
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRapid kinetic evaluation of homogeneous single-site metallocene catalysts and cyclic diene: how do the catalytic activity, molecular weight, and diene incorporation rate of olefins affect each other?†
Amjad Ali a,
Muhammad Nadeemb,
Jinwei Lua,
Jamile Mohammadi Moradiana,
Tahir Rasheedc,
Tariq Azizc,
Chanez Maouchea,
Yintian Guoc,
Muhammad Awaise,
Fan Zhiqiangd and
Li Quo*a
a,
Muhammad Nadeemb,
Jinwei Lua,
Jamile Mohammadi Moradiana,
Tahir Rasheedc,
Tariq Azizc,
Chanez Maouchea,
Yintian Guoc,
Muhammad Awaise,
Fan Zhiqiangd and
Li Quo*a
aResearch School of Polymeric Materials Science & Engineering, Jiangsu University, Zhenjiang, 212013, PR China. E-mail: liguo@ujs.edu.cn
bDepartment of Environmental Engineering, Wuhang University of Technology, Wuhan, 430223, PR China
cInterdisciplinary Research Center for Advanced Materials, King Fahd University of Petroleum and Minerals, Dhahran 31261, Saudi Arabia
dMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, PR China
eResearch Center of Fluid Machinery Engineering and Technology, Jiangsu University, Zhenjiang, 212013, PR China
First published on 27th September 2021
Abstract
The kinetics and mechanism of ethylene and cyclic diene 5-ethylidene-2-norbornene (ENB) copolymerization catalyzed by rac-Et(Ind)2ZrCl2/[Ph3C][B(C6F5)4]/triisobutylaluminium (TIBA) were investigated using a quench-labeling procedure using 2-thiophenecarbonyl chloride (TPCC). The E/ENB copolymers were characterized by gel permeation chromatography (GPC), differential scanning calorimetry (DSC), scanning electron microscopy (SEM), and 1H nuclear magnetic resonance (NMR) spectroscopy and sulfur analysis. To reduce the errors of the ethylene–diene copolymerization for the kinetics study, we selected E/ENB with steric and electronic features that permit us to elucidate the metallocene catalyst behavior against dienes. A quantitative approach of catalyst speciation, stereodynamics, and micro-kinetics assisted the resolution of mechanistic problems, such as the elastomeric synthesis of ethylene propylene diene monomer rubber (EPDM), the catalyst resting state nature, and how much ion-pairing occurs during polymerization. We report here the precise observation of metal–polymer species, explanation of the dynamics of their initiation, propagation, and termination, and ethylene ENB copolymer development. An approach based on acyl chloride was used to selectively quenched transition metal–polymer bonds to evaluate the polymeric catalyst in terms of its reaction rate, Rp, propagation rate content, kp, and mole fraction of active centers. It is noted that the decline in catalytic activity in the range of 1800 s, and the active center [Zr]/[*C] fraction significantly increased during the initial 1000 s and then tended towards a steady figure of 86%. It is suggested that nearly complete initiation of all olefins catalysts can be obtained after a sufficiently extended reaction. The quick increase in active sites in the first stage can be described by the immediate initiation of active sites positioned on the surfaces of catalyst particles. The initial polymerization rate, Rp, is high and the crystalline properties of the E/ENB copolymer are low due to the greater incorporation of ENB in the polymer backbone, and later the polymerization reaction rates remained stable with a lower mol% of ENB. The melting temperature (Tm) ranges from 108 to 127 °C, whereas the crystalline temperature ranges from 63 to 108 (J g−1). In the E–ENB copolymers, the value of kpE is much greater than that of kpENB; at 120 s, the kpE and kpENB values are 9115 and 431 L mol−1 s−1, respectively, implying smaller diffusion barriers in the early stages, which are close to the actual propagation rate constant.
1. Introduction
Polyethylenes (PEs), an emerging class of polymeric materials, are employed extensively in a number of fields globally. These materials may include low-density and high-density PE.1–4 Because of their readily accessible raw materials, more than 80 million tons of PEs are produced at an affordable cost every year. Industrial catalysts that are durable and recent polymerization methods are more efficient than ever before. PEs resins are used in product packaging, thin films, pipelines, tubes, and household appliances, among so many other things.5–9 Cyclic norbornene (NB) and its derivatives (ENB and VNB) have a ring strain in their structure and are easily polymerized via cationic, radical, and vinyl-type addition.10–12 Cyclic di-olefins prepared via vinyl-type addition show superior properties such as excellent optical transparency, chemical resistance, moisture absorption, and low birefringence.11,13 Because of their outstanding transparency and excellent chemical and thermal stability compared to PE and polypropylene (PP), ethylene/α-olefin copolymers and cyclic olefin copolymers (COCs) are suitable choices for optical devices and engineering plastics. Di-olefins typically have a low glass transition temperature (Tg), and can be controlled using different reaction parameters, co-catalyst, catalyst modification, and a suitable amount of these materials.14–17Since the discovery of olefin polymerization catalysts in the 1950s, they have been a source of fascination for scientists. Catalysts are essential in manufacturing ethylene/viny (ENB,VNB) derivatives (ENB, VNB) copolymers. The majority of catalysts in the literature are group 4 single-sight metallocenes, particularly ansa-zirconocenes, which are suitable polymerization catalysts for ethylene, and higher α-olefins.18 The potential of these catalysts to yield ethylene-based copolymers with homogeneous composition and insert higher olefins into the PE backbone is one of their most attractive features.7 Regardless of their renowned industrial accomplishments over the last 60 years, catalytic olefin polymerizations are still far from being completely understood.19,20 Surprisingly, there are no particularly well-defined rate laws for the basic initiation, propagation, and termination mechanisms for olefin polymerizations with beneficial productivity.20–22 Without understanding the concentration and speciation of catalytically active species, comparing empirical rate laws with molecular mechanisms is complicated. Since their discovery, it has been challenging to determine the concentration of [*C]/[Zr] active centers and the corresponding molecular kinetic constants in polyolefin catalysts.20,23,24 The first investigation, conducted in the late 1950s, involved adding a labeling agent to the end of propagating polyolefin chains. Several methods have been reported in the literature using different compounds, including CO, O2, I2, CS2, H, CH3COCl, and MeOH; however, they have disadvantages. In the case of carbon monooxide (CO), more than five insertions of the ligand exceed the capacity of the active site. Also, electrophilic and nucleophilic labeling compounds such as CS2, MeOH, and the oxidative labeling agents I2 and O2 are not precisely selective toward carbon–metal bonds and consequently also label “dead” polymer chains bound to (alkylaluminum MAO, TIBA, TEA) cocatalysts.21,25–28
In the 2020s, Bochmann and Landis et al. used the quenched-flow (often called “stopped-flow”) polymerization technique to develop a new way of measuring active centers and propagation rate constant (kp).23,28,29,32 The time correlation of polymer molecular weight (MW) and polymer yield have been well defined under controlled conditions, i.e., before chain transfer and termination processes become noticeable (characteristically shorter than 1 s).21,28–30 On the other hand, Landis et al. presented a method based on the employment of chromophore labeling agents.31 The growing polymer labelled chains formulated by MW, using this approach in combination with ultraviolet gel permeation chromatography (UV-GPC), might give insight into metallocene-catalyzed olefin polymerization mechanism theory. Due to a lack of conclusive evidence and the fact that the quench-labeling agent can re-join with the “stabilized alkylaluminum”, it is still unclear if the quenching agent is compatible with alkylaluminum.32,33
Herein, the efficiency of a symmetrical bridge zirconocenes catalyst in incorporating ethylene and propylene homo- and copolymerization is determined using a kinetics model developed in previous studies. By experimenting with different alkylaluminum materials, borates, and metallocene skeleton modification and substitutions, we explored the impact that symmetrical Cp ligands, co-catalyst, and polymerization time have on catalyst activation. To reduce the errors in the ethylene–diene copolymerization for kinetics study, we select E/ENB that has steric and electronic features that permit us to elucidate the metallocene catalyst behavior against the dienes. A quantitative approach of catalyst speciation, stereodynamics, and micro-kinetics could assist the resolution of mechanistic problems, such as the elastomeric synthesis of EPDM, the catalyst resting state nature, and how much ion-pairing occurs during polymerization.34 We report herein the precise observation of metal–polymer species; an explanation of the dynamics of their initiation, propagation, and termination; and ethylene ENB copolymer development and the clear distinction between irregular and continuous behavior. In addition, these results strongly confirm early concepts on metallocene deactivation in solution and why complete initiation of all olefins catalysts can be obtained after a sufficiently extended reaction.
2. Materials and methods
2.1. Materials
The polymeric zirconocene catalyst rac-Et(Ind)2ZrCl2 was bought from Sigma-Aldrich. Albemarle Co. supplied the cocatalyst triisobutylaluminum (TIBA) and the borate activator, (Ph3C)B(C6F5)4, was produced according to the literature.35,36 The quenching agent 2-thiophenecarbonyl chloride (TPCC, 98% purity) was obtained from J&K Scientific, PR China. Ethylene (standard polymerization grade, 99.9% purity) was bought from Zhejiang mixing Gas Co. (Hangzhou, PR China) then purified further by passing it through a gas purification column system. 5-Ethylidene-2-norbornene (ENB) was bought from Acros Organics Chemicals China, then further purified further through dehydration using 4 Å molecular sieves, collected via distillation, and kept under a nitrogen environment. Toluene, as a polymerization solvent (HPLC grade, Jiangsu Yonghua Fine Chemical Co., Ltd., China), was refluxed over sodium–benzophenone and further distilled under N2 gas before any experiments were carried out. An appropriate quantity of plain TIBA was injected into a sealed vial containing distilled n-heptane to make a solution of the cocatalyst TIBA (2 M in n-heptane). Before use, the TPCC was first distilled and then diluted to 2.0 M in distilled n-heptane. All moisture- and air-sensitive compounds were handled under a nitrogen environment in a glove box using standard Schlenk line techniques.2.2. Polymerization
All ethylene/ENB copolymer reactions were conducted in a 150 mL Schlenk round-bottom glass reactor. At 50 °C, the glass reactor was filled with approximately 50 mL of toluene, then saturated with 0.1 MPa ethylene. The reactor was initially injected with ENB, and then the TIBA (2 M) solution was added and the reaction was run for 5 minutes, followed by the addition of ansa-metallocene and borate activator in toluene solutions. The timing of the addition of the borate toluene solution was used to determine the beginning of the polymerization. During polymerization, ethylene was constantly supplied into the reactor at a pressure of 0.1 MPa. After a planned time, the copolymerization reaction was quenched by injecting TPPC (TPCC/Al = 2) for five minutes. Then, the additional volume of dehydrated ethanol containing 2% of hydrochloric acid was added to decompose the TIBA, borate, metallocene, and unreacted TPCC. The copolymer was precipitated in dehydrated ethanol. The obtained E/ENB copolymer samples were extensively purified in a multi-step procedure before being dried under vacuum.37–392.3. Characterization
A YHTS-2000 ultraviolet fluorescence sulfur analyzer was used to calculate the sulfur content of the purified E/ENB copolymer samples. Three sulfur (S) content measurements were prepared in parallel for each copolymer sample, and the average value was used to determine the S content. In contrast to the quench-labeled purified samples, which had an S concentration of 5–25 ppm, a blank E/ENB sample was produced under identical conditions without the quenching step with a sulfur concentration of practically 0 ppm.403. Results and discussion
The performance of ansa-metallocene catalysts generated with ethylene–ENB polymerizations was investigated, using the following protocol: after adding ENB, an appropriate quantity of TIBA solution (Al/Zr = 800) was allowed to react in polymerization solvent for 10 min at 50 °C with the ENB impurities and catalyst precursor. TPCC looks like a feasible quenching agent for measuring the number of active sites [Zr]/[*C] in polyolefin catalysts based on the facts and study (Fig. 1).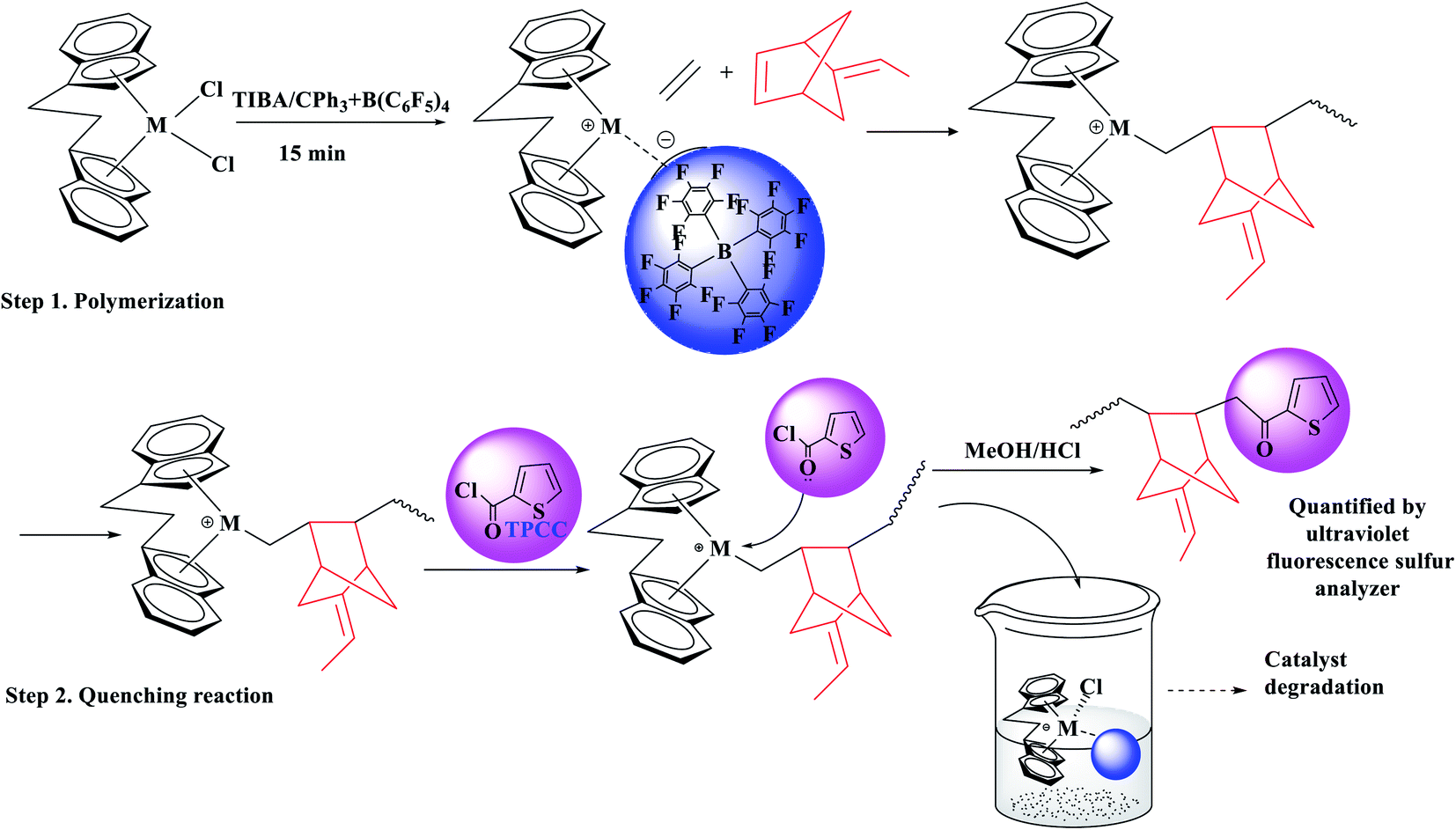 | ||
| Fig. 1 Schematic diagram of the quenching reaction of the active center [C*]/[Mt] by TPCC. | ||
At 50 °C, we performed a series of E–ENB copolymerization reactions using a fixed amount of ENB 0.06 mol L−1 in toluene, pre-contact time, polymerization time, and quenching time. The sulfur (S) content of the E/ENB copolymer was measured to calculate the concentration of the active centers using the equation [S] = [C], and the change in [C*]/[Zr] fraction with reaction time was calculated, with the results shown in Tables S1 and S2.† The rate equation Rp = kp[C*][M], which has been well established for most homo- and heterogeneous catalyzed olefins reactions, was used to determine the time-dependent change in the chain propagation rate constant (kp).40 The olefin polymerization rate (Rp) was calculated from differentiations of the curve of the obtained polymers against reaction time (see the ESI, Fig. S4 and S5†). According to the literature, the starting monomer concentration was taken as [M] for this system, which represents an equilibrium of the ethylene concentration in toluene under normal pressure (1 atm and 50 °C 0.085 mol L−1).40 Table 1 summarizes the copolymerization results conclusions, including the time-dependent [C*]/[Zr] fraction, ENB mol% in the polymer, molecular weight, molecular weight distribution, and thermal characteristics data.
| Run | Time (s) | Yield (g) | ENB in polb (mol%) | Activity (107 g poly per Mt per h) | *Cc (%) | MWd (g mol−1) | PDd | Tme (°C) | ΔHme (J g−1) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: catalyst (Et(Ind)2ZrCl2) = 1.25 μmol, borate (Ph3C)B(C6F5)4 = 2.50 μmol, ethylene pressure = 0.1 MPa, ENB = 0.06 mol L−1, solvent toluene = 50 mL, activator TIBA 1000 μmol, and TPCC 2000 μmol.b Determined by 1H NMR.c *C calculated from the quenching reaction of the active center [C*]/[Mt] by TPCC (see the ESI).d Determined by GPC.e Determined by DSC. | |||||||||
| 1.1 | 120 | 0.47 | 5.10 | 1.13 | 16.43 | 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 690 690 |
3.06 | 108.56 | 108.56 |
| 1.2 | 240 | 0.68 | 4.87 | 0.82 | 29.91 | 59543 |
3.87 | 109.03 | 65.60 |
| 1.3 | 360 | 0.87 | 4.61 | 0.70 | 45.38 | 84068 |
4.07 | 117.01 | 78.39 |
| 1.4 | 480 | 1.09 | 3.97 | 0.65 | 54.68 | 85067 |
3.88 | 111.00 | 63.38 |
| 1.5 | 840 | 1.44 | 3.43 | 0.49 | 75.42 | 86099 |
3.74 | 126.11 | 87.06 |
| 1.6 | 1200 | 1.59 | 2.98 | 0.38 | 79.2 | 89300 |
3.82 | 127.24 | 109.9 |
| 1.7 | 1500 | 1.7 | 2.87 | 0.33 | 82.67 | 97692 |
4.07 | 126.72 | 117.9 |
| 1.8 | 1800 | 1.76 | 2.83 | 0.28 | 86.06 | 100774 |
4.58 | 127.24 | 103.5 |
The influence that the metallocene/methylaluminoxane (MAO) catalyst system has on cyclic dienes is not fully understood, including the Et(Ind)2ZrCl2/TIBA/borate catalyst system. It is well known that for cyclic and linear dienes, increasing the diene content in the feed decreases the polymerization catalyst activity, as expected. The E/ENB copolymer activity decline in the reaction period of 120 to 1800 s, and active center fraction [Zr]/[*C] significantly increased in the initial 1000 s and subsequently tended towards a steady figure of 86%, which is higher than those of ethylene and propylene homopolymerization, but lower than their copolymerization, see Fig. 2. It is suggested that complete initiation of all olefin catalysts can be obtained after a sufficiently extended reaction. The fraction of active sites increased in two stages. In the range of 120–800 s, the active sites quickly reached 75%, and then little change was observed in the period of 800 s to 1800 s. The quick increase in the active sites in the first stage can be described by the immediate initiation of active sites positioned on the surfaces of catalyst particles. Subsequently, these active sites are easily reachable by the monomer and cocatalyst, they can be activated during the pre-contact process, and their initiation can be accomplished fast. The first steady level in the range of 120–800 s can be attributed to the initial induction-steady stage of active sites. In our previous study, we defined the development of the dormant or deactivated active site by the interaction of TMA with the metallocene to the incomplete activation of the metallocene complex. In addition, chain transfer with TIBA or another alkylaluminum will lead to saturated PE chain ends. The β-H transfer of a propagating chain results in a PE chain with a vinyl termination.
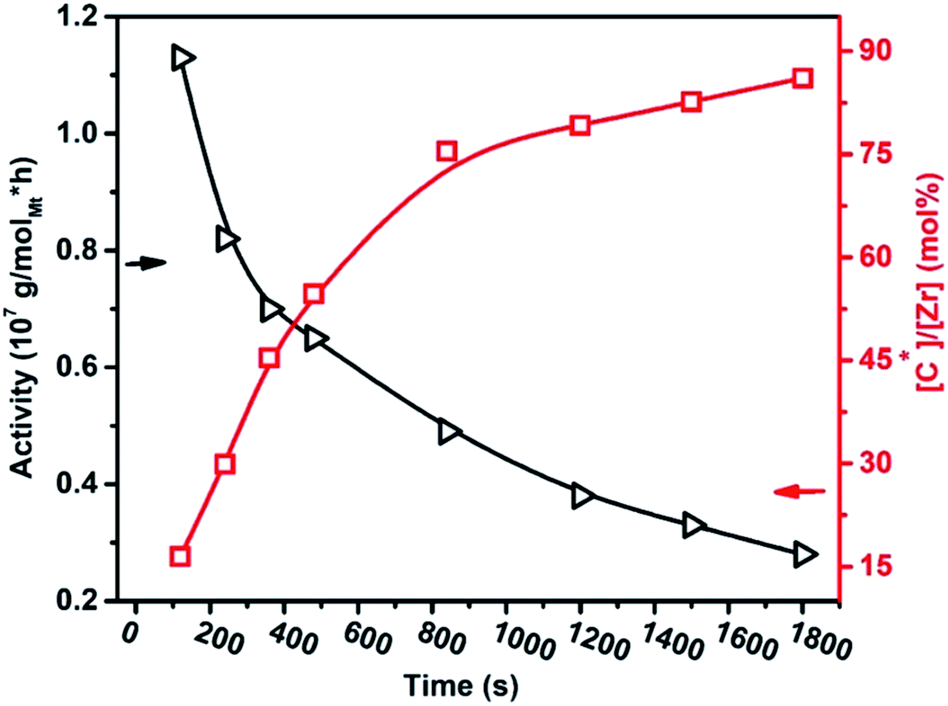 | ||
| Fig. 2 In ethylene/ENB copolymerization, the catalytic activity decreases, and the active center fraction increases with the reaction time. | ||
Fig. 3 illustrates the polymerization rate curve obtained using a metallocene/TIBA/borate catalyst system at 50 °C. We estimated the changes in ethylene and ENB consumption rate (RpE and RpENB, respectively) over time using the copolymer composition data and yield shown in Table 1. The following equations were used to calculate the rate constants for ethylene and ENB insertion separately:
| RpE = [C*][E] | (a) |
| RpENB = [C*][ENB] | (b) |
 | ||
| Fig. 3 The active centers and rate of chain propagation changes with reaction time in ethylene/ENB copolymerization. | ||
After introducing the borate cocatalyst, the polymerization rate slightly decreased in a reaction time of 1800 s (see Fig. 3). During this polymerization time, a significant change in the solvent viscosity was noted, and the construction of fibrous polymer entangled on the stirrer was also observed (see Fig. 4). Scanning electron microscopy (SEM) was used to examine the morphology using a Hitachi S-4800 system connected to an energy-dispersive X-ray spectroscopy (EDS) system. The growing active sites are evidently encapsulated in precipitated polymer throughout this phase, preventing monomers from diffusing into the active sites, resulting in declining rates.
 | ||
| Fig. 4 In ethylene/ENB copolymerization, the construction of polymer fibres in (a) and (b) at 120 s, (c) and (d) at 480 and (e) and (f) in the 1800 s range. | ||
In the situation that the initial polymerization rate is high and crystalline properties of the E/ENB copolymer are low due to the greater incorporation of ENB in the polymer backbone, later the polymerization reaction rates stay stable with a lower ENB mol%. As the polymerization time increases, the Rp and ENB mol% of the E–ENB copolymers decreases.
The molecular weight of the E–ENB copolymers was lower in the initial period and significantly increased with reaction time. When compared to the molecular weights of ethylene–propylene homopolymers and their copolymers produced with Mt-II/TIBA and Mt/MMAO under similar conditions, EP copolymers produced with lower molecular weight but PE molecular weights are comparable. This means that the chain transfer reaction of the active centers with TIBA in E/ENB copolymerization is less dominant than in the EP system. The molecular weight distributions (MWDs) of E/ENB copolymers were broad and became broader as the reaction progressed, MWD evidently larger than 2, the theoretical polydispersity index of a true single-site metallocene catalyst system, suggesting the presence of multiple active sites in the catalytic system (see Fig. S1†). Furthermore, 1H NMR spectroscopy was used to determine the integrated comonomer content. Fig. 5 shows the curve of ENB mol% in the copolymer against the active centers in the catalyst system. Surprisingly, the E/ENB copolymer system displays a higher reactivity of ENB than E/P/ENB with the same catalyst system in the 1200 s range.11 In addition, ENB is a non-conjugated cyclic diene and exhibits two different double bonds, one cis-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– and another vinyl bond.11
CH– and another vinyl bond.11
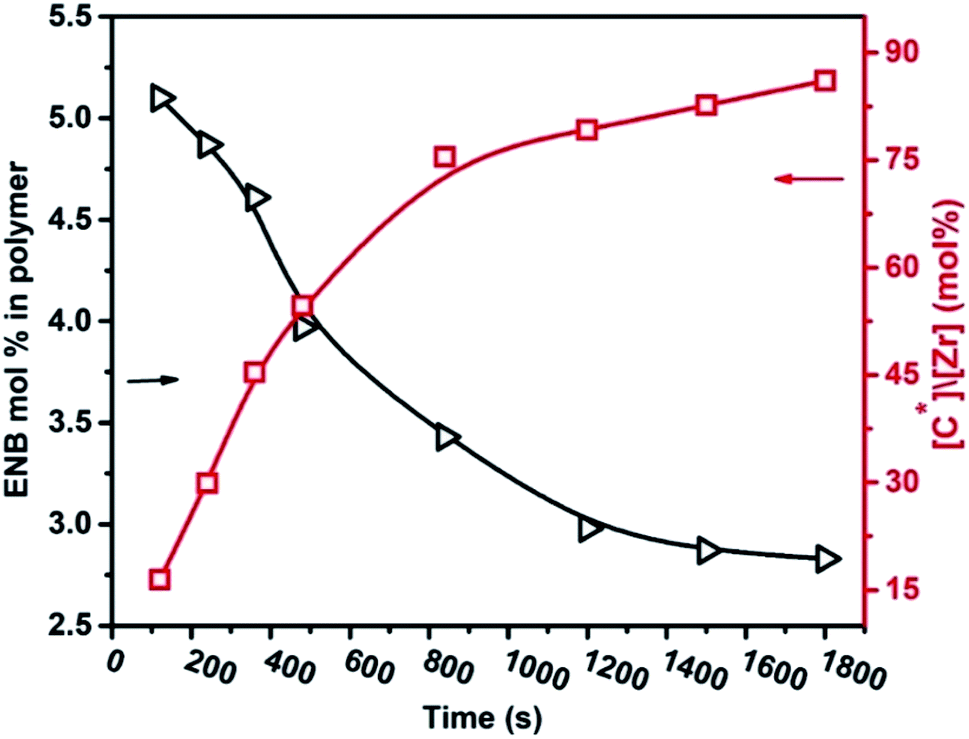 | ||
| Fig. 5 In ethylene/ENB copolymerization, the incorporation rate and active centers change with reaction time. | ||
However, the vinyl bond is nearly nonreactive with most of the Ziegler–Natta catalyst systems. ENB polymerizes as an α-olefin with a bulky alkyl group using an endocyclic double (cis-CHCH–) bond, and has more significant ring strain, leaving the vinyl group unreacted. These results look to be closely related to the comonomer's bulkiness.10,43–45
The above-discussed reaction has been confirmed from the peaks of the vinyl groups in the assembled polymer chain (see the ESI, Fig. S2†). In addition, the mol% of ENB in the E/ENB copolymers is higher at the initial time than later on. The active center also quickly increases over the first 120–480 s, meaning that the cyclic diene ENB quickly activates the active site in the initial time that was inactive for ethylene.
The DSC heating curve of the ethylene–ENB copolymers with time is shown in Fig. S3.† Usually, with an increase in ENB from 5 to 10 mol%, the ENB concentration and polymerization time substantially influence the crystallinity and melting temperature (Tm) because ENB is bulkier than ethylene, and propylene prevents the rotational movement of the polymer chains. The melting temperature (Tm) ranges from 108 to 127 °C, whereas the crystallinity temperature ranges from 63 to 108 (J g−1). These results appear to be closely related to the copolymer composition, implying that composition drift might have an unpredictably negative impact on thermal characteristics. In principle, the existence of ENB-rich segments and chains at the initial time of the reaction could indicate the presence of crystallizable segments with varying levels of ethylene sequences, such as chain segments with higher ENB content that melt at lower temperatures and blocks with a lower comonomer content that melt at higher temperatures.
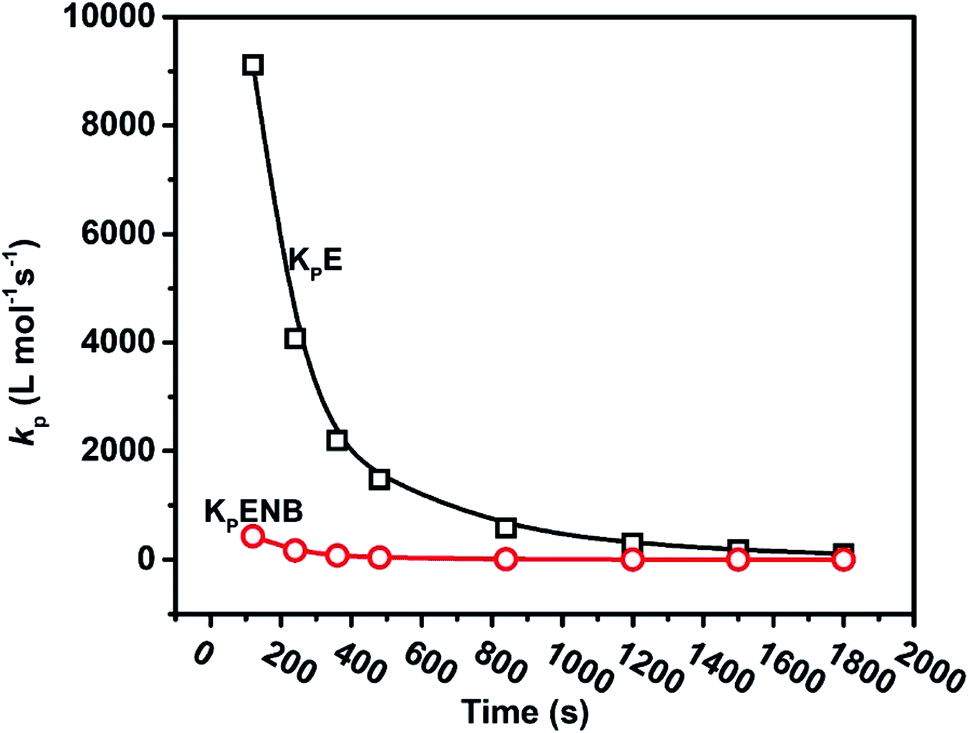
Table 2 summarizes the copolymerization results, including the time-dependent [C*]/[Zr] fraction, with kinetics data such as RpE, RpENB, kpE, and kpENB. As seen in Fig. 6 and 7, both the kpE and kpENB values moderately decreased with reaction time. The effects are comparable to those observed in E and P homo- and copolymerization systems. In the E–ENB copolymerization system, the value of kpE is much greater than that of kpENB. At 120 s, the kpE and kpENB values are 9115 and 431 L mol−1 s−1, respectively, implying more minor diffusion barriers in the early stages, which are close to the actual propagation rate constant.
| Run | Time/s | *Cb/% | RpEc/molcat molENB molcat−1 s−1 | RpENBc/molcat molENB molcat−1 s−1 | kpEd/L mol−1 s−1 | kpENBd/L mol−1 s−1) |
|---|---|---|---|---|---|---|
| a Reaction conditions: catalyst (Et(Ind)2ZrCl2) = 1.25 μmol, borate (Ph3C)B(C6F5)4 = 2.50 μmol, ethylene pressure = 0.1 MPa, ENB = 0.06 mol L−1, solvent toluene = 50 mL, activator TIBA 1000 μmol, and TPCC 2000 μmol.b *C calculated from the quenching reaction of the active center [C*]/[Mt] by TPCC (see the ESI).c RpE and RpENB in units of mmolpoly molMt−1 s−1.d The propagation constant of ethylene and ENB. | ||||||
| 1.1 | 120 | 16.43 | 62.9 | 3.21 | 9115 | 431 |
| 1.2 | 240 | 29.91 | 51.2 | 1.96 | 4076 | 165 |
| 1.3 | 360 | 45.38 | 41.8 | 1.14 | 2193 | 72 |
| 1.4 | 480 | 54.68 | 34 | 0.689 | 1480 | 38.6 |
| 1.5 | 840 | 75.42 | 18.5 | 0.145 | 584 | 6.79 |
| 1.6 | 1200 | 79.2 | 10 | 0.0228 | 301 | 0.987 |
| 1.7 | 1500 | 82.67 | 6.2 | 0.0075 | 179 | 0.322 |
| 1.8 | 1800 | 86.06 | 3.875 | 0.00228 | 107 | 0.0965 |
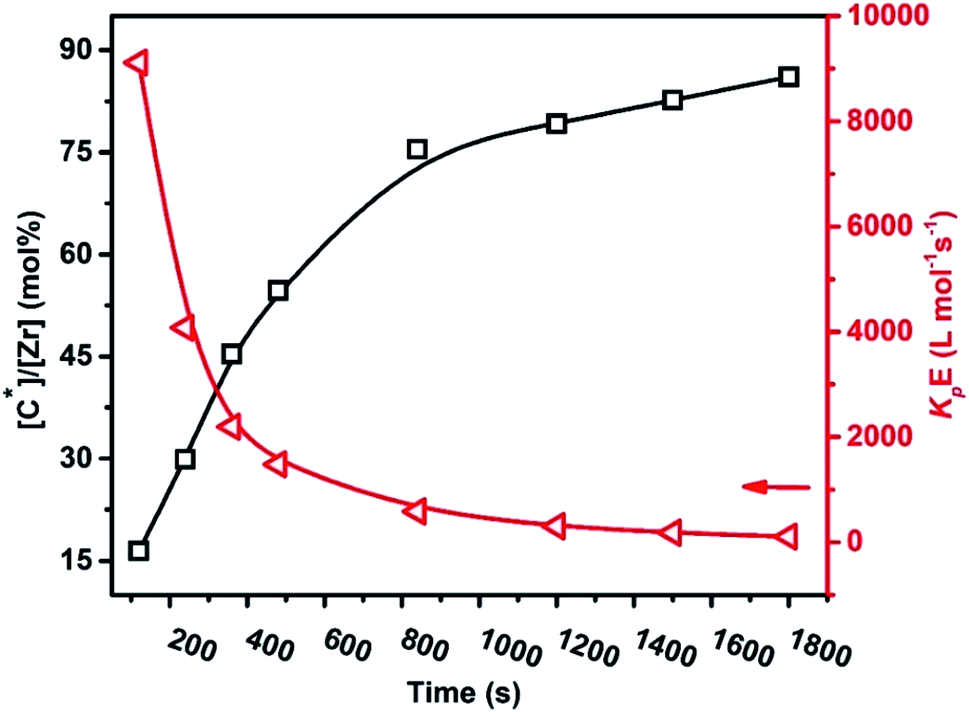 | ||
| Fig. 6 Variation of the propagation rate constant (kpE) and active centers [C*]/[Zr] with reaction time in ethylene/ENB copolymerization. | ||
 | ||
| Fig. 7 Variation of the propagation rate constant (kpENB) and active centers [C*]/[Zr] with reaction time in ethylene/ENB copolymerization. | ||
The previously reported initial kpE values of the ethylene homopolymerization and E/ENB copolymerization kpE are different from each other (4110 vs. 9115). This is because the active center [C*]/[Zr] ratio of E homopolymerization at the initial stage was significantly lower than that of the E/ENB copolymerization, meaning that the [C*]/[Zr] in these two systems must be quite different.12,37,40 The [C*]/[Zr] in the E/ENB copolymerization system that produces copolymers may be made up of loosely associated ion-pairs with large kp values, whereas [C*]/[Zr] in homopolymerization could be made up of a large number of contact ion-pairs with reduced kp values. To make comparisons among the kp values of the E, E/P, and E/ENB homo- and copolymerization systems that are more obvious, the curves of kp vs. the polymerization time are shown in the same graph in Fig. 8.37,42
 | ||
| Fig. 8 Comparison between the propagation rate constant (kpE) of ethylene homo-, ethylene/propylene co-, and ethylene–ENB copolymerization under the same catalyst and conditions. | ||
The initial value of kp for E/ENB copolymerization is higher than that for E/P copolymerization because the E/P copolymers are almost amorphous and may be dissolved in toluene, while E/ENB exhibits lower crystallinity, and this difference could be due to a higher diffusion barrier in the E/ENB copolymerization system.
The moderate decline in the kp values with polymerization time could also be attributed to the lower diffusion barrier in the E/ENB polymerization system (see Fig. 9). Although, the effect of the active center ratio on the kp should also be considered. The active center fraction in homopolymerization is lower than in E/ENB copolymerization, particularly in the later stages. The active centers generated later in the copolymerization process appear to have a lower kp value than those formed earlier. The active center fraction of the previously reported E and P homopolymerization and their copolymerization with E/ENB make it more interesting. It is well known that P is bulkier than E. The metallocene catalyst in the form of contact ion-pairs may be incapable of accepting P for further coordination, with the result of it becoming dormant or inactive in homopolymerization. In conclusion, in propylene polymerization, the [C*]/[Zr] ratio is lower than that in ethylene polymerization. Similarly, the active center fraction in E/ENB copolymerization is higher than that in ethylene propylene homopolymerization but lower than the copolymerization, meaning that ENB can also activate the active sites in the metallocene catalyst system that were inactive in the homopolymerization of E. According to Tsai et al., zirconocene complexes generally work as precatalysts that need to be activated by alkylaluminum and a borate cocatalyst. The active catalyst species were produced through the reaction of cocatalyst and precatalyst. Scheme 1 shows the ethylene insertion into the Zr–C bond and insertion of ENB into Zr–PE. Even though ENB is sterically more challenging than propylene, the π complexes disappear or are weaker, and the insertion barriers are more significant than for propylene insertions into Zr–C. The addition of ENB to the system resulted in a substantial decline in inactivity. In addition, the steric impact in ENB insertion is mainly caused by the repulsion between ENB and the precatalyst ligand.
 | ||
| Fig. 9 Comparison between the ethylene and ENB propagation rate constants in ethylene/ENB copolymerization. | ||
 | ||
| Scheme 1 Schematic diagram showing ethylene insertion into the Zr–C bond and the insertion of ENB into Zr–PE. | ||
In the proposed mechanistic model (Scheme 2), the catalyst active sites bearing a propagation chain (A) could undergo β-H migration, resulting in the generation of L2Zr–H species (B), which may be converted to species (C) via the insertion of ethylene into the L2Zr–H bond. As suggested by Kissin and co-workers, in their model, they clarified the influence of co-monomer activation in the ethylene with propylene and α-olefin copolymerizations using an industrialized heterogeneous polymeric Ziegler–Natta catalyst system.46,47 However, (C) is active for ethylene and higher α-olefin polymerization; it is also capable of reverting to a dormant state (D) due to robust β-agostic contacts between hydrogen on the methyl and Zr metal. The lower percentage of these inactive or dormant sites in the E/ENB copolymerization is the main reason for a higher fraction of active centers than in the previously reported ethylene homopolymerization. According to Fan et al., in E/P copolymerization, the insertion of P in the hydrogen zirconium bond (Zr–H) can bypass the chemical reaction, leading to an increased active center fraction, which is higher than their homopolymerization.20,47
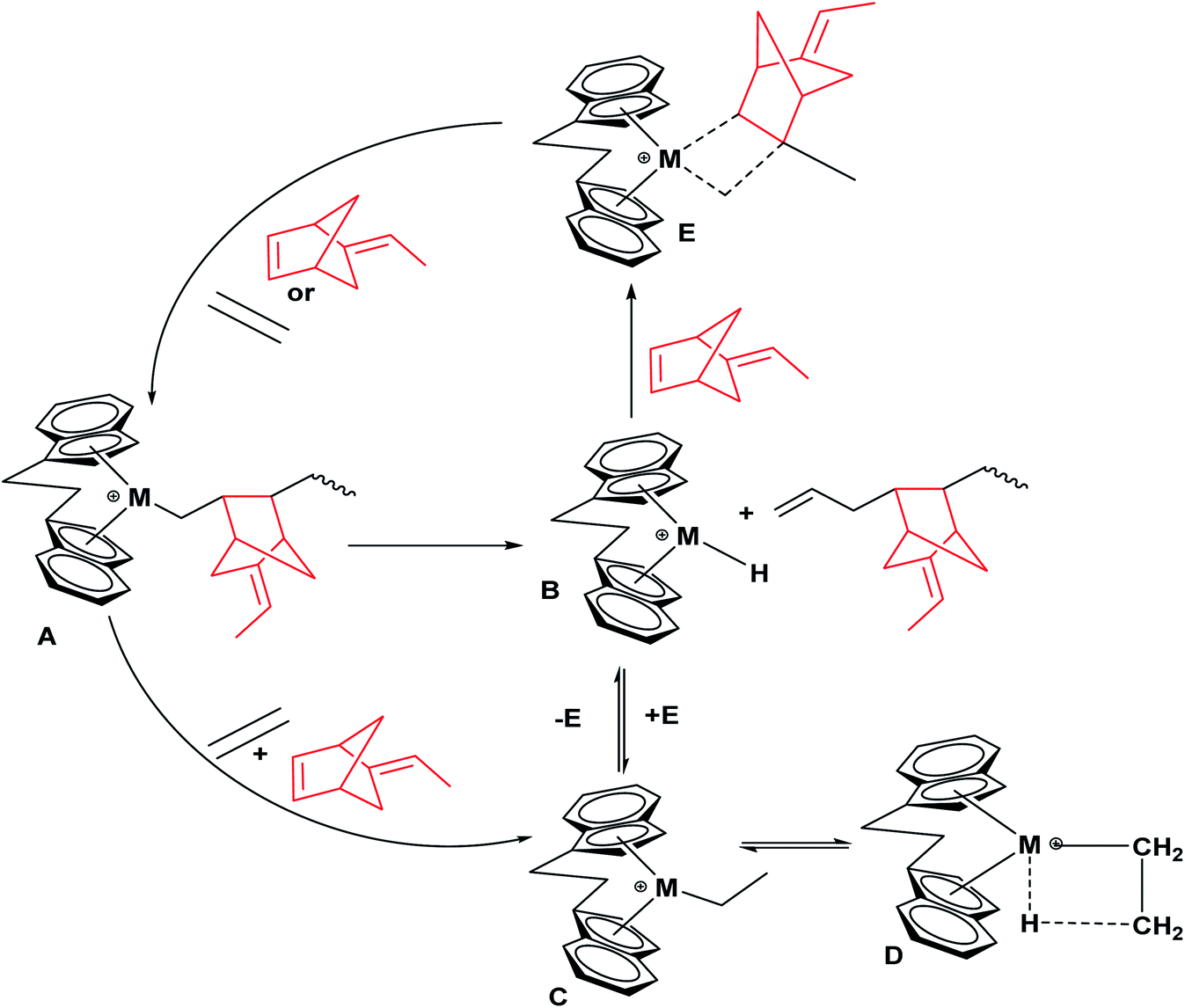 | ||
| Scheme 2 Schematic diagram showing a mechanistic model of the copolymerization of ethylene and ENB, how the inactive sites are formed, and how ENB can reactivate them. | ||
ENB polymerizes as an α-olefin with a bulky alkyl group using an endocyclic double (cis-CHCH–) bond and has more significant ring strain. Endocyclic double bond insertion of ENB in the hydrogen zirconium bond can sidestep the reactions, increasing the [C*]/[Zr] fraction ratio in the system through (E). The above-discussed information has been confirmed from the peaks of the vinyl groups in the assembled polymer chain (Fig. S1†). In addition, the formation of (D) from (C) may need suitable adjustments of zirconium and the alkyl (Zr–iPr) moiety, which require enough space among the cation and anion. It is logical to assume that [L2Zr–iPr] Cl–borate species with associated contact ion-pairs are hardly transferred in the dormant state than the loosely associated ion-pairs. This assumption was supported by a noticeable increase in the ENB incorporation rate during the initial 120–360 s of copolymerization (Table 1). As illustrated in Fig. 4, the incorporation rate of ENB is higher at the initial polymerization time, and the active center fraction reached 45%. Upon a further increase in the polymerization time, this fraction reached 85%. This indicates that the catalytic species activated during the initial 120–360 s can insert more ENB than those later activated catalytic species.
4. Conclusion
An approach based on acyl chloride selectively quenched transition metal–polymer bonds for evaluating polymeric catalysts in terms of the reaction rate Rp, propagation rate content kp, and mole fraction of active center ratio, was confirmed by investigations of Ziegler–Natta polymeric catalyst systems. These properties can now be evaluated in hours rather than days, making kinetic methodologies for the assessment of quantitative polymerization catalysts more feasible and practical. The catalytic activity decline in the reaction range of 1800 s, and the active center [Zr]/[*C] ratio significantly increased over the initial 1000 s and then tended to reach a steady figure of 86%. This suggests that almost complete initiation or activation of all olefins catalysts can be obtained after a sufficiently extended reaction. The quick increase in active sites in the first stage can be described by the immediate initiation of active sites positioned on the surfaces of catalyst particles. The initial polymerization rate is high and the crystalline properties of the E/ENB copolymer are low due to the greater incorporation of ENB in the polymer backbone, and later the polymerization reaction rates remain stable with a lower mol% of ENB. As the polymerization time increases, the Rp and ENB mol% values of the E–ENB copolymers decreased. The molecular weights of the E–ENB copolymers were initially lower and significantly increased with reaction time. At the initial polymerization time, the propagation rate content (kp) was higher than later, and this difference could be due to the higher mol% of ENB that converts to being amorphous and may be dissolved in toluene, resulting in a lower diffusion barrier in the E/ENB copolymer system. The active centers generated later in the copolymerization process appear to have a lower kp value than those formed earlier. The mol% of ENB in the E/ENB copolymers is higher initially rather than later. The active centers also quickly increase during the first 120–480 s, which means that cyclic diene ENB quickly activates the active sites early on that were inactive for ethylene.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The authors are thankful to the Natural Foundation China (51803081) for their financial support.References
- Y. Na, S. Dai and C. Chen, Macromolecules, 2018, 51, 4040–4048 CrossRef CAS.
- S. Saikrishnan, D. Jubinville, C. Tzoganakis and T. H. Mekonnen, Polym. Degrad. Stab., 2020, 182, 109390 CrossRef CAS.
- A. Khan, Y. Guo, Z. Zhang, A. Ali, Z. Fu and Z. Fan, J. Appl. Polym. Sci., 2018, 135, 46030 CrossRef.
- P. Lanzafame, S. Perathoner, G. Centi, S. Gross and E. Hensen, Catal. Sci. Technol., 2017, 7, 5182–5194 RSC.
- S. Song, X. Liu, H. Zhang, Z. Fu, J. Xu and Z. Fan, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 2715–2722 CrossRef CAS.
- Z.-Q. Zhang, J.-T. Qu, S. Zhang, Q.-P. Miao and Y.-X. Wu, Polym. Chem., 2018, 9, 48–59 RSC.
- F. Zaccaria, C. Zuccaccia, R. Cipullo, P. H. Budzelaar, A. Vittoria, A. Macchioni, V. Busico and C. Ehm, ACS Catal., 2021, 11, 4464–4475 CrossRef CAS.
- C. U. Pittman, J. Inorg. Organomet. Polym. Mater., 2005, 15, 33–55 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef.
- Y. Li, J. Yang, B. Wang and Y. Li, RSC Adv., 2016, 6, 59590–59599 RSC.
- A. Ali, M. K. Tufail, M. I. Jamil, W. Yaseen, N. Iqbal, M. Hussain, A. Ali, T. Aziz, Z. Fan and L. Guo, Molecules, 2021, 26, 2037 CrossRef CAS PubMed.
- A. Ali, M. A. Akram, Y. Guo, H. Wu, W. Liu, A. Khan, X. Liu, Z. Fu and Z. Fan, J. Macromol. Sci., Part A: Pure Appl. Chem., 2020, 57, 156–164 CrossRef CAS.
- S. I. Chowdhury, R. Tanaka, Y. Nakayama and T. Shiono, Polymer, 2020, 187, 122094 CrossRef CAS.
- A. Simanke, G. Galland, R. Baumhardt Neto, R. Quijada and R. Mauler, J. Appl. Polym. Sci., 1999, 74, 1194–1200 CrossRef CAS.
- Y.-M. Zhai, Y. Wang, W. Yang, B.-H. Xie and M.-B. Yang, J. Therm. Anal. Calorim., 2012, 110, 1389–1394 CrossRef CAS.
- F. Zhang, Q. Fu, T. Lü, H. Huang and T. He, Polymer, 2002, 43, 1031–1034 CrossRef CAS.
- J.-S. Yoon, D.-H. Lee, E.-S. Park, I.-M. Lee, D.-K. Park and S.-O. Jung, Polymer, 2000, 41, 4523–4530 CrossRef CAS.
- M. Ratanasak, J.-y. Hasegawa and V. Parasuk, New J. Chem., 2021, 45(18), 8248–8257 RSC.
- L. Resconi, L. Cavallo, A. Fait and F. Piemontesi, Chem. Rev., 2000, 100, 1253–1346 CrossRef CAS PubMed.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- Y. Yu, R. Cipullo and C. Boisson, ACS Catal., 2019, 9, 3098–3103 CrossRef CAS.
- M. Bochmann, J. Organomet. Chem., 2004, 689, 3982–3998 CrossRef CAS.
- M. D. Christianson, E. H. Tan and C. R. Landis, J. Am. Chem. Soc., 2010, 132, 11461–11463 CrossRef CAS PubMed.
- J. Huang and G. L. Rempel, Ind. Eng. Chem. Res., 1997, 36, 1151–1157 CrossRef CAS.
- C.-H. Chen, W.-C. Shih and C. Hilty, J. Am. Chem. Soc., 2015, 137, 6965–6971 CrossRef CAS PubMed.
- S. Collins, M. Linnolahti, M. G. Zamora, H. S. Zijlstra, M. a. T. Rodríguez Hernández and O. Perez-Camacho, Macromolecules, 2017, 50, 8871–8884 CrossRef CAS.
- M. Ratanasak, J.-y. Hasegawa and V. Parasuk, New J. Chem., 2021, 45, 8248–8257 RSC.
- X. Desert, J.-F. Carpentier and E. Kirillov, Coord. Chem. Rev., 2019, 386, 50–68 CrossRef CAS.
- F. Ghiotto, C. Pateraki, J. R. Severn, N. Friederichs and M. Bochmann, Dalton Trans., 2013, 42, 9040–9048 RSC.
- R. Kleinschmidt, Y. Van der Leek, M. Reffke and G. Fink, J. Mol. Catal. A: Chem., 1999, 148, 29–41 CrossRef CAS.
- D. L. Nelsen, B. J. Anding, J. L. Sawicki, M. D. Christianson, D. J. Arriola and C. R. Landis, ACS Catal., 2016, 6, 7398–7408 CrossRef CAS.
- F. Song, R. D. Cannon and M. Bochmann, J. Am. Chem. Soc., 2003, 125, 7641–7653 CrossRef CAS PubMed.
- B. M. Moscato, B. Zhu and C. R. Landis, Organometallics, 2012, 31, 2097–2107 CrossRef CAS.
- C. R. Landis, K. A. Rosaaen and D. R. Sillars, J. Am. Chem. Soc., 2003, 125, 1710–1711 CrossRef CAS PubMed.
- E. Ihara, V. G. Young Jr and R. F. Jordan, J. Am. Chem. Soc., 1998, 120, 8277–8278 CrossRef CAS.
- M. Lehmann, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2009, 48, 7444–7447 CrossRef CAS PubMed.
- A. Ali, X. Liu, Y. Guo, M. A. Akram, H. Wu, W. Liu, A. Khan, B. Jiang, Z. Fu and Z. Fan, J. Organomet. Chem., 2020, 922, 121366 CrossRef CAS.
- A. Ali, N. Muhammad, S. Hussain, M. I. Jamil, A. Uddin, T. Aziz, M. K. Tufail, Y. Guo, T. Wei and G. Rasool, Polymers, 2021, 13, 268 CrossRef CAS PubMed.
- B. Jiang, B. Zhang, Y. Guo, A. Ali, W. Guo, Z. Fu and Z. Fan, ChemCatChem, 2020, 12, 5140–5148 CrossRef CAS.
- A. Ali, M. I. Jamil, A. Uddin, M. Hussain, T. Aziz, M. K. Tufail, Y. Guo, B. Jiang, Z. Fan and L. Guo, J. Polym. Res., 2021, 28, 1–15 CrossRef.
- M. I. Jamil, Q. Wang, A. Ali, M. Hussain, T. Aziz, X. Zhan and Q. Zhang, Journal of Bionic Engineering, 2021, 18, 548–558 CrossRef.
- A. Ali, A. Uddin, M. I. Jamil, X. Shen, M. Abbas, T. Aziz, M. Hussain, S. Hussain, R. Fang and Z. Fan, J. Organomet. Chem., 2021, 121929 CrossRef CAS.
- G. Leone, G. Zanchin, R. Di Girolamo, F. De Stefano, C. Lorber, C. De Rosa, G. Ricci and F. Bertini, Macromolecules, 2020, 53, 5881–5894 CrossRef CAS.
- W. Kaminsky and M. Miri, J. Polym. Sci., Polym. Chem. Ed., 1985, 23, 2151–2164 CrossRef CAS.
- I. Tritto, L. Boggioni, J. C. Jansen, K. Thorshaug, M. C. Sacchi and D. R. Ferro, Macromolecules, 2002, 35, 616–623 CrossRef CAS.
- Y. V. Kissin, et al., Top. Catal., 1999, 7(1), 69–88 CrossRef CAS.
- Y. V. Kissin, J. Polym. Sci., Part A: Polym. Chem., 2003, 41(12), 1745–1758. CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06243c |
| This journal is © The Royal Society of Chemistry 2021 |