
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and antitumor effects of novel benzyl naphthyl sulfoxide/sulfone derivatives derived from Rigosertib†
Lin Tang a,
Tingting Chena,
Hongpeng Yangab,
Xiaoxue Wena,
Yunbo Suna,
Shuchen Liua,
Tao Peng*a,
Shouguo Zhang*a and
Lin Wang*ab
a,
Tingting Chena,
Hongpeng Yangab,
Xiaoxue Wena,
Yunbo Suna,
Shuchen Liua,
Tao Peng*a,
Shouguo Zhang*a and
Lin Wang*ab
aBeijing Institute of Radiation Medicine, Beijing 100850, P. R. China. E-mail: wanglin@bmi.ac.cn; wanglin07@sina.com; zhangshouguo1409@sina.com; pengtao@bmi.ac.cn; Tel: +86 010 66932239
bFaculty of Environment & Life, Beijing University of Technology, Beijing 100124, P. R. China
First published on 22nd November 2021
Abstract
In this work, a series of novel benzyl naphthyl sulfoxides/sulfones derived from Rigosertib were designed and synthesized as potential antitumor agents. The in vitro cytotoxicity against four human cancer cell lines (HeLa, MCF-7, HepG2 and SCC-15) and two normal human cell lines (HUVEC and 293T) indicated that some of the sulfones and sulfoxides possessed potent antineoplastic activity that reached nanomolar levels and relatively low toxicity to normal cells. Among them, (2-methoxy-5-((naphthalen-2-ylsulfonyl)methyl)phenyl)glycine (15b) was found to be a promising antitumor drug candidate that could significantly inhibit tumor cell migration and induce tumor cell apoptosis via the p53-Bcl-2-Bax signaling pathway at nanomolar concentrations.
1. Introduction
Rigosertib (Fig. 1) was developed by Onconova Therapeutics Inc. (Newtown, PA, USA) as a selective antitumor agent. It is a styryl benzyl sulfone and a multi-kinase inhibitor with significant activity against the polo-like kinases (PLKs) and phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathways.1–4 Both signaling pathways play important roles in tumorigenicity and progression of tumors, so Rigosertib exerts potent antineoplastic effects on various tumor cells. In vitro studies, Rigosertib showed strong cytotoxicity against 94 different cancer cell lines with IC50 values ranging from 50 to 250 nM. While for normal cells, such as HFL, PrEC, HMEC and HUVEC, Rigosertib had little or no anti-proliferative effect unless its concentration exceeded 5–10 μM.2,5 In mouse xenograft models using Bel-7402, MCF-7 and MIA-PaCa cells, Rigosertib (250 mg kg−1, i.p.) could significantly suppress tumor growth. In the mouse xenograft model using BT20 cells, Rigosertib (200 mg kg−1, i.p.) could also remarkably inhibit tumor growth.5,6 In multiple clinical trials, Rigosertib has revealed significant therapeutic effects for patients with solid tumors and hematological malignancies.7–16 A clinical study (phase I/II) of “Rigosertib Plus Nivolumab for KRAS + NSCLC Patients Who Progressed on First-Line Treatment” is currently in the stage of recruiting volunteers (registration time: 2020-02-20).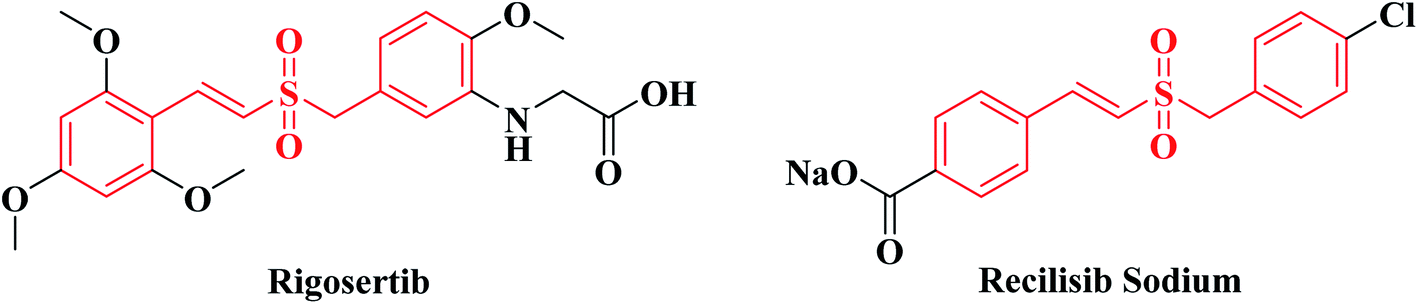 | ||
| Fig. 1 Structures of Rigosertib and Recilisib Sodium. | ||
Recilisib Sodium (synonyms: Ex-RAD; ON 01210.Na. Fig. 1), sharing the same core structure with Rigosertib, is a chloro styryl benzyl sulfone also developed by Onconova Therapeutics Inc. Different from Rigosertib, Recilisib Sodium has been used as a radioprotector and mitigator. It is a water soluble, non-toxic, synthetic molecule with potent radioprotective properties both in vitro and in vivo.17–19 Contrary to Rigosertib, Recilisib Sodium could activate the PI3K/Akt signaling pathway to prevent irradiated normal cells from apoptosis.20 By analyzing the structures of Rigosertib and Recilisib Sodium, we found that the introduction of an amino-group to the meta-position of benzene ring changed the styryl benzyl sulfone from a radioprotectant without cytotoxicity to a potent antineoplastic agent.
In our previous study, a series of benzyl naphthyl sulfoxide/sulfone derivatives (I in Fig. 2) derived from Recilisib Sodium were synthesized, and evaluated for their radioprotective effects. Several of these derivatives showed better in vitro biological activity than Recilisib Sodium, and one of them exhibited much more potent radioprotective properties than Recilisib Sodium both in vitro and in vivo.21 As the design strategies shown in Fig. 2, due to the trans-form of Recilisib Sodium had better biological activity, the styryl segment was cyclized to become a naphthyl to maintain the configuration. The design strategy was proved to be right, because we have got a series of novel small molecule radioprotective agents with better anti-radiation activity.
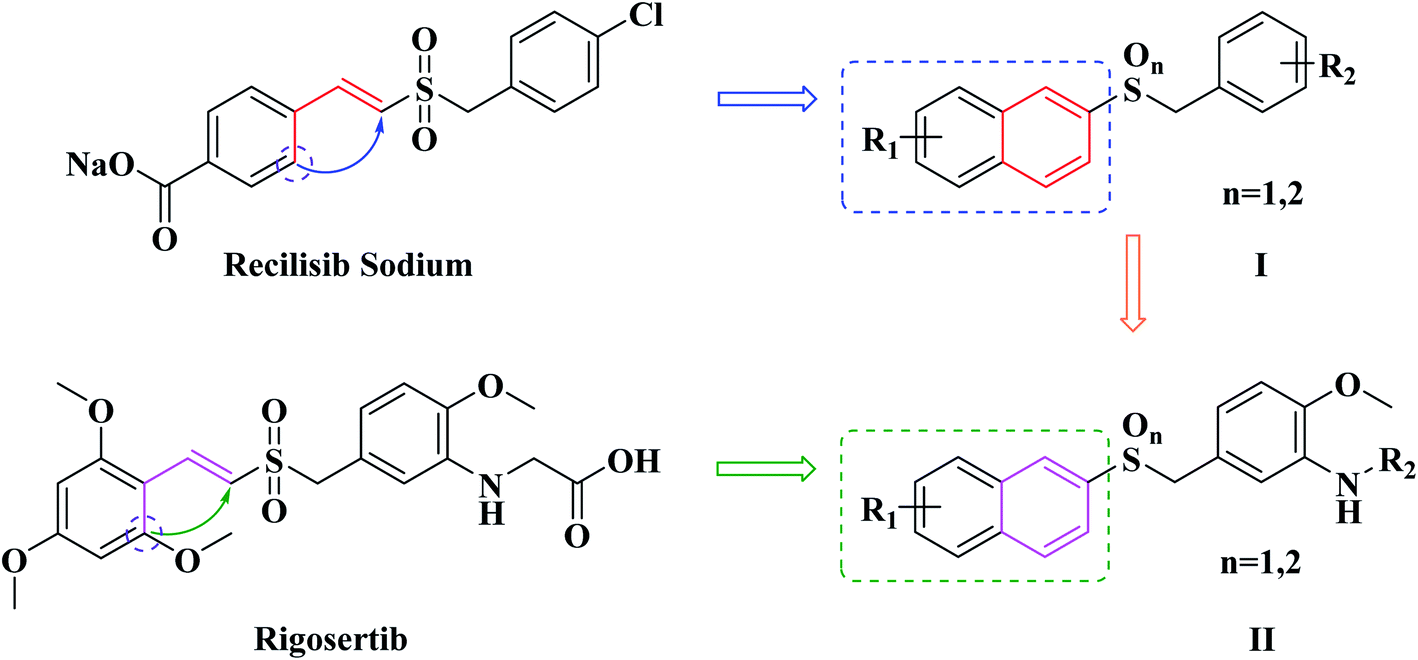 | ||
| Fig. 2 The design of novel benzyl naphthyl sulfoxide/sulfone derivatives (I and II). | ||
Inspired by our previous successful experience, as well as the trans-form of Rigosertib also possessed better antineoplastic activity, the same strategy was taken to modify Rigosertib (Fig. 2). Looking forward to obtain more effective antitumor agents, the second series of naphthyl benzyl sulfoxide/sulfone derivatives (II in Fig. 2) were designed rationally. Subsequently, eleven target compounds were prepared. The in vitro cytotoxicity of them was evaluated against both human cancer and normal cell lines. The antitumor mechanisms of the most effective compound were preliminary studied by flow cytometry and western blotting.
2. Results and discussion
2.1. Chemistry
The synthetic procedure for the target compounds is described in Scheme 1 in the literature.22–24 Intermediate 9 was prepared from anisic aldehyde (6) by nitration, reduction and bromination.25,26 Intermediates 10a–10b were synthesized from reactions of 5a–5b with 9 in the presence of NaOH.27 Next, the sulfides 10a–10b were oxidized to give the corresponding sulfoxides (11a, 11c) and sulfones (11b, 11d) with H2O2 in acetic acid.21 Compounds 12a–12e were prepared by nitro reduction of 11a–11d and 10a, while 12a–12d were further esterified, salified and followed by hydrolysis reactions to give compounds 13a–13c, 14a–14b and 15a–15b.28 All the synthesized compounds were purified by recrystallization or silica gel column chromatography, and all of the target compounds were characterized by 1H-NMR, 13C-NMR and HR-MS spectra analyses.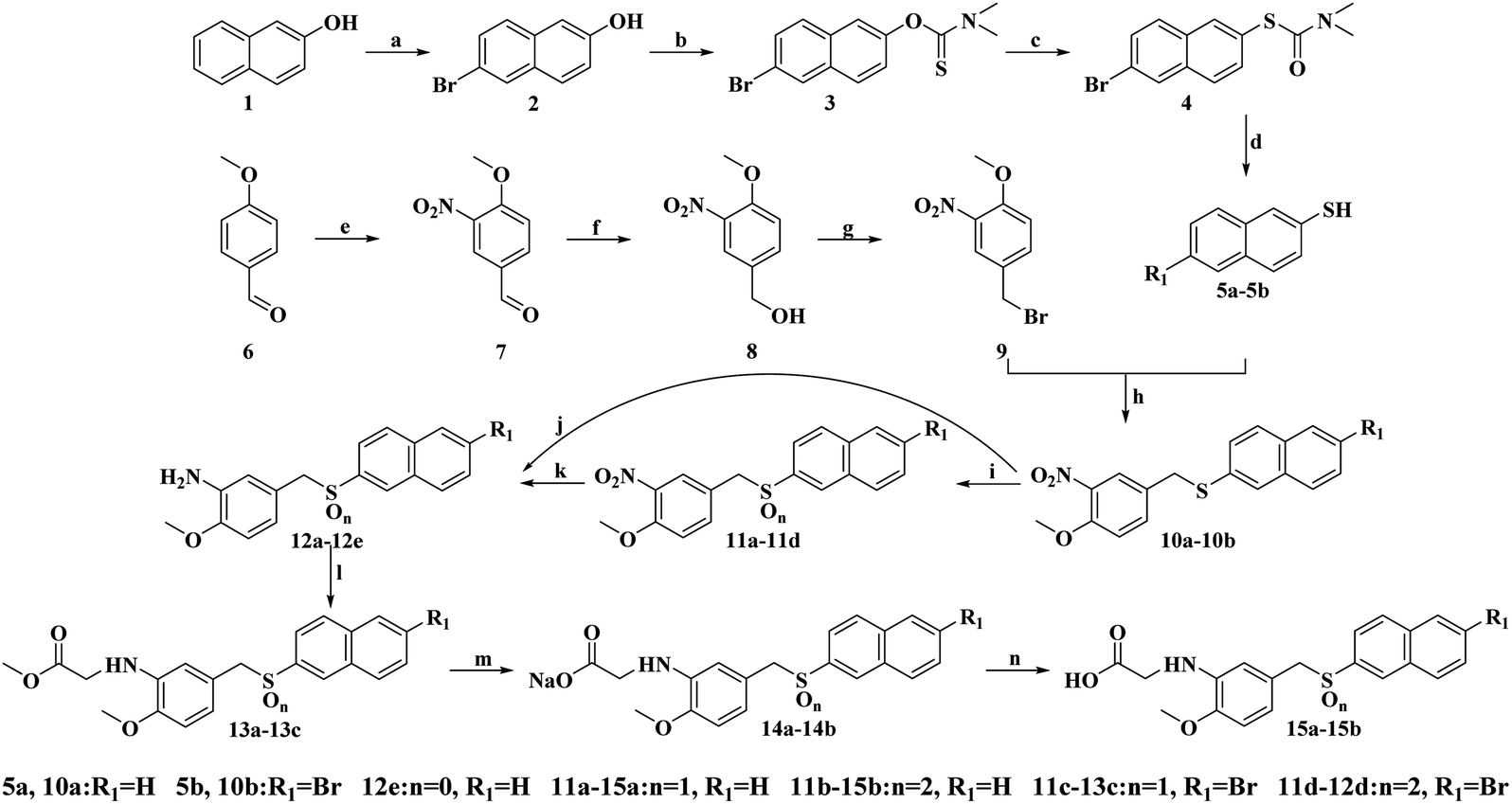 | ||
| Scheme 1 Reagents and conditions: (a) Br2, acetic acid, stannum, reflux, 3 h; (b) (I) NaH, DMF, 0 °C, 15 min, (II) dimethyl carbamoyl chloride, 80 °C (2 h), r.t. (15 h); (c) 220 °C, 6 h; (d) KOH, MeOH, 80 °C, 2.5 h; (e) 80% HNO3, 2 h, r.t.; (f) THF, EtOH, NaBH4, 2 h, 0 °C–r.t.; (g) PBr3, DCM, 0 °C, 2 h; (h) NaOH, EtOH, reflux, 3 h; (i) H2O2, acetic acid, r.t. or 50 °C, 3–5 h; (j) and (k) Na2S2O4, acetone, H2O, 50 °C, 1 h; (l) methyl bromoacetate, sodium acetate, MeOH, reflux, 4–6 h; (m) NaOH, EtOH, 60 °C, 6 h; (n) HCl, 30 min. | ||
2.2. In vitro cytotoxicity assays against cancer cells and normal cells
The in vitro cytotoxicity of the target compounds was firstly evaluated against four human cancer cell lines: human cervical carcinoma cell line (HeLa), human breast carcinoma cell line (MCF-7), human hepatoma carcinoma cell lines (HepG2) and human tongue carcinoma cell line (SCC-15). Besides, to determine the cytotoxicity of these compounds in human normal cell lines, human umbilical vein endothelial cells (HUVEC) and human embryonic kidney (293T) cells were selected for the study. The cytotoxicity of them was measured by MTS tetrazolium assay.29 Rigosertib was used as the positive control. The IC50 values for each compound are summarized in Table 1.| Comp. | IC50 (μM) | |||||
|---|---|---|---|---|---|---|
| HeLa | MCF-7 | HepG2 | SCC-15 | HUVEC | 293T | |
| 12e | 4.056 ± 0.316 | 17.50 ± 2.320 | 42.58 ± 3.810 | 2.209 ± 0.314 | 90.38 ± 4.720 | >100 |
| 12a | 0.173 ± 0.031 | 0.681 ± 0.048 | 0.434 ± 0.038 | 0.148 ± 0.027 | 5.596 ± 0.375 | 2.037 ± 0.091 |
| 12b | 0.030 ± 0.005 | 0.110 ± 0.014 | 0.043 ± 0.007 | 0.028 ± 0.006 | 0.416 ± 0.039 | 0.616 ± 0.051 |
| 12c | 0.092 ± 0.009 | 0.858 ± 0.088 | 1.307 ± 0.091 | 0.264 ± 0.027 | 2.504 ± 0.233 | 5.310 ± 0.486 |
| 12d | 0.064 ± 0.005 | 0.095 ± 0.008 | 0.402 ± 0.035 | 0.078 ± 0.002 | 1.816 ± 0.118 | 0.974 ± 0.112 |
| 13a | 0.974 ± 0.081 | 5.771 ± 0.438 | 4.288 ± 0.381 | 0.786 ± 0.081 | 11.40 ± 0.548 | >100 |
| 13b | 0.096 ± 0.009 | 0.140 ± 0.018 | 0.535 ± 0.036 | 0.154 ± 0.015 | 1.218 ± 0.150 | 0.409 ± 0.037 |
| 13c | 0.116 ± 0.006 | 1.734 ± 0.201 | 7.091 ± 0.661 | 3.251 ± 0.220 | 28.39 ± 3.110 | >100 |
| 14b | 0.112 ± 0.008 | 0.182 ± 0.017 | 0.993 ± 0.004 | 0.228 ± 0.004 | 3.810 ± 0.287 | 7.854 ± 0.417 |
| 15a | 0.230 ± 0.004 | 2.031 ± 0.073 | 5.080 ± 0.318 | 0.722 ± 0.029 | 6.772 ± 0.315 | 22.60 ± 1.180 |
| 15b | 0.064 ± 0.005 | 0.029 ± 0.001 | 0.390 ± 0.013 | 0.015 ± 0.002 | 1.055 ± 0.048 | 2.629 ± 0.151 |
| Rigosertib | 0.065 ± 0.005 | 0.022 ± 0.003 | 0.108 ± 0.017 | 0.035 ± 0.003 | 0.122 ± 0.015 | 0.036 ± 0.013 |
In the cytotoxic assay, all the target compounds showed apparent cytotoxic activity against the selected human tumor cells, with IC50 values ranging from 15 nM to 42.58 μM. In addition, they exhibited comparatively weaker cytotoxic activity against the selected human normal cells. The compounds containing sulfones (12b, 12d, 13b, 14b and 15b) possessed the best antineoplastic activity, followed by sulfoxide derivatives (12a, 12c, 13a, 13c and 15a), and that of the compound containing sulfide (12e) was the weakest. It is worth mentioning that, unlike reported in the literature, Rigosertib showed obviously toxicity in normal cell lines (IC50 values of 122 nM and 36 nM for HUVEC and 293T, respectively) in the study. Among these compounds, the in vitro antitumor activity of 12b, 12d and 15b was similar to Rigosertib, while the toxicity to normal cells was much lower. 15b was selected for further research because of its relatively lower toxicity to normal cells.
2.3. Apoptosis test
The potent antiproliferative activity of these sulfone derivatives raised a question whether the inhibition of growth of tumor cells was contributed by apoptosis. Consequently, as mentioned above, 15b was selected to assess apoptosis of HeLa cells by annexin V/7AAD assays. In early apoptosis, phosphatidylserine (PS) flipped from the inside of the cell membrane to the surface and could be identified and marked by annexin V FITC, which was shown in Q4 of the flow cytometry assay. In contrast, necrotic cells and late apoptotic cells were both stained with 7AAD, which was shown in Q1 and Q2, respectively. And lastly, living cells were shown in Q3. As shown in Fig. 3, compound 15b could efficiently induce apoptosis in HeLa cells in a dose dependent manner. After treatment with 15b for 24 h, 36 h and 48 h at the concentration of 100 nM, the apoptotic values (including the early and late apoptotic states) were 9.2%, 30.2%, and 53.2%, respectively. While in the control group, only 3.0% cells were in apoptotic states. The results revealed that the ability of 15b to mediate apoptosis closely correlates to its antitumor activity.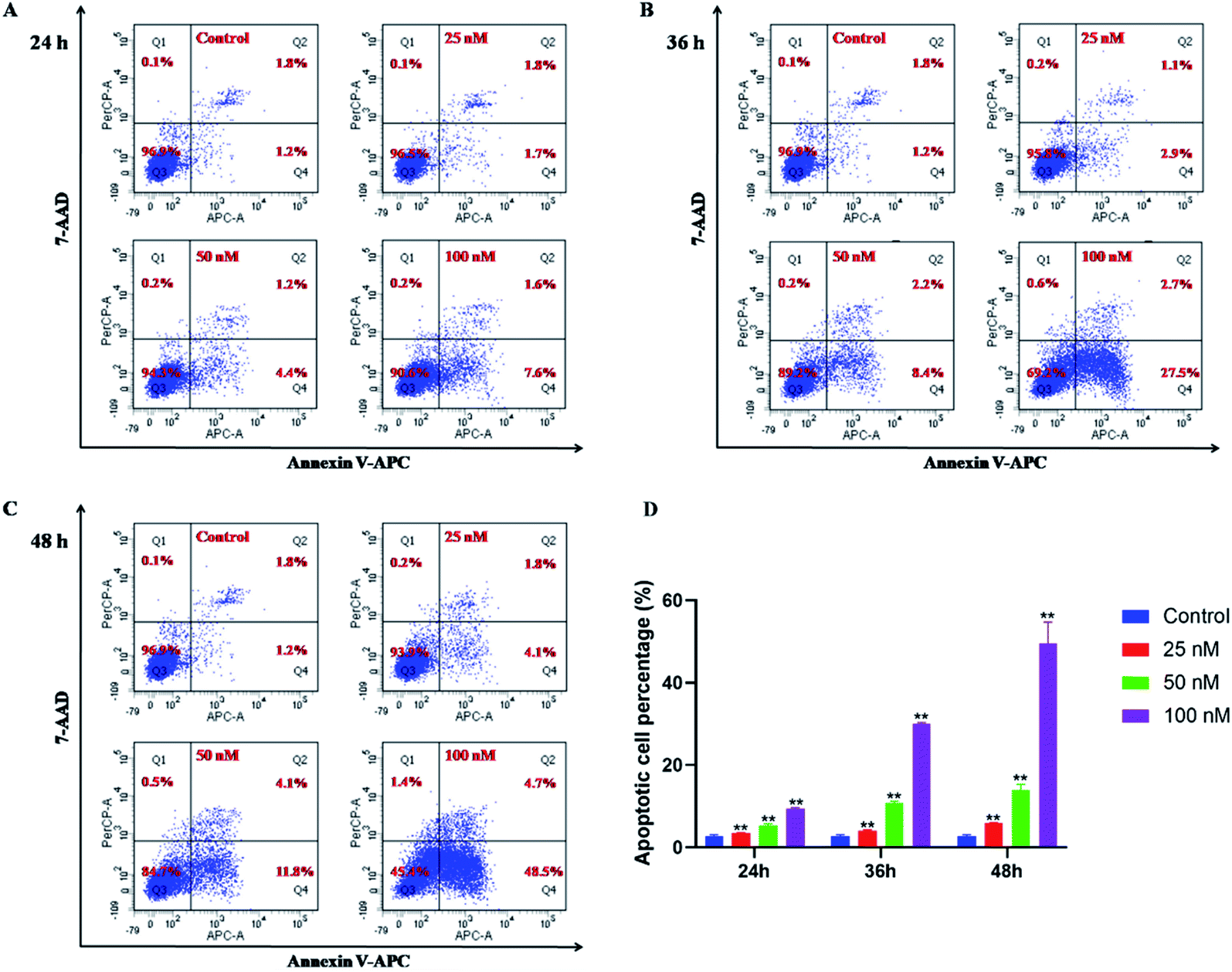 | ||
| Fig. 3 7-AAD/annexin V assay for the detection of apoptotic HeLa cancer cells after treatment with 15b (25 nM, 50 nM and 100 nM) for 24 h (A), 36 h (B) and 48 h (C), respectively. The results showed the percentage of living, apoptotic and dead cells. (D) The apoptotic cells were quantitated and shown as percentages. The values represent the mean ± SD of three independent experiments. **p < 0.01 significantly different from the control group. | ||
2.4. Wound healing assay
During the development of metastatic cancers, the important characteristic of them is migration and invasion of malignant cells. Therefore, the wound healing assay was employed to explore the ability of 15b to reduce the cancer cell migration and invasion. HeLa cells were scratched with pipette tips and co-incubated with control (DMSO) or 15b (25 nM, 50 nM and 100 nM) for 48 h. The cells were photographed after 0 h, 24 h and 48 h incubation. In order to avoid the influence of cell proliferation on the test results, the experiments were performed under serum starved conditions. Fig. 4 displayed the wound gaps at the time the wound was created, and 24 h, 48 h after treatment with samples. As illustrated in Fig. 4 and Table 2, compound 15b significantly inhibited the wound healing in a dose dependent manner. After treatment with DMSO for 24 h and 48 h, the wound healing rates of the control group were 46.4% and 70.8%, respectively. In marked contrast to the control group, with the increasing concentrations of 15b, the wound healing rates of 15b group reduced gradually, ranging from 20.0% and 32.0% to 3.9% and 4.4%. At the concentration of 100 nM, the cells barely migrated. These results indicated that 15b could efficiently suppress HeLa cells migration and further metastasis.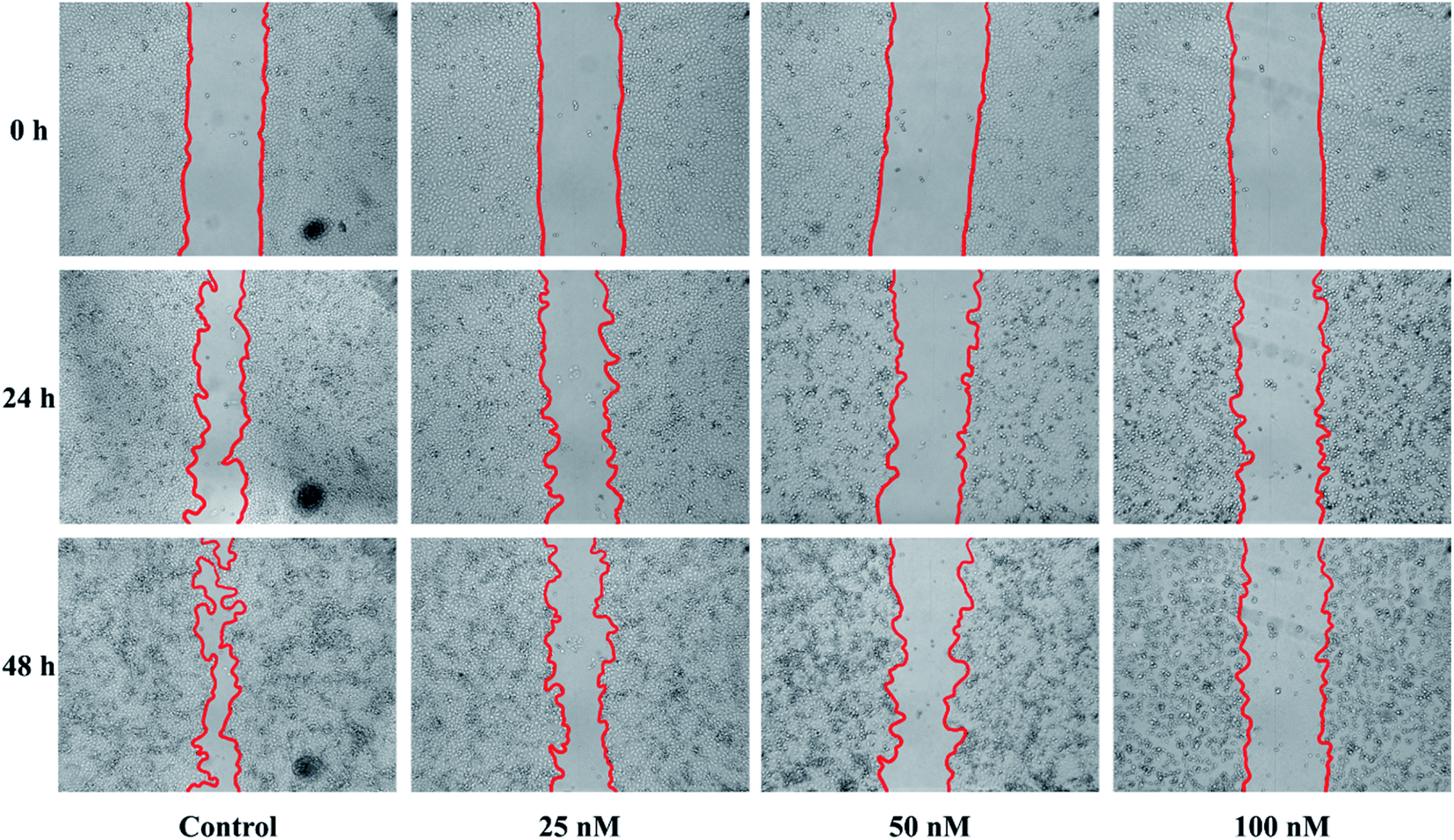 | ||
| Fig. 4 Compound 15b inhibited the migration of HeLa cells. The wounds were created by scratching with pipette tips and the cells were co-incubated with control (DMSO) and 15b (25 nM, 50 nM and 100 nM) for 48 h. The cells were photographed after 0 h, 24 h and 48 h of incubation. These figures are representative of one of three independent experiments with similar results. | ||
2.5. Western blotting study
The p53 protein has been found to be the most tumor-related gene up to now. And p53 is an essential protein that regulates cell apoptosis, together with its downstream regulatory proteins Bcl-2 and Bax. Hence, western blotting was used to investigate the antitumor mechanism of compound 15b on the expression of apoptosis-related proteins (p53, Bcl-2 and Bax) of HeLa cells. As shown in Fig. 5, the levels of the proapoptotic protein p53 increased significantly after treatment with 15b (25 nM, 50 nM and 100 nM) for 48 h, which led to the down-regulation of its downstream antiapoptotic protein Bcl-2 and the up-regulation of its downstream proapoptotic protein Bax. The western blot data indicated that 15b induced apoptosis by up-regulating p53 and altering the ratio of Bcl-2 to Bax.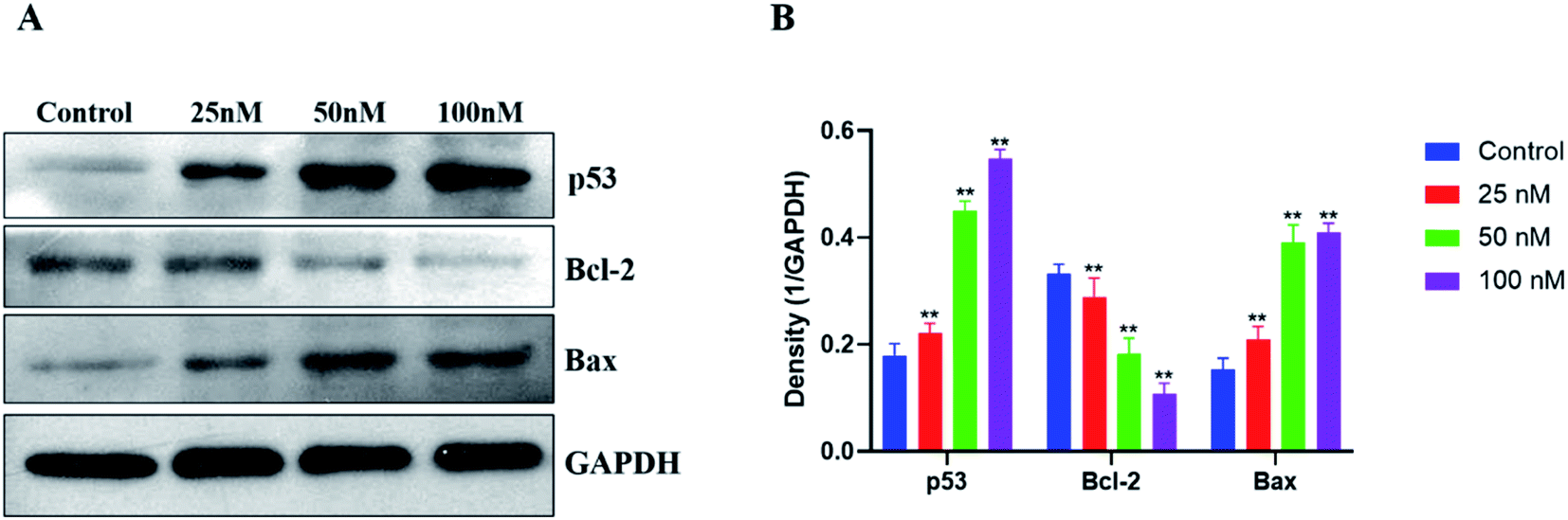 | ||
| Fig. 5 After 48 h treatment of HeLa cells with 15b (25 nM, 50 nM and 100 nM), (A) the expressions of p53, Bcl-2 and Bax were analyzed by western blotting, using GAPDH antibody as the reference control. (B) The protein expression levels of p53, Bcl-2 and Bax were quantitated and shown as percentages. The values represent the mean ± SD of three independent experiments. **p < 0.01 significantly different from the control group. | ||
3. Conclusions
Structure activity relationship (SAR) study revealed that the sulfones exhibited the best antineoplastic activity, followed the sulfoxides, and the sulfide was the weakest, suggesting that the sulfinyl and sulfonyl are the essential groups associated with antitumor activity, and the sulfonyl is more able to enhance the antitumor activity of the derivatives. For normal cell lines, the sulfide showed the weakest cytotoxicity, followed the sulfoxides, and the sulfones were the strongest. The introduction of ester groups and carboxylates to the side chain of benzene ring decreased the cytotoxicity to cancer and normal cell lines simultaneously. However, the carboxyl on the benzene ring decreased the cytotoxicity to normal cell lines while maintaining the antineoplastic activity. The introduction of halogen groups at 6-position of the naphthyl ring had no obvious influence with the biological activity.In summary, we report on the design and synthesis of several 2-naphthyl benzyl sulfoxide/sulfone derivatives for use as antitumor drugs. Evaluations of their in vitro cytotoxicity indicated that all of the sulfones and sulfoxides had potent antineoplastic activity that reached nanomolar levels and relatively lower toxicity to normal cells. Among them, 15b was selected for further study after comprehensive comparison. Results of flow cytometry assay revealed that 15b inhibited proliferation of cultured cancer cells by inducing apoptosis. Moreover, 15b also efficiently suppressed tumor cell migration in wound healing assay. Preliminary biological mechanism studies indicated that 15b could trigger the apoptosis of human cervical carcinoma HeLa cells via the p53-Bcl-2-Bax signaling pathway. The findings highlighted the potential of these derivatives as novel anticancer agents and 15b as a candidate for the treatment of cancer. Further detailed research will be conducted to evaluate the molecular mechanism underlying the anticancer activity of these compounds.
4. Experimental section
4.1. Chemistry
All chemicals and reagents were purchased from commercial suppliers, of reagents grade and used without further purification. Melting points were recorded in an open capillary tube and uncorrected. Reactions were monitored using TLC and performed on silica gel glass plates containing 60 GF-254. Visualization was achieved by UV light (λmax = 254 or 365 nm). Purification of compounds was performed by recrystallization or using silica gel (200–300 mesh) column chromatography. 1H-NMR and 13C-NMR spectra were recorded using Bruker (Palo Alto, CA, USA) AV-400 spectrometers in DMSO-d6 or CDCl3 with TMS as internal standard. Mass spectral data were obtained using electron spray ionization on a Micromass ZabSpec high-resolution mass spectrometer (Karlsruhe, Germany).Note: Only the synthesis and characterization of target compounds are presented in this article. The intermediates mentioned in Scheme 1 are described in ESI.†
4.1.1.1 (4-Methoxy-3-nitrobenzyl)(naphthalen-2-yl)sulfane (10a). Obtained in 97.5% yield, yellow solid, m.p. 102–105 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): δ 7.90 (d, J = 2.4 Hz, 1H), 7.87–7.73 (m, 4H), 7.64 (dd, J = 8.7, 2.3 Hz, 1H), 7.51–7.37 (m, 3H), 7.23 (d, J = 8.7 Hz, 1H), 4.36 (s, 2H), 3.83 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 151.0605, 138.7458, 134.7998, 133.3195, 132.9483, 131.2388, 130.2007, 128.3790, 127.6205, 126.9262, 126.7131, 126.1470, 125.7896, 125.1044, 114.3182, 56.6245, 34.7840. HRMS-ESI (m/z) calcd. for C18H16NO3S [M + H]+: 326.0851, found: 326.0844.
4.1.1.2 (6-Bromonaphthalen-2-yl)(4-methoxy-3-nitrobenzyl)sulfane (10b). Obtained in 98.5% yield, brown oil. The residue containing 10b was used for the next step without purification. HRMS-ESI (m/z) calcd. for C18H15BrNO3S [M + H]+: 403.9956, found: 403.9949.
4.1.2.1 2-((4-Methoxy-3-nitrobenzyl)sulfinyl)naphthalene (11a). Obtained in 97.4% yield, white solid, m.p. 134–136 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.05 (d, J = 8.0 Hz, 1H), 7.94–7.99 (m, 3H), 7.57–7.62 (m, 3H), 7.51 (s, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 4.43 (d, J = 12.0 Hz, 1H), 4.15 (d, J = 12.0 Hz, 1H), 3.83 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 151.8579, 140.2353, 138.4021, 136.5139, 133.8924, 132.2242, 128.8877, 128.3561, 127.9711, 127.7511, 127.2378, 126.6879, 124.7446, 122.5814, 120.5282, 113.9653, 59.0054, 56.6956. HRMS-ESI (m/z) calcd. for C18H16NO4S [M + H]+: 342.0800, found: 342.0795.
4.1.2.2 2-Bromo-6-((4-methoxy-3-nitrobenzyl)sulfinyl)naphthalene (11c). Obtained in 67.5% yield, white solid, m.p. 188–190 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.33 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.97–8.02 (m, 2H), 7.69–7.76 (m, 2H), 7.51 (s, 1H), 7.30 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 8.0 Hz, 1H), 4.48 (d, J = 12.8 Hz, 1H), 4.20 (d, J = 12.8 Hz, 1H), 3.87 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 152.2796, 141.3720, 138.7871, 136.9356, 135.4140, 131.1976, 130.9593, 130.6476, 130.3360, 128.5028, 127.0912, 125.2580, 122.7465, 122.2148, 121.5182, 114.4053, 59.1888, 57.1356; HRMS-ESI (m/z) calcd. for C18H15BrNO4S [M + H]+: 419.9905, found: 419.9900.
4.1.3.1 2-((4-Methoxy-3-nitrobenzyl)sulfonyl)naphthalene (11b). Obtained in 89.8% yield, white solid, m.p. 193–195 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.42 (s, 1H), 8.15–8.18 (m, 2H), 8.09 (d, J = 8.0 Hz, 1H), 7.68–7.78 (m, 4H), 7.46 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.0 Hz, 1H), 4.84 (s, 2H), 3.89 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 152.2269, 138.5213, 137.0822, 135.2169, 134.8205, 131.5711, 129.7516, 129.4743, 129.2910, 127.9321, 127.7947, 127.3707, 122.9275, 121.0484, 114.4053, 58.8725, 56.7895; HRMS-ESI (m/z) calcd. for C18H16NO5S [M + H]+: 358.0749, found: 358.0741.
4.1.3.2 2-Bromo-6-((4-methoxy-3-nitrobenzyl)sulfonyl)naphthalene (11d). Obtained in 64.1% yield, white solid, m.p. 232–235 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.45 (s, 1H), 8.41 (s, 1H), 8.14 (d, J = 8.0 Hz, 2H), 7.82 (t, J = 8.0 Hz, 2H), 7.67 (s, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.31 (d, J = 8.0 Hz, 1H), 4.85 (s, 2H), 3.89 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 152.6829, 138.9155, 137.5039, 136.3123, 136.0923, 132.0226, 131.2709, 130.5926, 130.3543, 130.2993, 128.9611, 127.7878, 124.5980, 123.4981, 121.3349, 114.8453, 59.2438, 57.2089; HRMS-ESI (m/z) calcd. for C18H15BrNO5S [M + Na]+: 457.9674, found: 457.9668.
4.1.4.1 2-Methoxy-5-((naphthalen-2-ylsulfinyl)methyl)aniline (12a). Obtained in 36.5% yield, white solid, m.p. 164–166 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.98–8.10 (m, 4H), 7.56–7.64 (m, 3H), 6.63 (d, J = 8.0 Hz, 1H), 6.53 (s, 1H), 6.27 (d, J = 8.0 Hz, 1H), 4.78 (s, 2H, NH2), 4.09 (d, J = 12.0 Hz, 1H), 3.89 (d, J = 12.0 Hz, 1H), 3.68 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 145.8633, 141.1702, 136.8622, 133.4524, 131.9125, 128.4294, 128.0077, 127.5678, 127.2378, 126.7611, 124.1030, 122.4897, 120.3082, 117.9067, 115.1935, 109.7856, 61.8469, 54.8624; HRMS-ESI (m/z) calcd. for C18H18NO2S [M + H]+: 312.1058, found: 312.1052.
4.1.4.2 2-Methoxy-5-((naphthalen-2-ylsulfonyl)methyl)aniline (12b). Obtained in 45.1% yield, white solid, m.p. 187–188 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.42 (s, 1H), 8.06–8.16 (m, 3H), 7.66–7.77 (m, 3H), 6.62 (d, J = 8.0 Hz, 1H), 6.56 (s, 1H), 6.24 (d, J = 8.0 Hz, 1H), 4.76 (s, 2H, NH2), 4.51 (s, 2H), 3.69 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 146.4499, 137.3938, 136.0945, 134.6830, 131.5963, 129.4285, 129.3414, 129.2223, 129.0115, 127.9138, 127.6388, 123.1314, 120.3632, 119.0204, 116.1010, 109.9620, 60.7332, 55.2015; HRMS-ESI (m/z) calcd. for C18H18NO3S [M + H]+: 328.1007, found: 328.1001.
4.1.4.3 5-(((6-Bromonaphthalen-2-yl)sulfinyl)methyl)-2-methoxyaniline (12c). Obtained in 65.2% yield, white solid, m.p. 170–174 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.30 (s, 1H), 8.11 (s, 1H), 7.93–8.05 (m, 2H), 7.65–7.75 (m, 2H), 6.62 (d, J = 8.0 Hz, 1H), 6.49 (s, 1H), 6.24 (d, J = 8.0 Hz, 1H), 4.73 (s, 2H, NH2), 4.10 (d, J = 12.6 Hz, 1H), 3.91 (d, J = 12.6 Hz, 1H), 3.68 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 146.1199, 142.0685, 136.9355, 134.7723, 130.6659, 130.4093, 129.9510, 129.7126, 127.9528, 127.8244, 124.3963, 122.4714, 121.7748, 120.7299, 118.2000, 115.4685, 109.9872, 61.8103, 55.0640; HRMS-ESI (m/z) calcd. for C18H17BrNO2S [M + H]+: 390.0163, found: 390.0158.
4.1.4.4 5-(((6-Bromonaphthalen-2-yl)sulfonyl)methyl)-2-methoxyaniline (12d). Obtained in 82.6% yield, white solid, m.p. 185–188 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.42 (s, 1H), 8.06–8.15 (m, 3H), 7.66–7.77 (m, 2H), 6.61 (d, J = 8.0 Hz, 1H), 6.48 (s, 1H), 6.23 (d, J = 8.0 Hz, 1H), 4.76 (s, 2H, NH2), 4.55 (s, 2H), 3.69 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 146.3216, 146.0466, 137.0272, 136.3672, 136.1655, 135.3589, 131.8759, 131.2159, 131.1609, 130.2809, 130.2259, 129.8593, 129.7859, 129.5110, 129.1627, 129.0343, 127.9527, 127.8427, 123.9380, 122.3798, 121.7198, 119.8132, 119.4649, 118.5850, 118.2917, 115.6335, 109.5472, 109.2356, 67.4720, 60.2520, 55.1923, 54.7890. HRMS-ESI (m/z) calcd. for C18H17BrNO3S [M + H]+: 406.0113, found: 406.0106.
4.1.4.5 2-Methoxy-5-((naphthalen-2-ylthio)methyl)aniline (12e). Obtained in 31.5% yield, white solid, m.p. 110 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.80–7.86 (m, 4H), 7.42–7.51 (m, 3H), 6.67–6.71 (m, 2H), 6.56 (d, J = 8.0 Hz, 1H), 4.73 (s, 2H, NH2), 4.19 (s, 1H), 3.71 (s, 3H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 145.4233, 137.3938, 134.5523, 133.2141, 130.7759, 128.7777, 127.9528, 127.4028, 126.6512, 126.4128, 126.0279, 125.2579, 124.6163, 116.5135, 113.9836, 109.9506, 55.0090, 36.2735; HRMS-ESI (m/z) calcd. for C18H18NOS [M + H]+: 296.1109, found: 296.1104.
4.1.5.1 Methyl(2-methoxy-5-((naphthalen-2-ylsulfinyl)methyl)phenyl)glycinate (13a). Obtained in 52.6% yield, white solid, m.p. 156–158 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.00–8.09 (m, 4H), 7.61–7.68 (m, 3H), 6.71 (d, J = 8.0 Hz, 1H), 6.39 (d, J = 8.0 Hz, 1H), 6.14 (s, 1H), 5.21 (t, J = 6.2 Hz, 1H, NH), 4.15 (d, J = 12.0 Hz, 1H), 4.00 (d, J = 12.0 Hz, 1H), 3.75 (s, 2H), 3.62–3.67 (m, 6H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 171.6201, 146.4683, 141.5369, 137.1189, 134.0757, 132.4808, 128.9977, 128.5761, 128.1728, 127.8428, 127.3845, 124.9280, 123.0031, 120.9866, 118.8417, 111.2155, 109.6756, 62.3786, 55.6140, 51.8926, 44.3947; HRMS-ESI (m/z) calcd. for C21H22NO4S [M + H]+: 384.1270, found: 384.1264.
4.1.5.2 Methyl(2-methoxy-5-((naphthalen-2-ylsulfonyl)methyl)phenyl)glycinate (13b). Obtained in 86.7% yield, white solid, m.p. 202–204 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.38 (s, 1H), 8.06–8.15 (m, 3H), 7.66–7.75 (m, 3H), 6.68 (d, J = 8.0 Hz, 1H), 6.36 (d, J = 8.0 Hz, 1H), 6.13 (s, 1H), 5.24 (t, J = 6.2 Hz, 1H, NH), 4.55 (s, 2H), 3.73 (s, 3H), 3.57–3.63 (m, 5H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 170.8867, 146.0466, 136.4405, 135.3773, 134.2590, 131.1425, 129.1443, 128.9610, 128.7960, 128.5577, 127.4761, 127.2194, 122.7464, 120.3265, 118.8783, 111.0871, 108.9789, 60.4537, 54.9357, 51.2143, 43.7714; HRMS-ESI (m/z) calcd. for C21H22NO5S [M + H]+: 400.1219, found: 400.1212.
4.1.5.3 Methyl(5-(((6-bromonaphthalen-2-yl)sulfinyl)methyl)-2-methoxyphenyl)glycinate (13c). Obtained in 46.0% yield, white solid, m.p. 128–130 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.33 (s, 1H), 8.05–8.08 (m, 2H), 7.99 (d, J = 8.0 Hz, 1H), 7.69–7.76 (m, 2H), 6.70 (d, J = 8.0 Hz, 1H), 6.37 (d, J = 8.0 Hz, 1H), 6.08 (s, 1H), 5.20 (t, J = 6.0 Hz, 1H, NH), 4.16 (d, J = 12.6 Hz, 1H), 4.01 (d, J = 12.6 Hz, 1H), 3.75 (s, 3H), 3.63–3.67 (m, 5H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 171.7851, 146.6700, 142.4719, 137.2839, 135.3957, 131.2343, 130.9593, 130.5743, 130.3177, 128.3745, 125.2213, 122.9665, 122.4531, 121.3532, 119.0984, 111.3988, 109.8589, 62.3236, 55.8156, 52.1309, 44.6147; HRMS-ESI (m/z) calcd. for C21H21BrNO4S [M + H]+: 462.0375, found: 462.0367.
4.1.6.1 Sodium(2-methoxy-5-((naphthalen-2-ylsulfinyl)methyl)phenyl)glycinate (14a). Obtained in 92.4% yield, beige solid. The purification of 14a was failed and it was not used for evaluation of biological activity. HRMS-ESI (m/z) calcd. for C20H19NNaO4S [M + H]+: 392.0932, found: 392.0925.
4.1.6.2 Sodium(2-methoxy-5-((naphthalen-2-ylsulfonyl)methyl)phenyl)glycinate (14b). Obtained in 99.5% yield, beige solid, m.p. 250–252 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.40 (s, 1H), 8.05–8.15 (m, 3H), 7.65–7.77 (m, 3H), 6.60 (d, J = 8.0 Hz, 1H), 6.26 (d, J = 8.0 Hz, 1H), 6.07 (s, 1H), 5.12 (t, J = 3.9 Hz, 1H, NH), 4.55 (s, 2H), 3.72 (s, 3H), 2.86 (d, J = 3.9 Hz, 2H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.3350, 146.6150, 138.1272, 136.3856, 135.1024, 132.0042, 129.9860, 129.8410, 129.6760, 129.3827, 128.3195, 128.0811, 123.6631, 121.1882, 118.0351, 112.0405, 109.2173, 61.4620, 55.6323, 47.9328; HRMS-ESI (m/z) calcd. for C20H19NNaO5S [M + H]+: 408.0882, found: 408.0876.
4.1.7.1 (2-Methoxy-5-((naphthalen-2-ylsulfinyl)methyl)phenyl)glycine (15a). Obtained in 59.5% yield, light grey solid, m.p. 199–202 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.97–8.05 (m, 4H), 7.56–7.63 (m, 3H), 6.67 (d, J = 8.0 Hz, 1H), 6.35 (d, J = 8.0 Hz, 1H), 6.17 (s, 1H), 4.11 (d, J = 12.0 Hz, 1H), 3.96 (d, J = 12.0 Hz, 1H), 3.72 (s, 3H), 3.54 (s, 2H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.2479, 146.2185, 141.4178, 137.0112, 133.8786, 132.2929, 128.8121, 128.3882, 127.9688, 127.6549, 127.1805, 124.7034, 122.9229, 120.7688, 118.4636, 111.0826, 109.3960, 62.3809, 55.4077, 44.2893; HRMS-ESI (m/z) calcd. for C20H20NO4S [M + H]+: 370.1113, found: 370.1108.
4.1.7.2 (2-Methoxy-5-((naphthalen-2-ylsulfonyl)methyl)phenyl)glycine (15b). Obtained in 66.2% yield, light grey solid, m.p. 163–165 °C. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 8.35 (s, 1H), 8.02–8.11 (m, 3H), 7.62–7.74 (m, 3H), 6.64 (d, J = 8.0 Hz, 1H), 6.32 (d, J = 8.0 Hz, 1H), 6.16 (s, 1H), 5.73 (s, 2H), 4.51 (s, 2H), 3.70 (s, 3H), 3.48 (s, 2H); 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.5367, 146.8350, 137.3572, 136.2573, 135.1024, 131.9859, 130.0060, 129.8044, 129.6577, 129.4194, 128.3011, 128.0628, 123.6264, 121.1516, 119.5567, 112.0771, 109.7306, 61.3520, 55.7973, 44.7063; HRMS-ESI (m/z) calcd. for C20H20NO5S [M + H]+: 386.1062, found: 386.1055.
4.2. Cell culture
The five human cell lines (HeLa, MCF-7, HepG2, SCC-15 and 293T) and the HUVEC cells were cultured respectively using Dulbecco's modified Eagle's medium (DMEM) and RPMI Medium 1640 supplemented with 10% (v/v) fetal bovine serum (FBS) and penicillin (100 units per mL)/streptomycin (100 mg mL−1), at pH 7.2 and 5% CO2 humidified atmosphere at 37 °C. After attaining 80% confluence, the cells were trypsinized with 0.25 trypsin–EDTA and diluted with media to a fixed number of cells.4.3. Cytotoxic activity assay
Cytotoxic activity was assessed using triplicate assay by the standard MTS method. The cells were seeded into 96-well plates containing the medium at the density of 4000–6000 cells per mL (100 μL per well). The tested compounds and the positive control (Rigosertib) were dissolved in DMSO to the concentration of 100 mM and diluted in the culture medium to the concentrations needed. After 24 h, the cultured cells were treated with multiple desired concentrations of target compounds for 48 h. After 48 h of incubation, the supernatant was replaced by fresh medium (100 μL per well), and 20 μL MTS reagent (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) was added to each well. The plate was further incubated for 3 h at 37 °C in 5% CO2. The optical absorbance in individual well was determined at 492 nm using a microplate reader. The inhibition rates were calculated using the following formula:| Inhibition rate (%) = (ODnegative control − ODsample)/(ODnegative control − ODblank) × 100%. |
The IC50 values were calculated using GraphPad Prism 5 (La Jolla, CA, USA). For each concentration, at least three wells were performed to calculate the average parameter.
4.4. Flow cytometry assay
Freshly trypsinized HeLa cells were seeded in 6-well microtiter plates with a density of 1 × 105 cells per mL (2 mL per well) and incubated overnight. The next day, compound 15b (final concentrations are 25 nM, 50 nM and 100 nM) were added to the medium and respectively incubated for 24 h, 36 h and 48 h. At the corresponding time, the supernatant was discarded, the cells were washed with pre-cooling PBS, trypsinized and centrifuged. After that, the cells were resuspended with 100 μL Annexin V-FITC Binding Buffer, treated with 2.5 μL Annexin V-FITC (BD, Pharmingen) and 2.5 μL 7-AAD, and incubated at room temperature in dark for 15 min. Lastly, 400 μL of Annexin V-FITC Binding Buffer was added to the tubes containing cells to be analyzed. The apoptosis of HeLa cells was measured by flow cytometer and the result was obtained with system software (Cell Quest; BD Biosciences).4.5. Wound healing assay
Freshly trypsinized HeLa cells were seeded in 6-well microtiter plates with a density of 2.5 × 105 cells per mL (2 mL per well) and incubated overnight. After attaining about 90% confluence, wounds were created with p10 micropipette tips. Then the cells were washed twice with fresh PBS and added fresh serum-free medium containing 15b (final concentrations are 25 nM, 50 nM and 100 nM). HeLa cells were co-incubated with 15b for 48 h under serum-starved conditions. The wound gaps were monitored and recorded photographically using inverted microscope (Nikon) under 10 magnification after 0 h, 24 h and 48 h of incubation. The wound areas were measured by ImageJ 1.51j8 (Wayne Rasband, National Institute of Health, USA). The wound healing rates were calculated according to the changes in wound areas.4.6. Western blotting assay
Freshly trypsinized HeLa cells were seeded in 6-well microtiter plates with a density of 1 × 105 cells per mL (2 mL per well) and incubated overnight. The next day, compound 15b (final concentrations are 25 nM, 50 nM and 100 nM) were added to the medium and incubated for 48 h. Then the cells were collected, centrifuged and washed twice with pre-cooling PBS. The cells harvested above were lysed with a lysis buffer on ice for 20 min and centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 5 min. Protein concentrations of the cell lysates were measured with the BCA protein assay kit (Cwbio, China) following the instructions of manufacture. Equal amount of total cellular protein extracts were separated by electrophoresis on SDS polyacrylamide gels and transferred to PVDF membranes. After blocking with 5% nonfat milk, the membranes were co-incubated with primary antibodies (anti-p53, anti-Bcl-2, anti-Bax and anti-GAPDH) at 4 °C overnight. Subsequently, the membranes were incubated with second antibodies conjugated with peroxidase for 2 h, washed three times with TBST buffer for 15 min and visualized with the chemiluminescence reagent (Thermo Fischer Scientifics Ltd).
000 rpm at 4 °C for 5 min. Protein concentrations of the cell lysates were measured with the BCA protein assay kit (Cwbio, China) following the instructions of manufacture. Equal amount of total cellular protein extracts were separated by electrophoresis on SDS polyacrylamide gels and transferred to PVDF membranes. After blocking with 5% nonfat milk, the membranes were co-incubated with primary antibodies (anti-p53, anti-Bcl-2, anti-Bax and anti-GAPDH) at 4 °C overnight. Subsequently, the membranes were incubated with second antibodies conjugated with peroxidase for 2 h, washed three times with TBST buffer for 15 min and visualized with the chemiluminescence reagent (Thermo Fischer Scientifics Ltd).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (Grant No. 81273431, 21102176, 21272273) for its financial support for this project.Notes and references
- F. Xu, Q. He and X. Li, et al., Rigosertib as a selective anti-tumor agent can ameliorate multiple dysregulated signaling transduction pathways in high-grade myelodysplastic syndrome, Sci. Rep., 2014, 4, 7310 CrossRef CAS PubMed.
- K. Gumireddy, M. V. Reddy and S. C. Cosenza, et al., ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent, Cancer Cell, 2005, 7(3), 275–286 CrossRef CAS PubMed.
- A. Prasad, I. W. Park and H. Allen, et al., Styryl sulfonyl compounds inhibit translation of cyclin D1 in mantle cell lymphoma cells, Oncogene, 2009, 28(12), 1518–1528 CrossRef CAS PubMed.
- P. Schöffski, Polo-like kinase (PLK) inhibitors in preclinical and early clinical development in oncology, Oncologist, 2009, 14(6), 559–570 CrossRef PubMed.
- M. V. Reddy, P. Venkatapuram and M. R. Mallireddigari, et al., Discovery of a clinical stage multi-kinase inhibitor sodium (E)-2-{2-methoxy-5-[(2′,4′,6′-trimethoxystyrylsulfonyl)methyl]phenylamino}acetate (ON 01910.Na): synthesis, structure-activity relationship, and biological activity, J. Med. Chem., 2011, 54(18), 6254–6276 CrossRef CAS PubMed.
- C. M. Chapman, X. Sun and M. Roschewski, et al., ON 01910.Na is selectively cytotoxic for chronic lymphocytic leukemia cells through a dual mechanism of action involving PI3K/AKT inhibition and induction of oxidative stress, Clin. Cancer Res., 2012, 18(7), 1979–1991 CrossRef CAS PubMed.
- A. Jimeno, J. Li and W. A. Messersmith, et al., Phase I study of ON 01910.Na, a novel modulator of the Polo-like kinase 1 pathway, in adult patients with solid tumors, J. Clin. Oncol., 2008, 26(34), 5504–5510 CrossRef CAS PubMed.
- S. Y. Cho, T. Ohnuma, L. R. Silverman, J. F. Holland and J. Roboz, Discontinuous drug binding to proteins: binding of an antineoplastic benzyl styryl sulfone to albumin and enzymes in vitro and in phase I clinical trials, Drug Metab. Dispos., 2010, 38(9), 1480–1485 CrossRef CAS PubMed.
- M. Seetharam, A. C. Fan and M. Tran, et al., Treatment of higher risk myelodysplastic syndrome patients unresponsive to hypomethylating agents with ON 01910.Na, Leuk. Res., 2012, 36(1), 98–103 CrossRef CAS PubMed.
- W. W. Ma, W. A. Messersmith and G. K. Dy, et al., Phase I study of Rigosertib, an inhibitor of the phosphatidylinositol 3-kinase and Polo-like kinase 1 pathways, combined with gemcitabine in patients with solid tumors and pancreatic cancer, Clin. Cancer Res., 2012, 18(7), 2048–2055 CrossRef CAS PubMed.
- M. J. Olnes, A. Shenoy and B. Weinstein, et al., Directed therapy for patients with myelodysplastic syndromes (MDS) by suppression of cyclin D1 with ON 01910.Na, Leuk. Res., 2012, 36(8), 982–989 CrossRef CAS PubMed.
- B. H. O'Neil, A. J. Scott and W. W. Ma, et al., A phase II/III randomized study to compare the efficacy and safety of Rigosertib plus gemcitabine versus gemcitabine alone in patients with previously untreated metastatic pancreatic cancer, Ann. Oncol., 2015, 26(9), 1923–1929 CrossRef PubMed.
- G. Garcia-Manero, P. Fenaux and A. Al-Kali, et al., Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): a randomised, controlled, phase 3 trial, Lancet Oncol., 2016, 17(4), 496–508 CrossRef CAS PubMed.
- A. Ottaiano, M. Capozzi and C. De Divitiis, et al., Gemcitabine mono-therapy versus gemcitabine plus targeted therapy in advanced pancreatic cancer: a meta-analysis of randomized phase III trials, Acta Oncol., 2017, 56(3), 377–383 CrossRef CAS PubMed.
- S. C. Navada, S. M. Fruchtman and R. Odchimar-Reissig, et al., A phase 1/2 study of Rigosertib in patients with myelodysplastic syndromes (MDS) and MDS progressed to acute myeloid leukemia, Leuk. Res., 2018, 64, 10–16 CrossRef CAS PubMed.
- S. C. Navada, G. Garcia-Manero and R. OdchimarReissig, et al., Rigosertib in combination with azacitidine in patients with myelodysplastic syndromes or acute myeloid leukemia: results of a phase 1 study, Leuk. Res., 2020, 94, 106369 CrossRef CAS PubMed.
- S. P. Ghosh, M. W. Perkins and K. Hieber, et al., Radiation protection by a new chemical entity, Ex-Rad: efficacy and mechanisms, Radiat. Res., 2009, 171(2), 173–179 CrossRef CAS PubMed.
- S. Suman, K. Datta and K. Doiron, et al., Radioprotective effects of ON 01210.Na upon oral administration, J. Radiat. Res., 2012, 53(3), 368–376 CrossRef CAS PubMed.
- S. P. Ghosh, S. Kulkarni and M. W. Perkins, et al., Amelioration of radiation-induced hematopoietic and gastrointestinal damage by Ex-RAD(R) in mice, J. Radiat. Res., 2012, 53(4), 526–536 CrossRef CAS PubMed.
- A. D. Kang, S. C. Cosenza, M. Bonagura, M. Manair, M. V. Reddy and E. P. Reddy, ON01210.Na (Ex-RAD®) mitigates radiation damage through activation of the AKT pathway, PLoS One, 2013, 8(3), e58355, DOI:10.1371/journal.pone.0058355 , Epub 2013 Mar 7, PMID: 23505494, PMCID: PMC3591351..
- L. Tang, T. Peng and G. Wang, et al., Synthesis and radioprotective effects of novel benzyl naphthyl sulfoxide (sulfone) derivatives transformed from Ex-RAD, Medchemcomm, 2018, 9(4), 625–631 RSC.
- J. K. Salunke, P. Sonar, F. L. Wong, V. A. Roy, C. S. Lee and P. P. Wadgaonkar, Pyrene based conjugated materials: synthesis, characterization and electroluminescent properties, Phys. Chem. Chem. Phys., 2014, 16(42), 23320–23328 RSC.
- M. Vilches-Herrera, J. Miranda-Sepúlveda and M. Rebolledo-Fuentes, et al., Naphthylisopropylamine and N-benzylamphetamine derivatives as monoamine oxidase inhibitors, Bioorg. Med. Chem., 2009, 17(6), 2452–2460 CrossRef CAS PubMed.
- S. Grunder, R. Huber, S. Wu, C. Schönenberger, M. Calame and M. Mayor, Novel cruciform structures as model compounds for coordination induced single molecule switches, Chimia, 2010, 64(3), 140–144 CrossRef CAS PubMed.
- A. Kamal, ChR. Reddy and M. V. Vishnuvardhan, et al., Synthesis and biological evaluation of cinnamido linked benzophenone hybrids as tubulin polymerization inhibitors and apoptosis inducing agents, Bioorg. Med. Chem. Lett., 2014, 24(10), 2309–2314 CrossRef CAS PubMed.
- S. Mutti, M. Lavigne and I. Malejonock, et al., Method for preparing combretastatins, WO2003084919 A2 2003-10-16, World Intellectual Property Organization, 2005 Search PubMed.
- K. P. Bryliakov and E. P. Talsi, Catalytic Enantioselective Oxidation of Bulky Alkyl Aryl Thioethers with H2O2 over Titanium–Salan Catalysts, Eur. J. Org. Chem., 2011, 2011(24), 4693–4698 CrossRef CAS.
- V. R. Pallela, M. R. Mallireddigari and S. C. Cosenza, et al., Hydrothiolation of benzyl mercaptan to arylacetylene: application to the synthesis of (E) and (Z)-isomers of ON 01910·Na (Rigosertib®), a phase III clinical stage anti-cancer agent, Org. Biomol. Chem., 2013, 11(12), 1964–1977 RSC.
- L. Tang, T. Peng and G. Wang, et al., Design, Synthesis and Preliminary Biological Evaluation of Novel Benzyl Sulfoxide 2-Indolinone Derivatives as Anticancer Agents, Molecules, 2017, 22(11), 1979, DOI:10.3390/molecules22111979 , PMID: 29144401, PMCID, PMC6150246..
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05226h |
| This journal is © The Royal Society of Chemistry 2021 |