirconium atoms forming a cubic lattice and carbon atoms occupying the octahedral interstitial sites. ZrCx has wide range of stoichiometry, with vacancies on up to ∼50% of the carbon sites.3,4 The mechanical and thermodynamic properties of ZrCx are strongly dependent on the stoichiometry and structural ordering,5 but much is unknown about the character of the vacancy ordering. As the variation of the thermophysical properties with vacancy concentration makes zirconium carbides a candidate for creating tuneable ceramics,6,7 it is necessary to more closely explore the origins and consequences of the structural vacancy ordering in order to successfully fabricate materials for a specific application.Historically, ZrCx has been generally treated as a single continuous solid solution phase where it is assumed that the vacancies are randomly distributed among all carbon sites.3 Various ordered phases have been intermittently reported in the experimental literature but there is no consensus regarding phase stability. Goretzki observed a Zr2C (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) superlattice;8 Obata and Nakazawa did not observe Zr2C, but observed Zr4C3 consistent with a Th4C3 prototype;9 de Novion and Maurice reported the Zr2C phase (Fdm);10 Karimov et al. observed Zr2C (Fdm);11 Khaenko et al. possibly observed a trigonal Zr8C5 (Rm);12 Hu et al. reported Zr2C (Fdm);13 Wei et al. observed Zr2C (Fdm);14 Zhou et al. fabricated Zr2C (Fdm);15 and Rana et al. report systematic vacancy ordering.4 A theoretical phase diagram from Gusev and Rempel16 determined by the Order Parameter Functional (OPF) method predicted the stability of several ordered zirconium carbide phases below 1217 K. In other theoretical studies, Zhang et al.17,18 used the Cluster Expansion Method (CEM), and Yu et al.19 and Xie et al.6 used the USPEX evolutionary algorithm, to obtain the convex hull of stable phases at 0 K, all predicting several ordered phases that are stable compared to the vacancy-disordered phase, but disagreeing upon which phases are stable. De Novion and Maurice10 proposed that the vacancy ordering in group IV transition metal carbides, including zirconium carbide, was driven by vacancies avoiding the second nearest neighbour (2nn) position with reference to other vacancies, slightly avoiding the first nearest neighbour (1nn) position, and preferring the third nearest neighbour (3nn) position. This was validated through theoretical calculations by Razumovskiy et al.20,21 and Zhang et al.17 who additionally demonstrated that the vacancy–vacancy interaction energy for the 2nn configuration was strongly repulsive and unfavourable compared to other pair configurations. Furthermore, Zhang et al. identified a “fingerprint” configuration of 3nn vacancy triplets that accounted for most vacancy arrangements within the stable ordered phases, and proposed that the energetic preference for different vacancy cluster configurations drove self-assembly of the vacancies into the long-range ordered phases.17
m) superlattice;8 Obata and Nakazawa did not observe Zr2C, but observed Zr4C3 consistent with a Th4C3 prototype;9 de Novion and Maurice reported the Zr2C phase (Fdm);10 Karimov et al. observed Zr2C (Fdm);11 Khaenko et al. possibly observed a trigonal Zr8C5 (Rm);12 Hu et al. reported Zr2C (Fdm);13 Wei et al. observed Zr2C (Fdm);14 Zhou et al. fabricated Zr2C (Fdm);15 and Rana et al. report systematic vacancy ordering.4 A theoretical phase diagram from Gusev and Rempel16 determined by the Order Parameter Functional (OPF) method predicted the stability of several ordered zirconium carbide phases below 1217 K. In other theoretical studies, Zhang et al.17,18 used the Cluster Expansion Method (CEM), and Yu et al.19 and Xie et al.6 used the USPEX evolutionary algorithm, to obtain the convex hull of stable phases at 0 K, all predicting several ordered phases that are stable compared to the vacancy-disordered phase, but disagreeing upon which phases are stable. De Novion and Maurice10 proposed that the vacancy ordering in group IV transition metal carbides, including zirconium carbide, was driven by vacancies avoiding the second nearest neighbour (2nn) position with reference to other vacancies, slightly avoiding the first nearest neighbour (1nn) position, and preferring the third nearest neighbour (3nn) position. This was validated through theoretical calculations by Razumovskiy et al.20,21 and Zhang et al.17 who additionally demonstrated that the vacancy–vacancy interaction energy for the 2nn configuration was strongly repulsive and unfavourable compared to other pair configurations. Furthermore, Zhang et al. identified a “fingerprint” configuration of 3nn vacancy triplets that accounted for most vacancy arrangements within the stable ordered phases, and proposed that the energetic preference for different vacancy cluster configurations drove self-assembly of the vacancies into the long-range ordered phases.17
In this study, first-principles calculations of isolated vacancies, vacancy pairs, and vacancy triplets are used to identify the local variation in bonding and electronic properties close to various vacancy configurations, in order to explain why certain vacancy arrangements are more stable than others. First-principles calculations of vacancy-ordered (superstructural) and vacancy-disordered phases are also performed to examine the phase stability and volume at 0 K. Special Quasirandom Structures (SQS)22 are used to model the vacancy-disordered phase.
2. Calculation method and models
2.1. First-principles calculations
The ground state energy at 0 K was determined via non-spin-polarised density functional theory (DFT) calculations using the Projector Augmented Wave (PAW) method23,24 in the GPU implementation25,26 of the Vienna Ab initio Software Package (VASP).27–29 Calculations were performed using both the Local Density Approximation (LDA)30 and the Generalized Gradient approximation (GGA) Perdew–Burke–Ernzerhof (PBE)31,32 for the exchange and correlation functionals. The cutoff energy and k-point Monkhorst–Pack mesh33 density were chosen as 700 eV and at least 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 k-points per supercell for consistency with previous calculations from Duff et al.34 and to ensure good convergence of all energy calculations. All vacancy-ordered structures were fully (ionic and volume) relaxed until all force components were smaller than 0.001 meV Å−1.
000 k-points per supercell for consistency with previous calculations from Duff et al.34 and to ensure good convergence of all energy calculations. All vacancy-ordered structures were fully (ionic and volume) relaxed until all force components were smaller than 0.001 meV Å−1.
Previously, Duff et al.34 performed fully anharmonic finite temperature calculations of ZrC up to the melting point using the TU-TILD approach. These finite temperature calculations found a significantly improved agreement with experimental observations at high temperature using the LDA compared to the GGA. Therefore, in this work, calculations using the GGA were performed for comparison with other studies, but calculations using the LDA were used to consider the properties of the calculated structures in more detail.
The enthalpy (energy) of formation with respect to the pure elements (hcp Zr and graphite C) at 0 K is defined as
| Eform = EZrCx − xZrEhcpZr − xCEgraphiteC |
The mixing enthalpy (energy) with respect to the fully occupied and empty carbon sublattice (fcc Zr and stoichiometric ZrC) at 0 K is defined as
| Emix = EZrCx − yCEZrC − yVaEfccZr |
The strength of the individual bonds between atomic pairs was investigated by considering the Crystal Orbital Hamiltonian Population (COHP)38 of each bond using the Local Orbital Basis Suite Towards Electronic-Structure Reconstruction (LOBSTER) analysis software.39–41 The projected COHP is obtained from the plane-wave basis sets used in the DFT calculation using the VASP27–29 output directly.42 The value of the integrated projected COHP (IpCOHP) up to the Fermi level is known to scale with the bond order43 and can be used to examine the relative strength of each pair bond in a given structure.39 The individual and average IpCOHP values for each bond type (e.g., C–Zr, Zr–Zr, etc. and first, second, etc. nearest-neighbour distance defined with reference to stoichiometric zirconium carbide) within 7 Å distance (according to the supercell dimensions) were examined for all structures to quantitatively understand the stability and structural trends from a local bonding perspective. For each COHP projection, the absolute charge spilling was reduced as much as possible,40 and was within 1.5% in all cases.
The charge density distribution was obtained using VASP and visualised using VESTA.44 Charge density differences arising from the presence of vacancies were calculated by considering different charge density distributions in unrelaxed structures (based on the pristine ZrC lattice) to match the lattices. The unrelaxed structure distributions of defective structures were compared with the relaxed structure distributions to confirm compatibility.
The electron localisation function (ELF) is a probabilistic measure of finding an electron close to a reference electron at a given point with the same spin.45–47 The ELF may be used to obtain detailed bonding information via the electron pair distribution and quantitatively characterise the electron interactions.48 The ELF may have values between 0 and 1, where 0 indicates fully delocalised electrons (metallic bonding), 1 indicates fully localised electrons (covalent bonding), and 0.5 is equivalent to a free electron gas.45 The ELF distribution was obtained using VASP and analysed using VESTA.
2.2. Ordered structures
All vacancy-ordered phases reported to be stable by Zhang et al.,17,18 Yu et al.,19 and Xie et al.6 are shown in Table 1. These theoretically predicted phases are consistent with experimental observations. The unit cells for each phase listed in Table 1, with vacancy sites indicated, are shown in Fig. 1.| Ordered structure | Space group | Space group number | Crystal system | Reference (previously reported stable by) | Formation energy (eV per atom) | Mixing energy (eV per atom) | Volume (Å3 per Zr atom) | Convex hull at 0 K |
|---|---|---|---|---|---|---|---|---|
| ZrC | Fmm |
225 | Cubic | 6 and 17–19 | −0.916 | 0.000 | 25.056 | Stable |
| Zr8C7 | P4332 | 212 | Cubic | 17 and 18 | −0.904 | −0.131 | 25.180 | Metastable |
| Zr7C6 | R |
148 | Trigonal | 6 and 19 | −0.907 | −0.154 | 25.161 | Stable |
| Zr6C5 | C2/m | 12 | Monoclinic | 17 and 18 | −0.894 | −0.168 | 25.160 | Metastable |
| Zr4C3 | C2/c | 15 | Monoclinic | 6, 18 and 19 | −0.864 | −0.230 | 25.130 | Stable |
| C2/m | 12 | Monoclinic | 17 | −0.860 | −0.225 | 25.140 | Metastable | |
| Zr3C2 | Fddd | 70 | Orthorhombic | 6, 17 and 18 | −0.818 | −0.273 | 25.043 | Stable |
| C2/m | 12 | Monoclinic | 19 | −0.815 | −0.270 | 25.066 | Metastable | |
| Zr2C | Fdm |
227 | Cubic | 6 and 17–19 | −0.684 | −0.310 | 24.916 | Stable |
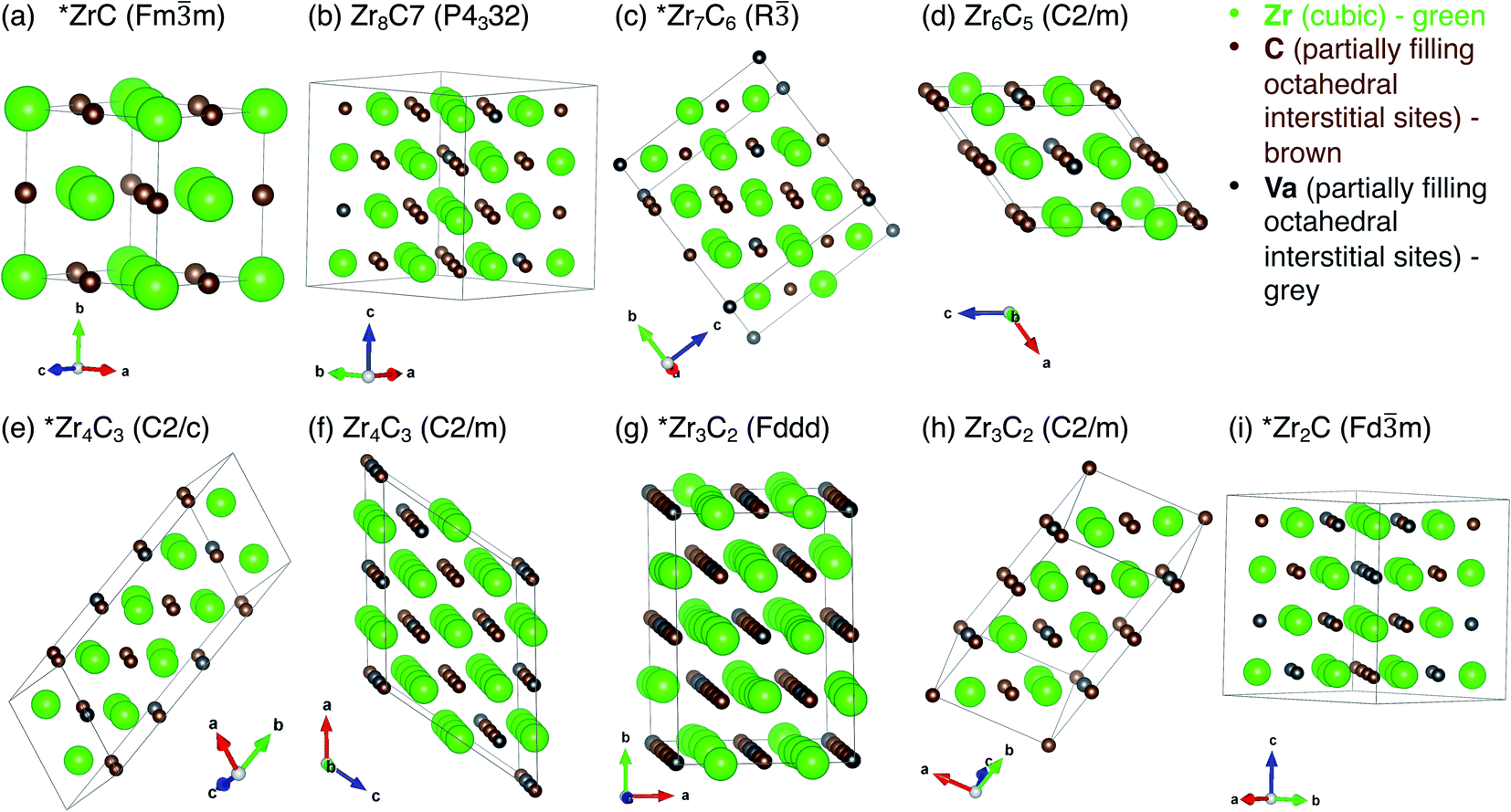 | ||
| Fig. 1 Unit cells for each of the superstructural ordered phases in Table 1, with vacant sites marked. (a) ZrC (Fmm), (b) Zr8C7 (P4332), (c) Zr7C6 (R), (d) Zr6C5 (C2/m), (e) Zr4C3 (C2/c), (f) Zr4C3 (C2/m), (g) Zr3C2 (Fddd), (h) Zr3C2 (C2/m), (i) Zr2C (Fdm). Structures marked with * are on the ground state convex hull of stable phases. | ||
The stable structures reported in previous theoretical studies are ZrC (Fmm, isotypic with NaCl), Zr8C7 (P4332, isotypic with V8C7), Zr7C6 (R, identified by Yu et al.19 and Xie et al.6), Zr6C5 (C2/m, isotypic with Ti6C5), Zr4C3 (C2/m, identified by Zhang et al.17), Zr4C3 (C2/c, isotypic with Hf4C3 predicted by Yu et al.19), Zr3C2 (Fddd, isotypic with Sc2S3), Zr3C2 (C2/m, isotypic with Ti3C2), and Zr2C (Fdm, isotypic with TiC2).
2.3. SQS models for disordered structures
SQS provide a structure that approximates the disordered state by choosing an atomic structure with atomic correlation functions that are close to the disordered state. SQS were generated at various compositions of Zr, C, and Va based on the rocksalt parent lattice using the mcsqs code in the Alloy Theoretic Automated Toolkit (ATAT) package.49 Zr atoms were fixed on a cubic lattice, with C and Va distributed among octahedral interstitial sites. SQS with 12.5% intervals in Va site fraction (vacancy content on the octahedral interstitial sites only) up to 50% Va site fraction were created. All SQS had 32 Zr atoms for consistency. The ground state energy of each structure at 0 K was obtained by the same method used for the ordered structures described above. During ionic relaxation, atomic size mismatch and presence of defects can cause distortions in the SQS lattice that cause a loss of symmetry.50 In such cases, the SQS can no longer be used to represent the disordered phase corresponding to the parent structure. The radial distribution function of the unrelaxed and fully relaxed structures was examined for each structure, to ensure that the character of the face-centred cubic rocksalt structure was retained during full (ionic and electronic) relaxation.3. Results and discussion
De Novion and Maurice10 and Zhang et al.17 determined that the long-range ordering of vacancies in zirconium carbide is driven by the relative stability of different vacancy pair and triplet configurations. In this work, in order to understand the local effects that determine the stability of each configuration, first-principles calculations of the energies and volumes of model structures were calculated, and local bonding and electronic states were analysed.3.1. Electronic structures of various vacancy-complex types
Fig. 2 shows the electronic structure of pristine ZrC. It can be seen from the electronic density of states (DOS) in Fig. 2(a) and (b) that the majority of the bonding arises from strong hybridisation between the Zr d-electrons and the C p-electrons, from the Femi energy to the deep energy band 5 eV below the Fermi energy. This is consistent with the IpCOHP for each 1nn and 2nn bond type (C–Zr, Zr–Zr, and C–C) shown in Fig. 2(c). As the IpCOHP is known to scale with the bond strength, the more negative the IpCOHP, the stronger the bond. The IpCOHP also generally decreases with increasing bond length, however, the 2nn C–C bond is stronger than the 1nn C–C bond, as it is enhanced by the electron cloud surrounding the Zr atom directly between the two carbon atoms. From Fig. 2(c), it can be seen that the strong 1nn C–Zr bonds (IpCOHP = −3.53 eV per bond) account for most of the bonding in zirconium carbide, with 1nn Zr–Zr bonds being the next largest contribution.
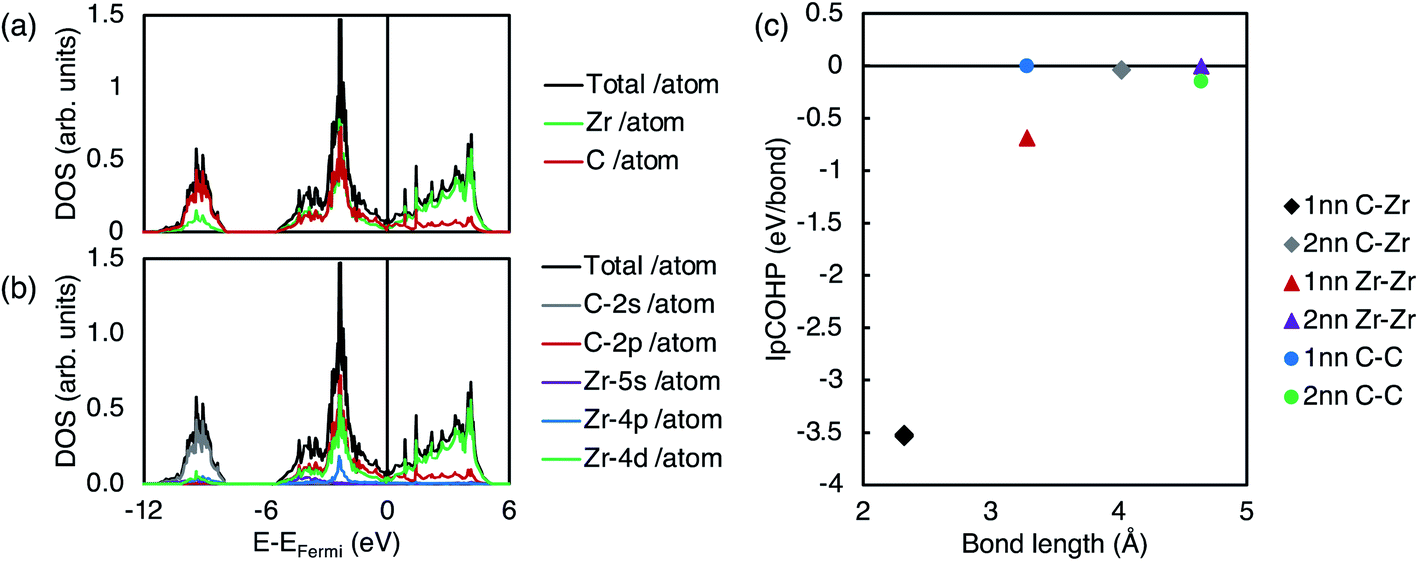 | ||
| Fig. 2 Density of states (DOS) and orbital characters of pristine stoichiometric ZrC. (a) Total DOS of ZrC and local DOS of each element. (b) Total and partial local DOS of ZrC. (c) IpCOHP vs. bond length for all 1nn and 2nn bond types (C–Zr, Zr–Zr, and C–C) in pristine stoichiometric ZrC. | ||
When all carbon atoms are removed from ZrC, the remaining structure is fcc Zr. In fcc Zr, the electronic charge is almost entirely localised at the Zr sites, which a maximum electronic charge density of 0.89983e per Bohr3. In ZrC, the formation of covalent C–Zr bonds causes partial delocalisation of the electrons, reducing the maximum charge density to 0.85464e per Bohr3. The electronic charge density distribution on the (001) plane in pristine stoichiometric ZrC is shown in Fig. 3(a).
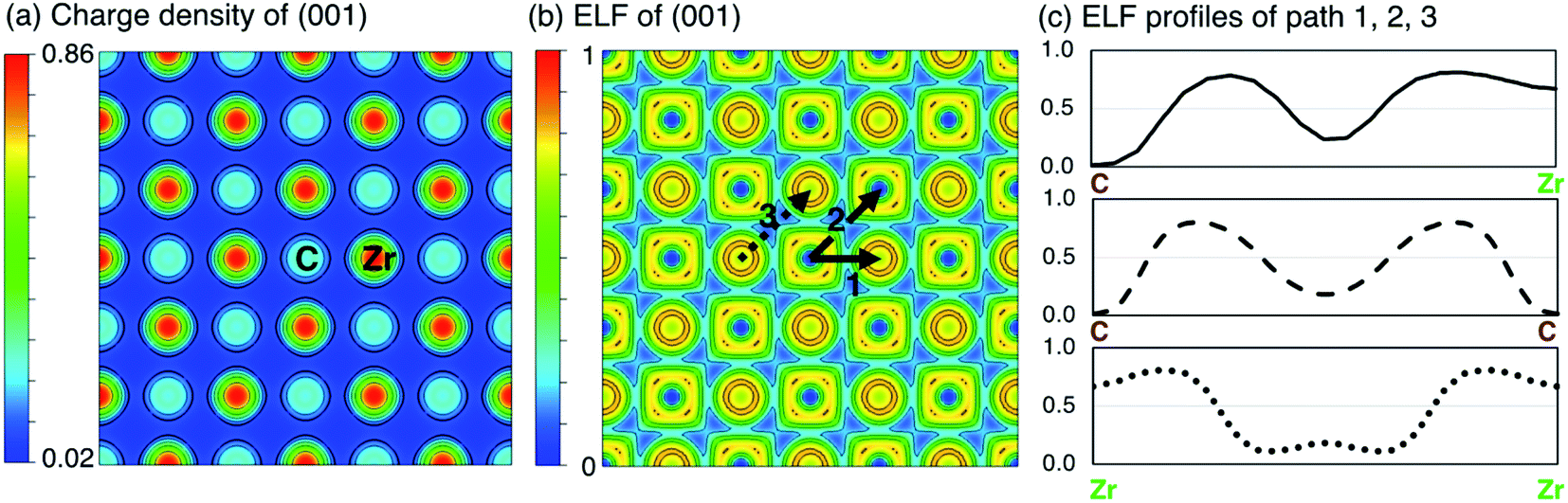 | ||
| Fig. 3 Charge density (e per Bohr3) and electron localisation function (ELF) of ZrC. (a) Charge density distribution on the (001) plane. (b) ELF on the (001) plane. (c) ELF profiles along the paths 1, 2, and 3 indicated in (b). | ||
A more detailed examination of the electronic structure and bonding character of ZrC is given in Fig. 3. Fig. 3(a) and (b) are the charge density distribution and ELF respectively in the (001) plane. Fig. 3(c) shows the ELF profiles along three paths marked in Fig. 3(b), corresponding to the C–Zr bond, C–C bond, and Zr–Zr bond. Along path 1 between C and Zr, a high ELF > 0.7 around Zr indicates the dominant covalent bonding of Zr–C, while the ELF attractor basins of 0.236 at the middle point of the path shows an evidence of mixed ionic bonding due to the small charge transfer from the Zr atom to the C atom, which verifies discussion by Li et al.54 It can also be seen that C–C bonds in ZrC combine slight metallic bonding features into the pure covalent bond found in pure C, and the Zr–Zr bonds in ZrC keep the metallic bonding seen in pure Zr with a slight covalent component.
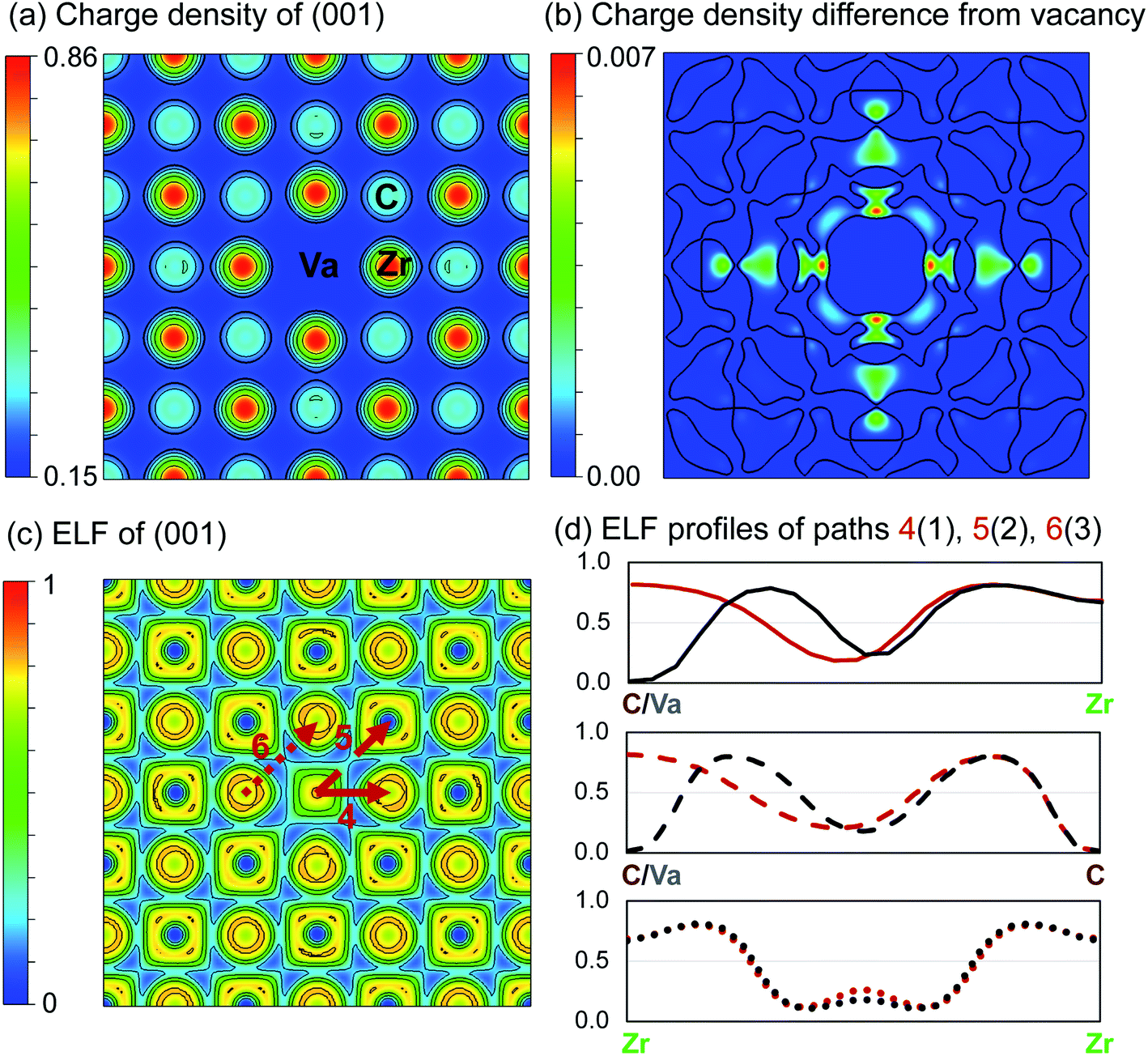 | ||
| Fig. 4 Charge density (e per Bohr3) and electron localisation function (ELF) of ZrC with single vacancy. (a) Charge density distribution of the (001) plane. (b) Charge density difference from pristine ZrC (positive only). Contours separate positive and negative regions. (c) ELF of the (001) plane. (d) ELF profiles along the paths 4, 5, and 6 indicated in (c), compared with profiles from Fig. 3. | ||
Fig. 4(a) shows the charge density and the ELF of the (001) plane, where there is a reduced charge density at the vacancy site, and an increased charge density at the nearest neighbour Zr sites, and nearest and next nearest neighbour C sites with respect to the vacancy.
Following removal of a carbon atom, the electrons involved in bonding (which primarily were donated from the nearest Zr atoms) are redistributed. Some are involved in the bonds between those Zr atoms and the remaining 1nn C–Zr bonds, and some remain localised at the Zr and vacancy sites. This results in an increased maximum charge density of 0.86053 e per Bohr3 at the Zr sites (compared to 0.85464 e per Bohr3 in pristine ZrC, a 0.7% increase), and a corresponding maximum −IpCOHP of 4.41 eV per bond in the 1nn C–Zr bonds (compared to 3.53 eV in the pristine case, a 25.1% increase). This indicates stronger local bonding surrounding the vacancy. The average −IpCOHP for 1nn C–Zr bonds in the whole structure is 3.57 eV, indicating that overall, the dominant 1nn C–Zr bonding is strengthened by the presence of the vacancy. This enhancement of bonding around the carbon vacancy can be observed in detail in Fig. 4(d) from the comparison of ELF profiles for C–Zr and vacancy-Zr. It can be seen a strong charge localisation occurs at the isolated vacancy site, with a maximum ELF of 0.819.
The Zr–Zr bonding among the six nearest neighbour Zr atoms surrounding the vacancy site is also enhanced by the electron cloud at the vacancy site, with 1nn Zr–Zr bonds having an average −IpCOHP of 0.818 eV per bond (an 18.7% increase compared to in the pristine case), and the 2nn Zr–Zr bonds that cross the vacancy site having an average −IpCOHP of 0.0456 eV per bond (14 times the pristine bond strength). However, the average −IpCOHP of the 1nn Zr–Zr bonds in the whole structure increases by only 1.3%, while the average −IpCOHP of the 2nn Zr–Zr bonds is decreased by 109%, becoming repulsive overall. The increased strength of the 1nn C–Zr bonds in the defective structure has a significantly larger effect than the changes to the strength of the Zr–Zr bonds.
The relaxation of the structure (allowing ionic displacement and volume change) stabilises the structure by 0.003 eV per atom compared the unrelaxed (pristine) structure containing a vacancy. Selective dynamics relaxations (allowing only certain atoms to move) allow this energy to be partitioned into stabilisation from relaxation of each nearest neighbour shell. 91.3% of the stabilisation can be accounted for by relaxation of the 1nn Zr, and 1nn and 2nn C relative to the vacancy site, where the stabilisation from each contributes for 77.1%, 5.1%, and 9.1% of the total stabilisation respectively. The 1nn and 2nn C relative to the vacancy share 1nn C–Zr bonds with the nearest neighbour Zr surrounding the vacancy, where the 2nn C are directly opposite the vacancy site. The local distortion can be represented well by considering the displacements of these 1nn and 2nn shells, which are shown schematically in Fig. 5(a). The repulsive force of the excess electrons in the vacancy site pushes the six 1nn Zr atoms, causing them to displace slightly away from the vacancy site (increasing the distance from the vacancy site from 2.322 Å to 2.410 Å (+3.8%), indicated in Fig. 5(a) as distance a). This displacement of ions is consistent with experimental measurements by de Novion and Maurice10 and calculations by Råsander and Delin.55
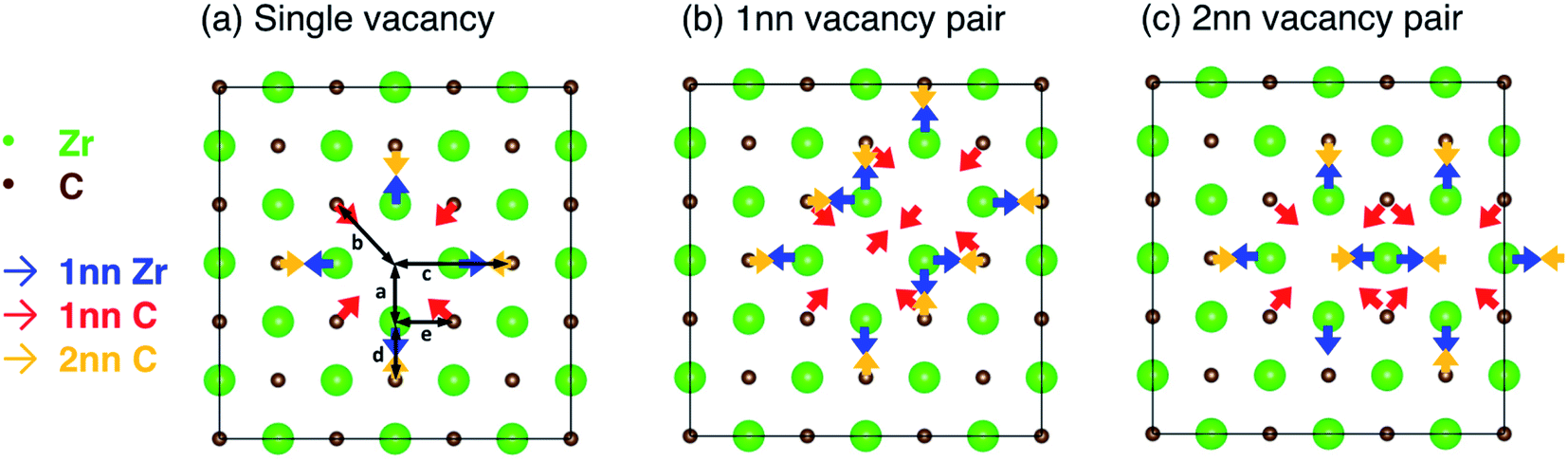 | ||
| Fig. 5 Schematic of the competing lattice distortion effects in the (001) plane from each vacancy in (a) an isolated vacancy, (b) 1nn pair configuration, (c) 2nn pair configuration. Blue arrows show the displacement direction of atoms in the 1nn Zr shell, red arrows show the displacement direction of atoms in the 1nn C shell, and yellow arrows show the displacement direction of atoms in the 2nn C shell. (a–e) Mark various interatomic distances (given in text). | ||
The twelve nearest neighbour C atoms (with respect to the vacancy site) are displaced towards the vacancy site, reducing the distance from the vacancy site from 3.284 Å to 3.268 Å (−0.5%), indicated in Fig. 5(a) as distance b. The six second nearest neighbour C atoms are also displaced towards the vacancy, reducing the distance from the vacancy site from 4.645 Å to 4.613 Å (−0.7%), indicated in Fig. 5(a) as distance c. The length of each 1nn C–Zr bond (involving the 1nn Zr atoms and the 1nn or 2nn C atoms with reference to the vacancy site, marked by distances d and e in Fig. 5) is reduced and the bonding is strengthened. The distances of atoms in farther layers to the vacancy are additionally accommodated according to the total effect of the redistribution of electrons. The unbalanced 1nn C–Zr bonding opposite the vacancy site results in shorter C–Zr bonds opposite the vacancies compared to the other 1nn C–Zr bonds involving the same Zr atom (2.203 Å compared to 2.317 Å, where the equivalent distance without any vacancies is 2.322 Å, so −5.1% and −0.2% respectively compared to the pristine bond length, indicated in Fig. 5(a) as distances d and e). These bonds are significantly strengthened, where the −IpCOHP for each is increased by 25.1% compared to pristine structure.
where EnVaZrC is the energy per atom of the ZrC supercell with n vacancies, and EZrC is the energy per atom of the pristine ZrC supercell. The formation energies and vacancy interaction energies associated with different vacancy pair configurations according to the vacancy–vacancy distance (1nn, 2nn, 3nn, 4nn) are shown in Fig. 6(a) and (b) (squares), together with the various vacancy triplet configurations (diamonds, discussed in the next section). It can be seen that these two energies with different physical meaning show similar relative trends for various vacancy configurations. Although longer distance pairs than shown here may be slightly more energetically favourable than some of the 1nn–4nn pairs shown,21 the non-dilute vacancy content in the ordered zirconium carbides necessarily leads to some clustering of these pair types. The 2nn vacancy pair is clearly much less stable than the other pair configurations, and the 3nn vacancy pair is the most stable and is the only pair configuration more stable than pristine ZrC or than isolated vacancies (which have zero vacancy interaction energy). At equilibrium, the vacancies will arrange into the configuration that is most energetically stable. This is consistent with observations by de Novion and Maurice,10 and calculation of vacancy–vacancy interactions by Razumovskiy et al.,20,21 and Zhang et al.17
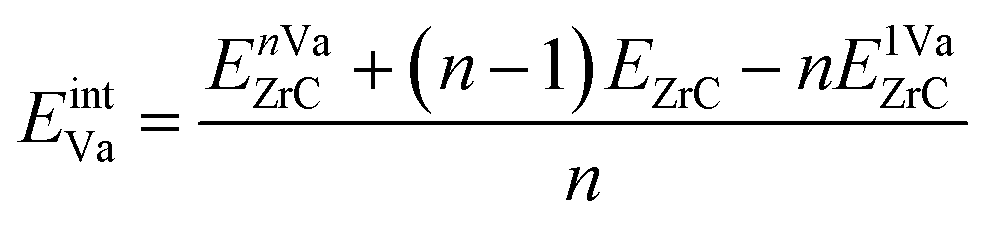
 | ||
| Fig. 6 (a) Formation energy and (b) vacancy interaction energy for vacancy pair and triplet configurations in a 3 × 3 × 3 ZrC supercell. “xnn” refers to the xth nearest neighbour distance of the vacancy pair. Vacancy triplets are defined by the pair lengths of each side of the triangle. The green line marks the formation energy of pristine ZrC. Formation energies below that of pristine ZrC and negative interaction energies are marked in red. | ||
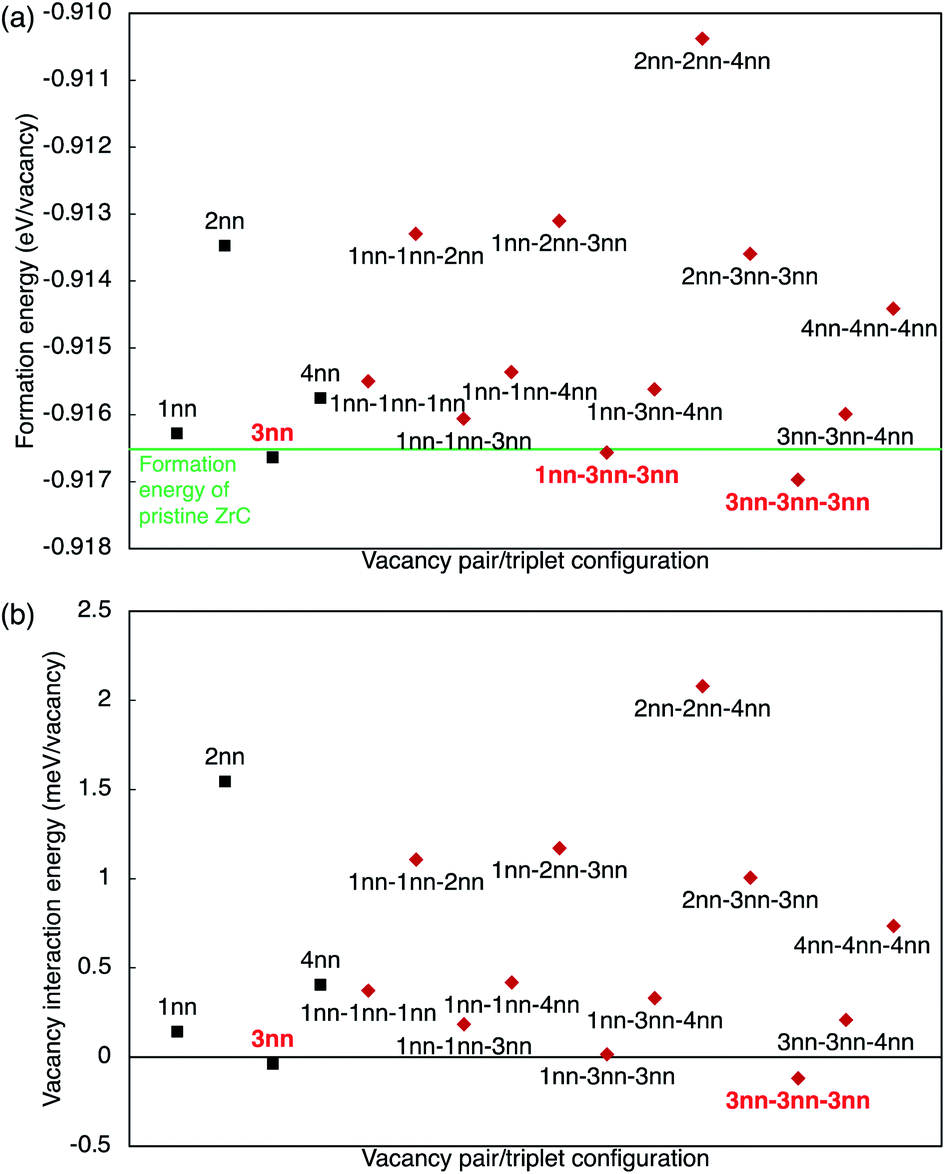
The vacancy formation energy (per vacancy) is defined as
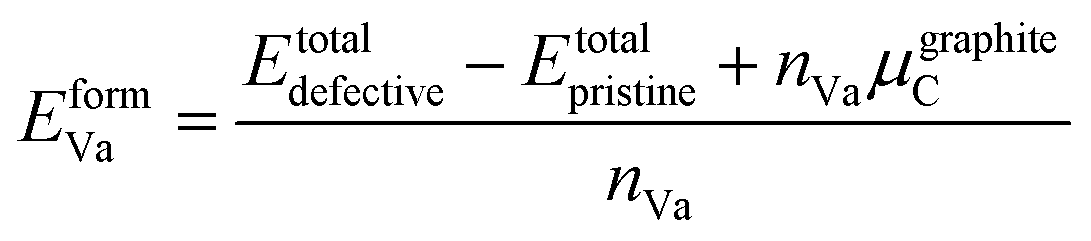
When more than one vacancy is present in the lattice, unless the vacancies are sufficiently separated, there will be superposition of the chemical effect (charge distribution) and size effect (lattice relaxation) from the vacancy complex, augmented by vacancy interactions. The combined effects depend on the configuration of the vacancy pairs within the lattice. Fig. 5 schematically shows the competing forces on each atom in the 1nn and 2nn Zr and C shells surrounding the single vacancy or the vacancy pairs in the 1nn and 2nn vacancy–vacancy configurations. The geometric arrangement of the vacancy pairs within the rocksalt lattice affects the possible ionic relaxation and electron redistribution. When vacancies are close together, some of the nearest neighbour Zr and C atoms that are displaced close to a vacancy (shown in Fig. 5(a)) are affected by displacive forces arising from both vacancies.
In the 1nn vacancy pair case, there are two Zr atoms within the 1nn Zr shell of both vacancies. Additionally, there are eight C atoms within the 1nn or 2nn shells of both vacancies, as well as each vacancy removing one C atom from the 1nn C shell of the other vacancy. In the 2nn vacancy pair case, there is one Zr atom within the 1nn shell of both vacancies, and there are also four C atoms within the 1nn or 2nn C shells surrounding both vacancies, as well as the two removed vacancies from those shells. When atoms are within the nearest neighbour shells surrounding both vacancies, there are displacive forces acting on them in two directions, as indicated in Fig. 5(b) and (c). The net displacement direction is determined by the integrated effect arising from each individual vacancy. In the 1nn vacancy pair case, the direction of forces on all atoms existing in both 1nn shells are not opposite to one another, resulting in an overall displacement in the average direction. Table 2 shows the average volume increase per vacancy for the isolated vacancy and each of the vacancy pair configurations, as well showing how many atoms are shared between vacancy shells in each configuration. The 1nn vacancy pair results in a net volume increase of 0.937 Å3 per vacancy, compared to 1.090 Å3 per vacancy for an isolated vacancy (an increase of 85.9% of the isolated vacancy volume).
| Vacancy configuration | Formation energy (eV per atom) | Average volume increase/vacancy (Å3) | Shared (trapped) nearest neighbour Zr (/12) | Shared (trapped) nearest and next neighbour C (/36) | Max. charge density (e per Bohr3) | Min. charge density at vacancy site (e per Bohr3) | Va–Va electronic interaction (chemical effect) (me Bohr3) | ELF at vacancy site | 1nn C–Zr bonds involving Zr atoms in 1nn shell and C atoms in the 2nn shell surrounding the vacancies | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max. | Min. | Average IpCOHP (eVper bond) | % total IpCOHP change from pristine | % total IpCOHP change from 1Va | Average bond length (Å) | % bond length change from pristine | % bond length change from 1Va | |||||||||
| ZrC (pristine) | −0.91652 | — | — | — | 0.85464 | — | — | — | — | −3.525 | — | — | 2.322 | — | — | |
| 1Va (isolated vacancy) | −0.91653 | 1.090 | — | — | 0.86053 | 0.015161 | — | — | 0.819 | −4.410 | 25.11 | — | 2.203 | −5.12 | — | |
| 2Va (vacancy pair) | 1nn | −0.91628 | 0.937 | 2(0) | 10(0) | 0.86630 | 0.015036 | 2.4844 | −1.1628 | 0.827 | −4.400 | 24.81 | −0.25 | 2.203 | −5.14 | −0.02 |
| 2nn | −0.91347 | 0.776 | 1(1) | 6(0) | 0.86203 | 0.014996 | 2.2444 | −4.6789 | 0.853 | −4.311 | 1.92 | −18.54 | 2.210 | −4.83 | 0.31 | |
| 3nn | −0.91664 | 1.039 | 0(0) | 4(0) | 0.86046 | 0.014998 | 0.3758 | −0.6583 | 0.820 | −4.429 | 25.63 | 0.42 | 2.201 | −5.20 | −0.08 | |
| 4nn | −0.91575 | 0.819 | 0(0) | 1(1) | 0.86047 | 0.015084 | 0.6410 | −0.7014 | 0.819 | −4.326 | 22.72 | −1.91 | 2.209 | −4.85 | 0.29 | |
However, in the 2nn vacancy pair configuration, the Zr atom directly between the two vacancies is trapped (has no net displacement), that is, no ionic relaxation of that Zr can occur. This results in a reduced net volume increase of 0.776 Å3 per vacancy (71.1% of the isolated vacancy volume). As this Zr atom is trapped between vacancies, the local displacement of that atom that stabilises the structure cannot take place, which makes this vacancy configuration unfavourable. The electronic distribution within the structure is also significantly affected. This trapping of atoms between two vacancies incurs an energy penalty, which destabilises that vacancy configuration. This results in the 2nn vacancy pair configuration having the highest formation and vacancy interaction energies within several pair configurations shown in Fig. 6.
The 4nn vacancy pair configuration also traps a C atom between the two vacancies, preventing full relaxation, and resulting in a net volume increase of 0.819 Å3 per vacancy (75.1% of the isolated vacancy volume). The energy penalty associated with an unrelaxed 1nn C ion is significantly less than a 1nn Zr ion (as the displacement of the Zr atom dominates the ionic relaxation), so the 4nn pair configuration is less destabilised than the 2nn pair, but the trapping of the C atom may be used to explain why the 4nn pair is less stable than the 3nn pair. The 3nn vacancy pair configuration has four carbon atoms shared between the 1nn and 2nn carbon shells, but none of them are trapped. This results in a net volume increase of 1.039 Å3 per vacancy (95.3% of the isolated vacancy volume), which is the largest volume expansion among the various vacancy pair configurations, and close to that of an isolated atom. This correlates with 3nn vacancy pair configuration being the most stable of all the vacancy pairs considered.
The distribution of electronic charge and the local bonding was examined to further understand the differing stability of each vacancy configuration. The maximum charge density (which occurs at the nearest neighbour Zr atoms) may also be understood by considering the Zr and C atoms that occur in the nearest neighbour shells of both vacancies. Fig. 7 shows the charge density distribution for 1nn, 2nn, 3nn, and 4nn vacancy pair configurations, as well as the charge density distribution difference from the pristine distribution, and the difference from the hypothetical ideal superposition of the charge density distributions created by isolated vacancies at each vacancy site. The difference from the charge density superposition of isolated vacancies allows visualisation of the electronic interactions between the two vacancies in Fig. 7(c). From the scales in the bottom row of Fig. 7 it can be seen that the charge density distribution may largely be described as a superposition of the charge concentration effects of two isolated single vacancies, and deviations (visualised in Fig. 7(c) and given numerically in Table 2) are relatively small.
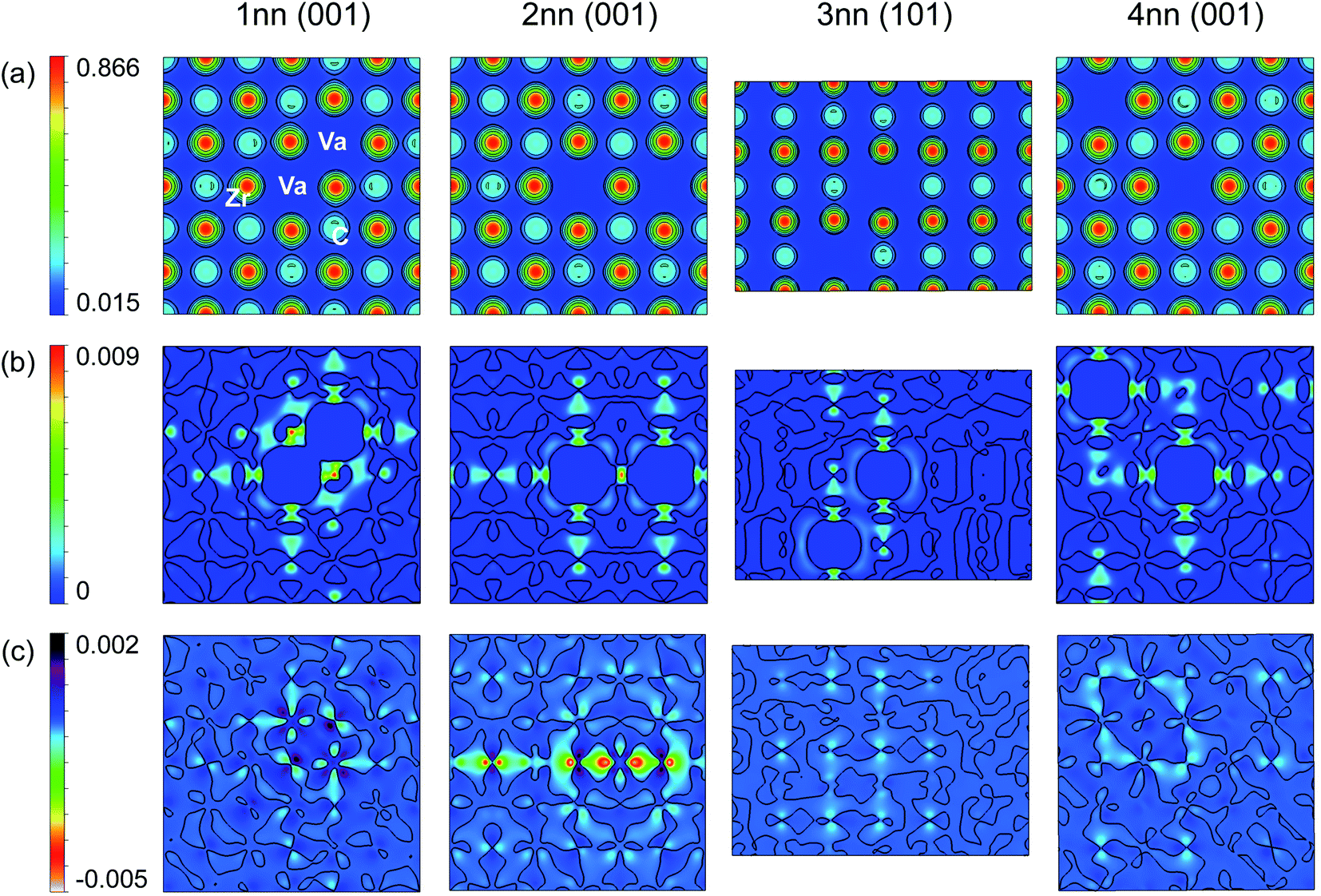 | ||
| Fig. 7 Charge density distribution (e per Bohr3) for 1nn, 2nn, 3nn, and 4nn vacancy pair configurations in the plane of the vacancies. (a) Top row: charge density distribution. (b) Middle row: the difference of the charge density distribution from the pristine case (colours show positive charge only). (c) Bottom row: the difference from the hypothetical charge density distribution arising from perfect superposition of isolated vacancy effects at each site. In (b) and (c) the contour lines separate positive and negative regions. | ||
Fig. 7(b) shows that the shared Zr atoms are more strongly charged than the unshared nearest neighbour Zr atoms in the 1nn and 2nn vacancy pair configurations. The isolated charge density distribution in Fig. 4 shows that the electron redistribution close to a vacancy results in more highly charged Zr atoms as well as stronger bonding between those Zr atoms and their nearest neighbour C atoms. In the 1nn vacancy pair case, this increased charge is approximately doubled (see values in Table 2) as each shared Zr loses two nearest neighbour C bonds compared to the pristine case. The minimum charge density at the vacancy sites is slightly lower than in the isolated vacancy case as a consequence of the vacancy–vacancy interaction, but the total charge (seen from Fig. 7(c)) and ELF at the vacancy site are higher, demonstrating that more electrons at the vacancy site are localised to that site and are less involved in the surrounding C–Zr covalent bonding. This is consistent with the average −IpCOHP for the C–Zr bonds involving the nearest neighbour Zr atoms and next nearest neighbour C atoms with reference to the vacancies. The average −IpCOHP is 4.40 eV per bond (−0.2% compared to the isolated vacancy case), demonstrating the strengthened bonding as a result of the electron cloud redistribution, but weakened compared to the isolated case by highly localised electrons at the vacancy site.
In the 2nn vacancy pair, the shared (trapped) Zr atom directly between the vacancies also has an increased charge density compared to the isolated vacancy arising from the overlapping electron clouds and the free electrons from the Zr that has lost two nearest neighbour C–Zr bonds. However, the vacancy–vacancy interactions (highly correlated to interference between competing relaxation directions) are strongest in the 2nn vacancy pair configuration (as shown in Table 2), reducing the minimum charge density at the Zr site and increasing the overall charge located at the vacancy site. The ELF at the vacancy site is also significantly increased in the 2nn vacancy pair configuration (0.853) compared to the isolated vacancy case (0.819) or any other of the considered vacancy pair configurations (see Table 2). This demonstrates that not only is there more charge at the vacancy site, but the electrons in that region are highly localised and not involved in the C–Zr bonding. The average −IpCOHP for the strengthened nearest neighbour C–Zr bonds involving the 1nn Zr shell is 4.31 eV per bond, −2.2% compared to the isolated vacancy case, but furthermore as the strengthened C–Zr bonds involve the 2nn C atom with reference to the vacancy, the number of those C–Zr bonds is reduced by the geometry of the vacancy pair. Consequently, the sum of the −IpCOHP for all 1nn C–Zr bonds involving Zr close to the vacancies is reduced by 18.5% compared to two isolated vacancies, which is significantly less than in all other pair configurations. Therefore, it can be said that the instability of the 2nn vacancy pair arises from its geometry in the rocksalt lattice.
The stability of the 3nn and 4nn vacancy pairs may also be explained by considering their geometric arrangement within the ZrC lattice. In the 3nn and 4nn vacancy pair configurations, the maximum charge density at the nearest neighbour Zr sites is very similar to the isolated vacancy case, as there are no Zr atoms within the nearest neighbour shells of both vacancies. The vacancy–vacancy interactions in the charge density are also much weaker in the 3nn and 4nn vacancy pair configurations compared to the 1nn and 2nn arrangements, however, the 4nn vacancy pair has greater interactions resulting from the trapped C atom directly between the vacancies. In the 3nn vacancy pair configuration, more of the electronic charge strengthens the closest C–Zr bonds, as seen from the low charge density, total charge, and ELF at the vacancy site compared to the isolated vacancy case. Correspondingly, the average −IpCOHP for the nearby strengthened C–Zr bonds is increased to 4.43 eV per bond, the strongest out of any pair configuration, and the only pair arrangement with stronger bonding than the isolated vacancy case (a 0.4% increase). As the bonding in zirconium carbide is dominated by the 1nn C–Zr bonds, the variation in the strength of such bonds close to the vacancies correlates with the overall stability of the structure. The increased −IpCOHP in the 3nn vacancy pair compared to the isolated vacancy case is consistent with the formation and vacancy interaction energies of each pair configuration shown in Fig. 6, where only the 3nn vacancy pair configuration is more stable than isolated vacancies. The 3nn vacancy pair configuration minimises the electronic interactions of the two vacancies through its geometric arrangement, and but the overlap of the electron clouds in this configuration also further stabilise it compared to the isolated vacancy case.
In the 4nn vacancy pair configuration, the average −IpCOHP of the strengthened C–Zr bonds is 4.33 eV per bond, 1.9% less than the isolated vacancy case, despite the electrons at the vacancy site having a similar ELF and charge density to the isolated vacancy case. The trapped C atom in this configuration becomes more highly charged, resulting in an overall weakening of the C–Zr bonding surrounding the vacancy. The trapped C atom in the nearest neighbour shells of the 4nn vacancy pair therefore destabilises that configuration over the 1nn configuration, where more C atoms are shared between shells, but none are trapped.
By examining the electronic structures and local bonding close to the vacancies in various arrangements, the differing stability of each configuration in Fig. 6 is understood in the context of the geometric features of the ZrC structure. The 2nn vacancy pair configuration traps a Zr atom between vacancies, leading to unfavourable electron redistribution, and weakened C–Zr bonding close to the vacancies compared to other vacancy arrangements. The 3nn vacancy pair configuration allows the local distortions of the lattice close to each vacancy to occur almost independently, which minimises the electronic interactions arising from two vacancies in close proximity. The electronic interactions serve to additionally stabilise the pair configuration by slightly increasing the strength of the C–Zr bonds close to the vacancies compared to the isolated vacancy case. This results in the small negative vacancy interaction energy in Fig. 6(b), that has been previously reported by Razumovskiy et al.20 and Zhang et al.17 This explanation for the differing stability of each vacancy pair configuration may also be applied to other point defects in similar rocksalt structures, although the electronic effects that arise may be material-specific.
) phases. At higher vacancy concentrations where it is no longer possible to exclusively form the fingerprint triplets, other vacancy configurations will form according to the lowest-energy arrangement possible at that concentration. The formation energy of the vacancy triplets may be broadly determined by the pair vacancy interaction energies, with vacancy–vacancy–vacancy interactions contributing less than 1 meV per vacancy in all cases, with 50% of the configurations being within 0.2 meV per vacancy, and 75% of the configurations within 0.5 meV per vacancy. All differences above 0.5 meV per vacancy arise in triplets containing 2nn pairs, where the longer range effects from the 2nn pair destabilise the other pairs in the triplet more than the sum of the individual pairs. Therefore, the stability of the ordered and disordered phases can be reasonably examined by considering the arrangements of vacancy pairs, as higher order interactions between the vacancies are relatively small.3.2. Ordered and disordered phases
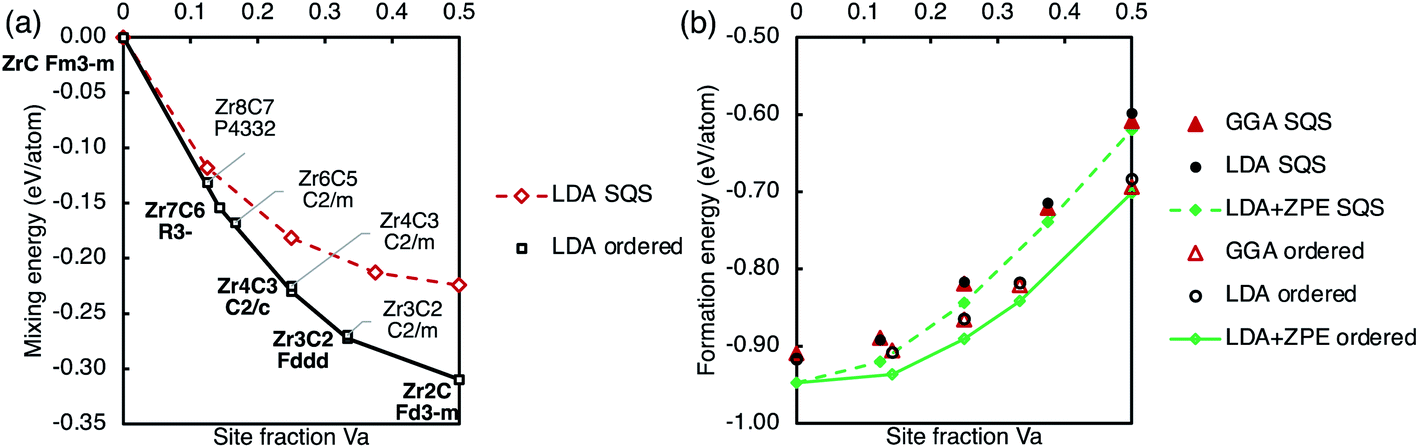 | ||
| Fig. 8 Ground state energies of the ordered phases and SQS representing the disordered phase. (a) Mixing energy (with reference to ZrC and fcc Zr) calculated with the LDA. (b) Formation energy (with reference to hcp Zr and graphite) calculated using the GGA, LDA, and LDA with ZPE included. Solid lines mark the convex hull of ordered phases, and dashed lines connect the SQS points to guide the eye. Numerical data are in Table 1. | ||
Fig. 8 also shows the mixing and formation energy of the SQS approximating the disordered phase, described above. Any scatter (deviation from a smooth trend) shown in the calculated energies of the disordered phase is an artefact of using SQS. Both ordered phases and disordered phases have same trends of the mixing energies and the formation energies becoming more negative and less negative respectively with increasing vacancy concentration. It can clearly be seen that the formation energy of the ordered phases is more negative than the disordered phase at all compositions considered, indicating stability of the ordered phases. The ordering energy, the difference between the formation energy of the ordered and disordered phases at a given composition, increases with vacancy site fraction.
A vacancy pair approximation was used to consider the average vacancy formation energy of the vacancy-ordered phases, based on the number of 1nn–4nn vacancy pairs in the structure, as done for the vacancy triplets. The vacancy formation energy derived using the pair approximation slightly underestimated the quantity calculated in the ordered phase structure and the difference varied linearly with vacancy concentration, suggesting that there are further higher-order vacancy cluster interactions yet to be considered that destabilise the structure compared to isolated vacancy clusters.
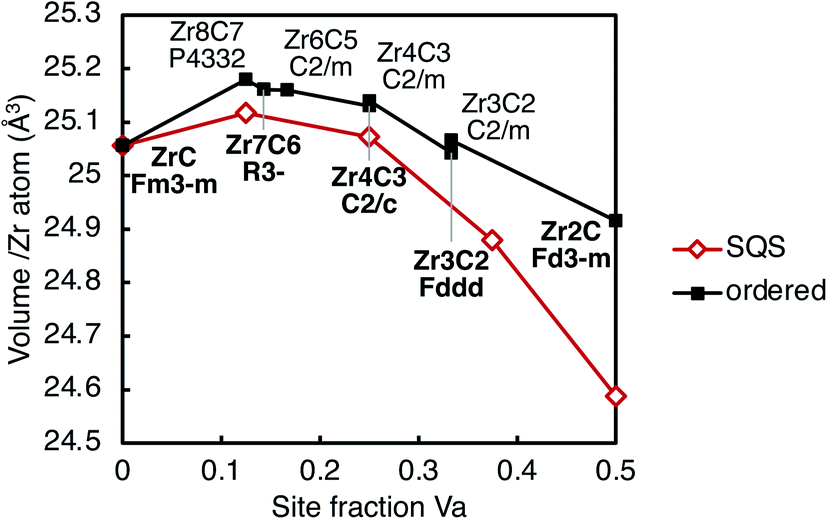 | ||
| Fig. 9 Volume per cation (Zr atom) as a function of vacancy site fraction in the ordered structures listed in Table 1 and the SQS at each vacancy concentration. Lines are shown to guide the eye only. | ||
For all ordered and SQS phases considered, the volumes have a trend of increasing with increasing vacancy concentration, up to ∼14% vacancy site fraction. At higher vacancy concentrations, the volumes decrease with increasing vacancy site fraction. This is consistent with experimental measurements of the lattice parameter, as summarised by Mitrokhin et al.56 and Gasparrini et al.57 The volumes of the sub-stoichiometric ordered structures are larger than the disordered phase (SQS) at the same composition, which is consistent with experimental measurements of the lattice parameter of Zr2C and disordered ZrCx at the same composition.13,58 The volume difference increases with vacancy site fraction. The origin of the volume variation with vacancy concentration is explained in next section in term of bond features.
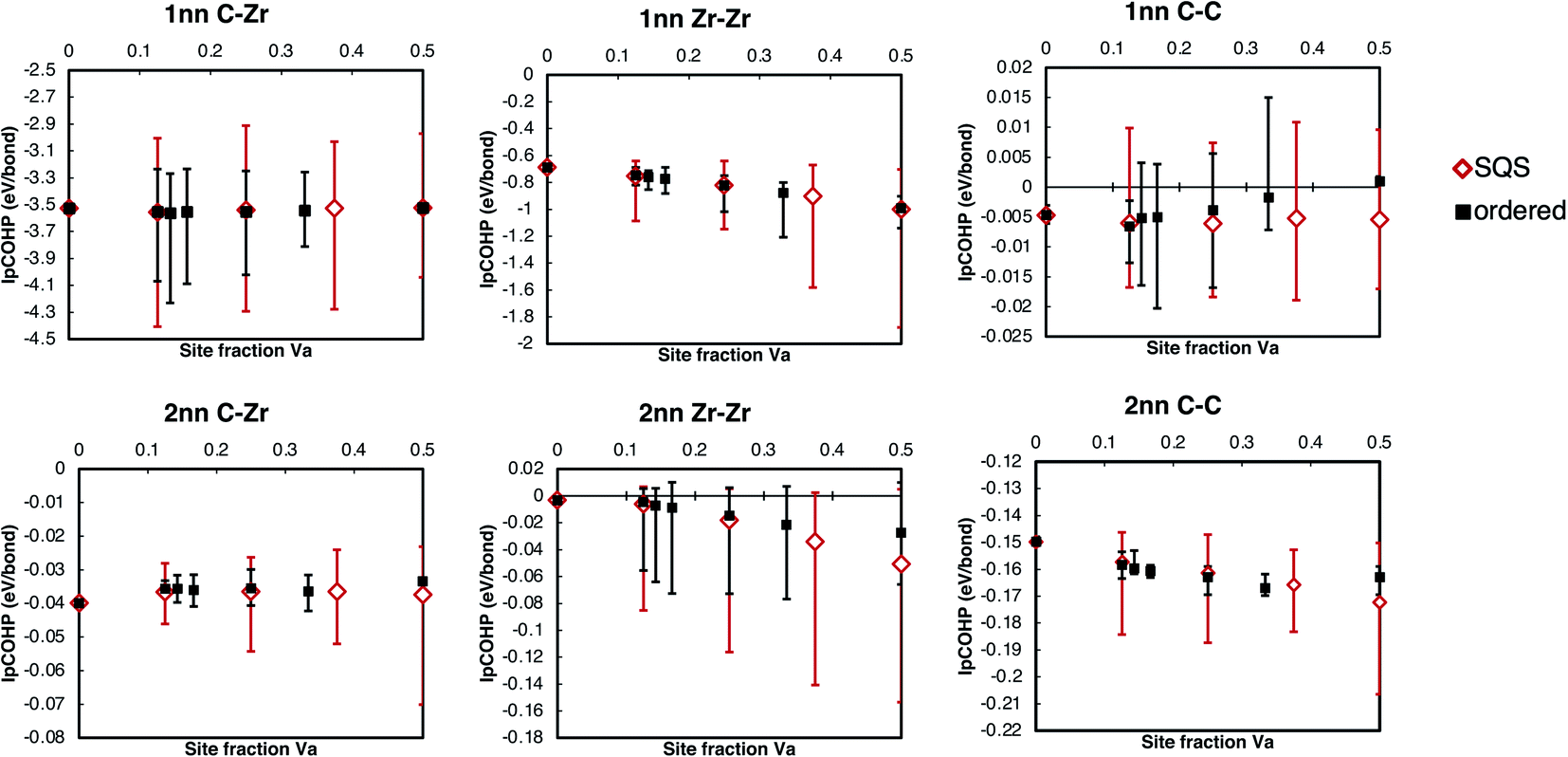 | ||
| Fig. 10 Average IpCOHP values for each bond type in vacancy-ordered and vacancy-disordered zirconium carbides with varying amounts of vacancies. The error bars indicate the highest and lowest values of each bond type found in the unit cell. | ||
Table 3 shows the number of all 1nn and 2nn bonds in each ordered and disordered (SQS) structure for pristine ZrC and the 12.5%, 25%, and 50% site fraction Va cases (normalised by the number of Zr atoms). Bonds at longer distances than this have a negligible contribution to the energy of the structure, and the 1nn C–Zr and the 1nn Zr–Zr bonds are the most significant. While the decreasing number of 1nn C–Zr bonds can explain the increasing formation energy with vacancy concentration, the disordered phases are not less stable than the ordered phase because of this effect. The number of C–X (X = Zr, C) bonds is dependent on the arrangement of vacancies in the structure. While the number of C–X bonds decreases as the vacancy site fraction increases, in each of the cases here, the number of each type of C–X bond is the same or greater in the disordered case than in the ordered structure with the same number of atoms.
| Structure | Vacancy site fraction | Number of bonds (/Zr atoms) | |||||
|---|---|---|---|---|---|---|---|
| 1nn C–Zr | 2nn C–Zr | 1nn Zr–Zr | 2nn Zr–Zr | 1nn C–C | 2nn C–C | ||
| ZrC | 0 | 6 | 8 | 6 | 3 | 6 | 3 |
| Zr8C7 (P4332) | 0.125 | 5.25 | 7 | 6 | 3 | 4.5 | 2.25 |
| 12.5% Va (SQS) | 0.125 | 5.25 | 7 | 6 | 3 | 4.59375 | 2.28125 |
| Zr4C3 (C2/c) | 0.25 | 4.5 | 6 | 6 | 3 | 3.25 | 1.5 |
| 25% Va (SQS) | 0.25 | 4.5 | 6 | 6 | 3 | 3.375 | 1.6875 |
| Zr2C (Fdm) |
0.5 | 3 | 3 | 6 | 3 | 1.5 | 0 |
| 50% Va (SQS) | 0.5 | 3 | 4 | 6 | 3 | 1.5 | 0.75 |
As the ordered phases are more stable than the disordered phase, these variations in the number of bonds arising from the local arrangement of vacancies do not explain why the ordered phases are more stable than the disordered. Instead, as expected from the isolated vacancy pair energies, the stability of each structure, ordered or disordered, is driven by the types of vacancy clusters appearing in each structure. Table 4 shows the number of each type of vacancy pair (normalised by the number of Zr atoms) in the ordered and quasirandom structures for 12.5%, 25%, and 50% site fraction Va cases, determined using Automatic FLOW for Materials Discovery software (AFLOW).59 A clear preference can be seen in the ordered structures for certain configurations of vacancy pairs, according to the formation energy of each vacancy pair configuration seen in Fig. 6, where no highly unstable 2nn vacancy pairs are present in any ordered structure.
| Structure | Vacancy site fraction | Number of vacancy pairs (/Zr atoms) | |||
|---|---|---|---|---|---|
| 1nn | 2nn | 3nn | 4nn | ||
| Zr8C7 (P4332) | 0.125 | 0 | 0 | 0.75 | 0 |
| 12.5% Va (SQS) | 0.125 | 0.1875 | 0.0625 | 0.375 | 0.1875 |
| Zr4C3 (C2/c) | 0.25 | 0.5 | 0 | 2 | 0.5 |
| 25% Va (SQS) | 0.25 | 0.75 | 0.375 | 1.5 | 1.5 |
| Zr2C (Fdm) |
0.5 | 3 | 0 | 6 | 6 |
| 50% Va (SQS) | 0.5 | 3 | 1.5 | 6 | 3 |
In Zr8C7, which has a low vacancy concentration (12.5% site fraction), all vacancies are at 3nn separation, with no other vacancy clusters present. By contrast, the SQS with the same concentration, which approximates a random distribution of vacancies on the carbon sublattice, has half as many 3nn vacancy pairs, as well as 1nn, 2nn, and 4nn pairs. As these vacancy pair configurations are less energetically favourable than the 3nn vacancy pair configuration, this results in Zr8C7 being more energetically stable than the disordered phase at the same composition.
At higher vacancy concentrations, it is not possible to arrange all vacancies at 3nn or greater distances. Repeating the same analysis for Zr4C3 (25% site fraction Va), it has 1nn pairs, 3nn, and 4nn vacancy pairs, while the corresponding disordered phase has 1nn, 2nn, 3nn, and 4nn vacancy pairs, where there are more 1nn and 4nn pairs and fewer 3nn pairs than in the ordered structure at the same composition. Therefore, based on the energies of the pair configurations, the disordered distribution of vacancies again has energetically unfavourable vacancy configurations compared to the ordered phase.
At even higher vacancy concentrations of 50% site fraction, the ordered phase Zr2C has 1nn, 3nn, and 4nn vacancy pairs (avoiding the 2nn vacancy pair), while the SQS has all four vacancy pair configurations, including three highly unstable 2nn vacancy pairs for every Zr atom present. The energy penalty associated with the formation of energetically unfavourable 2nn vacancy pairs explains the largest difference in formation energy between the vacancy-ordered and vacancy-disordered phases at this composition. Clearly, the more vacancies that are present, the more unfavourable bonds that will form in the disordered distribution, that can be avoided in the ordered distribution. Therefore, the ordering energy increases with increasing vacancy site fraction. As Zhang et al.17 proposed, when vacancies occupy more than half the carbon sublattice, 2nn vacancy pairs cannot be avoided, which limits the range of stoichiometry of the zirconium carbides.
The increasing bond strengths with increasing vacancy site fraction seen in Fig. 10 are a consequence of charge concentration near the clusters of vacancies. The C–C and Zr–Zr bonding when vacancies are present is enhanced by electron redistribution close to vacancies, as seen in the isolated vacancy and vacancy pairs. For the ordered phases, the maximum −IpCOHP (indicating the strongest local bonding) for the 1nn C–Zr bonds is consistent with the isolated and pair vacancy results above, where it is increased by the presence of vacancies, and is maximised when only 3nn vacancy pairs are present in the Zr8C7 structure.
The bond length variation and the structural properties of the different vacancy pair configurations (Table 2) may be used to explain the trend seen in the volume as a function of vacancy site fraction, for both ordered and disordered phases. At low vacancy concentration, the volume increases as the vacancy site fraction increases, but has a turning point at ∼14% site fraction of vacancies, beyond which the volume decreases as the vacancy site fraction increases, shown in Fig. 9. When a vacancy is present, there is a local expansion of the lattice close to the vacancy site, increasing the volume. When two non-isolated vacancies are present, the resulting volume increase depends on their configuration, where 3nn vacancy pairs have the largest volume increase, and 2nn vacancy pairs have the smallest.
At such low vacancy concentrations that all vacancies are isolated from other vacancies, the volume increase per vacancy is that of the isolated vacancy case. At slightly higher vacancy concentrations (up to ∼14%), vacancies may have pair interactions with only one other vacancy, in which case they are able to increase by the vacancy volume associated with that pair configuration, and therefore a higher vacancy concentration results in a larger volume. However, when more vacancies are present, competing displacements from interactions with other nearby vacancies will prevent the local expansion close to each vacancy from achieving the same volume as when vacancies are more isolated, resulting in a decreasing volume with increasing vacancy site fraction above ∼14% site fraction vacancies.
The volume increase per vacancy in each pair configuration (given in Table 2) is correlated to its energetic stability, that is, the 3nn vacancy pairs have the largest volume increase, then 1nn, 4nn, and 2nn, where the 2nn vacancy pairs have a significantly reduced vacancy volume compared to all other configurations. The ordered phases avoid the 2nn vacancy pair configuration and maximise the number of 3nn vacancy pairs, while the quasirandom structures have all types of pair configurations. In the low vacancy concentration ordered structures (<15% site fraction Va), the Zr8C7 and Zr7C6 phases have only 3nn vacancy pairs, which have the largest volume expansion. However, the quasirandom structure at the same composition comprises other vacancy pair configurations that have a smaller volume expansion than the 3nn pair. This results in the ordered phases having a larger volume than the disordered phase at the same concentration.
When the vacancy concentration is higher, there is more vacancy interaction. Fig. 11 shows the variation in IpCOHP and bond lengths for all the 1nn and 2nn bonds in ordered and quasirandom ZrCx with 25% site fraction vacancies. The IpCOHP results in Fig. 10 and 11 show that the variation of bond strengths is consistently larger in SQS than in ordered structures with the same composition. The ordered phases avoid 2nn vacancy pairs, which prevents close clusters of vacancies from forming. Such clusters of vacancies have greater vacancy interaction effects, resulting in regions of much higher and lower local charge density in the quasirandom structures compared to the ordered structures. Consequently, there is also more variation in the bond strengths and bond lengths, leading to more distortion in the structure, and consequently the volume of the disordered phase being significantly less than in the ordered structures. This is particularly evident when there are more than ∼25% site fraction disordered vacancies, where the lattice distortion arising from the vacancy interactions reduces the volume to less than that of the pristine ZrC structure.
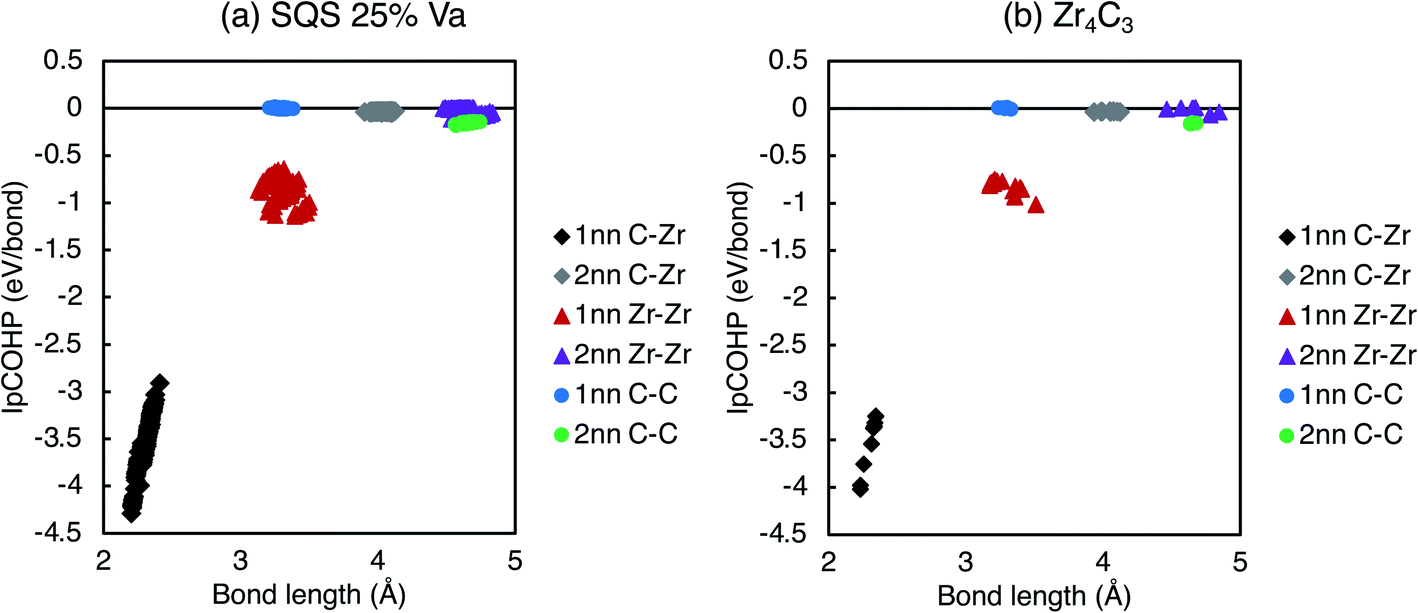 | ||
| Fig. 11 IpCOHP vs. bond length for all 1nn and 2nn bonds (C–Zr, Zr–Zr, C–C) in (a) vacancy-disordered and (b) vacancy-ordered ZrCx with 25% site fraction vacancies. | ||
4. Conclusions
First-principles calculations were used to determine the formation and mixing energies and structural properties of ordered phases and SQS representing the disordered phases between stoichiometric ZrC and ZrC0.5 where 50% of the carbon atoms are replaced by vacancies. At zero temperature, vacancy-ordered superstructural phases of zirconium carbide are found to be more stable than a random distribution of vacancies at the same composition, consistent with other theoretical works. The ordered phases are found to consistently have a larger volume than the disordered phase at the same composition. The variations in lattice parameter as a function of vacancy concentration reported in the experimental literature are generally consistent with the trends seen in the calculated disordered phase.Both the energetic stability and the volume differences between the ordered and disordered phases arise from the preference of certain vacancy configurations. Vacancies self-assemble into long-range ordered structures based on the preference of vacancy pair configurations that goes as 3nn > 1nn > 4nn > 2nn, where the 2nn vacancy pair configuration is avoided in all ordered structures. This preference for certain pair configuration is consistent with experimental observations.
The local bonding features, atomic displacement, and electron distribution were examined for isolated vacancies and various vacancy pair and triplet configurations, as well as the vacancy-ordered superstructural phases and SQS representing the disordered phase. The relative stability of each pair configuration is explained considering the geometric constraints on lattice relaxation and the electron distribution and local bonding. The preferred vacancy pair configuration is the most stable 3nn pair, which maximises the volume of each vacancy. The 3nn vacancy pair configuration allows near independent local ionic displacement and charge density distribution changes, although there is a slight effect of less charge being localised at the vacancy site resulting in stronger C–Zr bonding surrounding the vacancy complex. Consequently, the 3nn vacancy pair is slightly more stable than two independent, isolated vacancies. The 2nn vacancy pair is found to be far less stable than other pair configurations because of geometric trapping of a Zr atom directly between the vacancies, preventing local relaxation of the lattice and reducing the volume. The interactions between the vacancies in this configuration furthermore cause strong electron localisation at the vacancy sites, decreasing the strength of the bonding surrounding the vacancies, and destabilising the 2nn pair configuration. The relative stability of the 1nn and 4nn vacancy pair configurations is also explained using the same geometric and electronic arguments, and it is found that the properties of the vacancy triplets can be well described by a pair approximation. The stability and structural properties of the ordered and quasirandom phases are also well understood within this pair approximation framework.
As zirconium carbides are synthesised at high temperatures where some degree of vacancy-disordering is expected, the fully ordered structures are infrequently observed even after careful synthesis. At finite temperature, the configurational entropy contribution to the Gibbs energy of the vacancy-disordered phase will reduce the energy difference between the stable vacancy-ordered phases and the metastable disordered phase until the order-disorder transition temperature. In the fabrication process, zirconium carbides are heated to ∼2000 K, where it can be reasonably assumed that some vacancy disordering will occur. During cooling or annealing, vacancy (or carbon) diffusion drives the self-assembly of vacancies into ordered phases, however, this process is often slow and results in frozen-in disordered structures rather than the realisation of vacancy-ordered phases. The rate of vacancy diffusion is higher when more vacancies are present, hence the Zr2C ordered phase has been experimentally fabricated, whereas the near-stoichiometric phases have not yet. In addition to kinetic limitations in the fabrication processes, the presence of impurities such as oxygen is known to affect vacancy ordering,60 making fabrication of the near-stoichiometric ordered phases even more challenging. Knowledge of the stable and metastable ordered phases and their relaxed lattice distortions, ordering energies, ordering volumes, and underlying mechanisms may be helpful in selectively synthesising tuneable ordered or disordered structures in the future. Experimentally fabricated vacancy-ordered zirconium carbides are often partially disordered and coexisting with the vacancy-disordered phase. As such, recognising and solving their structures is challenging. Theoretical knowledge of the expected ordered phases can provide model data for comparison that could assist with phase or feature identification. This work also forms the basis for further studies examining the vacancy ordering at finite temperature or in the presence of impurities, that will provide theoretically predicted impurity and temperature thresholds for vacancy-ordered phase fabrication. Furthermore, the kinetic processes governing vacancy self-assembly are of great importance, and more work is needed to better understand the evolution between the ordered and disordered phases.
Data availability
The data used in this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP19K15273. The authors acknowledge the Center for Computational Materials Science of the Institute for Materials Research, Tohoku University, for the support of the supercomputing facilities.References
- R. V. Sara, The System Zirconium-Carbon, J. Am. Ceram. Soc., 1965, 48(5), 243–247, DOI:10.1111/j.1151-2916.1965.tb14729.x.
- H. F. Jackson and W. E. Lee, Properties and Characteristics of ZrC, in Comprehensive Nuclear Materials, Volume 2, Elsevier Inc., 1st edn, 2012, vol. 2, pp. 339–372 Search PubMed.
- A. Fernández Guillermet, Analysis of thermochemical properties and phase stability in the zirconium-carbon system, J. Alloys Compd., 1995, 217, 69–89, DOI:10.1016/0925-8388(94)01310-E.
- D. B. K. Rana, E. Z. Solvas, W. E. Lee and I. Farnan, An investigation of the long-range and local structure of sub-stoichiometric zirconium carbide sintered at different temperatures, Sci. Rep., 2020, 10, 3096, DOI:10.1038/s41598-020-59698-6.
- C. R. Weinberger and G. B. Thompson, Review of phase stability in the group IVB and VB transition-metal carbides, J. Am. Ceram. Soc., 2018, 101, 4401–4424, DOI:10.1111/jace.15768.
- C. Xie, A. R. Oganov, D. Li, T. T. Debela, N. Liu, D. Dong and Q. Zeng, Effects of carbon vacancies on the structures, mechanical properties, and chemical bonding of zirconium carbides: a first-principles study, Phys. Chem. Chem. Phys., 2016, 18, 12299–12306, 10.1039/c5cp07724a.
- Y. Zhou, W. G. Fahrenholtz, J. Graham and G. E. Hilmas, From thermal conductive to thermal insulating: Effect of carbon vacancy content on lattice thermal conductivity of ZrCx, J. Mater. Sci. Technol., 2021, 82, 105–113, DOI:10.1016/j.jmst.2020.11.068.
- H. Goretzki, “Neutron Diffraction Studies on Titanium-Carbon and Zirconium-Carbon Alloys, Phys. Status Solidi, 1967, 20(1), K141–K143, DOI:10.1002/pssb.19670200260.
- N. Obata and N. Nakazawa, Superlattice Formation in Zirconium-Carbon system, J. Nucl. Mater., 1976, 60, 39–42, DOI:10.1016/0022-3115(76)90115-X.
- C. H. De Novion and V. Maurice, Order and disorder in carbides and nitrides, J. Phys. Colloq., 1977, 38(C7), 211–220, DOI:10.1051/jphyscol:1977740.
- Y. Karimov, V. T. Ehm, Y. Khidirov and Y. S. Latergaus, Neutron diffraction study of ordering in titanium carbides and zirconium carbides, Izv. Akad. Nauk Uzb. SSR, Seriya Fiz. Nauk, 1979, 15(4), 81–83 Search PubMed.
- B. V. Khaenko, Ordering in cubic carbides and nitrides of group 4 and 5 transition metals, Izv. Akad. Nauk SSSR, Neorg. Mater, 1979, 15(11), 1952–1960 Search PubMed.
- W. Hu, et al., Superstructural nanodomains of ordered carbon vacancies in nonstoichiometric ZrC0.61, J. Mater. Res., 2012, 27(9), 1230–1236, DOI:10.1557/jmr.2012.72.
- B. Wei, et al., “Densification, mechanical and thermal properties of ZrC1 − x ceramics fabricated by two-step reactive hot pressing of ZrC and ZrH2 powders, J. Eur. Ceram. Soc., 2018, 38(2), 411–419, DOI:10.1016/j.jeurceramsoc.2017.09.027.
- Y. Zhou, T. W. Heitmann, E. Bohannan, J. C. Schaeperkoetter, W. G. Fahrenholtz and G. E. Hilmas, Carbon vacancy ordering in zirconium carbide powder, J. Am. Ceram. Soc., 2020, 103(4), 2891–2898, DOI:10.1111/jace.16964.
- A. I. Gusev and A. A. Rempel, Phase Diagrams of Metal-Carbon and Metal-Nitrogen Systems and Ordering in Strongly Nonstoichiometric Carbides and Nitrides, Phys. Status Solidi A, 1997, 163, 273–304, DOI:10.1002/1521-396X(199710)163:2%3C273::AID-PSSA273%3E3.0.CO;2-U.
- Y. Zhang, B. Liu and J. Wang, Self-assembly of Carbon Vacancies in Sub-stoichiometric ZrC(1-x), Sci. Rep., 2016, 5, 18098, DOI:10.1038/srep18098.
- Y. Zhang, B. Liu, J. Wang and J. Wang, Theoretical investigations of the effects of ordered carbon vacancies in ZrC1-x on phase stability and thermo-mechanical properties, Acta Mater., 2016, 111, 232–241, DOI:10.1016/j.actamat.2016.03.074.
- X. X. Yu, C. R. Weinberger and G. B. Thompson, Ab initio investigations of the phase stability in group IVB and VB transition metal carbides, Comput. Mater. Sci., 2016, 80, 341–349, DOI:10.1016/j.actamat.2014.07.070.
- V. I. Razumovskiy, A. V Ruban, J. Odqvist and P. A. Korzhavyi, Vacancy-cluster mechanism of metal-atom diffusion in substoichiometric carbides, Phys. Rev. B, 2013, 87, 054203, DOI:10.1103/PhysRevB.87.054203.
- V. I. Razumovskiy, M. N. Popov, H. Ding and J. Odqvist, Formation and interaction of point defects in group IVb transition metal carbides and nitrides, Comput. Mater. Sci., 2015, 104, 147–154, DOI:10.1016/j.commatsci.2015.03.042.
- A. Zunger, S.-H. Wei, L. G. Ferreira and J. E. Bernard, Special quasirandom structures, Phys. Rev. Lett., 1990, 65(3), 353–356, DOI:10.1103/PhysRevLett.65.353.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B, 1994, 50(24), 17953–17979, DOI:10.1103/PhysRevB.50.17953.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B, 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- M. Hacene, A. Anciaux-Sedrakian, X. Rozanska, D. Klahr, T. Guignon and P. Fleurat-Lessard, Accelerating VASP electronic structure calculations using graphic processing units, J. Comput. Chem., 2012, 33(32), 2581–2589, DOI:10.1002/jcc.23096.
- M. Hutchinson and M. Widom, VASP on a GPU: Application to exact-exchange calculations of the stability of elemental boron, Comput. Phys. Commun., 2012, 183(7), 1422–1426, DOI:10.1016/j.cpc.2012.02.017.
- G. Kresse and J. Hafner, “Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium, Phys. Rev. B, 1994, 49(20), 14251–14269, DOI:10.1103/PhysRevB.49.14251.
- G. Kresse and J. Furthmüller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B, 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- J. P. Perdew and A. Zunger, Self-interaction correction to density-functional approximations for many-electron systems, Phys. Rev. B, 1981, 23(10), 5048–5079, DOI:10.1103/PhysRevB.23.5048.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple- ERRATA, Phys. Rev. Lett., 1996, 78(7), 1396, DOI:10.1103/PhysRevLett.78.1396.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13(12), 5188–5192, DOI:10.1103/PhysRevB.13.5188.
- A. I. Duff, T. Davey, D. Korbmacher, A. Glensk, B. Grabowski, J. Neugebauer and M. W. Finnis, An improved method of calculating ab initio high-temperature thermodynamic properties with application to ZrC, Phys. Rev. B, 2015, 91, 214311, DOI:10.1103/PhysRevB.91.214311.
- S. Grimme, C. Mück-Lichtenfeld and J. Antony, Noncovalent interactions between graphene sheets and in multishell (hyper)fullerenes, J. Phys. Chem. C, 2007, 111(30), 11199–11207, DOI:10.1021/jp0720791.
- M. T. Yin and M. L. Cohen, Ground-state properties of diamond, Phys. Rev. B, 1981, 24(10), 6121–6124, DOI:10.1103/PhysRevB.24.6121.
- T. A. Mellan, A. I. Duff, B. Grabowski and M. W. Finnis, Fast anharmonic free energy method with an application to vacancies in ZrC, Phys. Rev. B, 2019, 100(2), 024303, DOI:10.1103/PhysRevB.100.024303.
- R. Dronskowski and P. E. Blöchl, Crystal orbital hamilton populations (COHP). Energy-resolved visualization of chemical bonding in solids based on density-functional calculations, J. Phys. Chem., 1993, 97, 8617–8624, DOI:10.1021/j100135a014.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Analytic projection from plane-wave and PAW wavefunctions and application to chemical-bonding analysis in solids, J. Comput. Chem., 2013, 34(29), 2557–2567, DOI:10.1002/jcc.23424.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, LOBSTER: A tool to extract chemical bonding from plane-wave based DFT, J. Comput. Chem., 2016, 37(11), 1030–1035, DOI:10.1002/jcc.24300.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, LOBSTER: Local orbital projections, atomic charges, and chemical-bonding analysis from projector-augmented-wave-based density-functional theory, J. Comput. Chem., 2020, 41(21), 1931–1940, DOI:10.1002/jcc.26353.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets, J. Phys. Chem. A, 2011, 115, 5461–5466, DOI:10.1021/jp202489s.
- R. Y. Rohling, I. C. Tranca, E. J. M. Hensen and E. A. Pidko, Correlations between Density-Based Bond Orders and Orbital-Based Bond Energies for Chemical Bonding Analysis, J. Phys. Chem. C, 2019, 123(5), 2843–2854, DOI:10.1021/acs.jpcc.8b08934.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44, 1272–1276, DOI:10.1107/S0021889811038970.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92(9), 5397–5403, DOI:10.1063/1.458517.
- B. Silvi and A. Savin, Classification of chemical bonds based on topological analysis of electron localization functions, Nature, 1994, 371(6499), 683–686, DOI:10.1038/371683a0.
- A. Savin, R. Nesper, S. Wengert and T. F. Fässler, ELF: The electron localization function, Angew. Chem., Int. Ed. Engl., 1997, 36(17), 1808–1832, DOI:10.1002/anie.199718081.
- K. Koumpouras and J. A. Larsson, Distinguishing between chemical bonding and physical binding using electron localization function (ELF), J. Phys. Condens. Matter, 2020, 32 DOI:10.1088/1361-648X/ab7fd8.
- A. van de Walle, et al., Efficient stochastic generation of special quasirandom structures, Calphad, 2013, 42, 13–18, DOI:10.1016/j.calphad.2013.06.006.
- D. Shin, R. Arróyave, Z.-K. Liu and A. Van de Walle, Thermodynamic properties of binary hcp solution phases from special quasirandom structures, Phys. Rev. B, 2006, 74, 024204, DOI:10.1103/PhysRevB.74.024204.
- K. K. Korir, G. O. Amolo, N. W. Makau and D. P. Joubert, First-principle calculations of the bulk properties of 4d transition metal carbides and nitrides in the rocksalt, zincblende and wurtzite structures, Diam. Relat. Mater., Feb. 2011, 20, 157–164, DOI:10.1016/j.diamond.2010.11.021.
- H. Fu, W. Peng and T. Gao, Structural and elastic properties of ZrC under high pressure, Mater. Chem. Phys., 2009, 115, 789–794, DOI:10.1016/j.matchemphys.2009.02.031.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A fast and robust algorithm for Bader decomposition of charge density, Comput. Mater. Sci., 2006, 36(3), 354–360, DOI:10.1016/j.commatsci.2005.04.010.
- J. Li, D. Liao, S. Yip, R. Najafabadi and L. Ecker, Force-based many-body interatomic potential for ZrC, J. Appl. Phys., 2003, 93(11), 9072–9085, DOI:10.1063/1.1567819.
- M. Råsander and A. Delin, Carbon vacancy formation in binary transition metal carbides from density functional theory, 2018, pp. 1–9, https://arxiv.org/abs/1808.07114 Search PubMed.
- V. A. Mitrokhin, R. A. Lyutikov and R. S. Yurkova, Change in the lattice constant of zirconium carbide in the region of homogeneity, Inorg. Mater., 1975, 11(6), 978–980 Search PubMed.
- C. Gasparrini, D.-S. Rana, N. Le Brun, D. Horlait, C. N. Markides, I. Farnan and W. E. Lee, On the stoichiometry of zirconium carbide, Sci. Rep., 2020, 10, 6347, DOI:10.1038/s41598-020-63037-0.
- B. Wei, Y. Wang, H. Zhang, D. Wang, S. Peng and Y. Zhou, Microstructure evolution of nonstoichiometric ZrC 0 . 6 with ordered carbon vacancies under ion irradiation, Mater. Lett., 2018, 228, 254–257, DOI:10.1016/j.matlet.2018.06.010.
- S. Curtarolo, et al., AFLOW: An automatic framework for high-throughput materials discovery, Comput. Mater. Sci., 2012, 58, 218–226, DOI:10.1016/j.commatsci.2012.02.005.
- T. Davey and Y. Chen, The effect of oxygen on the stability and volume of vacancy-ordered and -disordered ZrCx, 2021, in preparation.
Footnote |
| † Electronic supplementary information (ESI) available: The optimised POSCAR files for the ordered zirconium carbide phases, SQS, and vacancy clusters. See DOI: 10.1039/d1ra06362f |
| This journal is © The Royal Society of Chemistry 2021 |