
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformational adaptability determining antibody recognition to distomer: structure analysis of enantioselective antibody against chiral drug gatifloxacin†
Lanteng Wanga,
Wei Xieb,
Wenyang Jiaoa,
Chijian Zhanga,
Xiangmei Li a,
Zhenlin Xua,
Xin-an Huangc,
Hongtao Lei*a and
Xing Shen*a
a,
Zhenlin Xua,
Xin-an Huangc,
Hongtao Lei*a and
Xing Shen*a
aGuangdong Provincial Key Laboratory of Food Quality and Safety, College of Food Science, South China Agricultural University, Guangzhou 510642, China. E-mail: shenxing325@163.com; hongtao@scau.edu.cn
bMOE Key Laboratory of Gene Function and Regulation, State Key Laboratory for Biocontrol, School of Life Sciences, Sun Yat-Sen University, Guangzhou 510006, China
cTropical Medicine Institute & South China Chinese Medicine Collaborative Innovation Center, Guangzhou University of Chinese Medicine, Guangzhou 510405, China
First published on 13th December 2021
Abstract
Enantioselective antibodies have great potential to separate and detect chiral compounds. However, cross-reactivity of enantioselective antibodies to the distomer may limit the application. An in-depth understanding of interactions between antibodies and chiral drugs could be helpful to investigate antibody recognition to the distomer. In this study, a monoclonal antibody against chiral quinolone S-(−)-gatifloxacin (S-GAT) was produced and its Fab fragment was prepared by proteolysis. The S-GAT Fab exhibited 10% cross-reactivity against the R-enantiomer compared to that of the S-enantiomer in an indirect competitive enzyme-linked immunosorbent assay (icELISA). The crystal structures of the S-GAT Fab apo form and complex with S-GAT were analyzed, and molecular docking of the R-enantiomer was carried out. The ligand conformation was further studied using molecular dynamics simulations. The results showed that the distomer R-enantiomer could enter the chiral center recognition region of the antibody by adjusting the piperazine ring conformation. Meanwhile, the antibody binding cavity had obvious conformational adaptability during ligand binding. It demonstrated that conformational change of both ligand and antibody was the key reason why antibodies recognize the distomer. Restricting conformational adaptability could improve the enantioselective recognition ability of antibodies. This study provided a new explanation for the cross-reactivity of enantioselective antibodies to the distomer, and could help to modulate antibody enantioselectivity for immunoassay of chiral drugs.
1. Introduction
Chirality is an important characteristic of a large number of synthetic compounds, and the chiral center of chiral drugs is often the key to their physiological functions.1 Enantiomers may show different enantioselectivity when binding to their biological targets such as enzymes, receptors, and ion channels in organisms, so that it can result in different activities.2 To a specific target, the enantiomer with stronger affinity is usually called a eutomer, and the enantiomer with weaker affinity is called a distomer.3 Eutomers usually have better pharmacodynamic activity. For instance, the R-enantiomer of salbutamol is about 80 times stronger than the S-enantiomer for the treatment of bronchiectasis.4 The distomer can even lead toxic and side effects that the eutomer does not have. The S-enantiomer of ketamine is the eutomer for analgesia, while R-ketamine has side effects of hallucinations.5 When chiral drugs were used as racemates, the side effects of distomer would greatly affect human health. The most famous example was infant deformity caused by the thalidomide distomer.6 Therefore, in order to protect human health and accurately understand the efficacy of drugs, it is important to study chiral drugs at the enantiomeric level.As a kind of biomacromolecule receptor produced in vivo, antibody also has enantioselectivity for specific recognition to the enantiomers.7,8 At present, enantioselective antibodies were used as a chiral stationary phase for chiral drug separation and detection, and the absolute configuration of the drug could be directly determined.9–11 Enantioselective antibodies could also be used in the fields of immunoassays and immunosensors to determine the enantiomer of chiral drugs.12–14 Compared with traditional separation and detection methods such as chromatography techniques, capillary electromigration techniques and these techniques coupled to mass spectrometry,15 antibody-based methods had higher sensitivity and specificity.16 Meanwhile, in the European Union (EU) and China, immunoassay could be used as a rapid on-site screening method before using confirmatory method.17 In addition, the affinity, specificity and stability of the antibodies could be modulated by genetic engineering technology to adapt to different application requirements.18 Therefore, enantioselective antibodies had unique advantages and great potential in the research of chiral drugs.
In order to realize the application of enantioselective antibodies, high-quality and high-specificity antibodies should be prior to be obtained. The empirical method for preparing enantioselective antibodies was to use a single enantiomer as a hapten while exposing the chiral center as much as possible.7,19 However, the prepared enantioselective antibody often had cross-reactivity to distomer, and it indicated that the antibody could still recognize distomer in some extent.20–22 For example, the S-ibuprofen antibody still had more than 50% cross-reactivity to the R-enantiomer.20 Enantioselective antibodies had the ability to recognize the distomer, which would restrict the application of antibodies in the field of chiral drug separation and detection. It would cause false positives in analytical methods and decrease separation efficiency of separation methods. Therefore, it is necessary to study the enantioselective recognition mechanism of antibodies, which could be helpful to realize the further modulation of antibody enantioselective performance.
Some experimental and computer calculation methods have been used as a tool to study antibody enantioselective recognition, such as some spectroscopy methods and quantitative structure–activity relationship (QSAR) models.23,24 These methods could tell from various type of interactions such as hydrogen bonding or hydrophobic interaction. However, it is difficult to locate specific interaction sites on antibodies through these methods. In response to this problem, the homology modelling could build the three-dimensional protein conformation model and figure out the interaction between antibodies and chiral compounds through molecular docking.25,26 Furthermore, the protein crystallography methods could illustrate the refined structure and the real interaction of the antibodies, and it has been used in the study of enantioselective antibody recognition mechanisms.27,28 However, these studies from antibody structure aspects only elaborated the recognition of their own antibodies to corresponding eutomer and distomer, and have not demonstrated the common recognition characteristics between different antibodies to distomers. These could not provide a universal theory for the preparation of a new enantioselective antibody.
In our previous study, the recognition mechanism of antibodies to antibacterial drug quinolones was studied, and it was found that the most critical epitope for the recognition of quinolone antibodies was C-7 substituent of the quinolones.29–31 Gatifloxacin (GAT), a special quinolone drug, has a chiral center on the piperazine ring at C-7, and there are two enantiomers, S-(−)-gatifloxacin (S-GAT) and R-(+)-gatifloxacin (R-GAT) (Fig. S1†). This could ensure that the antibody obtained using gatifloxacin hapten would have a key recognition of the chiral center. Therefore, gatifloxacin could be an appropriate theoretical model for studying the recognition mechanism of enantioselective antibodies. Meanwhile, gatifloxacin has strong side effects that could decrease blood glucose level.32,33 It is necessary to detect and monitor the residue of gatifloxacin in environment samples (e.g. water samples) or food samples (e.g. animal products).34,35 In this study, an enantioselective monoclonal antibody (mAb) was produced against gatifloxacin hapten. And the mAb was digested to prepare Fab fragment which could be used in protein crystallography. The crystal structures of the Fab fragment were determined and used for the study of antibody enantioselectivity and recognition characteristic to distomer. Furthermore, the explanation of antibody recognition to the distomer could provide a theoretical basis for the preparation and performance modulation of enantioselective antibodies in chiral drug immunoassay.
2. Experimental
2.1 Preparation of hapten–protein conjugates and monoclonal antibody
2.2 Fab fragment preparation and purification
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (w/w). The final concentration of L-cysteine was 10 mM. The digestion time was 8 h for S-GAT mAb. Digestion reaction was carried out in a gravity column, and the hydrolysate and immobilized enzyme were separated by centrifugation (4000 rpm, 1 min) to terminate the reaction.
:1 (w/w). The final concentration of L-cysteine was 10 mM. The digestion time was 8 h for S-GAT mAb. Digestion reaction was carried out in a gravity column, and the hydrolysate and immobilized enzyme were separated by centrifugation (4000 rpm, 1 min) to terminate the reaction.2.3 Indirect competitive enzyme-linked immunosorbent assay (icELISA) procedure
Briefly, 96-well polystyrene microtiter plate was coated with coating antigen (100 μL per well), incubated at 37 °C overnight and then washed twice with PBST. 120 μL blocking buffer was then added into the wells, and the plates were incubated at 37 °C for 3 h and dried at 37 °C for 1 h. Gatifloxacin standards were added followed by the addition of S-GAT Fab dilution (50 μL per well). The wells were incubated at 37 °C for 40 min and then washed five times with PBST. After that, 100 μL of goat anti-mouse Fab-HRP (1:5000 in PBST) was added into each well and the microtiter plate was incubated at 37 °C for 30 min. The chromogenic substrate was added after washing the wells for five times. Finally, the reaction was stopped by addition of 50 μL of 2 M H2SO4. Absorbance of the reaction solution at 450 nm (A450) was recorded by Multiskan FC Microplate Reader (Thermo Scientific, Waltham, MA, U.S.A.). Mean A450 values of gatifloxacin standards were divided by mean A450 values of the blank control (in the absence of analyte), and the ratio was defined as B/B0 for calibration curves.36 The icELISA calibration curves were fitted with a four-parameter logistic function calculated by Origin software. IC50 is defined as the concentration of the analyte that resulted in 50% inhibition. The antibody enantioselectivity was measured with the cross-reactivity (CR) of the Rac-GAT (Meilunbio, Dalian, China), S-GAT and R-GAT (separated from DAICEL, Shanghai, China, ee >99%).
2.4 Fab fragment crystallization
The complex crystal of S-GAT Fab and corresponding drug was prepared by co-crystallization method with a molar ratio of 1:10. All the crystals were generated by the vapor diffusion method in sitting drops at 20 °C. Initial crystal screening was performed with 8 kits: JCSG core I, JCSG core II, JCSG core III, JCSG core IV, JCSG classic, JCSG +, PEGs I and PEGs II (QIAGEN, Hilden, Germany). The protein drops for initial crystal screening contained 0.1 μL Fab fragment and 0.1 μL reservoir solution were dispersed on 96-weill plates using Mosquito (TTP LabTech, Royston, U.K.). The crystals obtained by screening were determined as protein crystals by using Izit Crystal Dye (Hampton Research, Aliso Viejo, CA, U.S.A.), and then the corresponding conditions were optimized by adjusting the buffer pH and the precipitant concentration. The protein drops for optimization contained 1 μL Fab fragment and 1 μL reservoir solution, and crystals appeared after 30 d. The optimized reservoir condition of the S-GAT Fab apo form crystal contained 1.6 M ammonium sulfate, 24% glycerol and 0.8 M sodium acetate, pH 4.6. The optimized reservoir condition of S-GAT Fab crystal in complex with S-GAT (S-GAT Fab complex) contained 0.2 M sodium chloride, 1.4 M ammonium sulfate and 1 M sodium acetate, pH 4.9.
2.5 X-ray diffraction and data collection
The crystals were harvested by tiny nylon loops and quickly soaked into a drop of reservoir solution supplemented with 20% glycerol for a few seconds. Then the crystals were frozen and preserved in liquid nitrogen until use. The diffraction data of the crystals was collected on Beamline BL19U1 at the Shanghai Synchrotron Radiation Facility (SSRF, Shanghai, China) using MD2 diffractometer, Pilatus3 6M detector, Rigaku Actor robotic arm and LN2 Oxford700 Cryosystem. The exposure times were 0.2–0.5 s for per image, and images were collected per 1 degree rotation.2.6 Structure determination and refinement
The X-ray data was processed using the XDS program.37 The Fab fragment that exhibited the highest sequence homology with S-GAT Fab fragment was used as the search model (PDB: 1EGJ). The SWISS-MODEL program was used for the homologous modelling of the templates to replace the sequence with the S-GAT Fab fragment (https://swissmodel.expasy.org/).38 The structures were solved by molecular replacement using the PHASER program.39 The structures were manually modified using Coot and refined with PHENIX Refine for several cycles.40,41 Rwork and Rfree values were monitored during the refinement processing. All the figures that presented structural representations were prepared with the PyMOL 2.5, SYBYL-X 2.1.1 and Discovery Studio Visualizer 2016 software.2.7 Molecular docking between S-GAT Fab and R-GAT ligand
The recognition of S-GAT Fab on R-GAT ligand was analysed by molecular docking. The structure of R-GAT was obtained by chirality conversion from S-GAT ligand in the S-GAT Fab complex structure using Coot program. Gaussian 16 software was employed to optimize the R-GAT ligand structure and to minimize the energy.42 The B3LYP/6-311+G(2d,p) basis set was used to obtain the R-GAT ligand for molecular docking.4 The structure of S-GAT Fab in the complex crystal structure was used as the receptor in the molecular docking model, and the docking was performed by using Surflex-Dock program in SYBYL-X 2.1.1 software. The ligand in the S-GAT Fab complex structure was extracted, and the location of the original S-GAT ligand was used to generate the binding cavity by the Protomol program. The molecular docking method uses the ultra-high precision mode Surflex-Dock GeomX (SFXC), and the original S-GAT ligand extracted from the protein was used as the reference molecule. Ten molecular docking models were generated for evaluation, and the model with the highest total score was selected for analysis.2.8 Molecular dynamics simulation
The protonation states of titratable residues were assigned on the basis of pKa values using PROPKA software under pH 7.4.43 MD simulations were performed on the protein-ligand system using AMBER 20 software package.44 Simulations were carried out following the standard protocols. First, AMBER99 ff14sb force field was applied to the protein, GAFF force field was applied to the ligands. Then the complexes were placed in a truncated cube box of TIP3P water molecules, extending 12.0 Å along each dimension. Then, ions were added to the system as counterions to construct a neutral simulation system. After that, a step of energy minimization with Steepest Descent and Conjugate Gradient algorithms were performed. Afterwards, the system was gradually heated from 0 to 300 K at constant volume in 50 ps and then with a 50 ps equilibration at constant pressure and temperature (NPT). Finally, the production simulations with 200 ns were carried out in the absence of any restraint under NPT conditions. Trajectory protocol was used to analyze structural properties of a molecular dynamic trajectory. Root-mean-square deviations (RMSDs) for the backbone atoms of the protein and ligands are calculated, which reveal that the system has reached equilibrium. Average structures were calculated by CPPTRAJ to determine the average structure of the last 50 ns trajectories where the system has reached equilibrium. Single point energy of average S-GAT and R-GAT structures was calculated using the B3LYP/6-311+G(2d,p) basis set in Gaussian 16.3. Results & discussion
3.1 Monoclonal antibody preparation
Due to the uncertainty of using hapten to immunize experimental animals for antibody preparation,30 only an mAb against S-GAT enantiomer hapten (S-GAT mAb) have been produced. As gatifloxacin is a small molecule compound that has no immunogenicity, the gatifloxacin and BSA conjugate S-GAT-BSA was used as immunogen. Through the carboxyl terminal coupling protein, the distal chiral center could be exposed (Fig. S1†). The isotype of obtained S-GAT mAb was IgG1 and κ light chain determined by Pierce™ Rapid Antibody Isotyping Kit (Thermo Scientific, Waltham, MA, USA). Finally, S-GAT mAb was obtained from ascites production and purified using protein G affinity chromatography which could specifically bind to mouse IgG1. The purification effect was checked by SDS-PAGE (Fig. 1A). The band between 100 kDa and 150 kDa in the non-reduced SDS-PAGE showed the whole mAb. The reduced SDS-PAGE could destroy disulfide bonds between antibody chains. According to the theoretical molecular weight, the band near 50 kDa in the reduced SDS-PAGE was the heavy chain, and the band near 25 kDa was the light chain.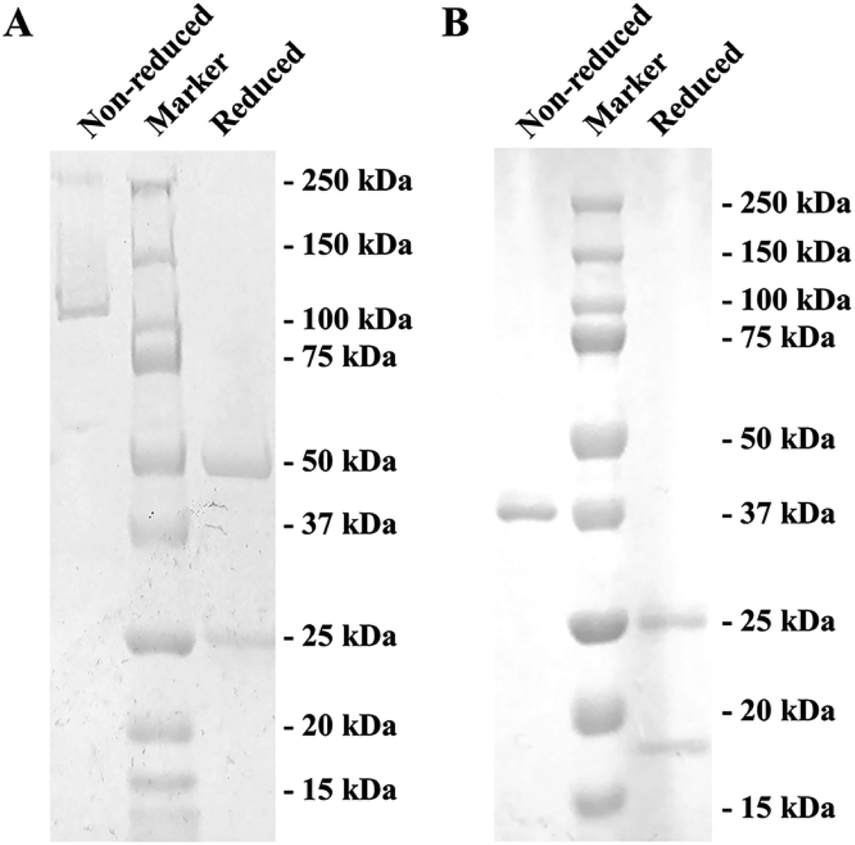 | ||
| Fig. 1 SDS-PAGE of the S-GAT mAb and S-GAT Fab under non-reduced and reduced conditions: (A) SDS-PAGE for S-GAT mAb after purification by protein G affinity chromatography. (B) SDS-PAGE for S-GAT Fab after purification by protein L affinity chromatography and Superdex 200 gel filtration chromatography. Non-reduced SDS-PAGE showed the bands of whole mAb and Fab, and reduced SDS-PAGE showed the bands of heavy chain and light chain, respectively. | ||
3.2 Fab fragment preparation and characterization
After papain digestion, the obtained Fab fragment of S-GAT mAb (S-GAT Fab) had obvious band near 37 kDa on non-reduced electrophoresis (Fig. 1B). Under the reduced pattern, the S-GAT Fab showed two bands. The band near 25 kDa was still the light chain, and the band between 15 and 20 kDa was the digested heavy chain. The S-GAT Fab band was identified as mouse antibody Fab fragments by peptide mass fingerprinting, and the matching protein in Swiss-Prot 2016 Database was KV3AG_MOUSE (Ig kappa chain V-III region PC 7210 OS = Mus musculus PE = 1 SV = 1) with matching score of 148 and expect of 2.7 × 10−11. The IC50 of S-GAT Fab to S-GAT, racemic gatifloxacin (Rac-GAT) and R-GAT were 4.70 ng mL−1, 7.56 ng mL−1 and 45.23 ng mL−1 determined by icELISA method, respectively (Fig. 2). The cross-reactivity of R-GAT was 10.4%, thus S-GAT Fab showed enantioselectivity to S-enantiomer.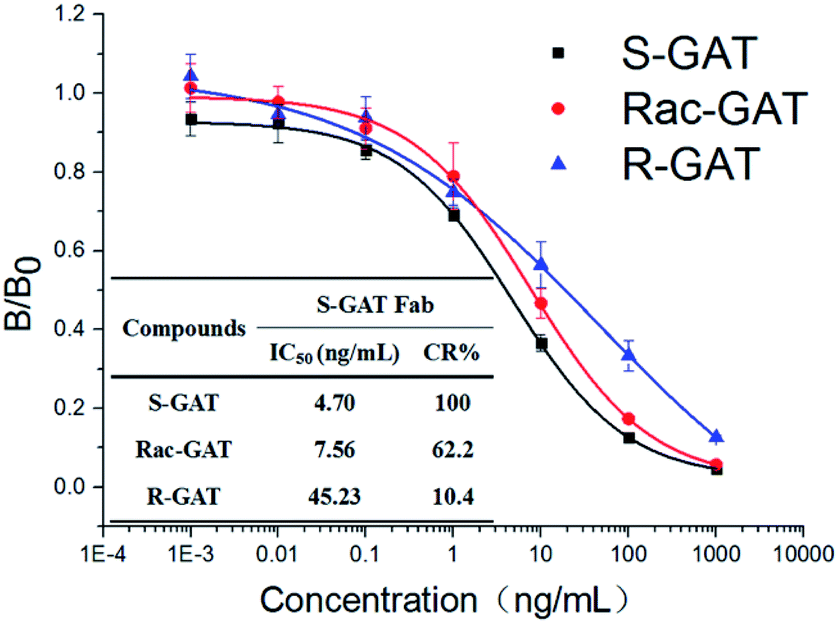 | ||
| Fig. 2 icELISA standard curves and cross-reactivity (CR) of S-GAT Fab to different GAT enantiomers (n = 3). The error bars represent standard deviations from three repeated experiments. | ||
3.3 Crystal structure determination
The apo form and drug-complex crystals for S-GAT Fab were optimized and screened for data collection. Finally, two structures including S-GAT Fab apo form (PDB: 7F2S) and S-GAT Fab in complex with S-GAT ligand (S-GAT Fab complex) (PDB: 7F35) were solved. The complete statistics of data processing and refinement were presented in Table 1. The resolutions of S-GAT Fab apo form crystal, S-GAT Fab complex crystal were 2.62 Å and 2.60 Å, respectively.| Data statistics | S-GAT Fab apo form (PDB: 7F2S) | S-GAT Fab complex (PDB: 7F35) |
|---|---|---|
| a The data in brackets were all for the highest resolution shell (outer shell).b Rmerge = ΣΣi|I(h)i − 〈I(h)〉|/ΣΣi|I(h)i|, where 〈I(h)〉 is the mean equivalent intensity.c Rwork = Σ|Fo − Fc|/Σ|Fo|, where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.d Rfree = Σ|Fo − Fc|/Σ|Fo|. This value was calculated using a test data set comprising 5% of the total data that was randomly selected from the observed reflections. | ||
| Data collection | ||
| Synchrotron source | SSRF BL19U1 | SSRF BL19U1 |
| Wavelength (Å) | 0.9793 | 0.9793 |
| Space group | P 6522 | P 42212 |
| Cell dimensions (a, b, c) (Å) | 76.1, 76.1, 378.9 | 184.1, 184.1, 100.7 |
| Angles (α, β, γ) (°) | 90, 90, 120 | 90, 90, 90 |
| Resolution (Å) | 47.36–2.62 (2.74–2.62)a | 48.55–2.60 (2.68–2.60) |
| Rmerge (%)b | 9.5 (223.2) | 10.2 (193.6) |
| Rpim (%) | 2.1 (49.3) | 2.8 (52.4) |
| CC1/2 | 1.000 (0.883) | 1.000 (0.715) |
| I/σ(I) | 33.6 (2.3) | 29.2 (2.2) |
| Redundancy | 36.5 (39.3) | 26.4 (27.7) |
| Completeness (%) | 100 (99.9) | 100 (100) |
| No. of unique reflections | 20813 (2452) |
53623 (4571) |
|
||
| Refinement | ||
| Reflections in refinement | 20679 |
53536 |
| Rworkc/Rfreed | 0.247/0.278 | 0.201/0.237 |
| Rmsd bond lengths (Å) | 0.011 | 0.010 |
| Rmsd bond angles (°) | 1.20 | 1.22 |
|
||
| B-factors (Å2) | ||
| Average | 112.77 | 85.24 |
| Macromolecules | 112.94 | 85.47 |
| Ligands | 115.18 | 77.18 |
| Solvent | 91.66 | 69.07 |
|
||
| Ramachandran plot (%) | ||
| Most favorable | 94.5 | 98.0 |
| Generously allowed | 5.5 | 1.7 |
| Disallowed | 0 | 0.3 |
There was only one copy in the asymmetric unit of S-GAT Fab apo form crystal (Fig. S2A†). The S-GAT Fab was assembled by light chain and heavy chain, which were composed of the variable region and the constant region, and these regions mainly contained the anti-parallel β-sheet and a few α-helix. Four disulfide bonds were formed for the antibody structure stability between L-Cys23 and L-Cys92 residues, L-Cys138 and L-Cys198 residues, H-Cys22 and H-Cys96 residues, and H-Cys143 and H-Cys198 residues (L means light chain; H means heavy chain). The six complementarity-determining regions (CDRs) for antigen binding of S-GAT Fab were shown in Fig. S2A.† Because the electron density was not clear for L-CDR2, part of the L-CDR2 structure was missing (Residue 56–58, LKS). S-GAT Fab complex crystal had three copies in the asymmetric unit (Fig. S2B†). The electron density of ligand could be seen clearly in the S-GAT Fab complex crystal structure near CDRs (Fig. S3†). The missing part of the L-CDR2 structure was still missing, but the electron density of binding region with the ligand in L-CDR2 (L-Tyr51) was clear. The structures of each copy were similar, so the Fab fragment structure with the best electron density assembled by chain C (light chain) and chain D (heavy chain) was chosen for analysis.
3.4 Molecular docking model of R-GAT ligand binding to S-GAT Fab
In some previous researches, molecular docking based on crystal structure was used to study the antibody recognition to small molecule, and could provide accurate interaction information.28,45 Therefore, molecular docking method was used to study the interaction between S-GAT Fab and R-GAT ligand, since no drug-complex crystal for S-GAT Fab and R-GAT ligand was obtained.The crystal structure of S-GAT Fab complex was employed here to carry out molecular docking. The B3LYP/6-311+G(2d,p) basis set was selected to minimize the energy of the ligand structure using Gaussian 16 software, and the initial ligand structure for molecular docking were obtained. The Surflex-Docking method in SYBYL-X 2.1.1 was used to obtain the top 10 molecular docking models with total score (−log(Kd); Kd, dissociation equilibrium constant) (Table S2†). The total score of the optimal docking model of R-GAT ligand was 5.5597, and the total score higher than 5 in the Surflex-Docking method indicated that the interaction was exited between the protein and the ligand. The similarity between the R-GAT ligand in the optimal docking model and the S-GAT ligand in S-GAT Fab complex as reference molecule was 0.822 (maximum value is 1), and it demonstrated that the correct position of the ligand binding had already been found. The optimal docking model would be used for interaction analysis.
3.5 Interaction between S-GAT Fab and S-enantiomer
The electron density of the ligand near CDR regions in the S-GAT Fab complex crystal could match the S-GAT enantiomer (Fig. S3†). S-GAT ligand was inserted in a vertical and narrow cavity of S-GAT Fab formed by four CDRs, including CDR1 in light chain (L-CDR1) and three CDR regions in heavy chain (H-CDR1, H-CDR3 and H-CDR3) (Fig. 3A). The quinolone parent ring, two six-membered aromatic rings was embedded in the middle of the binding cavity, and the piperazine ring with chiral center on S-GAT was anchored at the bottom of the binding cavity. By analyzing the hydrophobic surface of the binding cavity (Fig. 3B), it showed obvious hydrophobicity near the cyclopropyl group of S-GAT at the entrance and the methyl group on chiral center at the bottom. Because the cyclopropyl, methyl and aromatic rings in S-GAT also have hydrophobicity, strong hydrophobic interactions between S-GAT Fab and S-GAT ligand were recognized.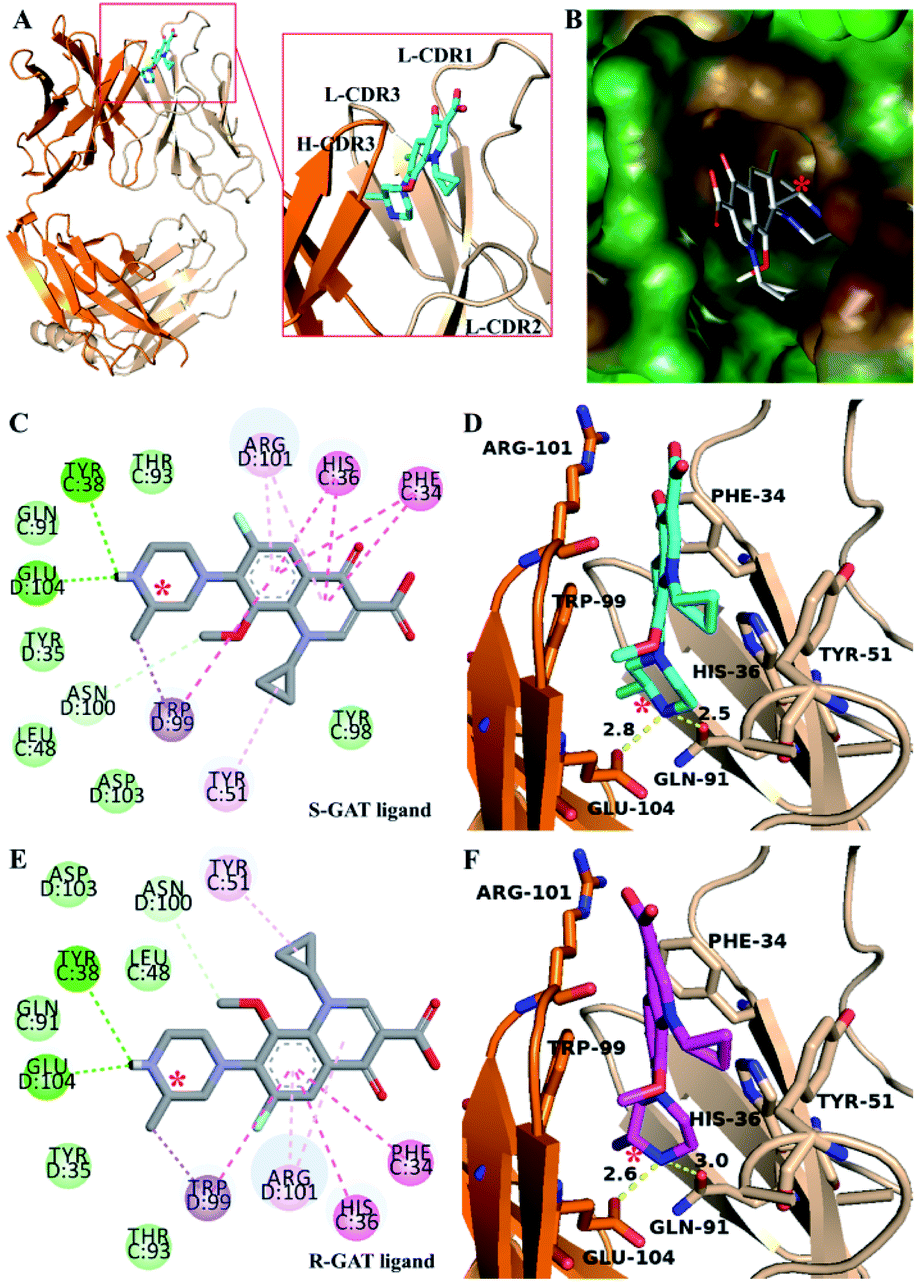 | ||
| Fig. 3 Structure of S-GAT Fab complex crystal and molecular docking model of R-GAT ligand to S-GAT Fab: (A) structure of S-GAT Fab in complex with S-GAT ligand (cyan, O atom for red, N atom for blue and F atom for gray) assembling by chain C (light chain, wheat color) and chain D (heavy chain, orange). (B) The hydrophobic surface of the S-GAT Fab binding cavity. The hydrophobicity gradually increased from blue (hydrophilic) to brown (hydrophobic), and green color meant neutral. (Cand E) Two dimensional interaction diagrams of S-GAT Fab to S-GAT ligand (C) and R-GAT ligand (E). The interactions were noted as π–π stacked or T-shaped interaction (dark pink), π–alkyl interaction (light pink), hydrogen bond (dark green), carbon hydrogen bond (light green with dotted line) and π–σ interaction (purple). The other residues were van der Waals interaction. (D and F) Key amino acid residues for interaction between S-GAT Fab and S-GAT ligand (D) or R-GAT ligand (F). The yellow dotted lines were hydrogen bonds. Asterisks (*) indicated the chiral center of GAT ligands. The images were drawn by PyMOL 2.5, SYBYL-X 2.1.1, and Discovery Studio 2016 Visualizer software. | ||
The weak intermolecular interaction other than hydrophobic interaction was analyzed (Fig. 3C and D). Hydrogen bonding was formed between the piperazine ring of S-GAT ligand and the H-Glu104 and L-Gln91 residues of S-GAT Fab, and the bond lengths were 2.8 Å and 2.5 Å, respectively. The hydrogen bonding residues were the key recognition sites for S-GAT ligand recognition. In addition to hydrogen bonding, the π–π interaction formed by aromatic rings is another important interaction. In this complex, the L-His36 and H-Trp99 residues of S-GAT Fab formed the π–π stacked interaction to quinolone parent ring of S-GAT, and the L-Phe34 residue formed π–π T-shaped interaction. The three π–π interactions immobilized the ligand from three directions at the six-membered aromatic rings (Fig. 3D). In addition, there were π–σ interactions, π–alkyl interactions between S-GAT Fab and ligand.
3.6 Enantioselective recognition characteristic of S-GAT Fab
The activity of S-GAT Fab showed significant enantioselectivity to S-enantiomer, whose IC50 was ten times lower than R-GAT (Fig. 2). By analysing the surface of the chiral recognition region (Fig. 4A), the S-GAT chiral center was embedded at the bottom of the binding cavity, which formed a small cavity in the deepest to contain the methyl group of the chiral center. Strong hydrophobic interactions with the S-GAT chiral center in this small cavity that provided by H-Tyr35, H-Trp99 and L-Tyr98 were observed (Fig. 4B). The three residues all contain large aromatic side chains and form hydrophobic interactions with the methyl group. In addition, the methyl group pointed to the H-Trp99 residue, which formed a π–σ interaction between them. The special conformation in the chiral center recognition region might hinder the distomer R-GAT binding.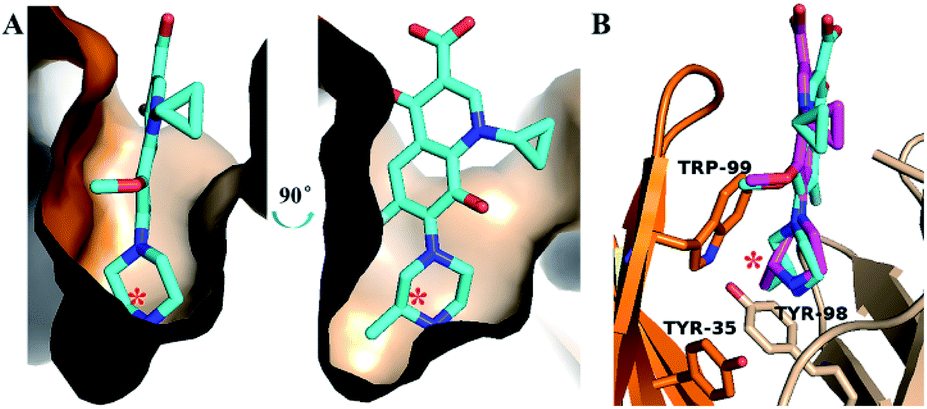 | ||
| Fig. 4 Recognition region for chiral center of S-GAT Fab: (A) binding cavity surface of S-GAT Fab. (B) Key residues for recognition of chiral center. The colors were shown as follows: S-GAT ligand for cyan, R-GAT ligand for magenta, Fab light chain for wheat color, Fab heavy chain for orange, O atom for red, N atom for blue and F atom for grays. Asterisks (*) indicated the chiral center of GAT ligands. The images were drawn by PyMOL 2.5 software. | ||
The molecular docking of the R-GAT ligand was operated using the binding cavity information of the S-GAT Fab complex crystal structure, the interaction between the S-GAT Fab and the R-GAT ligand was analyzed using the optimal docking model. It could be found that the position of the R-GAT ligand was basically consistent with the S-GAT ligand (Fig. 4B), and the types of weak intermolecular interactions were the same as S-GAT ligand (Fig. 3E). However, the π–π interactions of the L-Phe34 and L-His36 residues to the R-GAT ligand had made some difference. The π–π interactions were simultaneously acted on the two aromatic rings of the quinolone parent ring of the S-GAT ligand, but there was only the benzene ring of the R-GAT ligand participated in the formation of the π–π interactions. Meanwhile, the bond lengths of hydrogen bonds between the R-GAT ligand and the H-Glu104 and L-Gln91 residues were 2.6 Å and 3.0 Å, respectively (Fig. 3F), and the interaction of the L-Gln91 residue was weakened. Due to the differences in π–π and hydrogen bond interactions, the binding activity to the R-GAT ligand decreased, resulting in the enantioselectivity of the S-GAT Fab to S-GAT enantiomer.
3.7 Conformational adaptability of ligands for enantioselective binding
Since the S-GAT Fab was produced by the S-GAT hapten, the binding cavity of the S-GAT Fab should only have spatial complementarity with the conformation of the S-GAT ligand. However, the C–C bond connecting the quinolone parent ring and the piperazine ring could rotate. Even if the dimensional orientation of the methyl group on the chiral center changed, the R-GAT ligand could still adjust and rotate the conformation of the piperazine ring to make the position of the methyl group on the S-GAT and R-GAT ligand basically coincide (Fig. 4B). And the methyl group on the R-GAT ligand could enter the chiral center recognition region. The conformational adaptability of the piperazine ring could cause the quinolone parent ring and the N atom formed hydrogen bonds of the R-GAT ligand to shift to the heavy chain, so as to weaken the corresponding interactions.Molecular dynamics simulations were used to further analyze the conformational adaptability of ligands. The S-GAT Fab complex crystal structure was used as the initial model of the S-GAT ligand simulation, and the best docking model was used as the initial model for the R-GAT ligand. The RMSDs for the backbone atoms of the protein and ligands tended to be stable with 200 ns of simulation, and it demonstrated that the system has reached equilibrium (Fig. S4†). The RMSDs of the ligands in equilibrium system were basically the same as the RMSDs of the initial conformation (Fig. 5A and B), and it demonstrated that the docking model of the R-GAT ligand was accurate. The dihedral angles between quinolone parent ring and piperazine ring (C1–C2–N3–C4 in Fig. 5A and B) of ligands during the simulation were analyzed, and it could be found that the dihedral angles of the S-GAT ligand fluctuated in the range of 30–160° in the equilibrium system (Fig. 5C). It indicated that the conformation of the piperazine ring could be rotated in the antibody binding cavity within a certain range. However, the dihedral angles of the R-GAT ligand stabilized around 120° after the first 20 ns of change (Fig. 5D). It can be seen from the average structure (Fig. 5B) that the piperazine ring of the R-GAT ligand was almost in the same plane as the quinolone parent ring in order to make the methyl group enter the chiral center binding region. On the contrary, the piperazine ring of the S-GAT ligand was perpendicular to the parent ring (Fig. 5A). The single point energy of the average structures was calculated by Gaussian 16, and ΔG of the R-GAT ligand was 8.0 kcal mol−1 taking the S-GAT ligand as a reference (ΔG, 0 kcal mol−1). In summary, the R-GAT ligand could enter the chiral center binding region through the rotation of the piperazine ring, but the piperazine ring was fixed at a distortion position with high energy. This was also a reason for the lower affinity of the R-GAT ligand. The binding cavity provided space for the conformational adjustment of the piperazine ring of the R-GAT ligand. Molecular dynamics simulation confirmed that the conformational adaptability of the ligand caused the binding of the antibody to the distomer, even if their spatial complementarity was poorer.
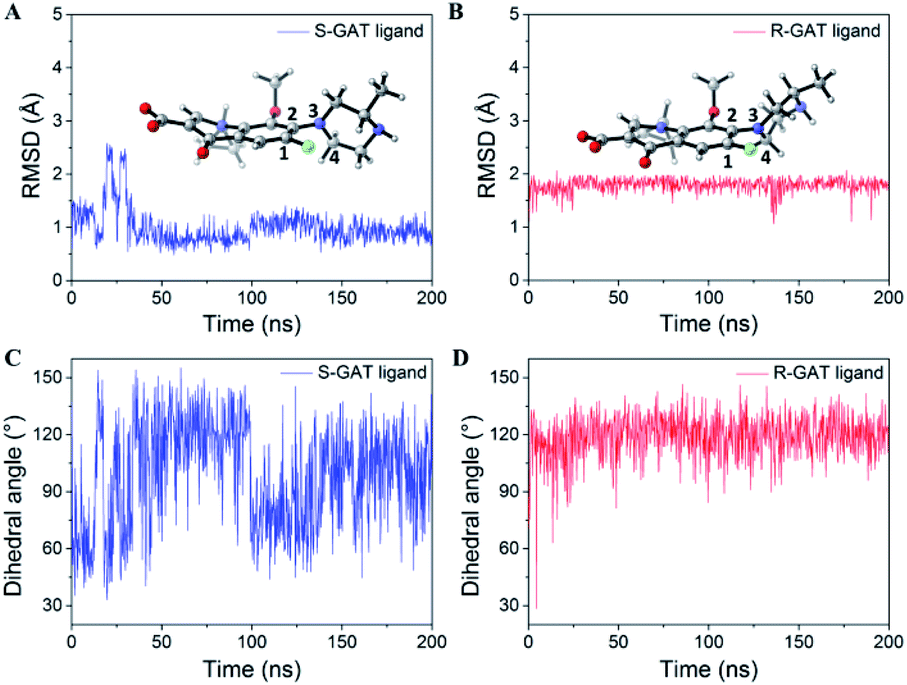 | ||
| Fig. 5 Data statistics of molecular dynamics simulation: (A and B) RMSDs for S-GAT ligand (A) and R-GAT ligand (B) during 200 ns of simulation. The 3D structures showed that the average structures of the last 50 ns trajectories. (C and D) Dihedral angles between quinolone parent ring and piperazine ring (C1–C2–N3–C4) of S-GAT ligand (C) and R-GAT ligand (D) during 200 ns of simulation. | ||
Interestingly, we also found that some ligands had conformational adaptability in other enantioselective antibody researches, but these studies themselves did not realize the impact of the conformational adaptability. This may indicate that conformational adaptability is a common factor affecting antibody enantioselectivity. The relevant ligand structure information and recognition characteristics in previous studies have been summarized in Table S3.† The affinity of finrozole antibody ENA11His to the finrozole eutomer was twice as much as the distomer.27 The conformation adaptability of the flexible chain with chiral centers stabilized by the three aromatic rings, and the hydrogen bonding differences between the central hydroxyl group and the H-Asp95 residue caused different affinity of the finrozole enantiomers (Fig. S5A†). The antibody AZ28 exhibited enantioselective for the transition state analogue (TSA) of the oxy–cope reaction, and the TSA was from the substituted hexadiene.26 Similarly, the positions of two benzene rings of TSA were relatively fixed, and the chiral central hydroxyl groups of different enantiomers were combined with different residues because of the orientation difference (Fig. S5B†). It demonstrated that when some other bulky and rigid substituents (such as aromatic ring) were fixed, the conformational adaptability of flexible structure near chiral center made the distomer enter the antibody binding cavity. However, the poor spatial complementarity would reduce the activity of the distomer.
However, in some cases, enantioselective antibodies had no affinity towards the distomer. For example, antibody C2 could only recognize the R-enantiomer of BINOL derivative.46 The naphthalene ring with two benzene ring substituents of eutomer S-enantiomer was completely fixed inside the antibody binding cavity, and the other naphthalene ring on the outside of the binding cavity has a longer side chain (Fig. S5C†). After the axis chirality was changed, the long side chain of distomer would shift to the completely opposite side. Due to the molecular volume and the rigidity of the naphthalene ring, the long side chain could not reach the current binding position of R-enantiomer through the conformational adaptability. Similarly, the anti-L-AA 80.1R antibody could not recognize D-phenylalanine, and the flexible chain with a chiral center of eutomer L-phenylalanine was tightly fixed at the bottom of the binding cavity (Fig. S5D†).47 The conformation of the flexible chain could not be adjusted, so the benzene ring on distomer would clash with the binding cavity in the other direction. The anti-D-AA 67.36 antibody which could not recognize L-phenylalanine had the similar binding mode with anti-L-AA 80.1R.48
In summary, if there was enough space around the chiral center of the ligand, the distomer could adjust the conformation of the chiral center region to adapt the antibody binding cavity. In this situation, the antibody would display cross-reactivity to the distomer. On the contrary, the distomer conformation was difficult to adjust in a narrow cavity, which made the antibody unable to recognize the distomer. Therefore, it is suggested that the enantioselectivity of antibody could be improved by limiting the conformation of the chiral center region of eutomer in antibody binding cavity.
3.8 Conformational adaptability of the antibody during the ligand binding
Comparing the antigen binding regions of S-GAT Fab apo form crystal structure and S-GAT Fab complex crystal structure (Fig. 6), it could be found that the H-CDR3, L-CDR1 and L-CDR3 β-sheet regions of the S-GAT antibody Fab fragment apo form were closer together, and the entrance of the binding cavity was narrower than the S-GAT Fab complex (Fig. 6A). Comparing the key amino acid residues in the binding cavity, the distance between H-Trp99 and L-His36 residues at the narrowest part of binding cavity in the S-GAT Fab complex structure was 7.8 Å, while the distance between the H-Trp99 and L-His36 residues of the S-GAT Fab apo form structure was only 3.7 Å (Fig. 6B). It showed that when the ligand did not bind to the S-GAT Fab, the binding cavity was in a closed state, so the binding cavity of the S-GAT Fab apo form was shallow.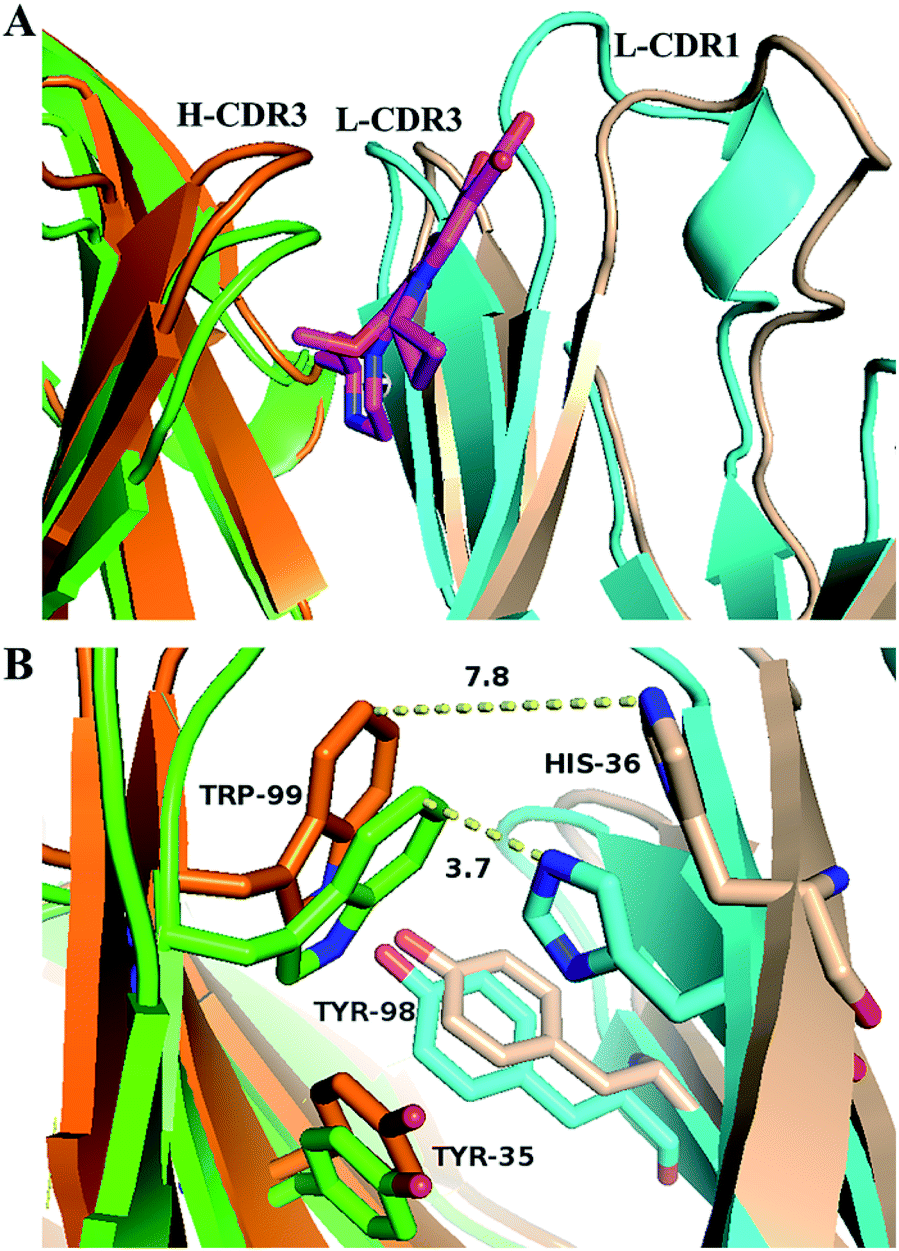 | ||
| Fig. 6 Conformation changes of S-GAT Fab binding cavity during the S-GAT ligand binding: (A) structure alignment of binding cavity for S-GAT Fab fragment apo form and S-GAT Fab complex crystal structures. (B) Conformation changes of key residues in binding cavity. The yellow dotted lines showed the distances between H-Trp99 and L-His36 residues, and the S-GAT ligand structure was hidden. The colors were shown as follows: S-GAT ligand for magenta, light chain of S-GAT Fab apo form for cyan, light chain of S-GAT Fab complex for wheat color, heavy chain of S-GAT Fab apo form for green, heavy chain of S-GAT Fab complex for orange. The images were drawn by PyMOL 2.5 software. | ||
This relative displacement of amino acid residues in the binding cavity caused an “induced fit” of the antibody to the ligand, rather than a “key and lock fitt”.49 The H-Trp99 and L-His36 residues of the S-GAT Fab were the key amino acid residues that modulated the conformation of the binding cavity, and the two residues could control the chiral recognition cavity open or close at the bottom of binding cavity. When the ligand was bound, the binding cavity was opened to allow the entry of GAT molecules. During the S-GAT ligand binding, the H-Trp99 residues moved 2.9 Å, and L-His36 residues moved 4.2 Å. However, the H-Trp99 and L-His36 residues are located in the β-sheet region of the CDRs, which did not have too much flexibility like the loop region. During ligand binding, the β-sheet conformations of the CDR region on the light chain and the heavy chain were separated from each other (Fig. 6A), thereby adjusting the conformation to expand the space of the binding cavity.
The three key amino acid residues of the chiral recognition cavity were H-Tyr35, H-Trp99 and L-Tyr98 residues. In addition to the H-Trp99 residue analyzed above, the conformations of the H-Tyr35 and L-Tyr98 residue also have changed. The moving distances were 1.4 Å for H-Tyr35 and 1.0 Å for L-Tyr98 (Fig. 6B). These two residues in the complex structure moved closer to the chiral center of the GAT ligand, and the residues in the apo form structure were farther from the binding region. It demonstrated that the S-GAT Fab could adjust the steric hindrance and hydrophobic interaction of the chiral center recognition region by modulating the conformation of the three amino acid residues, and it also caused the distomer R-GAT ligand to smoothly enter the chiral recognition cavity of S-GAT Fab. The conformational adaptability of the antibody to the ligand might make the binding cavity more compatible with the distomer, thereby enhancing the affinity of the antibody to the distomer. This was similar to the conformational adaptability of ligands, which need to be limited to improve the enantioselectivity of antibodies.
3.9 Co-action of antibody and ligand conformational adaptability
Although the antibody and the ligand might have the conformational adaptability at the same time during the antibody binding, it seemed that the ligand had a greater role. The reason was that not all antibodies were like the S-GAT Fab with a big change of the conformation. For example, by analyzing the crystal structure of apo form and its complex, there was no obvious conformation change of the finrozole antibody ENA11His before and after binding ligand.27 The recognition of ENA11His to distomer only came from the conformational adaptability of finrozole ligand. It is worth noting that the flexibility of the piperazine ring of the GAT ligand was less than the carbon chain of the finrozole, where the chiral centers were located. Therefore, due to the limited flexibility of the piperazine ring, the conformational adaptability of the S-GAT Fab itself could further assist the binding of distomer. It demonstrated that the conformational adaptability of the ligand was the key factor for the antibody to recognize the distomer, and the conformational adaptability of the antibody could help enhance the recognition to some less flexible distomers. This could provide theoretical guidance for the preparation and performance modulation of enantioselective antibodies in the future.4. Conclusion
The conformational adaptability of both the antibody and the ligand would weaken the enantioselectivity of the antibody. The ligand conformational adaptability affected by steric hindrance was the key factor for the antibody recognition to the distomer. The antibody binding cavity needed to enclose the flexible chiral center region on the ligand and limit the space near the chiral center. Thus the conformational adaptability could be limited, and the enantioselectivity of the antibody to the eutomer would be increased. In contrast, reducing the steric hindrance near the chiral center or even exposing the chiral center outside the binding cavity could increase the affinity of the antibody to the distomer. The conformational adaptability was a common factor affecting enantioselectivity of different antibodies. This research studied the cross-reactivity of enantioselective antibodies to distomer from a new perspective, and also could provide guidance for the preparation and performance modification of enantioselective antibodies.Ethical statement
All animal experiments are conducted at Laboratory Animal Center of South China Agricultural University (Guangzhou, China). Animal experiments conformed to the 3R principle, and the ethical approval of laboratory animals has been approved by the Ethics Committee of South China Agricultural University (protocol code: 2020B035 and date of approval: July 5, 2020).Author contributions
Lanteng Wang: investigation, validation, formal analysis, visualization, writing – original draft, writing – review & editing. Wei Xie: conceptualization, resources. Wenyang Jiao: investigation, validation. Chijian Zhang: investigation, validation. Xiangmei Li: project administration, funding acquisition. Zhenlin Xu: conceptualization. Xin-an Huang: software, resources. Hongtao Lei: conceptualization, supervision, funding acquisition, writing – review & editing. Xing Shen: methodology, project administration, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the National Natural Science Foundation of China [grant number 31871883, 31701703, 32072316]. Thanks for the Shanghai Synchrotron Radiation Facility (SSRF), the crystal data collection was finished using SSRF Beamline BL19U1. Thanks Dr Jie Ni for the help in molecular dynamics simulation and the AMBER software license of Warshel Institute for Computational Biology, School of Life and Health Sciences, The Chinese University of Hong Kong (Shenzhen).References
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- B. Zhao, P. A. Oroskar, X. Wang, D. House, A. Oroskar, A. Oroskar, C. Jameson and S. Murad, Langmuir, 2017, 33, 11246–11256 CrossRef CAS PubMed.
- R. Seifert and S. Dove, Mol. Pharmacol., 2009, 75, 13–18 CrossRef CAS PubMed.
- L. Wang, W. Jiang, X. Shen, X. Li, X.-a. Huang, Z. Xu, Y. Sun, S.-W. Chan, L. Zeng, S. A. Eremin and H. Lei, Anal. Chem., 2018, 90, 2716–2724 CrossRef CAS PubMed.
- P. F. White, J. Ham, W. L. Way and A. J. Trevor, Anesthesiology, 1980, 52, 231–239 CrossRef CAS PubMed.
- C. Álvarez, J. A. Sánchez-Brunete, S. Torrado-Santiago, R. Cadórniga and J. J. Torrado, Chromatographia, 2000, 52, 455–458 CrossRef.
- H. Hofstetter and O. Hofstetter, TrAC, Trends Anal. Chem., 2005, 24, 869–879 CrossRef CAS.
- B. A. Baldo, Monogr. Allergy, 1990, 28, 1–10 CAS.
- E. J. Franco, G. J. Sonneson, T. J. DeLegge, H. Hofstetter, J. R. Horn and O. Hofstetter, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 177–186 CrossRef CAS PubMed.
- S. Wei, H. Guo and J.-M. Lin, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2006, 832, 90–96 CrossRef CAS PubMed.
- A. Vuolanto, M. Leisola and J. Jokela, Biotechnol. Prog., 2004, 20, 771–776 CrossRef CAS PubMed.
- Z. He, S. Zang, Y. Liu, Y. He and H. Lei, Biosens. Bioelectron., 2015, 73, 85–92 CrossRef CAS PubMed.
- K. A. Gräfe and H. Hoffmann, Die Pharmazie, 2000, 55, 286–292 Search PubMed.
- M. Cao, M. Li, X. Yan and M. Wang, Anal. Methods, 2012, 4, 534–538 RSC.
- A. E. Schwaninger, M. R. Meyer and H. H. Maurer, J. Chromatogr. A, 2012, 1269, 122–135 CrossRef CAS PubMed.
- Y. Zhang, L. Wang, X. Shen, X. Wei, X. Huang, Y. Liu, X. Sun, Z. Wang, Y. Sun, Z. Xu, S. A. Eremin and H. Lei, J. Agric. Food Chem., 2017, 65, 4830–4838 CrossRef CAS PubMed.
- L. Wang, J. Wang, A. Zhang, X.-a. Huang and H. Lei, Food Chem., 2022, 371, 131071 CrossRef CAS PubMed.
- T. K. Nevanen, L. Söderholm, K. Kukkonen, T. Suortti, T. Teerinen, M. Linder, H. Söderlund and T. T. Teeri, J. Chromatogr. A, 2001, 925, 89–97 CrossRef CAS PubMed.
- P. A. Got and J. M. Scherrmann, Pharm. Res., 1997, 14, 1516–1523 CrossRef CAS PubMed.
- H. Ito, S. Ishiwata, T. Kosaka, R. Nakashima, H. Takeshita, S. Negoro, M. Maeda and S. Ikegawa, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 806, 11–17 CrossRef CAS PubMed.
- I. Morita, H. Oyama, Y. Kanda, M. Yasuo, A. Ito, M. Toyota, Y. Hayashi, T. Yokoyama and N. Kobayashi, Biol. Pharm. Bull., 2018, 41, 123–131 CrossRef CAS PubMed.
- S. Shen, F. Zhang, S. Zeng, Y. Tian, X. Chai, S. Gee, B. D. Hammock and J. Zheng, Anal. Chem., 2009, 81, 2668–2677 CrossRef CAS PubMed.
- H. Mu, Z. Xu, Y. Liu, Y. Sun, B. Wang, X. Sun, Z. Wang, S. Eremin, A. V. Zherdev, B. B. Dzantiev and H. Lei, Spectrochim. Acta, Part A, 2018, 194, 83–91 CrossRef CAS PubMed.
- H. Mu, H. Lei, B. Wang, Z. Xu, C. Zhang, L. Ling, Y. Tian, J. Hu and Y. Sun, J. Agric. Food Chem., 2014, 62, 7804–7812 CrossRef CAS PubMed.
- M. Geva, M. Eisenstein and L. Addadi, Proteins, 2004, 55, 862–873 CrossRef CAS PubMed.
- S. Martí, J. Andrés, V. Moliner, E. Silla, I. Tuñón and J. Bertrán, J. Phys. Chem. A, 2006, 110, 726–730 CrossRef PubMed.
- T. Parkkinen, T. K. Nevanen, A. Koivula and J. Rouvinen, J. Mol. Biol., 2006, 357, 471–480 CrossRef CAS PubMed.
- L. Zheng, U. Baumann and J. L. Reymond, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3387–3392 CrossRef CAS PubMed.
- J. Chen, L. Wang, L. Lu, X. Shen, X.-a. Huang, Y. Liu, X. Sun, Z. Wang, S. A. Eremin, Y. Sun, Z. Xu and H. Lei, Anal. Chem., 2017, 89, 6740–6748 CrossRef CAS PubMed.
- H. Zeng, J. Chen, C. Zhang, X.-a. Huang, Y. Sun, Z. Xu and H. Lei, Anal. Chem., 2016, 88, 3909–3916 CrossRef CAS PubMed.
- J. Chen, N. Lu, X. Shen, Q. Tang, C. Zhang, J. Xu, Y. Sun, X. A. Huang, Z. Xu and H. Lei, J. Agric. Food Chem., 2016, 64, 2772–2779 CrossRef CAS PubMed.
- C. Yip and A. J. Lee, Clin. Ther., 2006, 28, 1857–1866 CrossRef CAS PubMed.
- M. Bito, T. Tomita, M. Komori, T. Taogoshi, Y. Kimura and K. Kihira, Biol. Pharm. Bull., 2013, 36, 31–35 CrossRef CAS PubMed.
- C. Zhao, W. Liu, H. Ling, S. Lu, Y. Zhang, J. Liu and R. Xi, J. Agric. Food Chem., 2007, 55, 6879–6884 CrossRef CAS PubMed.
- P. Dramou, D. Xiao, H. He, T. Liu and W. Zou, J. Sep. Sci., 2013, 36, 898–906 CrossRef CAS PubMed.
- X. Tao, M. Chen, H. Jiang, J. Shen, Z. Wang, X. Wang, X. Wu and K. Wen, Anal. Bioanal. Chem., 2013, 405, 7477–7484 CrossRef CAS PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- A. Waterhouse, M. Bertoni, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, W296–W303 CrossRef CAS PubMed.
- A. J. McCoy, R. W. Grosse-Kunstleve, L. C. Storoni and R. J. Read, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 458–464 CrossRef PubMed.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Blolino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Oligaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Ragavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision A.03, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- M. H. Olsson, C. R. Søndergaard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2011, 7, 525–537 CrossRef CAS PubMed.
- D. A. Case, H. M. Aktulga, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, G. A. Cisneros, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, C. Jin, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O'Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xue, D. M. York, S. Zhao and P. A. Kollman, AMBER 20, University of California, San Francisco, 424 CA, 2021 Search PubMed.
- K. He, X. Du, W. Sheng, X. Zhou, J. Wang and S. Wang, J. Agric. Food Chem., 2016, 64, 2627–2634 CrossRef CAS PubMed.
- B. S. Rasmussen, J. M. Pedersen, J. Sørensen, J. Egebjerg, B. Schiøtt, K. K. Mortensen and T. Skrydstrup, Chembiochem, 2007, 8, 1974–1980 CrossRef CAS PubMed.
- D. I. Ranieri, H. Hofstetter and O. Hofstetter, J. Sep. Sci., 2009, 32, 1686–1695 CrossRef CAS PubMed.
- D. I. Ranieri, D. M. Corgliano, E. J. Franco, H. Hofstetter and O. Hofstetter, Chirality, 2008, 20, 559–570 CrossRef CAS PubMed.
- T. N. Bhat, G. A. Bentley, T. O. Fischmann, G. Boulot and R. J. Poljak, Nature, 1990, 347, 483–485 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra07143b |
| This journal is © The Royal Society of Chemistry 2021 |