
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTreatment of electrochemical plating wastewater by heterogeneous photocatalysis: the simultaneous removal of 6:2 fluorotelomer sulfonate and hexavalent chromium†
Hak-Hyeon Kim,
Seyfollah Gilak Hakimabadi and
Anh Le-Tuan Pham *
*
Department of Civil and Environmental Engineering, University of Waterloo, Waterloo, ON N2L 3G1, Canada. E-mail: anh.pham@uwaterloo.ca; Tel: +1-519-888-4567 ext. 30337
First published on 22nd November 2021
Abstract
6:2 fluorotelomer sulfonate (6:2 FtS) is being widely used as a mist suppressant in the chromate (Cr(VI)) plating process. As a result, it is often present alongside Cr(VI) in the chromate plating wastewater (CPW). While the removal of Cr(VI) from CPW has been studied for decades, little attention has been paid to the treatment of 6:2 FtS. In this study, the removal of Cr(VI) and 6:2 FtS by Ga2O3, In2O3, and TiO2 photocatalysts was investigated. In the Ga2O3/UVC system, over 95% of Cr(VI) was reduced into Cr(III) after only 5 min. Simultaneously, 6:2 FtS was degraded into F− and several perfluorocarboxylates. The predominant reactive species responsible for the degradation of 6:2 FtS in the Ga2O3 system were identified to be hVB+ and O2˙−. In addition, it was observed that the presence of Cr(VI) helped accelerate the degradation of 6:2 FtS. This synergy between Cr(VI) and 6:2 FtS was attributable to the scavenging of eCB− by Cr(VI), which retarded the recombination of eCB− and hVB+. The In2O3/UVC system was also capable of removing Cr(VI) and 6:2 FtS, although at significantly slower rates. In contrast, poor removal of 6:2 FtS was achieved with the TiO2/UVC system, because Cr(III) adsorbed on TiO2 and inhibited its reactivity. Based on the results of this study, it is proposed that CPW can be treated by a treatment train that consists of an oxidation–reduction step driven by Ga2O3/UVC, followed by a neutralization step that converts dissolved Cr(III) into Cr(OH)3(S).
1 Introduction
Owing to their simultaneous lipophobicity and hydrophobicity, high chemical and thermal stability, and surfactant property, per- and polyfluorinated alkyl substances (PFAS) are being used in a wide variety of industrial and commercial products, including in fire-fighting foams, flame retardants, mist suppressants, adhesives, and surface coatings, among others.1 The extensive use of PFAS and the release of PFAS-containing wastes into the environment have led to their detection in air,2,3 surface water,4,5 groundwater,6–9 drinking water,10,11 soils6,8 and biosolids.12–14 Exposure to PFAS has been linked to various health problems, such as increased blood cholesterol, liver damage, kidney and testicular cancer, and thyroid dysfunction.15–18 Therefore, guidelines and cleanup criteria in the low part-per-trillion levels are being proposed by jurisdictions around the world. For example, the US Environmental Protection Agency has recently established a health advisory level of 70 ng L−1 for perfluorooctanoate (PFOA) and perfluorooctane sulfonate (PFOS).19 Health Canada has proposed drinking water screening values of 200–600 ng L−1 for several PFAS compounds.20,21 As our knowledge about the occurrence, fate, and toxicity of PFAS continues to grow, the guidelines for these compounds may become increasingly more stringent. Therefore, there is an urgent need to develop effective technologies for the treatment of PFAS-containing wastes to meet these future guidelines and to protect ecosystem and human health.This study concerns the treatment of PFAS in chromate plating wastewater (CPW). During electrochemical plating, the evolution and burst of gas bubbles produced by electrolysis generates chromic acid mists, which pose significant health risks to workers on site. Therefore, a surfactant is usually added to the plating bath to suppress mist formation as well as to decrease surface wettability. Certain PFAS compounds are uniquely suited as mist suppressants in electrochemical plating because of their high aqueous solubility and resistance to electrochemical degradation. PFOS was traditionally the most popular mist suppressant in the electroplating industry, but this compound has been gradually replaced by 6:2 fluorotelomer sulfonate (6:2 FtS),22 a less recalcitrant and bioaccumulative compound.23,24 The concentration of 6:2 FtS in electrochemical plating baths is typically tens of mg L−1, resulting in a concentration of 6:2 FtS in CPW ranging up to hundreds of μg L−1.22
While the treatment of CPW has been investigated for decades, previous efforts focused primarily on removing hexavalent chromium (Cr(VI)) and other metals,25 with little/no attention being paid to the removal of 6:2 FtS. However, it can be expected that conventional CPW treatment processes such as chemical reduction and coprecipitation are unlikely effective at removing 6:2 FtS. This is because like most PFAS, 6:2 FtS does not adsorb to metal hydroxides to an appreciable extent. Also, 6:2 FtS is generally resistant to chemical degradation; recent studies have shown that PFAS can only be degraded by harsh treatment processes such as thermal treatment,26–28 plasma-based oxidation and reduction,29 and reduction by hydrated electrons.30–32
Several studies have reported that PFAS can be degraded by heterogeneous photocatalysis. For example, it has been shown that PFOA was destroyed by photogenerated valence band holes (hVB+) produced when wide-bandgap semiconductors such as In2O3,33,34 Ga2O3,35 and In modified-Ga2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 were irradiated with ultraviolet (UV) light. It has also been shown that PFOA could be degraded by the widely used TiO2 photocatalyst,33,34 although the degradation rate was relatively slow since PFOA did not react appreciably with hydroxyl radical (˙OH) (i.e., the predominant reactive species in the TiO2/UV system). In a recent study, an excellent removal of PFOA was achieved with a commercial boron nitride (BN) photocatalyst.37 The degradation of PFOA in this system was attributable to multiple reactive species, including hVB+, ˙OH, and superoxide (O2˙−). In addition to oxidative degradation, it appears that PFOA can also be reductively destroyed via reactions with photogenerated conduction band electrons (eCB−)38 or by surface hydrogen.39 While the generation of a suite of different reactive species capable of destroying PFAS makes heterogeneous photocatalysis appealing, a major drawback of this approach is that hVB+ and eCB− usually recombine rapidly, resulting in inefficient production of reactive species. This limitation can be overcome by the addition of a scavenger (e.g., H2O2, IO4−, HS−, I−) that react with either hVB+ or eCB−, thereby extending the lifetime of the other species.40
36 were irradiated with ultraviolet (UV) light. It has also been shown that PFOA could be degraded by the widely used TiO2 photocatalyst,33,34 although the degradation rate was relatively slow since PFOA did not react appreciably with hydroxyl radical (˙OH) (i.e., the predominant reactive species in the TiO2/UV system). In a recent study, an excellent removal of PFOA was achieved with a commercial boron nitride (BN) photocatalyst.37 The degradation of PFOA in this system was attributable to multiple reactive species, including hVB+, ˙OH, and superoxide (O2˙−). In addition to oxidative degradation, it appears that PFOA can also be reductively destroyed via reactions with photogenerated conduction band electrons (eCB−)38 or by surface hydrogen.39 While the generation of a suite of different reactive species capable of destroying PFAS makes heterogeneous photocatalysis appealing, a major drawback of this approach is that hVB+ and eCB− usually recombine rapidly, resulting in inefficient production of reactive species. This limitation can be overcome by the addition of a scavenger (e.g., H2O2, IO4−, HS−, I−) that react with either hVB+ or eCB−, thereby extending the lifetime of the other species.40
We hypothesize that heterogeneous photocatalysis is a promising approach for the treatment of CPW, owing to its potential to remove both Cr(VI) and 6:2 FtS in a single treatment step. Specifically, we envision in that in heterogeneous photocatalysis Cr(VI) will be reduced by eCB− into Cr(III), which then can be separated as Cr(OH)3(s) by precipitation. Simultaneously, 6:2 FtS will be oxidized by hVB+ and/or ˙OH. Moreover, we hypothesize that there could be a synergistic effect between the removals of Cr(VI) and 6:2 FtS—that is the removal of each contaminant will be enhanced in the presence of the other. This is because the scavenging of eCB− by Cr(VI), and of hVB+ by 6:2 FtS could potentially mitigate the recombination of hVB+ and eCB− as discussed above.
| Photocatalyst + hν → hVB+ + eCB− | (1) |
| eCB− + Cr(VI) →→→ Cr(III) | (2) |
| hVB+ + 6:2 FtS → byproducts | (3) |
| hVB+ + H2O → ˙OH + H+ | (4) |
| ˙OH + 6:2 FtS → byproducts | (5) |
To test the hypotheses above, the removals of Cr(VI) and 6:2 FtS were investigated using three commercially available photocatalysts, namely Ga2O3, In2O3, and TiO2. A series of experiment with probe compounds were conducted to gain insights into the reactive species responsible for degradation of 6:2 FtS. In addition, the 6:2 FtS degradation byproducts were measured throughout the experiment to close mass balance and elucidate the degradation pathway. Finally, the reusability of Ga2O3 (which turned out to be the most promising among the studied materials) was investigated through a repeated contaminant removal experiment.
2 Materials and methods
2.1 Materials
The photocatalysts Ga2O3 (≥99.99%), In2O3 (99.9%), and TiO2 (Degussa P25, 99.5%) were purchased from Sigma Aldrich and were used as received. The surface morphology and elemental composition of these materials were characterized by scanning electron microscopy (SEM)/energy dispersive X-ray spectrometry (EDS), while their surface area was determined using the Brunauer–Emmett–Teller (BET) N2 physisorption method. The results of these characterizations are available in the ESI (Table S1 and Fig. S1†). 6:2 FtS (2-(perfluorohexyl)ethane-1-sulfonic acid sodium salt, >98%) was purchased from Toronto Research Chemicals (Toronto, ON), while PFAS analytical native and mass-labelled standards were purchased from Wellington Laboratories (Guelph, ON). All other chemicals were purchased at the highest purity available from Sigma Aldrich or Fisher Scientific. A stock solution of 5 mg L−1 6:2 FtS was prepared by dissolving 6:2 FtS in an aqueous solution consisting of 15 mM methanol. Other stock solutions, namely Cr(VI) (100 mg L−1 as Cr), Cr(III) (100 mg L−1 as Cr), p-benzoquinone (BQ, 100 mM) and bromate (BrO3−, 500 mM), were prepared by dissolving K2Cr2O7, CrCl3·6H2O, BQ, and KBrO3 in 18.2 MΩ cm MilliQ water (Millipore).2.2 Photolysis experiments
All experiments were carried out in a reactor chamber that was equipped with 6 × 4 W UVC lamps (Fig. S2 in the ESI†). A quartz container, which held the reaction suspension, was placed at the center of the chamber. In most experiments, the reaction suspension was prepared by adding a photocatalyst and aliquots containing 6:2 FtS and Cr(VI) from their respective stock solutions to MilliQ water such that [catalyst] = 0.5 g L−1, [6:2 FtS]0 = 100 or 500 μg L−1, and [Cr(VI)]0 = 0 or 1 mg L−1. Although the concentration of Cr(VI) in chromium plating wastewaters can be in the order of tens of mg L−1, the concentration of Cr(VI) employed in most experiments was kept low to avoid damaging the liquid chromatograph mass spectrometer (LC/MS/MS). However, in one experiment, the initial concentrations of Cr(VI) and 6:2 FtS were 20 mg L−1 and 2 mg L−1, respectively. The samples collected from this experiment were diluted 400 times prior to LC/MS/MS analysis. In some experiments, the reaction suspension also contained formic acid (HCOOH, a hVB+ scavenger),40 tert-butyl alcohol (t-BuOH, an ˙OH scavenger),41 BQ (a O2˙− scavenger),42 or BrO3− (a eCB− scavenger).43 The initial pH of the reaction suspension was adjusted to pH = 3 ± 0.1 using either 1 M H2SO4 or 1 M NaOH. The pH of the suspension varied by less than 0.1 unit throughout the entire course of each experiment.Preliminary experiments showed that approximately 10% of the initial 6:2 FtS was lost by adsorption on the photocatalysts, and that adsorption equilibrium was attained within 30 min. As such, the suspension was vigorously mixed in the dark for 1 hour. Subsequently, photochemical reactions were initiated by turning the UVC lamps on. At predetermined time intervals, aliquots of suspension were withdrawn and subjected to analyses for dissolved Cr, fluoride (F−), as well as 6:2 FtS and its degradation products.
As will be presented in the Results and discussion sections, Ga2O3 was observed to be the photocatalyst most efficient at removing 6:2 FtS and Cr(VI). Therefore, the reusability of Ga2O3 was evaluated over 3 experimental cycles wherein at the end of each cycle Ga2O3 particles were separated and added to a fresh reaction solution containing 100 μg L−1 6:2 FtS and 1 mg L−1 Cr(VI). Subsequently, the photolysis experiment was repeated.
All experiments were carried out at least in triplicate, and the average values are presented along with one standard deviation. The evolution of the concentrations of different species in the solution is expressed against energy flux (J cm−2) received by the reaction suspension, which was determined based on ferrioxalate actinometry.
2.3 Analytical methods
For the analysis of F−, Ga2O3 particles were separated from the suspension by centrifugation, and F− in the supernatant was measured spectrophotometrically using the modified SPADNS method (limit of quantitation of 50 μg L−1).44 For the analysis of dissolved Cr, the suspension was first acidified to pH < 2 to dissolve particulate Cr(III) (which was formed due to the photoreduction of Cr(VI) to Cr(III)). Subsequently, Ga2O3 particles were separated by centrifugation, and Cr(VI) and total Cr (i.e., Cr(VI) + Cr(III)) in the acidified supernatant were measured spectrophotometrically using the 1,5-diphenylcarbazide method.45 The limits of quantitation by this method are 30 μg L−1 for Cr(VI), and 50 μg L−1 for total Cr. The concentration of Cr(III) in the sample was calculated based on the difference between the concentrations of total Cr and Cr(VI).For the analysis of PFAS by LC/MS/MS, each sub-aliquot was diluted 9 times in a solution consisting of 50 v% methanol and 50 v% 5 mM ammonium acetate in water. The purpose of this dilution was threefold: (a) to desorb PFAS from the catalyst in order to enable closing mass balance, (b) to precipitate Cr(III) from the sample in order to minimize the amount of dissolved Cr introduced into the LC/MS/MS, and (c) to obtain a sample consisting of ca. 50/50 v/v methanol/water for analysis by LC/MS/MS. Following dilution, the suspension was centrifuged to separate Ga2O3 and Cr(OH)3, and the supernatant was added to an HPLC vial. Prior to LC/MS/MS analysis, mass-labelled PFAS, which served as internal standards, were spiked into each vial. Details about the LC/MS/MS operating condition and the list of PFAS compounds analysed in this study can be found in the ESI (Tables S2 and S3†).
3 Results
Photolysis without a catalyst did not result in 6:2 FtS or Cr(VI) removal (data not shown). Also, both contaminants were not removed appreciably by adsorption in the absence of light (Fig. S3†). In contrast, both contaminants were removed when Ga2O3, In2O3, or TiO2 was present, although the reaction rates varied significantly (Fig. 1). In the Ga2O3 system, the concentration of Cr(VI) decreased to around the method quantitation limit after only 5 min of irradiation (Fig. 1a). Simultaneously, the concentration of 6:2 FtS gradually decreased throughout the course of the experiment, with 90% 6:2 FtS removed after 2 hours (equivalent to a fluence of 19 J cm−2, Fig. 1d). Interestingly, the 6:2 FtS degradation rate was over twice slower when Cr(VI) was absent (kobserved = 0.34 h−1, versus kobserved = 0.73 h−1 in the presence of Cr(VI)). Excellent removals of Cr(VI) and 6:2 FtS were also observed in the experiment wherein the initial concentrations of the contaminants were increased by 20 times (i.e., [Cr(VI)]0 = 20 mg L−1, [6:2 FtS] = 2 mg L−1; Fig. S4†).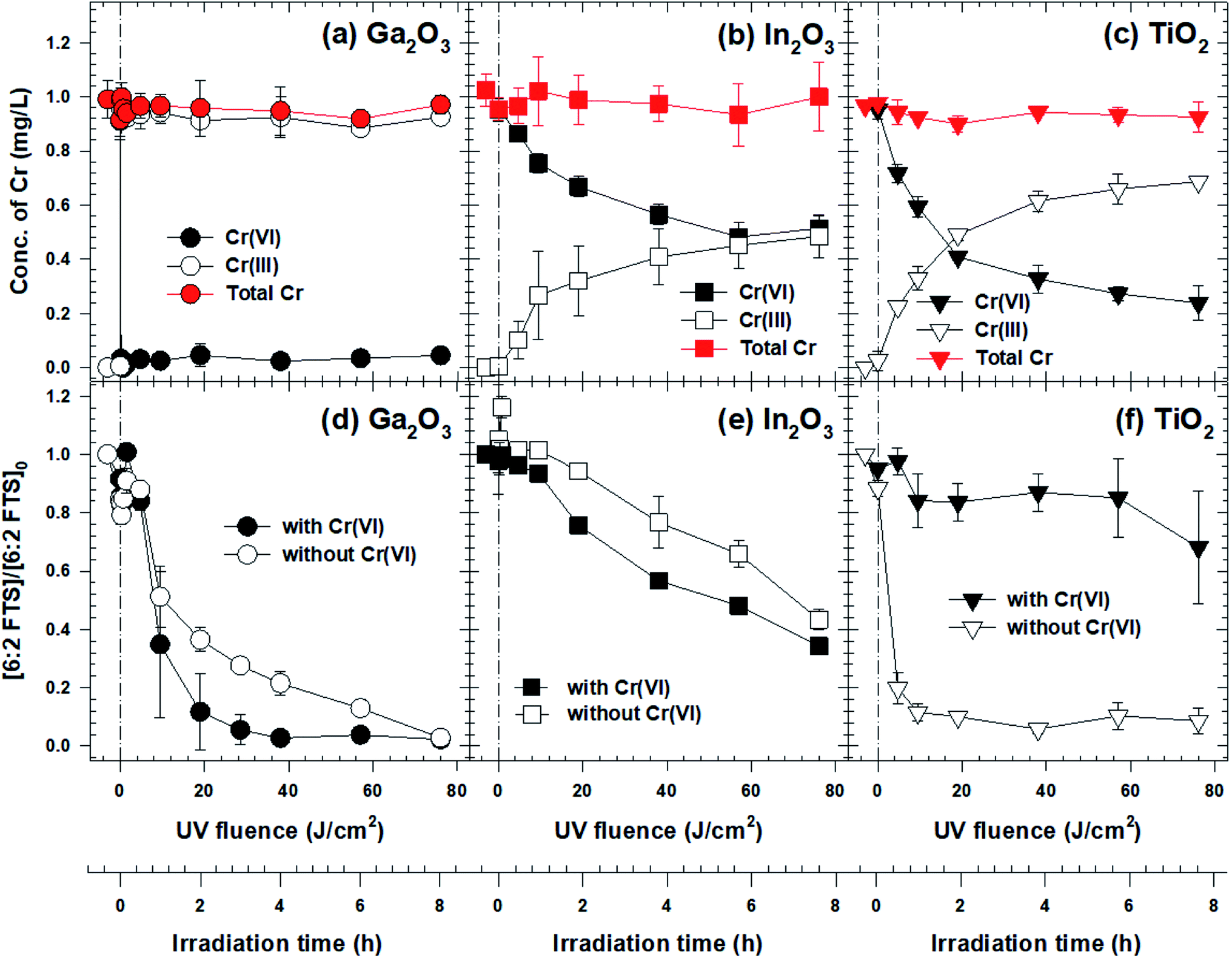 | ||
| Fig. 1 Removals of Cr(VI) and 6:2 FtS by Ga2O3 (a) and (d), In2O3 (b) and (e) and TiO2 (c) and (f) photocatalysts. [Catalyst] = 0.5 g L−1, [6:2 FtS]0 = 100 μg L−1, [Cr(VI)]0 = 1 mg L−1, pH = 3.0 ± 1. The solution was vigorously mixed in the dark for 1 hour. Subsequently, photolysis was initiated by turning UV-C lamps on at t = 0 h. | ||
Cr(VI) and 6:2 FtS were also simultaneously removed in the In2O3 system, although at significantly slower rates. After 8 hours, the concentrations of Cr(VI) and 6:2 FtS decreased by only 50% and 35%, respectively (Fig. 1b and e). As in the Ga2O3 system, the presence of Cr(VI) enhanced the rate of 6:2 FtS degradation (kobserved = 0.12 h−1 and 0.10 h−1 in the presence and absence of Cr(VI), respectively). On the contrary, the presence of Cr(VI) inhibited the degradation of 6:2 FtS in the TiO2 system: whereas 80% of 6:2 FtS was removed within the first 20 min in the absence of Cr(VI), only 30% removal was achieved in the presence of Cr(VI) even after 8 hours (kobserved = 0.04 h−1) (Fig. 1f). If the 6:2 FtS degradation was very slow, the reduction of Cr(VI) in the TiO2 system was slightly faster than that in the In2O3 system (Fig. 1c).
The varying reactivity among the materials discussed above is attributable to their inherent photocatalytic reactivity rather than to the difference in their surface area. In the absence of Cr(VI), the 6:2 FtS degradation rate with TiO2 (kobserved = 2.39 h−1) was over seven times faster than that with Ga2O3 (kobserved = 0.34 h−1), although the surface area of TiO2 (51.9 m2 g−1) is only three times greater than that of Ga2O3 (19.5 m2 g−1). In the presence of Cr(VI), the 6:2 FtS degradation rate with Ga2O3 (kobserved = 0.73 h−1) was over six times greater than that of In2O3 (kobserved = 0.12 h−1), even though the surface area of the latter (7.1 m2 g−1) is only 2.7 times less than that of the former.
Not only did the contaminant removal rates vary but also the reactive species responsible for the degradation of 6:2 FtS appeared to be significantly different in the three photocatalyst systems. With In2O3, 6:2 FtS did not degrade appreciably in the presence of either 10 mM HCOOH or 10 mM t-BuOH (Fig. S5a†). With TiO2, the degradation of 6:2 FtS was also significantly inhibited by HCOOH, but was only partially inhibited by t-BuOH (Fig. S5b†). Somewhat surprisingly, neither of these two scavengers had any effect on the degradation of 6:2 FtS by Ga2O3 (Fig. 2a). However, the degradation was partially inhibited when BrO3− was present, and was completely inhibited in the presence of HCOOH and BrO3−, or HCOOH and BQ (Fig. 2a).
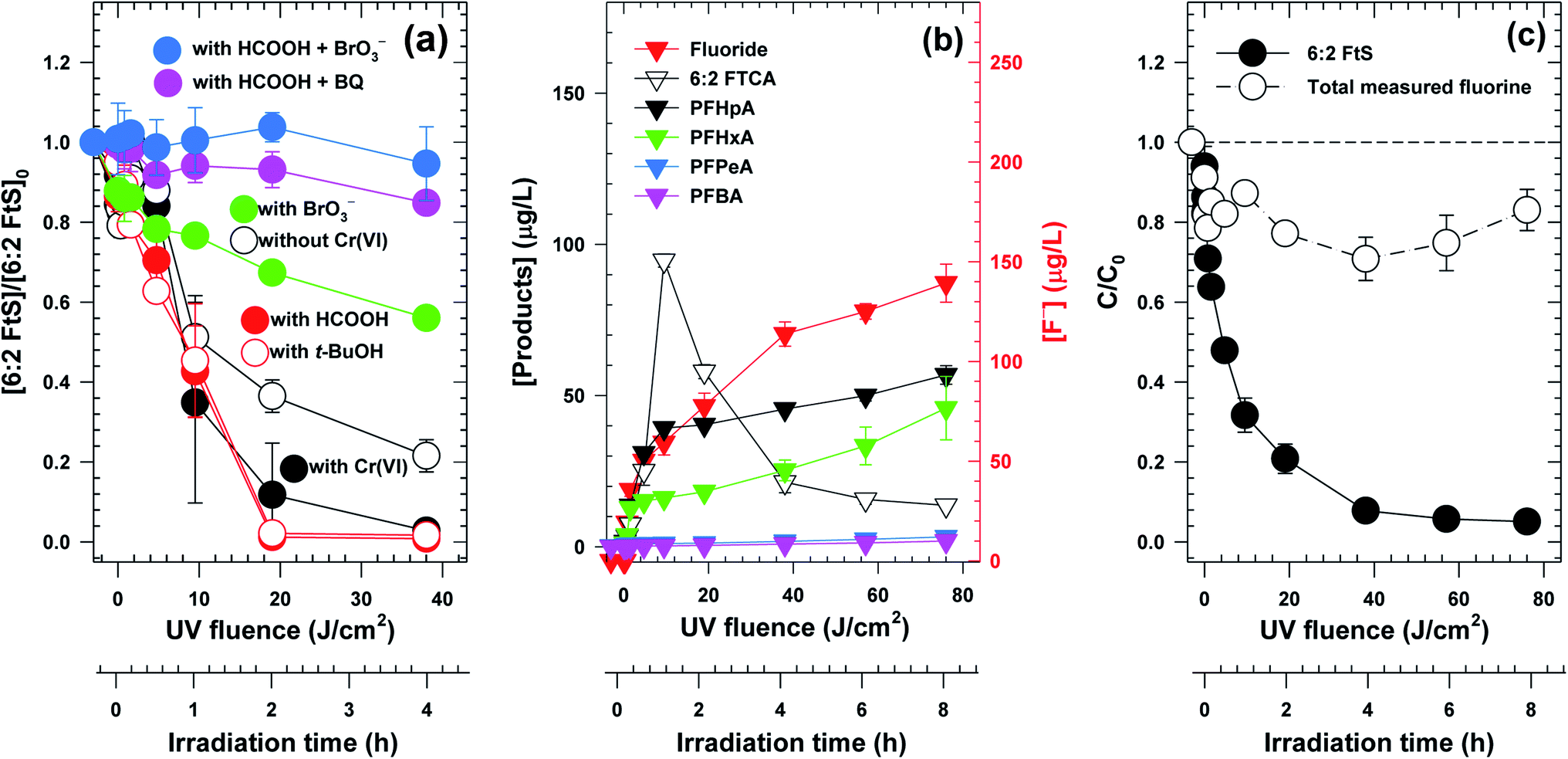 | ||
| Fig. 2 (a) Effect of various scavengers on the degradation of 6:2 FtS; (b) evolution of byproducts; (c) F mass balance. [Ga2O3]0 = 0.5 g L−1, [6:2 FtS]0 = 100 μg L−1 in (a) 500 μg L−1 in (b) and (c), [Cr(VI)] = 1 mg L−1, [t-BuOH]0 = [HCOOH]0 = [BrO3−]0 = 10 mM, [BQ]0 = 1 mM, pH = 3.0 ± 1. It is noted that Cr(VI) was not added in the experiments wherein other scavengers (i.e., HCOOH, BQ, BrO3−, and t-BuOH) were present. | ||
Because Ga2O3/UVC was effective at removing both Cr(VI) and 6:2 FtS, the evolution of the 6:2 FtS degradation byproducts in this system was further investigated to gain insight into the reaction pathway. Besides F−, a total of 5 byproducts, i.e., 6:2 fluorotelomer carboxylate (6:2 FtCA), perfluoroheptanoate (PFHpA), perfluorohexanoate (PFHxA), perfluoropentanoate (PFPeA), perfluorobutanoate (PFBA), were observed as 6:2 FtS was degraded (Fig. 2b). The fluorine mass balance indicates that the sum of F−, the F in the 5 byproducts, and the F in the remaining 6:2 FtS accounted for 80% of the total F initially added as 6:2 FtS (Fig. 2c). This result suggests that there must be other byproducts that were not detected by the LCMS analytical method used in this study. Among the measured byproducts, 6:2 FtCA exhibited a two-phase concentration–time profile wherein its concentration first increased and subsequently decreased (Fig. 2b). For the other 4 byproducts, their concentrations increased gradually (Fig. 2b). By the end of the experiment, the concentration of F− was 135 μg L−1 (equivalent to approximately 50% of the total F added).
In the Ga2O3 reusability experiment, excellent removals of both Cr(VI) and 6:2 FtS were observed over three cycles (Fig. 3a). In each cycle, more than 95% of Cr(VI) was reduced into Cr(III), 80% of which can be separated by Cr(OH)3(s) from the treated stream by raising the solution pH to 7.5 (Fig. 3b). The concentration of dissolved Cr after pH adjustment and Cr(OH)3(s) separation (by centrifugation) was 0.2 mg L−1, well below the US EPA's discharge limit of 2–4 mg L−1.46 Meanwhile, the fraction of 6:2 FtS degraded in each cycle ranged between 86 and 92%. The slightly lower 6:2 FtS removal in cycle 3 could be due to the adsorption of Cr species on Ga2O3, thereby diminishing its photocatalytic reactivity.
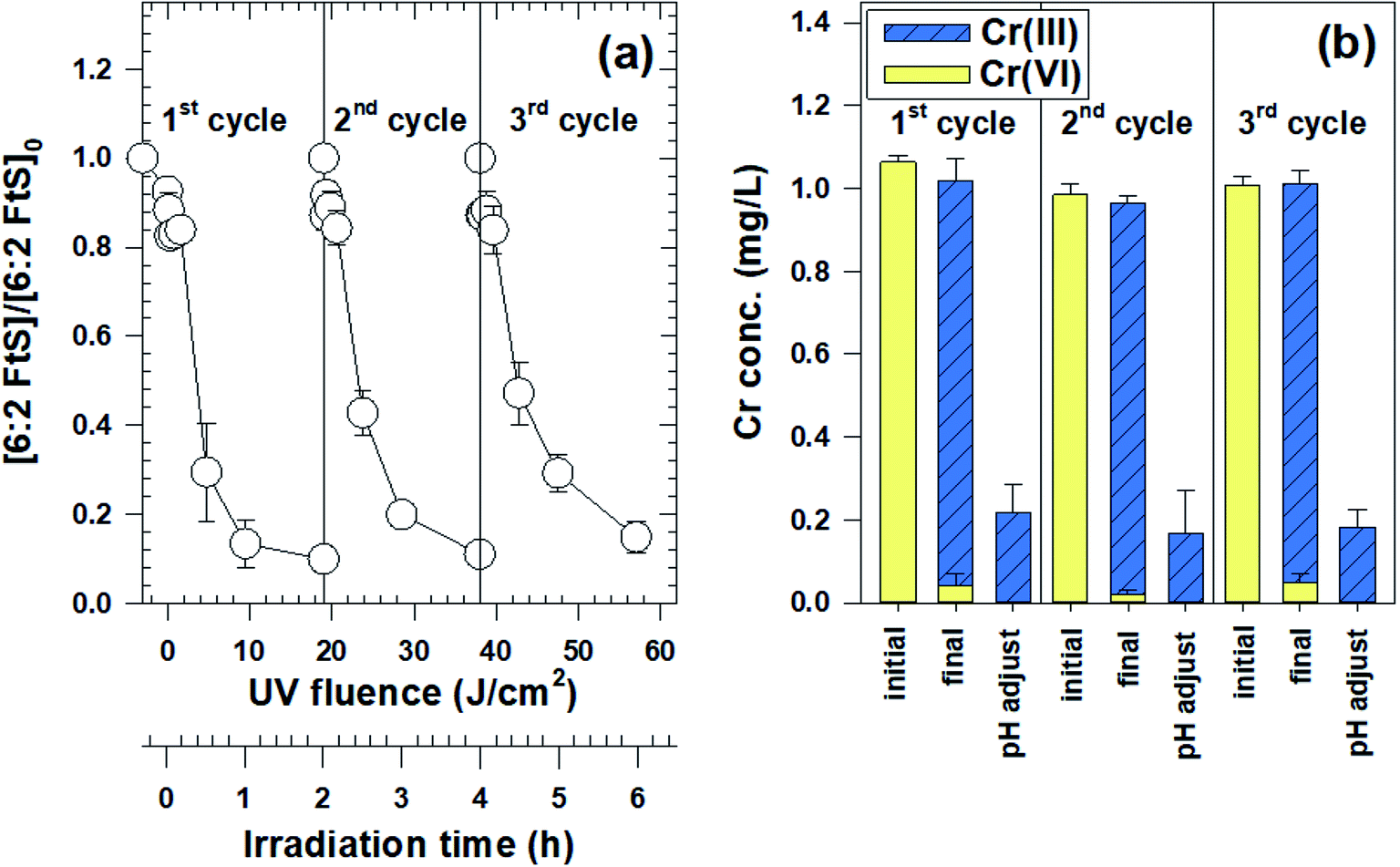 | ||
| Fig. 3 (a) Degradation of 6:2 FtS by Ga2O3/UV-C over three cycles. The concentration of Ga2O3 was 0.5 g L−1. At the beginning of each cycle, the spent solution was replaced with a fresh solution containing [6:2 FtS]0 = 100 μg L−1 and [Cr(VI)]0 = 1 mg L−1; (b) the concentrations of dissolved Cr(VI) and Cr(III) at the beginning of each cycle (labelled as “initial”), after photolysis (labelled as “final”), and after adjusting pH to 7.5 and separating Cr(OH)3(s) (labelled as “pH adjust”). | ||
4 Discussion
4.1 Reactive species responsible for contaminant removal
The Cr mass balance and the stoichiometric production of Cr(III) in all three photocatalytic systems suggest that Cr(VI) was removed via reactions with eCB− (reaction (2)). (The reduction of Cr(VI) via direct reactions with 6:2 FtS can be ruled out as Cr(III) was not produced in the absence of photocatalysts. The production of Cr(III) from direct reactions between Cr(VI) and the byproducts of 6:2 FtS can also be ruled out, since 6:2 FtS was not degraded appreciably in the first 5 min in the Ga2O3 system (Fig. 1a)). The much faster Cr(VI) reduction in the Ga2O3 compared to that in the other two systems is attributable to Ga2O3's higher conduction band position (ECB,Ga2O3 = −2.95 eV versus ECB,TiO2 = −4.21 eV and ECB,In2O3 = −4.30 eV), resulting in eCB− that possesses a greater reduction power.47–49 Despite a rapid removal at the beginning, the concentration of Cr(VI) in the Ga2O3 system never decreased to zero. Instead, a steady state concentration of Cr(VI) of ca. 30 μg L−1 was maintained in the solution throughout the course of the experiment. As such, it is hypothesized that there was a redox cycle between Cr(VI) and Cr(III) in this system, driven by reaction (2) and the oxidation of Cr(III) back to Cr(VI):| hVB+ + Cr(III) →→→ Cr(VI) | (6) |
The role of the Cr(VI)–Cr(III) redox cycling toward sustaining the enhanced 6:2 FtS degradation (compared to when Cr was absent) in the Ga2O3 system will be discussed further in Section 4.2.
Whereas eCB− was the only reactive species responsible for Cr(VI) removal, a few different reactive species appears to be involved in the degradation of 6:2 FtS. In the In2O3 system, the predominant oxidant likely was ˙OH, although hVB+ also could have had a partial contribution, since the 6:2 FtS degradation was somewhat further inhibited by HCOOH (a hVB+ scavenger, which is also expected to inhibit reaction (4)) (Fig. S5a†). In the TiO2 system, both hVB+ and ˙OH played an important role because HCOOH and t-BuOH appreciably retarded the degradation of 6:2 FtS (Fig. S5b†). Because hVB+ and ˙OH can oxidize substrates via hydrogen abstraction,40 it is proposed that the oxidation of 6:2 FtS proceeded as follows:
| C6F13C2H4SO3− + hVB+ → C6F13CH2˙CHSO3− + H+ | (7) |
| C6F13C2H4SO3− + hVB+ → C6F13˙CHCH2SO3− + H+ | (8) |
| C6F13C2H4SO3− + ˙OH → C6F13CH2˙CHSO3− + H2O | (9) |
| C6F13C2H4SO3− + ˙OH → C6F13˙CHCH2SO3− + H2O | (10) |
Previous studies have reported that ˙OH played a minor role in the degradation of PFOA by TiO2- and In2O3-based photocatalysts.33,34 This is because PFOA, a fully fluorinated compound, does not react with ˙OH to an appreciable extent. The second-order rate constant for the reaction between PFOA and ˙OH is thought to be k˙OH < 106 M−1 s−1, although the precise value is not available in the literature.50 For 6:2 FtS, the second-order rate constant with ˙OH was estimated by density functional theory to be 1.0 × 106 (reaction (9)) and 3.0 × 106 M−1 s−1 (reaction (10)).51 It is generally agreed that contaminants with a k˙OH < 108 M−1 s−1 will not be removed appreciably by ˙OH-based processes, due to the ˙OH scavenging by HCO3− and other solutes. However, since CPW is acidic (pH < 3), the concentration of HCO3− in CPW should be very small (H2CO3 has a pKa1 = 6.5). In case where the scavenging of ˙OH by other background solutes is significant, 6:2 FtS could still be degraded by hVB+ (reactions (7) and (8)).
It has been well established that the irradiation of Ga2O3 by UVC generates hVB+ and ˙OH. As mentioned previously, however, neither HCOOH nor t-BuOH had any appreciable effect on the oxidation of 6:2 FtS (Fig. 2a). The rate of 6:2 FtS removal remained the same even when the concentration of HCOOH was increased to 50 mM (Fig. S6†). At a first glance, these results seem to suggest that hVB+ and ˙OH played a minimal role in the degradation of 6:2 FtS in the Ga2O3 system. This hypothesis appears to be further corroborated by the experiment in the presence of BrO3−, which indicates that 6:2 FtS was degraded by reductants such as eCB− and/or superoxide radical:42,52
| eCB− + O2 → O2˙− | (11) |
| eCB− + 6:2 FtS → byproducts | (12) |
| O2˙− + 6:2 FtS → byproducts | (13) |
| eCB− + BrO3− → BrO3˙− | (14) |
However, if eCB− and O2˙− were the only reactive species involved in the degradation of 6:2 FtS, it would not have been possible to explain why the presence of HCOOH together with BrO3− further inhibited the degradation (Fig. 2a). In fact, the inhibitory effect of HCOOH + BrO3− suggests that the involvement of hVB+ cannot be ruled out. To reconcile these seemingly conflicting experimental observations, the degradation of 6:2 FtS by the Ga2O3/UVC system is proposed to have occurred as follows. First, both hVB+ and eCB− should be important to the degradation of 6:2 FtS. It is noted that the degradation of 6:2 FtS is determined not only by the rate at which hVB+ and eCB− were produced, but also by the rate at which these species recombined. Thus, although the presence of HCOOH would on the one hand suppress the oxidation of 6:2 FtS, the scavenging of hVB+ by HCOOH would on the other hand mitigate the recombination of hVB+ and eCB−, thereby extending the lifetime of the latter. As a result, while the removal of 6:2 FtS by reactions (7)–(10) would be retarded, the removal by reactions (12) and (13) would be enhanced, resulting in no net change in the removal rate. On the contrary, hVB+ and eCB− would be scavenged when both HCOOH and BrO3− were present, resulting in the inhibition of reactions (7)–(13). It is further hypothesized that the involvement of ˙OH (i.e., reactions (9) and (10)) is likely minimal, since t-BuOH (which is not expected to affect the production or recombination of hVB+ and eCB−) did not influence the rate of 6:2 FtS degradation. Finally, it appears that O2˙− played a greater role than eCB− in the reductive degradation 6:2 FtS. This hypothesis is supported by two lines of evidence. Firstly, the 6:2 FtS degradation profile in the presence of HCOOH + BQ was similar to that in the presence of HCOOH + BrO3−. (BQ only scavenges O2˙− but not eCB−). Secondly, removing O2 from the solution (by sparging N2) also retarded the degradation to the same extent as BrO3− did (Fig. S7†).
4.2 The effect of Cr(VI) on 6:2 FtS removal
In agreement with the hypothesis that a synergistic effect may exist between Cr(VI) and 6:2 FtS, the 6:2 FtS degradation rate was enhanced in the Ga2O3 and In2O3 systems when Cr(VI) was present. In the In2O3 system, reaction (2) would mitigate the hVB+/eCB− recombination, thereby making hVB+ more available to H2O—as discussed above, ˙OH, which is generated from the reaction between hVB+ and H2O (reaction (4)), is likely the primary species involved in the degradation of 6:2 FtS in the In2O3 system. In the Ga2O3 system, the presence of Cr(VI) can on the one hand enhance reactions (7) and (8) (by retarding the hVB+/eCB− recombination), but on the other hand suppress reaction (11) (and thereby suppressing reaction (13)). However, it is hypothesized that the production of O2˙− by reaction (11) was not appreciably affected by Cr(VI), since the concentration of dissolved O2 (0.25 mM) in the solution were over 400 times greater than that of Cr(VI) (30 μg L−1, or 0.57 × 10−3 mM). (As the rate constants for reactions (2) and (11) are not known, the exact proportions of eCB− that react with O2 and Cr(VI) cannot be estimated).Unlike the other two photocatalysts, TiO2 was not effective at removing 6:2 FtS when Cr(VI) was present (Fig. 1c). As has been established above, eCB− and HO2˙ are not involved in the degradation of 6:2 FtS in the TiO2 system. Therefore, the inhibitory effect of Cr(VI) cannot be ascribed to the scavenging of eCB− by reaction (2). Instead, the most plausible explanation could be that the reactivity of TiO2 was severely reduced by the deposition of Cr(III) on the surface of TiO2. Whereas the color of In2O3 and Ga2O3 did not change throughout the experiment, TiO2 rapidly turned grey/black when photolysis started (Fig. S8†). Surface elemental analysis by EDS revealed that the surface of the spent TiO2 catalyst consisted of 3.7 wt% of Cr (this number was 0.22 wt% and 0.44 wt% for the spent Ga2O3 and In2O3, respectively) (Fig. S9†). Therefore, it is hypothesized that the Cr(III) adsorbed on TiO2 suppressed the degradation of 6:2 FtS by inhibiting the reactive sites on TiO2 and/or competing with 6:2 FtS for hVB+ (reaction (6)). A similar hypothesis was invoked in several studies to explain for the reactivity decrease of TiO2.53,54 In the Ga2O3 system, essentially all Cr remained in the solution by the end of the experiment (Fig. 3b), suggesting that the adsorption of Cr on Ga2O3 was minimal, which is consistent with SEM/EDS results (Fig. S9a†).
4.3 The transformation pathway of 6:2 FtS in the Ga2O3/UV system
According to the discussion above about the reactive species involved and the concentration–time profiles of the byproducts, the transformation of 6:2 FtS in the Ga2O3/UV system is proposed to take place as follows (Scheme 1). Upon reacting with hVB+ (reactions (7) and (8)), 6:2 FtS is oxidized into intermediate radicals IM1a and IM1b. 6:2 FtS can also be reduced by O2˙− into IM2 (reaction (13))—this nucleophilic substitution reaction is thought to occur in a way similar to the dehalogenation of alkyl halides by O2˙−.42,55 Next, IM1a, IM1b and IM2 are oxidized into 6:2 FtCA. It is noted that the exact mechanism through which 6:2 FtCA is produced is unclear at this point. It is also noted that IM1a, IM1b could also have been converted into other intermediates that were not detected by the LCMS analytical method used in this study. For example, by employing suspected screening-high resolution mass spectrometry, Zhang et al.51 observed that sulfonate byproducts such as C6F13C2H2OSO3−, C6F13C2H2OSO3−, and C6F13C2H4O2SO3− were produced when ˙OH reacted with 6:2 FtS (˙OH was generated using Co2+/peroxymonosulfate). Whether these sulfonate byproducts can be produced via reactions with hVB+ remains to be explored. However, as was discussed earlier, approximately 20% of F was unaccounted for in the fluorine mass balance. This missing F fraction could be that associated with sulfonate intermediate products, as well as other short-chain fluorinated compounds such as trifluoroacetic acid.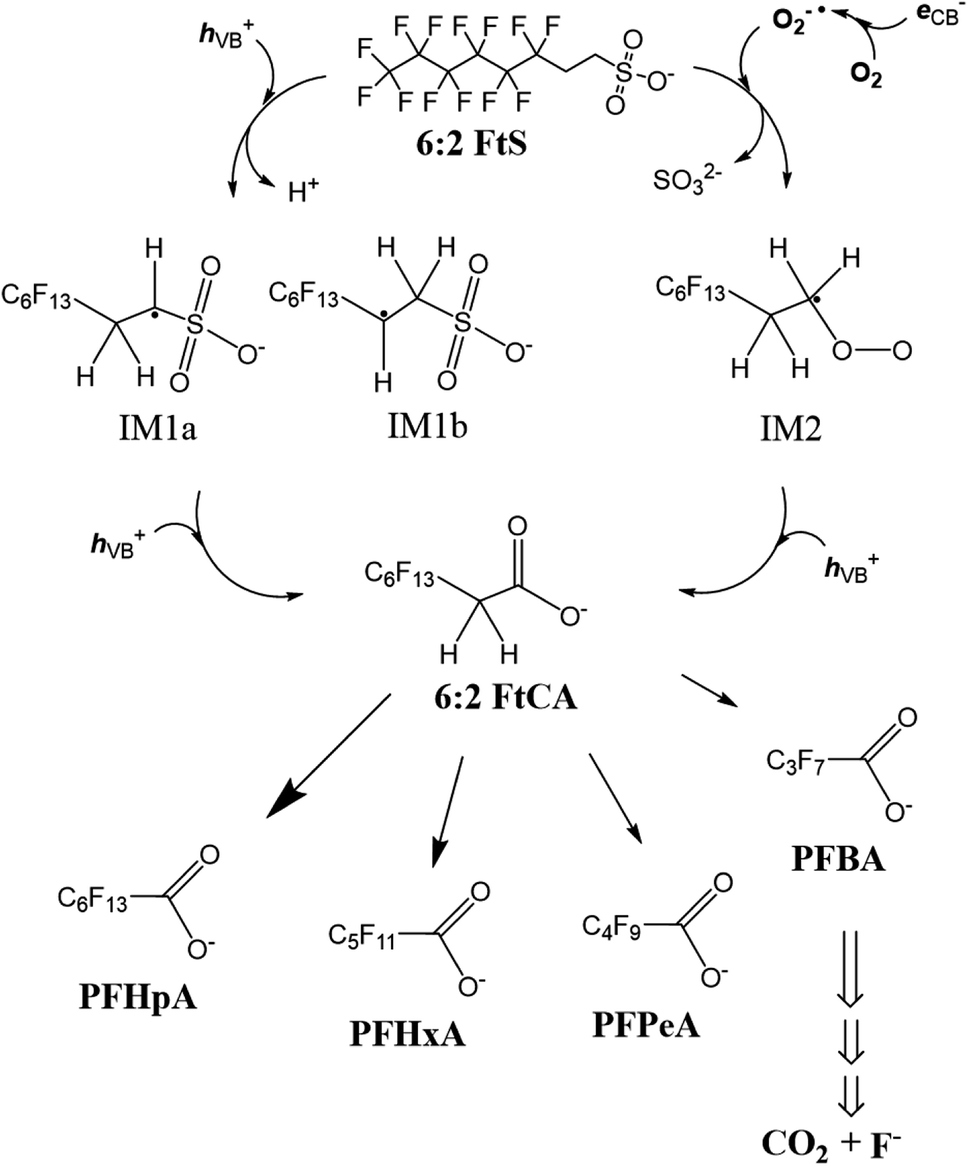 | ||
| Scheme 1 Proposed 6:2 FtS degradation pathway. | ||
Most previous studies have proposed that the degradation of PFOA by hVB+ and ˙OH takes place via a stepwise decarboxylation mechanism PFOA → PFHpA → PFHxA → PFPeA → PFBA.56,57 This stepwise mechanism implies that the concentration of the preceding byproduct will rise and fall before the subsequent byproduct is generated. However, it was observed that PFHpA, PFHxA, PFPeA and PFBA were generated simultaneously, and that none of these byproduct exhibited a two-phase concentration–time profile (Fig. 2b). Therefore, we argue that PFHpA, PFHxA, PFPeA and PFBA were generated directly from 6:2 FtCA. Additional research is needed to investigate the mechanism through which this transformation occurs.
4.4 Proposed treatment train for the removal of 6:2 FtS and Cr(VI) in CPW
Based on the results of this study, it is proposed that Cr(VI) and 6:2 FtS in CPW can be removed using a treatment train that consists of an oxidation–reduction step driven by Ga2O3/UVC, followed by a neutralization step (Scheme 2). The neutralization step not only will convert dissolved Cr(III) into Cr(OH)3(S), which can be separated (e.g., by filtration, centrifugation, or sedimentation), but also will raise the pH to the range that would comply with discharge regulations (typically pH = 6–8). If required, the effluent from this treatment system can be further polished using ion exchange or activated carbon to enhance the removal of Cr species as well as the fluorinated byproducts. Because conventional CPW treatment systems usually already consist of a neutralization step and a polishing step, existing systems can be readily upgraded by placing the Ga2O3/UVC unit in the front. Although existing systems with ion exchange or activated carbon could also remove 6:2 FtS, the upfront degradation by Ga2O3/UVC will significantly reduce the loading of fluorinated compounds on the polishing step. Recall that under the condition employed in this study, 50% of the initial organic fluorine was converted into F−. While F− could be toxic to aquatic life,58 its concentration in the treated CPW is not expected to exceed discharge limits (which is typically above 1 mg L−1),59,60 since raw CPW only contains up to a few hundreds μg L−1 of 6:2 FtS.22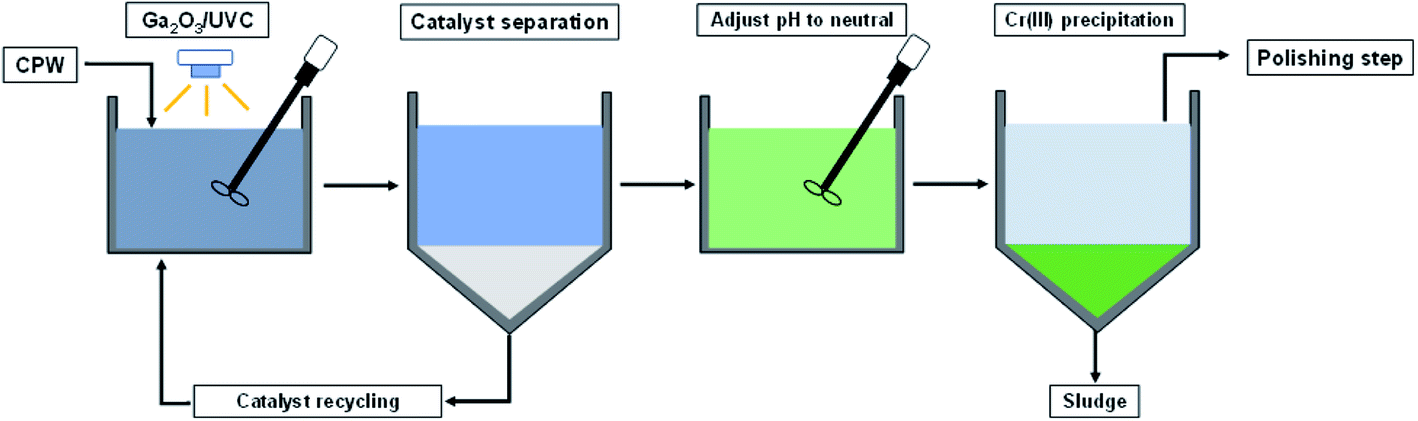 | ||
| Scheme 2 Process flow chart for CPW treatment including the oxidation–reduction step using Ga2O3/UVC system. | ||
5 Conclusions
Heterogeneous photocatalysis is a promising approach for the treatment of complex waste streams, owing to its ability to generate several types of oxidative and reductive species, namely hVB+, ˙OH, eCB−, and O2˙−. This study showed that the two commercial wide-band gap photocatalysts In2O3 and Ga2O3 were capable of simultaneously removing Cr(VI) and 6:2 FtS in chromate plating wastewater. With a significantly higher reactivity, Ga2O3 appeared to be superior to In2O3. Through a detailed investigation of how different probe compounds influence 6:2 FtS removal, hVB+ and O2˙− were identified to be the primary reactive species involved in the 6:2 FtS degradation in the Ga2O3/UVC system. It was also found that there was a synergistic effect between Cr(VI) and 6:2 FtS. The faster degradation of 6:2 FtS in the presence of Cr(VI) was attributable to the scavenging of eCB− by Cr(VI), which prevented the recombination hVB+ and eCB−.While this study has successfully demonstrated how Cr(VI) and 6:2 FtS can be simultaneously removed using photocatalysis, additional research is needed to improve the efficiency of this approach. For example, a recent study reported that In-doped Ga2O3 is significantly more reactive than Ga2O3. Future research should explore if In-doped Ga2O3 as well as other highly reactive catalysts (e.g., BN) can be employed for the treatment of CPW. Future studies should also employ real CPW samples to explore how factors such as the catalyst dosage, the solution pH, the concentration of Cr(VI) and 6:2 FtS, and the presence of other contaminants typically present in CPW (e.g., heavy metals) may affect the treatment efficacy. Additionally, future research should investigate if photocatalysis can be used to treat other types of PFAS used in electrochemical plating, such as PFOS and F-53B (i.e., ClC6F12OCF2CF2SO3−).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this research was provided by Natural Sciences and Engineering Research Council of Canada (Discovery Grant #2015-04850), and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (Grant NRF-2021R1A6A3A03044333).References
- ITRC, History and Use of Per- and Polyfluoroalkyl Substances (PFAS), 2017 Search PubMed.
- J. A. Padilla-Sánchez, E. Papadopoulou, S. Poothong and L. S. Haug, Investigation of the best approach for assessing human exposure to poly- and perfluoroalkyl substances through indoor air, Environ. Sci. Technol., 2017, 51, 12836–12843 CrossRef PubMed.
- C. Rauert, T. Harner, J. K. Schuster, A. Eng, G. Fillmann, L. E. Castillo, O. Fentanes, M. Villa Ibarra, K. S. B. Miglioranza, I. Moreno Rivadeneira, K. Pozo and B. H. Aristizábal Zuluaga, Atmospheric concentrations of new persistent organic pollutants and emerging chemicals of concern in the group of Latin America and Caribbean (GRULAC) region, Environ. Sci. Technol., 2018, 52, 7240–7249 CrossRef CAS PubMed.
- M. K. So, Y. Miyake, W. Y. Yeung, Y. M. Ho, S. Taniyasu, P. Rostkowski, N. Yamashita, B. S. Zhou, X. J. Shi, J. X. Wang, J. P. Giesy, H. Yu and P. K. S. Lam, Perfluorinated compounds in the Pearl river and Yangtze river of China, Chemosphere, 2007, 68, 2085–2095 CrossRef CAS PubMed.
- S. F. Nakayama, M. J. Strynar, J. L. Reiner, A. D. Delinsky and A. B. Lindstrom, Determination of perfluorinated compounds in the upper Mississippi river basin, Environ. Sci. Technol., 2010, 44, 4103–4109 CrossRef CAS PubMed.
- E. F. Houtz, C. P. Higgins, J. A. Field and D. L. Sedlak, Persistence of perfluoroalkyl acid precursors in AFFF-impacted groundwater and soil, Environ. Sci. Technol., 2013, 47, 8187–8195 CrossRef CAS PubMed.
- K. A. Barzen-Hanson, S. C. Roberts, S. Choyke, K. Oetjen, A. McAlees, N. Riddell, R. McCrindle, P. L. Ferguson, C. P. Higgins and J. A. Field, Discovery of 40 classes of per- and polyfluoroalkyl substances in historical aqueous film-forming foams (AFFFs) and AFFF-impacted groundwater, Environ. Sci. Technol., 2017, 51, 2047–2057 CrossRef CAS PubMed.
- F. Xiao, M. F. Simcik, T. R. Halbach and J. S. Gulliver, Perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in soils and groundwater of a U.S. metropolitan area: migration and implications for human exposure, Water Res., 2015, 72, 64–74 CrossRef CAS PubMed.
- M. Murakami, K. Kuroda, N. Sato, T. Fukushi, S. Takizawa and H. Takada, Groundwater pollution by perfluorinated surfactants in Tokyo, Environ. Sci. Technol., 2009, 43, 3480–3486 CrossRef CAS PubMed.
- X. C. Hu, D. Q. Andrews, A. B. Lindstrom, T. A. Bruton, L. A. Schaider, P. Grandjean, R. Lohmann, C. C. Carignan, A. Blum, S. A. Balan, C. P. Higgins and E. M. Sunderland, Detection of poly- and perfluoroalkyl substances (PFASs) in U.S. drinking water linked to industrial sites, military fire training areas, and wastewater treatment plants, Environ. Sci. Technol. Lett., 2016, 3, 344–350 CrossRef CAS PubMed.
- Y. L. Mak, S. Taniyasu, L. W. Y. Yeung, G. Lu, L. Jin, Y. Yang, P. K. S. Lam, K. Kannan and N. Yamashita, Perfluorinated compounds in tap water from China and several other countries, Environ. Sci. Technol., 2009, 43, 4824–4829 CrossRef CAS PubMed.
- R. Kim Lazcano, Y. J. Choi, M. L. Mashtare and L. S. Lee, Characterizing and comparing per- and polyfluoroalkyl substances in commercially available biosolid and organic non-biosolid-based products, Environ. Sci. Technol., 2020, 54, 8640–8648 CrossRef CAS PubMed.
- A. K. Venkatesan and R. U. Halden, National inventory of perfluoroalkyl substances in archived U.S. biosolids from the 2001 EPA national sewage sludge survey, J. Hazard. Mater., 2013, 252–253, 413–418 CrossRef CAS PubMed.
- R. J. Letcher, S. Chu and S.-A. Smyth, Side-chain fluorinated polymer surfactants in biosolids from wastewater treatment plants, J. Hazard. Mater., 2020, 388, 122044 CrossRef CAS PubMed.
- CDC, An Overview of Perfluoroalkyl and Polyfluoroalkyl Substances and Interim Guidance for Clinicians Responding to Patient Exposure Concerns, 2016, available from https://stacks.cdc.gov/view/cdc/77114/ Search PubMed.
- U.S. EPA, Basic Information on PFAS, 2021, available from https://www.epa.gov/pfas/basic-information-pfas/ Search PubMed.
- J. M. Braun, A. Chen, M. E. Romano, A. M. Calafat, G. M. Webster, K. Yolton and B. P. Lanphear, Prenatal perfluoroalkyl substance exposure and child adiposity at 8 years of age: the HOME study, Obesity, 2016, 24, 231–237 CrossRef CAS PubMed.
- Y. Liu, N. Li, G. D. Papandonatos, A. M. Calafat, C. B. Eaton, K. T. Kelsey, A. Chen, B. P. Lanphear, K. M. Cecil, H. J. Kalkwarf, K. Yolton and J. M. Braun, Exposure to per- and polyfluoroalkyl substances and adiposity at age 12 years: evaluating periods of susceptibility, Environ. Sci. Technol., 2020, 54, 16039–16049 CrossRef CAS PubMed.
- U.S. EPA, Drinking Water Health Advisories for PFOA and PFOS, 2016 Search PubMed.
- Health Canada, Guidelines for Canadian Drinking Water Quality Guideline Technical Document Perfluorooctanoic Acid (PFOA), 2018 Search PubMed.
- Health Canada, Guidelines for Canadian Drinking Water Quality Guideline Technical Document Perfluorooctane Sulfonate (PFOS), 2018 Search PubMed.
- German Environment Agency, Use of PFOS in chromium plating – characterisation of closed-loop systems, use of alternative substances, 2017 Search PubMed.
- Y. Bao, S. Deng, G. Cagnetta, J. Huang and G. Yu, Role of hydrogenated moiety in redox treatability of 6:2 fluorotelomer sulfonic acid in chrome mist suppressant solution, J. Hazard. Mater., 2021, 408, 124875 CrossRef CAS PubMed.
- J. A. Field and J. Seow, Properties, occurrence, and fate of fluorotelomer sulfonates, Crit. Rev. Environ. Sci. Technol., 2017, 47, 643–691 CrossRef CAS.
- F. Fu and Q. Wang, Removal of heavy metal ions from wastewaters: a review, J. Environ. Manage., 2011, 92, 407–418 CrossRef CAS PubMed.
- F. Xiao, P. C. Sasi, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and D. Soli, Thermal stability and decomposition of perfluoroalkyl substances on spent granular activated carbon, Environ. Sci. Technol. Lett., 2021, 8, 343–350 Search PubMed.
- F. Xiao, P. C. Sasi, B. Yao, A. Kubátová, S. A. Golovko, M. Y. Golovko and D. Soli, Thermal decomposition of PFAS: response to comment on “thermal stability and decomposition of perfluoroalkyl substances on spent granular activated carbon”, Environ. Sci. Technol. Lett., 2020, 7, 364–365 Search PubMed.
- M. Altarawneh, M. H. Almatarneh and B. Z. Dlugogorski, Thermal decomposition of perfluorinated carboxylic acids: kinetic model and theoretical requirements for PFAS incineration, Chemosphere, 2021, 131685 Search PubMed.
- R. K. Singh, S. Fernando, S. F. Baygi, N. Multari, S. M. Thagard and T. M. Holsen, Breakdown products from perfluorinated alkyl substances (PFAS) degradation in a plasma-based water treatment process, Environ. Sci. Technol., 2019, 53, 2731–2738 CrossRef CAS PubMed.
- J. Cui, P. Gao and Y. Deng, Destruction of per- and polyfluoroalkyl substances (PFAS) with advanced reduction processes (ARPs): a critical review, Environ. Sci. Technol., 2020, 54, 3752–3766 CrossRef CAS PubMed.
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: structural dependence and implications to PFAS remediation and management, Environ. Sci. Technol., 2019, 53, 3718–3728 CrossRef CAS PubMed.
- Y. Gu, T. Liu, H. Wang, H. Han and W. Dong, Hydrated electron based decomposition of perfluorooctane sulfonate (PFOS) in the VUV/sulfite system, Sci. Total Environ., 2017, 607–608, 541–548 CrossRef CAS PubMed.
- Z. Li, P. Zhang, T. Shao and X. Li, In2O3 nanoporous nanosphere: a highly efficient photocatalyst for decomposition of perfluorooctanoic acid, Appl. Catal., B, 2012, 125, 350–357 CrossRef CAS.
- X. Li, P. Zhang, L. Jin, T. Shao, Z. Li and J. Cao, Efficient photocatalytic decomposition of perfluorooctanoic acid by indium oxide and its mechanism, Environ. Sci. Technol., 2012, 46, 5528–5534 CrossRef CAS PubMed.
- T. Shao, P. Zhang, L. Jin and Z. Li, Photocatalytic decomposition of perfluorooctanoic acid in pure water and sewage water by nanostructured gallium oxide, Appl. Catal., B, 2013, 142–143, 654–661 CrossRef CAS.
- X. Tan, G. Chen, D. Xing, W. Ding, H. Liu, T. Li and Y. Huang, Indium-modified Ga2O3 hierarchical nanosheets as efficient photocatalysts for the degradation of perfluorooctanoic acid, Environ. Sci.: Nano, 2020, 7, 2229–2239 RSC.
- L. Duan, B. Wang, K. Heck, S. Guo, C. A. Clark, J. Arredondo, M. Wang, T. P. Senftle, P. Westerhoff, X. Wen, Y. Song and M. S. Wong, Efficient photocatalytic PFOA degradation over boron nitride, Environ. Sci. Technol. Lett., 2020, 7, 613–619 CrossRef CAS.
- B. Zhao and P. Zhang, Photocatalytic decomposition of perfluorooctanoic acid with β-Ga2O3 wide bandgap photocatalyst, Catal. Commun., 2009, 10, 1184–1187 CrossRef CAS.
- D. Huang, L. Yin and J. Niu, Photoinduced hydrodefluorination mechanisms of perfluorooctanoic acid by the SiC/graphene catalyst, Environ. Sci. Technol., 2016, 50, 5857–5863 CrossRef CAS PubMed.
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Environmental applications of semiconductor photocatalysis, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (˙OH)/(˙O−) in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Superoxide ion: generation and chemical implications, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- X. Zhang, T. Zhang, J. Ng, J. H. Pan and D. D. Sun, Transformation of bromine species in TiO2 photocatalytic system, Environ. Sci. Technol., 2010, 44, 439–444 CrossRef CAS PubMed.
- E. Bellack and P. J. Schouboe, Rapid photometric determination of fluoride in water. Use of sodium 2-(p-Sulfophenylazo)-1,8-dihydroxynaphthalene-3,6-disulfonate-zirconium lake, Anal. Chem., 1958, 30, 2032–2034 CrossRef CAS.
- B. E. Saltzman, Microdetermination of chromium with diphenylcarbazide by permanganate oxidation, Anal. Chem., 1952, 24, 1016–1020 CrossRef CAS.
- U.S. EPA, Guidance Manual for Electroplating and Metal Finishing Pretreatment Standards, 1984 Search PubMed.
- A. Walsh, C. R. A. Catlow, K. H. L. Zhang and R. G. Egdell, Control of the band-gap states of metal oxides by the application of epitaxial strain: the case of indium oxide, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 161202 CrossRef.
- M.-G. Ju, X. Wang, W. Liang, Y. Zhao and C. Li, Tuning the energy band-gap of crystalline gallium oxide to enhance photocatalytic water splitting: mixed-phase junctions, J. Mater. Chem. A, 2014, 2, 17005–17014 RSC.
- Y. Xu and M. A. A. Schoonen, The absolute energy positions of conduction and valence bands of selected semiconducting minerals, Am. Mineral., 2000, 85, 543–556 CrossRef CAS.
- C. D. Vecitis, H. Park, J. Cheng, B. T. Mader and M. R. Hoffmann, Kinetics and mechanism of the sonolytic conversion of the aqueous perfluorinated surfactants, perfluorooctanoate (PFOA), and perfluorooctane sulfonate (PFOS) into inorganic products, J. Phys. Chem. A, 2008, 112, 4261–4270 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, A. Moores and S. Ghoshal, Transformation of 6:2 fluorotelomer sulfonate by cobalt(II)-activated peroxymonosulfate, Environ. Sci. Technol., 2020, 54, 4631–4640 CrossRef CAS PubMed.
- G. V. Buxton and F. S. Dainton, The radiolysis of aqueous solutions of oxybromine compounds; the spectra and reactions of BrO and BrO2, Proc. R. Soc. London, Ser. A, 1968, 304, 427–439 CAS.
- G. Chen, J. Feng, W. Wang, Y. Yin and H. Liu, Photocatalytic removal of hexavalent chromium by newly designed and highly reductive TiO2 nanocrystals, Water Res., 2017, 108, 383–390 CrossRef CAS PubMed.
- W. Liu, J. Ni and X. Yin, Synergy of photocatalysis and adsorption for simultaneous removal of Cr(VI) and Cr(III) with TiO2 and titanate nanotubes, Water Res., 2014, 53, 12–25 CrossRef CAS PubMed.
- R. A. Johnson, E. G. Nidy and M. V. Merritt, Superoxide chemistry. Reactions of superoxide with alkyl halides and alkyl sulfonate esters, J. Am. Chem. Soc., 1978, 100, 7960–7966 CrossRef CAS.
- B. N. Nzeribe, M. Crimi, S. Mededovic Thagard and T. M. Holsen, Physico-chemical processes for the treatment of per- and polyfluoroalkyl substances (PFAS): a review, Crit. Rev. Environ. Sci. Technol., 2019, 49, 866–915 CrossRef.
- M. Trojanowicz, A. Bojanowska-Czajka, I. Bartosiewicz and K. Kulisa, Advanced oxidation/reduction processes treatment for aqueous perfluorooctanoate (PFOA) and perfluorooctanesulfonate (PFOS) – a review of recent advances, Chem. Eng. J., 2018, 336, 170–199 CrossRef CAS.
- Michigan Department of Environmental Quality, Aquatic Life Fact Sheet for Fluoride: (Aquatic Life – Acute Concentration), 2017 Search PubMed.
- EPA, Aluminum, Copper, and Nonferrous Metals Forming & Metal Powders Pretreatment Standards: A Guidance Manual, 1989 Search PubMed.
- The City of Ottawa, Guide for Discharging Wastewater from Industrial Facilities, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Text S1, Tables S1–S3 and Fig. S1–S9. See DOI: 10.1039/d1ra06235b |
| This journal is © The Royal Society of Chemistry 2021 |