
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodegradable antimicrobial agents − synthesis, photodegradation, and biological evaluation†
Vebjørn Eikemoa,
Leiv K. Sydnesb and
Magne O. Sydnes *a
*a
aDepartment of Chemistry, Bioscience and Environmental Engineering, Faculty of Science and Technology, University of Stavanger, NO-4036 Stavanger, Norway. E-mail: magne.o.sydnes@uis.no
bDepartment of Chemistry, University of Bergen, NO-5007 Bergen, Norway
First published on 30th September 2021
Abstract
Multi-drug resistant (MDR) bacteria are already a significant health-care problem and are making the combat of infections quite challenging. Here we report the synthesis of several new compounds containing an ethanolamine moiety, of which two exhibit promising antimicrobial activity (at the 6 μM level). All the compounds are degraded when exposed to light and form inactive products.
Introduction
Alexander Fleming's discovery of penicillin in 1928![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 started a revolution in the treatment of infections that over the years has saved millions of lives.2,3 However, the extensive use of penicillin and other antibiotics is about to make them less effective due to the development of multi-drug resistant (MDR) bacteria.4–6 Such bacteria are already a widespread problem, and several public-health organisations describe the situation as critical and predict catastrophic consequences with an annual death toll in the millions by 2050.2,7–10 Thus, there is a need for new antibiotics that both combat existing MDR bacteria and prevent formation of new ones.11
1 started a revolution in the treatment of infections that over the years has saved millions of lives.2,3 However, the extensive use of penicillin and other antibiotics is about to make them less effective due to the development of multi-drug resistant (MDR) bacteria.4–6 Such bacteria are already a widespread problem, and several public-health organisations describe the situation as critical and predict catastrophic consequences with an annual death toll in the millions by 2050.2,7–10 Thus, there is a need for new antibiotics that both combat existing MDR bacteria and prevent formation of new ones.11
There are several causes for the MDR-bacteria problems,2,7,8 but a most important factor is the excessive use, both in human health care and agriculture,4 of antibiotics, which enter the environment more or less metabolized through sewage and run-off from farmland.12–15 A proof of the alarming state of affairs is the increasing levels of antibiotic residues in drinking water, wastewater, ground water, coastal waters, and marine organisms.12,16–20 In order to stop this development, it is necessary to develop antibiotics based on scaffolds with a structural motif that will enable their degradation under ambient conditions so that accumulation in the biosphere does not take place.
Most antibiotics act inside the body, so the stability of conceivable new agents must necessarily be significant under aqueous and physiological conditions. Our search for degradable moieties was therefore turned to motifs that are known to react with light, another reagent present in abundance in the environment. Light has already been applied repeatedly to turn compounds into derivatives for medical application,21–23 and irradiation of a carbamate group has successfully been utilized by Lee et al. to facilitate the decomposition of the β-lactam moiety embedded in cephalosporanic-acid antibiotic (1) (Scheme 1) in a cascade reaction resulting in fragments with no antibiotic activity.24
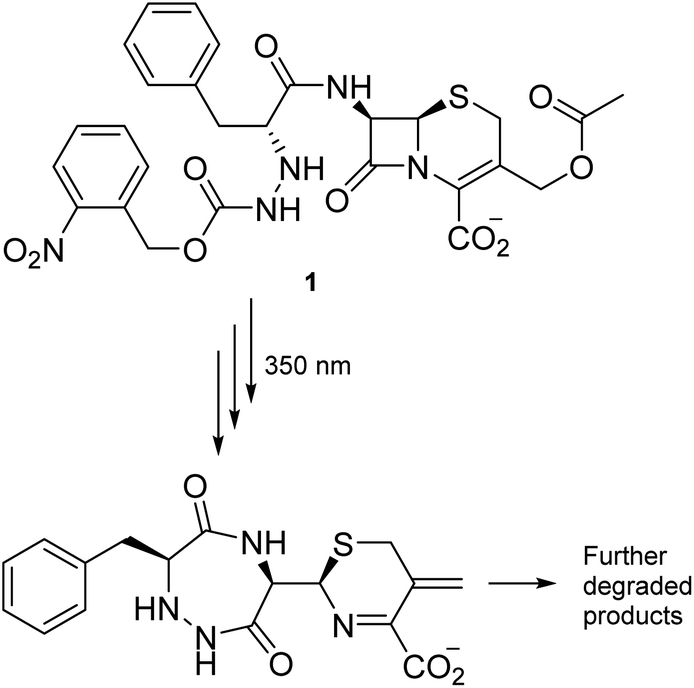 | ||
| Scheme 1 Light-induced degradation of a cephalosporanic acid analogue. | ||
The decomposition illustrated in Scheme 1 appeared to be made possible by the nitrobenzyl group, which has a well-documented reactivity in the excited state.25 This reactivity has been utilized by Wan and Muralidharan to carry out photo-retro-aldol reactions with a variety of 1-(4-nitrobenzyl)-1-alkanols, the result of which was fragmentation and formation of 4-nitrotoluene, 4,4′-dinitrobibenzyl, and the corresponding alkanal in yields that were pH dependent (Scheme 2).26 We therefore speculated if such alkanols have the potential to function as scaffolds for new photodegradable antibiotics provided substituents that induce biological activity are attached in appropriate positions (Fig. S1†).
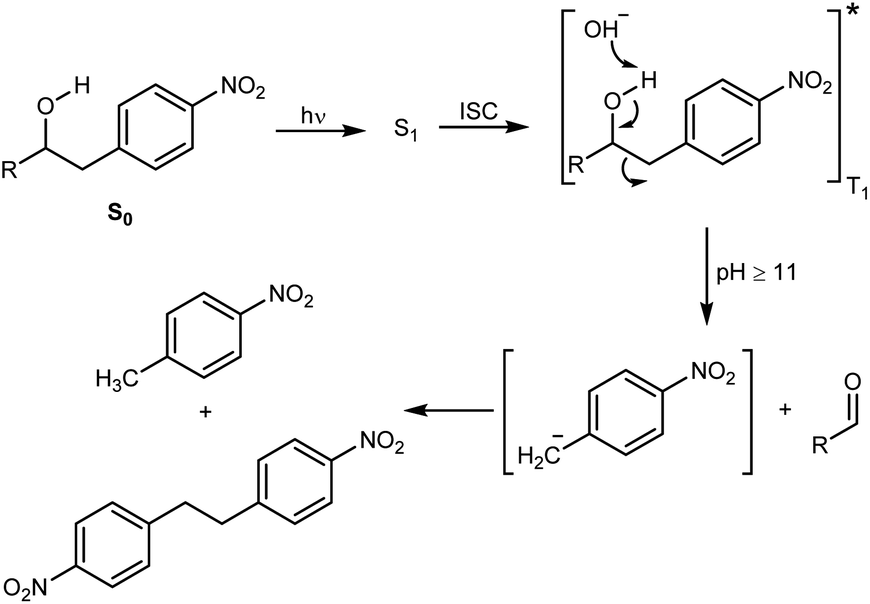 | ||
| Scheme 2 The photo-retro-aldol type reaction of nitrophenethyl alcohols. R = Ph, CH3. Irradiation with a 254, 300, or 350 nm lamp yields the first excited singlet state (S1), which undergoes intersystem crossing (ISC) and formation of triplet-excited (T1) alkanols that suffer fragmentation. | ||
Synthesis
The substituents we had in mind were halogenated anilines that were present in some biologically active compounds prepared and tested for other purposes (Fig. S2†). In order to make the alkanols as biocompatible as possible, we opted for 2-(arylamino)-1-(nitrobenzyl)ethanol derivatives (2–5), which were prepared in quite satisfactory yields by application of standard coupling and oxidation reactions (Scheme 3). | ||
| Scheme 3 Synthesis of aminols 2–5. Reagents and conditions: (i) Pd(PPh3)4, CsF, AllylBpin, THF, reflux;27 (ii) mCPBA, DCM, rt; (iii) 5 M LPDE, 40 °C; (iv) H2SO4, HNO3, 0 °C; (v) Pd(PPh3)4, Bu3SnAllyl, DMF, 110 °C. | ||
To this end, a Suzuki–Miyaura cross-coupling reaction with allylboronic acid pinacol ester, using a method described by Kotha and co-workers,27 yielded the allylated product (Scheme 3). Subsequent treatment with mCPBA gave the corresponding epoxide and a Lewis acid-promoted epoxide ring-opening reaction using 5 M lithium perchlorate-diethyl ether (LPDE) solution gave aminol 2. The remaining three compounds were prepared by a selective meta nitration followed by a Stille cross-coupling reaction with allyltributylstannane yielding allylbenzene. Subsequent epoxidation with mCPBA gave the required epoxide and a Lewis acid-promoted epoxide ring opening with anilines A, B, and C gave aminols 3–5.
Photodecomposition
The photodegradation of compounds 2–5 was studied at pH 8 and 13 following the procedure reported by Wan and Muralidharan26 using a low-pressure mercury-vapor lamp emitting light mainly at 254 nm (Fig. S3†). The reaction was monitored by proton NMR spectroscopy, which proved that almost complete conversion was achieved after 24 hours, for compounds 2–4 at pH 13 and compounds 3 and 5 at pH 8 (Table S1†). Control experiments show that compounds 2–5 are stable for more than 24 hours in the dark at pH 8 and compound 2 is even stable at pH up to 13 for 24 hours. The changes that took place are illustrated by three spectra associated with photolysis of aminol 2. Sharp, intense peaks in the spectrum of 2 (A in Fig. 1) are essentially absent in the spectrum of the reaction mixture after irradiation for 24 hours (B in Fig. 1), which on the other hand, contains strong peaks due to aniline A (Scheme 3; C in Fig. 1) formed during the degradation of 2 (see also Fig. S4†). MS analysis of the decomposition mixture also disclosed a mass equivalent to para-nitrobenzoic acid, which must have derived from the corresponding aldehyde formed under the photodecomposition.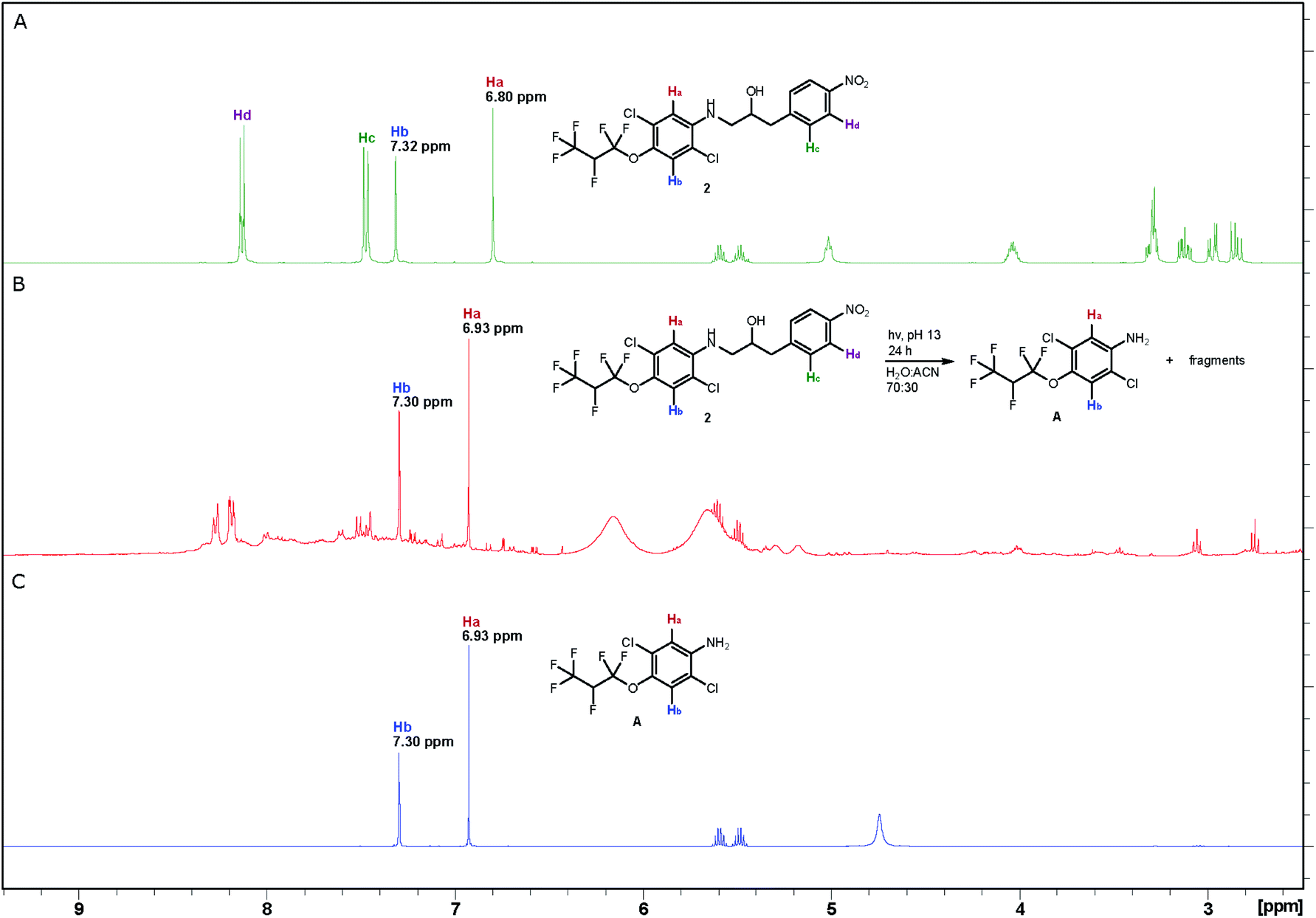 | ||
| Fig. 1 1H NMR spectra (A) compound 2 (0.7 mM) before irradiation; (B) reaction mixture after photolysis of compound 2 for 24 hours; (C) decomposition product (A). The NMR analyses were recorded in CD3CN at 400 MHz. | ||
Antibacterial activity
With compounds 2–5 and the corresponding crude degradation mixtures (2d–5d) at hand, the minimum inhibitory concentration (MIC) was determined by screening the compounds against several laboratory strains of Gram-positive and Gram-negative bacteria, including E. faecalis, E. coli, P. aeruginosa, S. aureus, S. agalactiae, and S. epidermidis (Tables 1 and S2†). As the results in Table 1 show, compounds 2 and 5 inhibited the growth of S. agalactiae at 6.3 μM while compounds 3 and 4 inhibited bacteria growth at 50 μM. The corresponding degradation product mixtures exhibited no antimicrobial activity whatsoever and showed no toxicity towards MRC5 and HepG2 cell lines either (Table 1). However, the two most active antimicrobial agents, viz. compounds 2 and 5, were somewhat toxic (Table 1), but at a much higher concentration then that required to reveal antimicrobial activity.| Compound | MIC | Tox | |||
|---|---|---|---|---|---|
| S. epidermidis | S. aureus | S. agalactiae | MRC5 | HepG2 | |
| a I = inactive at the tested concentrations. | |||||
| 2 | I | I | 6.3 | 50 | 75 |
| 3 | 50 | 50 | 50 | 50 | 75 |
| 4 | I | 50 | I | 50 | 50 |
| 5 | I | I | 6.3 | 25 | 50 |
| 2d | I | I | I | I | I |
| 3d | I | I | I | I | I |
| 4d | I | I | I | I | I |
| 5d | I | I | I | I | I |
Experimental
General information
IR spectra were recorded on an Agilent Cary 630 FT-IR spectrophotometer equipped with an attenuated total reflectance (ATR) attachment. Samples were analysed neat on a ZnSe crystal and the absorption frequencies are given in wave numbers (cm−1).UV-vis spectra were obtained on an Agilent 8453 single-beam UV-vis spectrophotometer with a deuterium-discharge lamp for the UV range and a tungsten lamp for the visible wavelength range. Samples were analysed in an Agilent open-top UV quartz cell (10 mm, 3.0 mL) with ethanol as solvent. The wavelengths are reported in nm and molar attenuation coefficients in M−1 cm−1.
NMR spectra were recorded on a Bruker Ascend™ 400 spectrometer (400.13 MHz for 1H, 100.61 MHz for 13C, 376.46 MHz for 19F) or a Bruker Ascend™ 850 spectrometer (850.13 MHz for 1H and 213.77 MHz for 13C). Coupling constants (J) are given in Hz and the multiplicity is reported as singlet (s), doublet (d), triplet (t), sextet (sxt), multiplet (m), and broad singlet (bs). The chemical shift are reported in ppm in the order downfield to upfield and calibration is done using the residual solvent signals for chloroform-d (1H 7.26 ppm; 13C 77.16 ppm) or acetonitrile-d3 (1H 1.94 ppm; 13C 1.32 ppm).28 Calibration for 19F NMR is done using α,α,α-trifluorotoluene as internal standard in chloroform-d (−62.61 ppm) and acetonitrile-d3 (−63.10 ppm).29
High-resolution mass spectra were obtained on a JEOL AccuTOF™ T100GC mass spectrometer. The instrument was operated with an orthogonal electrospray ionization source (ESI), an orthogonal accelerated time of flight (TOF) single stage reflectron mass analyzer and a dual micro channel plate (MCP) detector at the following instrumental settings/conditions; ionization mode: positive, desolvating temperature/ion source temperature = 250 °C, needle voltage = 3000 V, desolvation gas flow = 2.0 L min−1, nebulising gas flow = 1.0 L min−1, orifice1 temperature = 120 °C, orifice1 voltage = 24 V, ring lens voltage = 12 V, orifice2 voltage = 6 V, ion guide peak voltage = 800 V, detector voltage = 2300 V, acquisition range = 4–1000 m/z, spectral recording interval = 0.5 s, wait time = 0.03 ns and data sampling interval = 0.5 ns. Mass calibration were performed using the internal standard method and mass drift compensation was performed in each acquisition.
Thin-layer chromatography (TLC) was carried out with silica gel (60 F254) on aluminium sheets with solvent systems consisting of various mixtures of petroleum ether, ethyl acetate, and dichloromethane. Visualization was achieved with either UV light (254 and/or 365 nm) or a potassium permanganate stain. Flash chromatography was performed with a hand pump and 230–400 mesh silica gel or an Interchim Puriflash 215 autoflash chromatography system with Biotage Snap Ultra HP-Sphere™ 25 μm silica-gel columns.
General procedure
:DCM, 3:7), and concentration of the relevant fractions yielded the nitrophenethyl alcohols.:EtOAc, 80:20 v/v)) yielded 1-allyl-4-nitrobenzene (0.35 g, 52%) as a slightly yellow liquid. The spectroscopic data were in accordance with previously reported data.30IR (neat): νmax 3091, 3017, 2918, 1593, 1502; 1H NMR (400.13 MHz, CDCl3): δ 8.15 (d, J = 8.8 Hz, 2H), 7.34 (d, J = 8.8 Hz, 2H), 5.99–5.89 (m, 1H), 5.18–5.09 (m, 2H), 3.49 (d, J = 6.7 Hz, 2H); 13C NMR (100.61 MHz, CDCl3): δ 147.9, 146.7, 135.6, 129.5, 123.8, 117.5, 40.0.
:EtOAc:DCM, 93:2:5 → 40:55:5) and concentration of the relevant fractions (Rf = 0.26 (pet. ether:DCM, 50:50 v/v)) yielded 2-(4-nitrobenzyl)oxirane as a yellow oily liquid (0.20 g, 52%). The spectroscopical data were in full accord with the previously reported data.31IR (neat): νmax 2957, 2923, 2853, 1599, 1516, 1345; 1H NMR (400.13 MHz, CDCl3): δ 8.18 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 8.5 Hz, 2H), 3.20–3.16 (m, 1H), 3.06 (dd, J = 14.8 Hz, 4.2 Hz, 1H), 2.90 (dd, J = 14.8 Hz, 6.4 Hz, 1H), 2.84 (dd, J = 4.7 Hz, 4.0 Hz, 1H), 2.55 (dd, J = 4.7 Hz, 2.6 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 147.1, 145.0, 130.0, 123.9, 51.7, 46.8, 38.6.
:DCM, 3:7)) as a white solid (87 mg, 50%, mp. 121–123 °C) along with 40% recovery of epoxide.IR (neat): νmax 3507, 3420, 3376, 2980, 2918, 2857, 1599; UV/vis (EtOH): λmax 255 nm (ε 18339 M−1 cm−1); 1H NMR (850.13 MHz, CD3CN): δ 8.14 (d, J = 8.7 Hz, 2H), 7.49 (d, J = 8.7 Hz, 2H), 7.33 (s, 1H), 6.81 (s, 1H), 5.55 (dsxt, J = 42.8 Hz, 5.8 Hz, 1H), 5.03 (t, J = 5.6 Hz, NH), 4.06–4.02 (m, 1H), 3.30 (ddd, J = 13.3 Hz, 6.4 Hz, 3.9 Hz, 1H), 3.27 (d, J = 4.4 Hz, OH), 3.15–3.11 (m, 1H), 2.98 (dd, J = 13.8 Hz, 4.5 Hz, 1H), 2.85 (dd, J = 13.8 Hz, 8.4 Hz, 1H); 13C NMR (213.77 MHz, CD3CN): δ 148.2, 147.7, 145.2, 134.5, 131.5, 127.9, 125.6, 124.3, 121.3 (qd, J = 281 Hz, 25 Hz), 119.1 (td, J = 271 Hz, 23 Hz), 117.7, 112.6, 85.4 (dsxt, J = 198 Hz, 35 Hz), 70.8, 49.7, 41.7; 19F NMR (376.46 MHz, CD3CN): δ −75.6–75.7 (m, 3F), −78.4–80.4 (m, 2F), −213.3 (sxt, J = 12 Hz, 1F); HRMS: (ESI/TOF) m/z: [M + Na]+ calcd for C18H14Cl2F6N2O4Na+ 529.01270; found 529.01286.
IR (neat): νmax 3099, 1916, 1591, 1530; 1H NMR (400.13 MHz, CDCl3): δ 8.12 (ddd, J = 9.4 Hz, 8.0 Hz, 5.5 Hz, 1H), 7.14 (ddd, J = 9.4 Hz, 7.0 Hz, 2.0 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 163.1 (dd, J = 260 Hz, 3 Hz), 154.3 (dd, J = 267 Hz, 5 Hz), 134.8, 126.2 (dd, J = 10 Hz, 2 Hz), 112.1 (dd, J = 24 Hz, 4 Hz), 101.3 (dd, J = 25 Hz, 24 Hz); 19F NMR (376.46 MHz, CDCl3): δ −92.0 (d, J = 9.5 Hz), −104.4 (d, J = 9.5 Hz); HRMS: (EI/TOF) m/z: [M]+ calcd for C6H2BrF2NO2+ 236.92315; found 236.92306.
:EtOAc, 99:1) and concentration of the relevant fractions (Rf = 0.32 (pet. ether:EtOAc, 95:5, v/v)) yielded 2-allyl-1,3-difluoro-4-nitrobenzene as a colourless liquid (194 mg, 27%).IR (neat): νmax 3087, 2957, 2925, 2855, 1623, 1596; 1H NMR (400.13 MHz, CDCl3): δ 8.01 (ddd, J = 9.0 Hz, 8.5 Hz, 5.7 Hz, 1H), 7.04–6.99 (m, 1H), 5.95–5.85 (m, 1H), 5.13–5.08 (m, 2H), 3.51–3.48 (m, 2H); 13C NMR (100.61 MHz, CDCl3): δ 164.2 (dd, J = 258 Hz, 8 Hz), 155.2 (dd, J = 266 Hz, 10 Hz), 134.5–134.4 (m), 132.9, 125.4 (dd, J = 11 Hz, 2 Hz), 118.8 (dd, J = 22 Hz, 19 Hz), 117.4, 111.7 (dd, J = 25 Hz, 4 Hz), 26.8 (t, J = 3 Hz); 19F NMR (376.46 MHz, CDCl3): δ −102.5 (d, J = 14.1 Hz), −116.6 (d, J = 14.1 Hz); HRMS: (EI/TOF) m/z: [M]+ calcd for C9H7F2NO2+ 199.04394; found 199.04399.
:1 sat. aq. NaHCO3:10% Na2S2O3 solution (30 mL). The phases were separated and the aqueous layer was extracted with DCM (3 × 15 mL). The combined organic phases were washed with a 1:1 sat. aq. NaHCO3:10% Na2S2O3 solution (30 mL), sat. aq. NaHCO3 (30 mL), water (30 mL), dried (MgSO4), filtered and concentrated under reduced pressure to yield 2-(2,6-difluoro-3-nitrobenzyl)oxirane as a slightly yellow oily liquid (183 mg, 88%). The product was used without further purification.IR (neat): νmax 3104, 3000, 2926, 1728, 1624; 1H NMR (400.13 MHz, CDCl3): δ 8.05 (ddd, J = 9.2 Hz, 8.5 Hz, 5.7 Hz, 1H), 7.07–7.02 (m, 1H), 3.22–3.17 (m, 1H), 3.16–3.11 (m, 1H), 3.03–2.97 (m, 1H), 2.81–2.79 (m, 1H), 2.56 (dd, J = 4.8 Hz, 2.5 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 164.5 (dd, J = 259 Hz, 8 Hz), 155.5 (dd, J = 266 Hz, 9 Hz), 134.5, 126.1 (dd, J = 11 Hz, 1 Hz), 115.8 (dd, J = 22 Hz, 20 Hz), 111.8 (dd, J = 25 Hz, 4 Hz), 50.1, 46.9, 25.8 (t, J = 2 Hz); 19F NMR (376.46 MHz, CDCl3): δ −101.5 (d, J = 13.6 Hz), −115.7 (d, J = 13.6 Hz); HRMS; (EI/TOF) m/z: [M–C2H2O]+ calcd for C7H4F2NO2+ 172.02046; found 172.02080.
:7 (Rf = 0.36)) as a sticky colourless oil (60 mg, 42%) along with 7% recovery of epoxide.IR (neat): νmax 3565, 3425, 3101, 2926, 2857, 1597; UV/vis (EtOH): λmax 254 nm (ε 16575 M−1 cm−1); 1H NMR (400.13 MHz, CD3CN): δ 8.05 (ddd, J = 9.3 Hz, 8.5 Hz, 5.7 Hz, 1H), 7.14 (ddd, J = 9.3 Hz, 8.5 Hz, 1.9 Hz, 1H), 6.73–6.69 (m, 2H), 4.93 (bs, NH), 4.06–3.98 (m, 1H), 3.34 (d, J = 5.3 Hz, OH), 3.32 (ddd, J = 13.5 Hz, 6.7 Hz, 4.0 Hz, 1H), 3.20–3.13 (m, 1H), 2.99–2.87 (m, 2H); 13C NMR (100.61 MHz, CD3CN): δ 165.5 (dd, J = 256 Hz, 8 Hz), 156.2 (dd, J = 263 Hz, 10 Hz), 146.8 (d, J = 239 Hz), 139.9 (d, J = 12 Hz), 135.5, 130.5 (d, J = 4 Hz), 126.7 (dd, J = 12 Hz, 2 Hz), 121.4 (d, J = 16 Hz), 118.6 (dd, J = 22 Hz, 19 Hz), 116.5 (d, J = 1 Hz), 112.6 (dd, J = 25 Hz, 4 Hz), 111.6 (d, J = 4 Hz), 69.3, 49.5, 29.1; 19F NMR (376.46 MHz, CD3CN): δ −103.0 (d, J = 13.6 Hz, 1F), −117.5 (d, J = 13.6 Hz, 1F), −141.7 (s, 1F); HRMS: (ESI/TOF) m/z: [M + H]+ calcd for C15H12Cl2F3N2O3+ 395.01716; found 395.01724.
:7)) as a white solid (45 mg, 33%, mp. 110–112 °C) along with 46% recovery of epoxide.IR (neat): νmax 3433, 3382, 3103, 2935, 2857, 1625; UV/vis (EtOH): λmax 242 nm (ε 16289 M−1 cm−1); 1H NMR (400.13 MHz, CD3CN): δ 8.04 (ddd, J = 9.2 Hz, 8.6 Hz, 5.7 Hz, 1H), 7.13 (ddd, J = 9.2 Hz, 8.5 Hz, 1.8 Hz, 1H), 6.79 (dd, J = 8.6 Hz, 7.1 Hz, 1H), 4.72 (bs, NH), 4.05–3.97 (m, 1H), 3.34 (d, J = 5.3 Hz, OH), 3.92 (ddd, J = 13.4 Hz, 6.6 Hz, 4.1 Hz, 1H), 3.16–3.10 (m, 1H), 2.98–2.87 (m, 2H); 13C NMR (100.61 MHz, CD3CN): δ 165.5 (dd, J = 256 Hz, 8 Hz), 156.2 (dd, J = 263 Hz, 10 Hz), 146.9 (dd, J = 242 Hz, 1 Hz), 146.3 (dd, J = 237 Hz, 2 Hz), 135.7 (dd, J = 12 Hz, 3 Hz), 135.4 (dd, J = 8 Hz, 3 Hz), 126.7 (d, J = 12 Hz), 118.5 (dd, J = 22 Hz, 20 Hz), 117.3 (dd, J = 18 Hz, 4 Hz), 112.6 (dd, J = 25 Hz, 4 Hz), 111.4–111.0 (m, 1C), 111.2 (d, J = 4 Hz), 69.4, 49.7, 29.1; 19F NMR (376.46 MHz, CD3CN): δ −103.0 (d, J = 14.0 Hz, 1F), −117.5 (d, J = 14.0 Hz, 1F), −133.9 (d, J = 4.0 Hz, 1F), −137.0 (d, J = 4.0 Hz, 1F); HRMS: (ESI/TOF) m/z: [M + H]+ calcd for C15H11Cl2F4N2O3+ 413.00774; found 413.00790.
:7)) as a sticky colourless oil (104 mg, 42%) along with 16% recovery of epoxide.IR (neat): νmax 3550, 3411, 3103, 2937, 1624; UV/vis (EtOH): λmax 254 nm (ε 25248 M−1 cm−1); 1H NMR (850.13 MHz, CD3CN): δ 8.06–8.04 (m, 1H), 7.33 (s, 1H), 7.15–7.13 (m, 1H), 6.86 (s, 1H), 5.55 (dsxt, J = 42.7 Hz, 5.8 Hz, 1H), 5.04 (t, J = 5.7 Hz, NH), 4.07–4.04 (m, 1H), 3.39 (d, J = 5.2 Hz, OH), 3.36 (ddd, J = 13.3 Hz, 6.4 Hz, 4.1 Hz, 1H), 3.22–3.19 (m, 1H), 2.97 (dd, J = 13.9 Hz, 4.8 Hz, 1H), 2.93 (dd, J = 13.9 Hz, 8.3 Hz, 1H); 13C NMR (213.77 MHz, CD3CN): δ 165.5 (dd, J = 256 Hz, 8 Hz), 156.2 (dd, J = 263 Hz, 10 Hz), 145.2, 135.5 (d, J = 5 Hz), 134.5, 127.9, 126.7 (d, J = 12 Hz), 125.6, 121.3 (qd, J = 281 Hz, 25 Hz), 119.1 (td, J = 271 Hz, 23 Hz), 118.5 (dd, J = 22 Hz, 19 Hz), 117.7, 112.73–112.60 (m, 1C), 112.71, 85.4 (dsxt, J = 198 Hz, 35 Hz), 69.3, 49.6, 29.2; 19F NMR (376.46 MHz, CD3CN): δ −75.6–75.7 (m, 3F), −78.5–80.4 (m, 2F), −102.9 (d, J = 14.0 Hz, 1F), −117.4 (d, J = 14.0 Hz, 1F), −213.3 (sxt, J = 11.2 Hz, 1F); HRMS: (ESI/TOF) m/z: [M + H]+ calcd for C18H13Cl2F8N2O4+ 543.01191; found 543.01196.
Photodegradation reactions at ∼0.7 mM for NMR analysis
All reactions were carried out in a 75 mL gas inlet flask (∼0.25–0.35 mM) under a constant stream of nitrogen with a quartz well water-cooled cold finger for 24 hours. The appropriate aminol (∼0.02 mmol) was dissolved in acetonitrile (23 mL), diluted with distilled water adjusted to pH 8 with sodium hydroxide (52 mL), and photolyzed using a 254 nm low-pressure mercury lamp. The aqueous solution was extracted with DCM (3 × 20 mL) and the combined organic layers were dried (MgSO4), filtered, and concentrated to yield a black residue, which was analysed by 1H NMR spectroscopy.Conclusions
In conclusion, two compounds based on a new structural scaffold, 2 and 5, possess both antimicrobial activity at the 6 μM level and the ability to decompose upon exposure to light. The decomposition products do not exhibit antimicrobial activity or toxicity. This opens for the development of antibiotics that when released to the environment after treatment will be removed and not contribute to the evolution of MDR bacteria. Both 2 and 5 were not active towards Gram-negative bacteria, but work by Richter et al. outlines techniques that can be applied to expand their capability to encompass Gram-negative bacteria as well.32 Work directed towards this is ongoing in these laboratories.Author contributions
LKS and MOS had the initial idea; VE, LKS, and MOS developed the idea; VE performed the experimental work; VE drafted the manuscript; VE, LKS, and MOS revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by University of Stavanger, the Plogen program (IN-12440), and Norsk Hydros fond til vitenskapelig forskning (IN-12634). Thanks are also due to Dr B. Holmelid and Dr Jarl Underhaug, University of Bergen, for skilful performance of HRMS and 850 MHz NMR analyses, respectively. Dr M. Albrigtsen and Professor J. H. Andersen at Marbio, UiT the Arctic University of Norway are thanked for performing the biological assays.Notes and references
- A. Fleming, Br. J. Exp. Pathol., 1929, 10, 226 CAS.
- C. L. Ventola, Pharmacol. Ther., 2015, 40, 277 Search PubMed.
- K. C. Nicolaou and S. Rigol, J. Antibiot., 2018, 71, 153 CrossRef CAS PubMed.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336 CrossRef CAS PubMed.
- C. Manyi-Loh, S. Mamphweli, E. Meyer and A. Okoh, Molecules, 2018, 23, 795 CrossRef PubMed.
- World Health Organization, Prioritization of pathogens to guide discovery, research and development of new antibiotics for drug-resistant bacterial infections, including tuberculosis, 2017, https://www.who.int/medicines/areas/rational_use/PPLreport_2017_09_19.pdf, accesed July 9, 2021 Search PubMed.
- R. Laxminarayan, A. Duse, C. Wattal, A. K. M. Zaidi, H. F. L. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057 CrossRef PubMed.
- V. K. Visvanathan, Gut Microbes, 2014, 5, 3 CrossRef PubMed.
- M. McKenna, Nature, 2020, 584, 338 CrossRef CAS PubMed.
- M. May, Nat. Med., 2021, 27, 358 CrossRef CAS PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089 CrossRef CAS PubMed.
- M. C. Danner, A. Robertson, V. Behrends and J. Reiss, Sci. Total Environ., 2019, 664, 793 CrossRef CAS PubMed.
- K. Kümmerer, J. Antimicrob. Chemother., 2004, 54, 311 CrossRef PubMed.
- S. Berkner, S. Konradi and J. Schönfeld, EMBO Rep., 2014, 15, 740 CrossRef CAS PubMed.
- A. Anadón, M. R. Martínez-Larranaga, J. Iturbe, M. A. Martínez, M. J. Díaz, M. T. Frejo and M. Martínez, Res. Vet. Sci., 2001, 71, 101 CrossRef PubMed.
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319 CrossRef CAS PubMed.
- K. Kümmerer, Chemosphere, 2009, 75, 417 CrossRef PubMed.
- C. Y. Ojemaye and L. P. Petrik, Environ. Rev., 2019, 27, 151 CrossRef.
- I. Sanseverino, A. N. Cuenca, R. Loos, D. Marinov and T. Lettieri, JRC Technical Reports, State of the Art on the Contribution of Water to Antimicrobial Resistance-State of the Art on the Contribution of Water to Antimicrobial Resistance, 2018, file:///C:/Users/2908143/Downloads/amr__jrc_technical__report_final_online_15jan.2019.pdf, accesed July 9, 2021 Search PubMed.
- I. T. Carvalho and L. Santos, Environ. Int., 2016, 94, 736 CrossRef PubMed.
- M. J. Fuchter, J. Med. Chem., 2020, 63, 11436 CrossRef CAS PubMed.
- W. Velema, J. P. van der Berg, M. Hansen, W. Szymanski, A. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924 CrossRef CAS PubMed.
- W. A. Velema, M. J. Hansen, M. M. Lerch, A. J. M. Driessen, W. Szymanski and B. L. Feringa, Bioconjugate Chem., 2015, 26, 2592 CrossRef CAS PubMed.
- W. Lee, Z.-H. Li, S. Vakulenko and S. Mobashery, J. Med. Chem., 2000, 43, 128 CrossRef CAS PubMed.
- P. Wang, Asian J. Org. Chem., 2013, 2, 452 CrossRef CAS.
- P. Wan and S. Muralidharan, J. Am. Chem. Soc., 1988, 110, 4336 CrossRef CAS.
- S. Kotha, M. Behara and V. Shah, Synlett, 2005, 1877 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176 CrossRef CAS.
- C. P. Rosenau, B. J. Jelier, A. D. Gossert and A. Togni, Angew. Chem., Int. Ed. Engl., 2018, 57, 9528 CrossRef CAS PubMed.
- M. O. Akram, P. S. Mali and N. T. Patil, Org. Lett., 2017, 19, 3075 CrossRef CAS PubMed.
- D. J. Bartkovitz, X.-J. Chu, Q. Ding, N. Jiang, A. J. Lovey, J. A. Moliterni, J. G. Mullin, B. T. Vu and P. M. Wovkulich, Preparation of (4-Aminopyrimidin-5-yl)[(hetero)aryl]methanones as cyclin-dependent kinase inhibitors for treating solid tumors, WO, 2004/069139A2, 2004 Search PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures for antimicrobial activity and cytotoxicity testing and NMR spectra of new compounds. See DOI: 10.1039/d1ra06324c |
| This journal is © The Royal Society of Chemistry 2021 |