
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhich fraction of stone wool fibre surface remains uncoated by binder? A detailed analysis by time-of-flight secondary ion mass spectrometry and X-ray photoelectron spectroscopy
Sabine
Hirth
 *,
Hubert
Waindok
and
Wendel
Wohlleben
*,
Hubert
Waindok
and
Wendel
Wohlleben
BASF SE, Carl-Bosch-Straße 38, 67056 Ludwigshafen am Rhein, Germany. E-mail: sabine.hirth@basf.com
First published on 13th December 2021
Abstract
Biodurability of man-made vitreous fibres (MMVF) is often measured on naked fibres, i.e. fibres devoid of the phenol-urea-formaldehyde (PUF) binder that is sprayed and baked on the commercial product to reduce dustiness and to provide mechanical strength to fibre mats. This simplification of the hazard assessment relies on the assumption that the binder would not actually coat the entire fibre surface, but would occur only at the touching points where two fibres are glued together. We challenged this assumption by using surface analysis by X-ray photoelectron spectroscopy (XPS) and Time-of-Flight Secondary Ion mass spectrometry (ToF-SIMS). We analysed commercial stone wool MMVF sourced from Denmark, United Kingdom and Germany. XPS as well as ToF-SIMS-mapping combined with gas-cluster-ion-sputtering revealed that all mineral fibres investigated show a complete layer of organics over the surface of the fibres with only a few defects: before sputtering, organic components (PUF binder and oils) uniformly cover the spatial structures; only after sputtering, the inorganic components of the stone wool emerge on the visible surfaces. A preferential localisation of PUF binder on fibre-to-fibre touching points or as droplets was not observable. We finally explored the correlation to dissolution rates, but found that total PUF binder content and the experimentally determined thickness of the PUF binder layer are not sufficient to predict dissolution rates, which instead must consider chemical composition and other properties. In summary, none of the investigated stone wool fibre surfaces were uncoated by the PUF binder.
Introduction/motivation
The effect of inhaled fibres on the lung depends very much on their biodurability. The paradigm of fibre inhalation toxicity regards the dissolution rate as most critical property, next to the fibre dimension and rigidity.1–5Although abiotic dissolution rate is considered an appropriate parameter to predict fibre biodurability6 little work has yet been performed to justify the choice of testing man-made vitreous fibre (MMVF) dissolution either with or without fibre coatings e.g., the oils and binder that are applied to the fibres in the process of their production.4,6 In particular, it is debated if the binder is irrelevant to dissolution,7,8 or if instead it modulates the gel formation and slows down dissolution.9,10 A majority of studies on the abiotic dissolution and on in vivo inhalation clearance used uncoated fibres (or fibres after thermal removal of the binder) to evaluate the effects in respiratory studies.4 More recent studies on the dissolution of stone wool MMVF in lysosomal simulating fluids found significant differences in the presence of fibre coatings,9,10 indicating that they might cause an unwanted increase in biopersistence, although they are required components of the commercial product to reduce dustiness and to add mechanical stability.
Dissolution is governed by bulk composition but can be modulated by surface treatments. Therefore, the question must be raised whether the binder forms a closed layer on the fibre surface or if major fractions of stone wool fibre surface remain uncoated, because the binder might have preferentially accumulated only at the touching points between two fibres, where it is needed to hold fibres together. Barly et al. opined that the binder is not uniformly distributed on the fibre surface, and thus they argued that binder cannot modulate biodissolution.8 They provided evidence by low-energy Scanning Electron Microscopy SEM analysis, showing patchy structures on the fibres. Another contribution by Okhrimenko et al. investigated the surface wetting behaviour on fibres with PUF binder and could fit their results rather well with the assumption of a complete surface coverage.11 Here we employed the surface sensitive techniques of X-Ray Photoelectron Spectroscopy (XPS) and spatially resolved Time-of-Flight Secondary-Ion-Mass-Spectrometry (TOF-SIMS) to investigate the distribution of coatings on the fibres. By comparing the spatial distribution of metals or organic species that are characteristic of the core MMVF fibre and of the binder respectively, we aim to provide a spatial map of fibre fractions that may have accumulated binder, or that may have remained uncoated. We finally aim to employ sputtering techniques to establish a depth profile of the binder.
Materials and methods
Sample description and sample preparation
Eight samples of commercially available MMVF from different manufacturers were obtained. The sample set contained five products from Denmark (abbreviated in the sample list as DK1 to DK5), two samples from German manufacturers (named DE1 and DE2) and one sample from the UK (UK1).No prior knowledge was available as to the composition of the binder but the presence of nitrogen on all samples points to a Phenol-Formaldehyde-Urea (PUF) resin that is commonly used for such types of fibre.
X-ray measurement conditions
The XPS analyses were carried out with a Versa Probe 5000 (Physical Electronics, USA) spectrometer using monochromatic Al Kα radiation (hν = 1486.6 eV) at a base pressure of 1.5 × 10−7 Pa. The instrument work function was calibrated to give a binding energy (BE) of 84.00 eV for the Au 4f7/2 line of metallic gold and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The built in Physical Electronics charge neutralizer system was used on all specimens. To minimize the effects of differential charging, all samples were mounted insulated from the ground.Survey scan analyses were carried out with an analysis spot of area 100 μm × 1400 μm (meaning that we probe some hundred fibres with every position for good statistics about a multitude of fibres on every sample), a pass energy of 117 eV and an energy step size of 0.5 eV. High resolution analyses were carried out on the same analysis area with a pass energy of 23.5 eV and an energy step size of 0.1 eV. X-ray source power was 96.5 W and take off-angle was 45°.
All samples have been measured on three different non-overlapping locations to obtain information about their homogeneity. Spectra have been charge-corrected to the main line of the carbon C 1s spectrum set to 285.0 eV.
Resulting survey and detail spectra were analysed using CasaXPS software (Casa Software Ltd, Teignmouth, UK) version 2.3.22PR1.0 using Shirley background subtraction for all peaks.
Detail spectra of carbon were fitted with five contributions. Curve fitting of C 1s peaks were performed using the same initial conditions and peak separation constraints for each spectrum. A Voigt function of the Shape LA(1,1,900) as provided by the Software. A constant spacing was used for all peaks as stated in Table 1 of the ESI.† FWHM for all carbon-peaks was set equal to the width of the hydrocarbon peak at 285.0 eV.
Nitrogen spectra were fitted with three contributions although the overall spectral quality of the nitrogen signal was low due to low concentration of the element and the results are only evaluated qualitatively.
ToF-SIMS measurement conditions
Static ToF-SIMS spectra were recorded using a ToF.SIMS 5 spectrometer (IonTOF GmbH, Münster Germany). A pulsed mass-filtered primary ion beam of 50 keV doubly charged triple bismuth clusters (Bi32+) was used. This primary ion beam was raster scanned over an area of 500 μm × 500 μm to record spectra and images of positive secondary ions (128 × 128 pixels). This resolution was selected on purpose because the high surface roughness of the fibrous samples leads to very low count rates in each pixel of the mappings when higher lateral resolution is used.To prevent charging of the sample surface, a low-electron energy flood gun (∼20 eV) was used. Mass resolution was limited by the roughness of the surfaces and was in the range of M/ΔM ∼ 1000 at 55 u.
The resulting spectra were analysed using SurfaceLab 7.0 provided by the instrument manufacturer after mass calibration by using the hydrocarbon peaks CH3+ (15 u), C2H3+ (27 u), C3H5+ (41 u), C3H7+ (43 u) and C4H7+ (55 u).
Depth profiles were acquired by means of gas-cluster-ion-sputtering with a 10 keV, Ar1500+ cluster beam of area 700 μm × 700 μm, intermittently sputtering in 1 second intervals between the measurements for 80–140 seconds.
PCA analysis was performed with the built-in module of the SurfaceLab 7.0. Analysis software using the default settings of the program (Poisson scaling, peaks from automatic peak search in a m/z-range of 1–200 u).
Dissolution measurement conditions
Dissolution rates in pH 4.5 lysosomal conditions are partially reproduced from our earlier publications,9,10 and partially original results. The methods are described there, and reproduce the EURIMA guideline6 except for two well-supported choices: we measured the MMVF as-commercialised (with their binder) and we chose the PSF pH 4.5 lysosomal simulant fluid.12 It has been debated which pH 4.5 lysosomal simulant fluid is best adapted to predict in vivo biopersistence,13 but the PSF fluid is one of the established options,13 is recommended by ISO19057,14 and was experimentally confirmed by comparison to alternative simulant fluids recently.10 In any case, for the present paper only the relative ranking between different materials is being correlated to PUF binder properties, and not the absolute predictivity of the dissolution method. In short, 50 mg of MMVF are held in a continuous flow system at 37 °C, and the ions that elute during 14 days with intermediate samplings are quantified by inductively coupled-plasma optical-emission-spectroscopy (ICP-OES). The method was consistently employed for all data shown here, and was completely described elsewhere.9,10Results and discussion
XPS-elemental composition of the coated fibres
The elemental composition of the MMVF-fibre samples varied considerably among measurement positions, as is visible from the error bars in Fig. 1 and this is especially true for UK1 but also for DK1 and DK2. However, all samples show a very high amount of carbon and some nitrogen. Parts of the substrate were detectable by the presence of aluminium, calcium, sodium, sulphur, magnesium in the photoelectron spectra.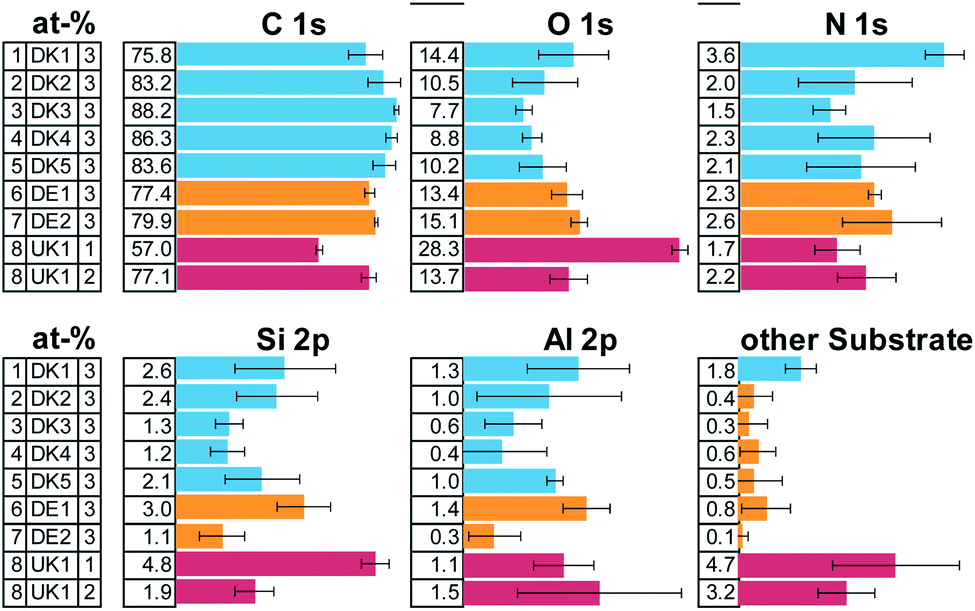 | ||
| Fig. 1 Result of the XPS-quantification of the survey spectra given in atom-%. The error bars describe the standard-deviation across the three measurement positions. Note: one measurement position of UK1 showed extremely low carbon amounts and is therefore presented as a separate measurement. | ||
All samples show high amounts of carbon and oxygen on the surface. From the amount of carbon, we can directly deduce that the coverage by organic compounds on the fibres is least in case of sample [1] DK1 and greatest in case of sample [3] DK3 with all other samples falling in-between. Only on one of the samples, UK1, we found one measurement position with exceptionally low carbon concentration on the surface. This can be indication of 1 occasional “patchy deposition” as interpreted by Barly et al.8 In the other 21 measurement spots of the present analysis, the C/O ratio remained on the order of eight, in very good agreement with the XPS analysis of PUF coating on MMVF fibres and MMVF flat wafers by Okhrimenko et al.11
The fits of the N 1s detail spectra indicate several types of nitrogen in the form of ammonium, imine and, as expected, urea (see ESI Fig. 1 and 3†). This indicates that PUF was used as a binder in all the samples. Elements from the fibres show up in variable amounts due to changes in coating thickness and bulk composition of the fibres.
The overall composition of the carbon and nitrogen signals is very similar for all samples. Most of the surface of all fibres is covered by unfunctionalized carbon (CH, CC) and the overall amount differs only slightly across samples. Also, similar functional groups are found in all cases in quite comparable amounts, indicating a rather similar composition of the coating (see ESI Fig. 2 and 3†).
Analysis of the background shape of XPS-spectra
It is a well investigated fact, that the non-uniform distribution of elements on a surface not only crucially influences the resulting XPS-quantification but also that the background shape of the inelastically scattered electrons from a given element changes with its distribution thereby yielding important information about the distribution of the said element on/in the surface.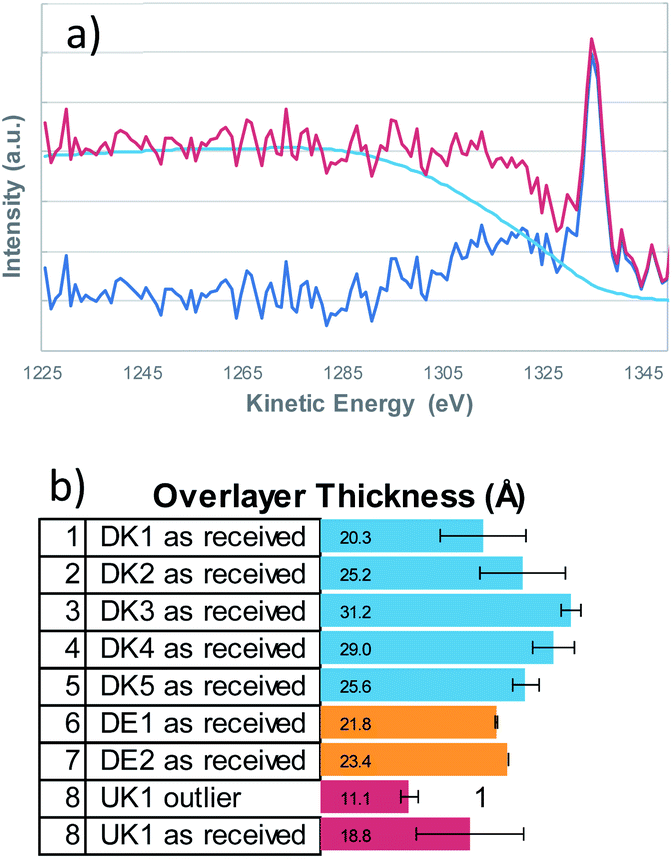 | ||
| Fig. 2 The buried layer model was used to obtain the film thicknesses from the Si 2s signals with QUASES Analyze 7.04 (IMFP 3.5 nm, angle of emission 45°)- (a) example fit of DK1, (b) resulting coating thicknesses that show considerable spread in correlation to the amounts of carbon and nitrogen that were detected on the fibres respectively. | ||
Qualitatively speaking, a rising background in the first 100 eV of the high binding energy side of the XPS-peak, indicates that the element is buried, hence covered by another element. We therefore determined the elemental distribution by means of a Tougaard analysis with QUASES Analyse 7.04 (QUASES-Tougaard Inc., Kobenhavn; Denmark) like the approach given in previous publications.15 To obtain this information we used the spectral region of the survey spectra covering Si 2s, Si 2p, Al 2s and Al 2p from a kinetic energy range of 1220 to 1428 eV. The spectra were pre-treated according to Tougaard's algorithm, and the background was fitted iteratively to the buried layer model (IMFP 3.5 nm, angle of emission 45°, fit region of background from 1285–1235 eV) in all cases (Fig. 2). However, as overall signal intensity from the substrate signals was quite low, the error was within ±20% of layer thickness and small deviations from a closed layer might be not visible from the fit. Additional systematic errors might be induced from the fact that silicon might be also present in the binder of the fibres, leading to an underestimation of the coating thickness.
Therefore, an additional estimation of the film thickness was performed based on the atomic concentration of carbon as described by Smith.16 With this method, the film thickness was a bit smaller but a very good correlation between the two methods exists (data shown in the ESI†). Even though the coating is not solely composed of hydrocarbons but also contains oxygen and nitrogen, the correlation is in fact remarkably good.
From the Tougaard background analysis as well as the correlation with the simple model for adventitious carbon we can therefore conclude that the coating covers the fibres completely in all cases.
ToF-SIMS
Although the XPS-results already strongly indicate that a uniform layer is present, we decided to investigate the distribution further by means of a laterally resolving surface technique, namely ToF-SIMS.In a first analysis, we compared the composition of the surfaces of the fibres as received across all MMVF by means of a principle-component-analysis of the spectra after a nominal mass binning in the range of 0–600 m/z to account for the high roughness of the sample. The sample set was mostly described by two factors that accounted for 99.8% of the total variance. Again, as in the XPS-experiments on the sample set, all samples are chemically quite similar. This is also mirrored by the extremely high amount of variation that is captured by Factor 1.
The loadings-plots in Fig. 3 show dominating contributions by signals from sodium (m/z = 23), potassium (m/z = 39) and various contributions from hydrocarbons (m/z = 41/43/55/57/67/69/79/81/95/97/109) in the coating in accordance with the results found previously.17 These fragments are related mostly to the oil and indicate that petroleum destillates (aka “white oil”) was used in the manufacturing of all samples.
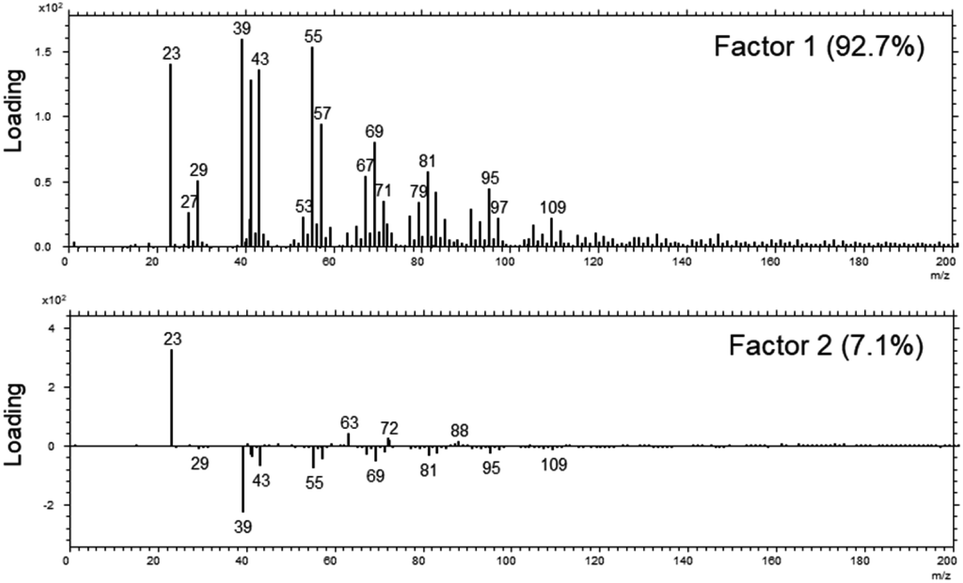 | ||
| Fig. 3 The loadings of Factor 1 (upper panel) and Factor 2 (lower panel) of the PCA analysis across the set of MVVF investigated in the range of 0–200 m/z. | ||
The DE1 and DE2 samples differed most from the rest of the sample set and they are mostly discriminated by variations in the amount of sodium and potassium in Factor 2 as well as a slight change in the amount of binder vs. oil (signals from 63/72/88 vs. signals from 69/81/95/109).
The aromatic contributions of the PUF binder that were found by Zafar et al.17 at masses of m/z = 77/91/128/152/165/178/189 in pure PUF binder samples and that originated from the phenol groups in the binder, were also present on the samples investigated here, although the intensity was very low in the range of 3% of the total surface signal (data not shown).
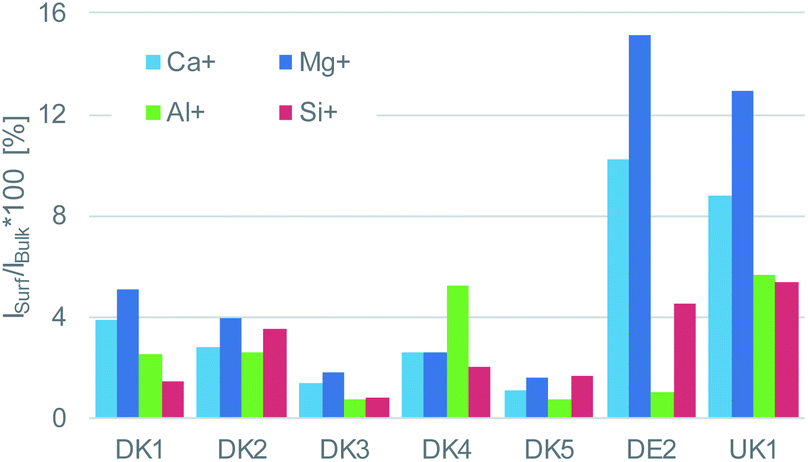 | ||
| Fig. 4 Intensity ratio of the elements calcium, magnesium, aluminium, and silicon on the surface referenced to their respective bulk intensity that was obtained after GCIB-sputtering to remove the coating. The intensities of said elements on the surface are very low for DK1 to DK5 and higher by a factor of 2–3 for UK1 and DE1, but still on a low level. For all materials, only a fraction between 1% and 15% may be considered as uncoated. | ||
Even more relevant than the chemical composition is the distribution of the organic layer on top of the inorganic fibre. The results of the ToF-SIMS-mapping are shown in Fig. 5 and 6. They very clearly demonstrate that the surface (as represented by the Al+ signal to prevent possible influences of a silane signal from the coating15) is completely covered by a homogenous (in the level of lateral resolution of ToF-SIMS) overlayer that can be removed to a big extent by gas-cluster-ion-beam-sputtering (GCIB-sputtering).
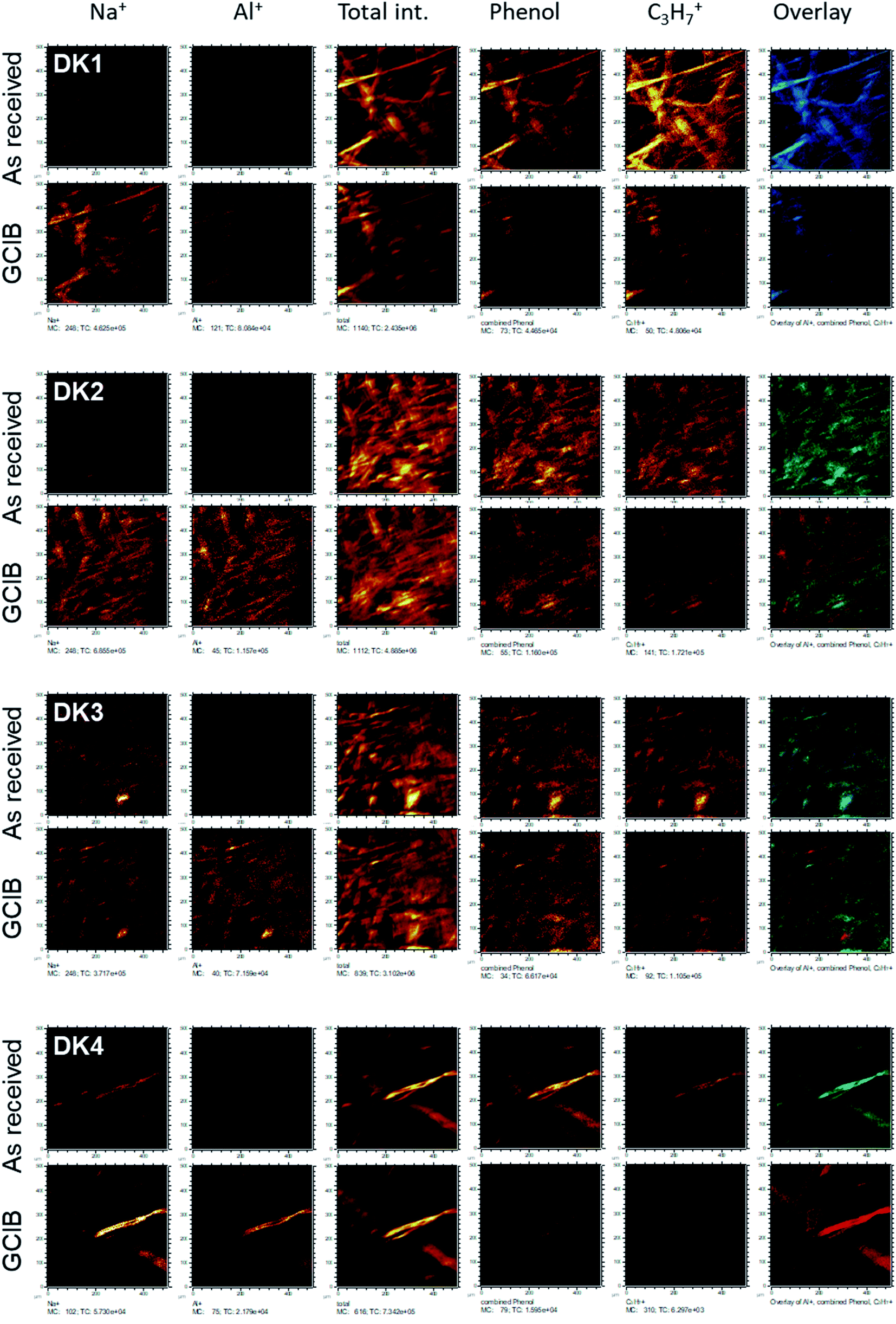 | ||
| Fig. 5 ToF-SIMS-imaging results of the fibres DK1 to DK4 as received (upper line) and after GCIB-sputtering (lower line). Elements shown are (from left to right): Na+, Al+, total ion intensity, sum of all phenolic signals as stated in the text, C3H7+ as signal from hydrocarbon, RGB-overlay. The RGB overlays (rightmost panels) allow to check for the colocalization of Al+ (red), aromatics (phenol, in green) and C3H7+ (blue). All images are scaled in such a way, that the signal range is the same for as received and sputtered samples. | ||
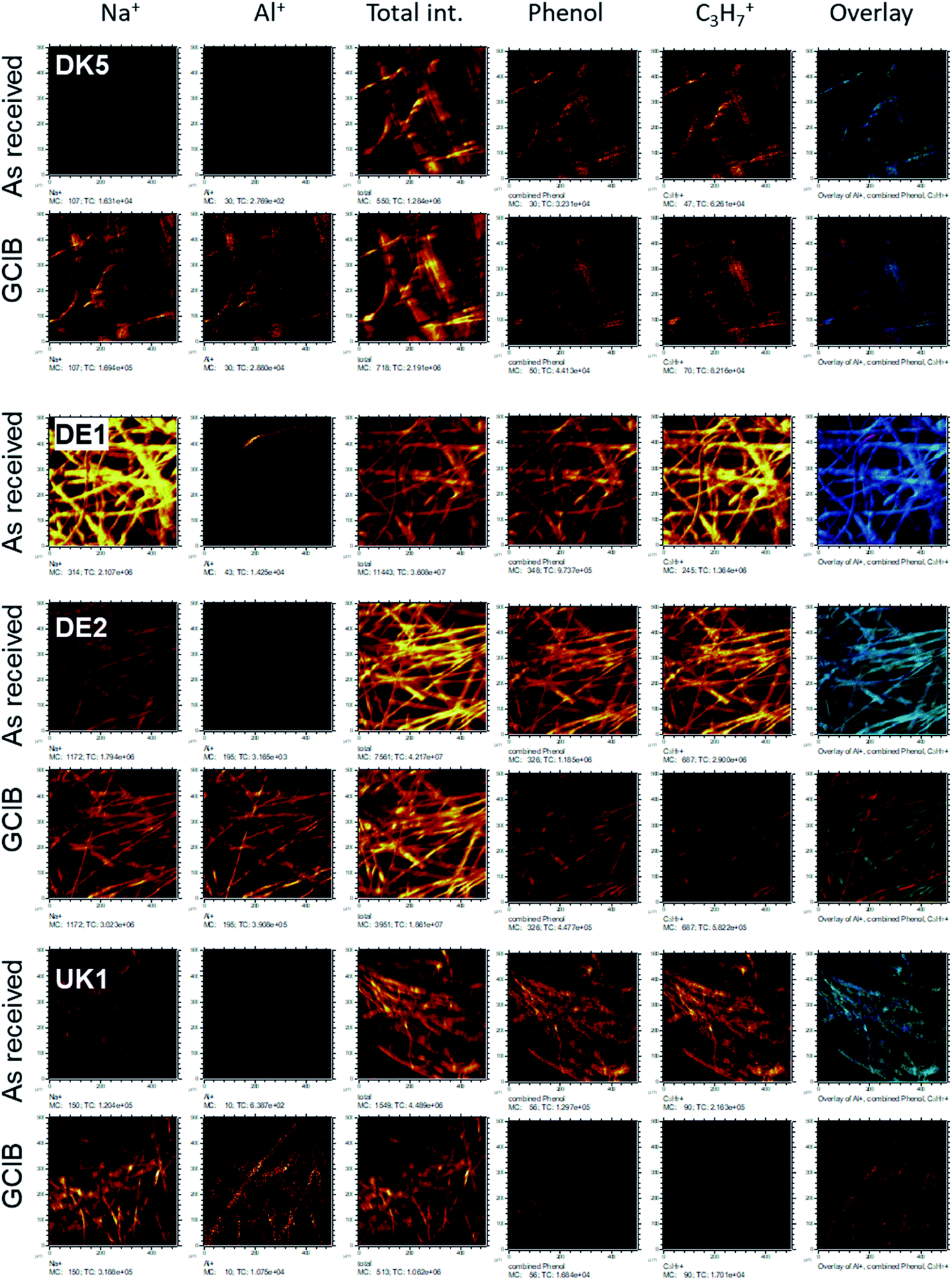 | ||
| Fig. 6 ToF-SIMS-imaging results of the fibres DK5, DE1, DE2, and UK1 as received (upper line) and after GCIB-sputtering (lower line). Elements shown are (from left to right): Na+, Al+, total ion intensity, sum of all phenolic signals as stated in the text, C3H7+ as signal from hydrocarbon, RGB-overlay. The RGB overlays (rightmost panels) allow to check for the colocalization of Al+ (red), aromatics (phenol, in green) and C3H7+ (blue). All images are scaled in such a way, that the signal range is the same for as received and sputtered samples. For DE1 no sputter profile was obtained. | ||
This coverage causes a strong decrease in the intensity of the signals representing the mineral fibre (Ca+, Al+, Mg+, Si+) by a factor of around ∼10 to 25 on the surface when the organic overlayer is present (Fig. 4). While again, DK1 to DK5 are very similar, DE2 and UK1 differ from the set by the fact that higher amounts of divalent ions as well as Si+ are visible on the surface. This might indicate that the coating exhibits more defects, or that these elements are part of the overlayer. However, as the maps in Fig. 6 show, there is also a higher signal from aluminium visible for sample UK1. This indicates that indeed, defects in the coating can be found for this sample, but on a low level: 95% of the surface are still coated. Based on the elements that constitute the majority oxide network, Al and Si, 95 to 9 9% of the surface are coated for all materials. If one considers the minor elements Ca and Mg, similar coating fractions are found for the DK fibres, and at most a fraction of 15% may be considered as uncoated for Mg in the DE2 material.
Correlation with dissolution rates
Since the presence of binder was shown to modulate the dissolution behaviour both in terms of the quantitative dissolution rate9 and in terms of the qualitative dissolution morphologies,10 evidenced also by gel formation in the presence of any binder content from 2.3% to 6.4%, but not at 0% binder,8 one might ask how the dissolution rates scales with the thickness of the coating. For this purpose, we assessed the dissolution of the different MMVF fibres in PSF pH 4.5 lysosomal simulant fluid in the CFS system and compare the dissolution rates against (a) the binder percentage from TGA and (b) against the overlayer thickness from XPS (Fig. 7). We found that total PUF binder content (Fig. 7a) and the experimentally determined thickness of the PUF binder layer (Fig. 7b) are not sufficient to predict dissolution rates, since the R2 remains limited to values around 0.01 to 0.3. We suggest that instead of only the thickness of the binder, one must consider the chemical composition of MMVF and binder, among other properties, to explain the complex gels, pits, bubbles, and deposits that are promoted by the presence of binder during CFS dissolution testing. Also, the choice of the physiological simulant medium is critical because the modulation of wetting, local supersaturation and organic–inorganic complexes should not be suppressed to create a robust dissolution test but should simulate physiological conditions. The near-complete coating by the binder is one of the relevant factors,9,19 and is supported by the re-analysis of MMVF surfaces by XPS after dissolution testing: Barly et al. found that, after dissolution testing, the surfaces assimilated from any initial content of binder to a C/Si ratio of 7–10 (compared to a value ≪1 measured on the MMVF without binder),8 which we interpret as indication of a universal organic–inorganic composite gel formation that is triggered by the binder, but is not directly scaling with binder content, consistent with our findings.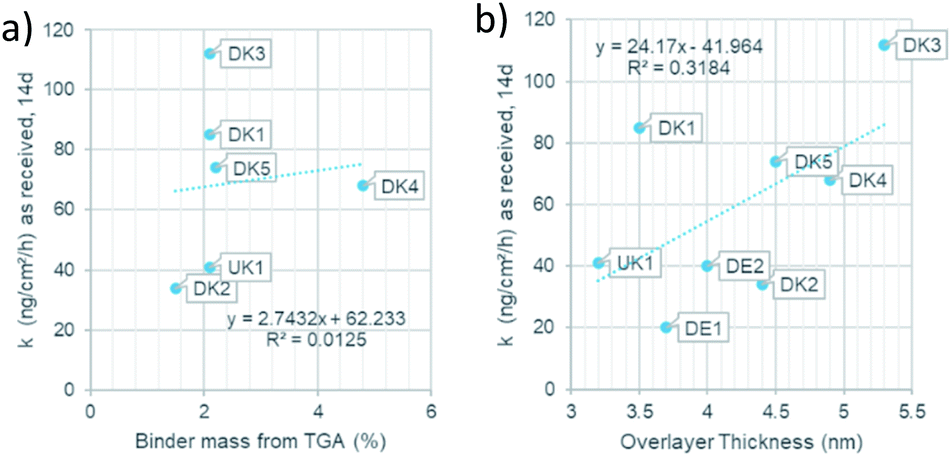 | ||
| Fig. 7 (a): Relation of the binder mass to the dissolution rate. (b): Relation of the overlayer thickness to dissolution rate. | ||
Conclusions
One must refute the assumption that the binder would not actually coat the entire fibre surface and would occur only at the touching points where two fibres are glued together. Instead, all eight samples, regardless of manufacturer or country of origin, showed the presence of nearly complete or complete layers, composed of oils and nitrogen-containing-binders, most likely Poly-Urea-Formaldehyde-binder (PUF binder). Before sputtering, organic components (binder and oils) uniformly cover the spatial structures; only after sputtering, the inorganic components of the stone wool emerge on the visible surfaces. A preferential localisation of binder on fibre-to-fibre touching points was not observable.The present data is sufficient to conclude that, although there might be patches where the binder is thicker than elsewhere, the binder is everywhere. Overlayer thicknesses of the coating vary between 1.8 nm and 4.4 nm in the set of investigated samples. This interpretation is consistent with both the Scanning Electron Microscopy analysis by Barly et al.,8 and with our ToF-SIMS analysis when considering the different information depth of the three analysis techniques. While XPS has an information depth of 5–10 nm and ToF-SIMS in the range of 1–2 nm, typical scanning electron microscopy probes 100 nm and even several 100 nm. One notes that also earlier studies on the surface wetting behaviour on fibres with PUF binder could fit their XPS results rather well with the assumption of a complete surface coverage.11
Because none of the investigated stone wool fibre surfaces remained uncoated, hazard assessment based on binder-free MMVF is questionable. The results support that both abiotic dissolution and in vivo clearance half-life should be determined on the commercial products with their binder. This was also the consensus that emerged as common point of departure for hazard assessment from a debate about critical choices in the measurement of MMVF stone wool biodurability.18–20
Follow-up experiments may advance the safer-by-design balance between mechanical robustness, durability, low dustiness, and sufficiently fast dissolution, guided by a mechanistic understanding of the binder composition and distribution.
Author contributions
Design of the XPS- and ToF-SIMS-analysis, analysis, and interpretation of data: Sabine Hirth. Provision of the topic, assessment of dissolution rates: Wendel Wohlleben. Discussion and relation to prior SEM-work: Hubert Waindok.Conflicts of interest
All authors are employees of BASF SE, a competitor in the market of insulation materials.Acknowledgements
BASF SE received funding and material from Kingspan Insulation Ltd, Pembridge (UK) to support the conduct of the experiments presented herein (no grant number).Notes and references
- K. Donaldson, F. Murphy, R. Duffin and C. Poland, in Particle and Fibre Toxicology, 2010 Search PubMed.
- K. Donaldson, Toxicol. Lett., 1994, 72, 299–305 CrossRef CAS PubMed.
- G. Oberdörster, Int. Arch. Occup. Environ. Health, 2000, 73, S60–S68 CrossRef PubMed.
- A. Andersen, C. Axten, D. M. Bernstein, P. Brochard, V. Castranova, K. Donaldson, P. Dumortier, J. I. Everitt, P. Gustavsson, T. W. Hesterberg, M. C. Jaurand, A. B. Kane, G. M. Marsh, Y. Morimoto, H. Muhle, G. Oberdörster, S. Olin, K. M. Savolainen and T. Schneider, Man-made Vitreous Fibres – IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, IARC Press, Lyon, France, 2002, pp. 1–433 Search PubMed.
- D. M. Bernstein, J. M. R. Sintes, B. K. Ersboell and J. Kunert, Inhalation Toxicol., 2001, 13, 851–875 CrossRef CAS PubMed.
- K. Sebastian, J. Fellman, R. M. Potter, J. Bauer, A. Searl, A. de Meringo, B. Maquin, A. de Reydellet, G. Jubb, M. Moore, R. Preininger, B. Zoitos, P. Boymel, T. Steenberg, A. L. Madsen and M. Guldberg, Glass Sci. Technol., 2002, 75, 263–270 CAS.
- R. M. Potter and N. Olang, Part. Fibre Toxicol., 2013, 10, 13 CrossRef CAS PubMed.
- S. H. Q. Barly, D. V. Okhrimenko, M. Solvang, Y. Yue and S. L. S. Stipp, Chem. Res. Toxicol., 2020, 33(2), 657–668 Search PubMed.
- W. Wohlleben, H. Waindok, B. Daumann, K. Werle, M. Drum and H. Egenolf, Part. Fibre Toxicol., 2017, 14, 29 CrossRef PubMed.
- U. G. Sauer, K. Werle, H. Waindok, S. Hirth, O. Hachmöller and W. Wohlleben, Chem. Res. Toxicol., 2021, 34(3), 780–792 Search PubMed.
- D. V. Okhrimenko, A. Budi, M. Ceccato, D. B. Johansson, D. Lybye, K. Bechgaard and S. L. S. Stipp, Polym. Degrad. Stab., 2021, 183, 109431 CrossRef CAS.
- A. B. Stefaniak, R. A. Guilmette, G. A. Day, M. D. Hoover, P. N. Breysse and R. C. Scripsick, Toxicol. In Vitro, 2005, 19, 123–134 CrossRef CAS PubMed.
- E. Innes, H. H. Yiu, P. McLean, W. Brown and M. Boyles, Crit. Rev. Toxicol., 2021, 1–32 Search PubMed.
- ISO/TR19057, ISO/TR, 2017, 19057.
- D. V. Okhrimenko, A. B. Thomsen, M. Ceccato, D. B. Johansson, D. Lybye, K. Bechgaard, S. Tougaard and S. L. S. Stipp, Polym. Degrad. Stab., 2018, 152, 86–94 CrossRef CAS.
- G. C. Smith, J. Electron Spectrosc. Relat. Phenom., 2005, 148, 21–28 CrossRef CAS.
- A. Zafar, J. Schjødt-Thomsen, R. Sodhi, R. Goacher and D. de Kubber, Polym. Degrad. Stab., 2013, 98, 339–347 CrossRef CAS.
- U. G. Sauer, K. Werle, H. Waindok, S. Hirth, O. Hachmöller and W. Wohlleben, Chem. Res. Toxicol., 2021, 34, 780–792 Search PubMed.
- U. G. Sauer, K. Werle, H. Waindok, S. Hirth, O. Hachmöller and W. Wohlleben, Chem. Res. Toxicol., 2021, 34(7), 1697–1698 Search PubMed.
- C. Koch, D. V. Okhrimenko, M. Solvang, A. Aznar, E. Pezennec, E. Chaudan, J. Magrane Francesch, P. Lindberg, Q. Herault and A. Alami Badissi, Chem. Res. Toxicol., 2021, 34(7), 1695–1696 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06251d |
| This journal is © The Royal Society of Chemistry 2021 |