
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improvement of thermal stability of highly active species on SiO2 supported copper-ceria catalysts
Gonzalo Aguila *a,
Rafael Called,
Sichem Guerrerob,
Patricio Baezac and
Paulo Arayad
*a,
Rafael Called,
Sichem Guerrerob,
Patricio Baezac and
Paulo Arayad
aDepartamento de Ciencias de la Ingeniería, Facultad de Ingeniería, Universidad Andres Bello, Antonio Varas 880, Santiago, Chile. E-mail: gonzalo.aguila@unab.cl
bFacultad de Ingeniería y Ciencias Aplicadas, Universidad de los Andes, Monseñor Álvaro del Portillo, Las Condes, Santiago, 12455, Chile
cInstituto de Química, Pontificia Universidad Católica de Valparaíso, Casilla 4059, Valparaíso, Chile
dDepartamento de Ingeniería Química, Biotecnología y Materiales, Facultad de Ciencias Físicas y Matemáticas, Universidad de Chile, Beauchef 851, Santiago, Chile
First published on 11th October 2021
Abstract
CuO–CeO2/SiO2 catalysts lose activity when they are calcined at 600 °C and temperatures above. This loss of activity was related to a decrease in the amount of highly dispersed Cu species interacting with Ce (CuO–CeO2 interface) over the SiO2 support. These species are highly active in CO oxidation, so this reaction was selected to conduct this study. In order to avoid the activity loss in CuO–CeO2/SiO2 catalysts, the effect of high Ce loads (8, 16, 24, and 36%) on the thermal stability of these catalysts was studied. The results reveal that when increasing calcination temperature from 500 to 700 °C, the catalysts with Ce load equal to or higher than 24% increase the formation of highly dispersed Cu interacting with Ce and therefore the activity (90% of CO conversion at 120 °C). In catalysts with Ce load below 24%, Cu species agglomerate and decrease the activity (less than 5% of CO conversion at 120 °C).
Introduction
Since the pioneering work of the Flytzani-Stephanopoulos group,1–3 who showed the high activity of the CuO–CeO2 system in the oxidation reaction of CO with O2, this system has also been shown to be highly active in other reactions such as the Water Gas-Shift (WGS) reaction,4 and more recently the decomposition of N2O.5–8In all these reactions, the interfacial CuO–CeO2 sites are responsible for the high activity of these catalysts. Thus, Martinez-Arias et al.9 studying the oxidation of CO, propose that CO will react with oxygen in the CuO–CeO2 interface to produce CO2. The controversy on the exact nature of the active sites, the oxidation state of copper, and the reaction mechanism continues.10,11 In the case of the WGS, the redox mechanism proposed in the literature assumes that the oxygen of the H2O molecule is abstracted in an oxygen vacancy at the CuO–CeO2 interface releasing H2. The oxygen atom is then removed by CO, allowing the continuation of the catalytic cycle.4–12 Finally, the decomposition of N2O would occur by the abstraction of the oxygen atom in a vacancy site (Cu1+) of the CuO–CeO2 interface.5 It seems that the higher the dispersion of copper and ceria, the higher the interaction of both metals, which in turn favors the catalytic activity.13
Most of the work published in the literature, deals with CuO/CeO2 supported catalysts or CuO–CeO2 mixed oxides. Some works have been published using Cu and Ce supported on traditional supports like SiO2 or alumina14–21 in these reactions, and the nature and activity of the CuO–CeO2 interface generated over these support is less known, although some works point to the role of labile oxygen species, surface defects, and oxygen vacancies.14,15,22–29 Several works dealing with the CuO–CeO2 supported catalysts have been done in our laboratory.19–21 The oxidizing activity of CO with O2 of 2% Cu and 8% Ce systems supported on SiO2, Al2O3, and ZrO2 was compared and we found that the most active system was generated by using SiO2 as support.19 The greater activity of the CuO–CeO2/SiO2 catalyst was attributed to the fact that the inert character of the silica would favor the formation of the CuO–CeO2 interface and thus, improving the interaction between both phases.16,22,23,25,26,28–30 Considering the high activity of CuO–CeO2 supported on SiO2 catalysts, the optimum Cu/Ce load ratio and the total Cu + Ce load of catalysts supported on SiO2 were studied in a second work of our group.20 The results showed that the optimum Cu/Ce load ratio of catalysts supported on SiO2 is very similar to that used by Park et al.18 for catalysts supported on alumina. In a latter work, Luo et al.17 found the same optimum of Ce content for Cu–Ce supported on SiO2 catalysts calcined at 500 °C. In a third work we studied the thermal stability of catalysts with constant loading of 2% Cu and 8% Ce supported on SiO2 changing the calcination temperature in the range of 500–700 °C.21 It was found that calcining the catalyst at temperatures from 600 °C generated a noticeable loss of activity and almost a complete deactivation after calcining at 700 °C. Characterization studies on the catalyst calcined at 700 °C showed that the CuO–CeO2 interface had decreased substantially due to the migration and segregation of the CuO and CeO2 particles on the surface of the silica.21
With the purpose of improving the thermal stability of highly dispersed Cu species interacting with Ce (CuO–CeO2 interface) over SiO2 support, the present work studies the effect of increasing the Ce load above the 8% used in our previous work,21 maintaining the Cu load constant at 2%. The hypothesis is that as the Cu species migrate due to the temperature increase, the existence of larger regions of the support covered with CeO2 should increase the possibility of contact between CuO and CeO2 on the surface of the silica. The oxidation of CO at low temperature has been selected as a test reaction because it is highly sensitive to the formation of the CuO–CeO2 interface. Obviously, this reaction does not require high temperatures, so a study of the thermal stability at temperatures above 500 °C does not seem necessary. However, in other reactions like the decomposition of N2O, the catalyst must resist reaction temperatures between 600 °C (tail process option) and 850 °C (gas process option),31 justifying this study of the thermal stability of CuO–CeO2 interface on SiO2 support.
Results and discussion
The specific surface area (SSA) of the catalysts is shown in Table 1, where it is seen that, at each calcination temperature, the SSA decreases as the Ce content increases. This effect is more pronounced at 900 °C, where the SSA of the catalysts decreases from 110 m2 g−1 (2Cu–8Ce-900 catalyst) to 80 m2 g−1 (2Cu–36Ce-900 catalyst). However, due to the high thermal stability of silica support (nominal SSA of 130 m2 g−1), the SSA of the catalysts are relatively high in all the samples, so it seems reasonable to assume that this is not a key parameter in the possible variations of the CuO–CeO2 interface.| Catalyst | Specific surface area (m2 g−1) | H2 consumption ratio | CeO2 particle size (Å) |
|---|---|---|---|
| 2Cu–8Ce-500 | 128 | 1.43 | 58 |
| 2Cu–16Ce-500 | 127 | 1.66 | 65 |
| 2Cu–24Ce-500 | 111 | 1.79 | 73 |
| 2Cu–36Ce-500 | 108 | 1.87 | 94 |
| 2Cu–8Ce-700 | 120 | 1.16 | 58 |
| 2Cu–16Ce-700 | 111 | 1.29 | 73 |
| 2Cu–24Ce-700 | 119 | 1.24 | 97 |
| 2Cu–36Ce-700 | 115 | 1.44 | 113 |
| 2Cu–8Ce-900 | 110 | 0.99 | 97 |
| 2Cu–16Ce-900 | 91 | 1.04 | 117 |
| 2Cu–24Ce-900 | 83 | 1.03 | 146 |
| 2Cu–36Ce-900 | 80 | 0.98 | 219 |
The XRD analyses of the different samples are shown in Fig. 1. In the catalysts calcined at 500 °C and 700 °C, independently of Ce load, only the peaks of the fluorite phase of CeO2 are seen (JCPDS Card N° 81-0792). It is not possible to see the peaks of CuO, so it can be assumed that the Cu phase is highly dispersed at those temperatures. When the catalysts are calcined at 900 °C, the peaks of CuO at 35.5° and 38.7° (JCPDS Card N° 45-0937) are visible in all the catalysts, reflecting the sintering of the Cu species. The intensity of CuO peaks decreases as Ce content increases due to the diluting effect of Ce. Table 1 reports the diameters of CeO2 crystalline particles calculated by means of the Scherrer equation, showing that, as expected, at each calcination temperature, the size of the CeO2 particles increases as the Ce loading increases. This effect is clearly more important at higher temperatures. In fact, at 900 °C, the CeO2 particle diameter increases almost 225% when Ce load increases from 8% to 36%, while at 500 °C, the increment in particle size is only of 162%.
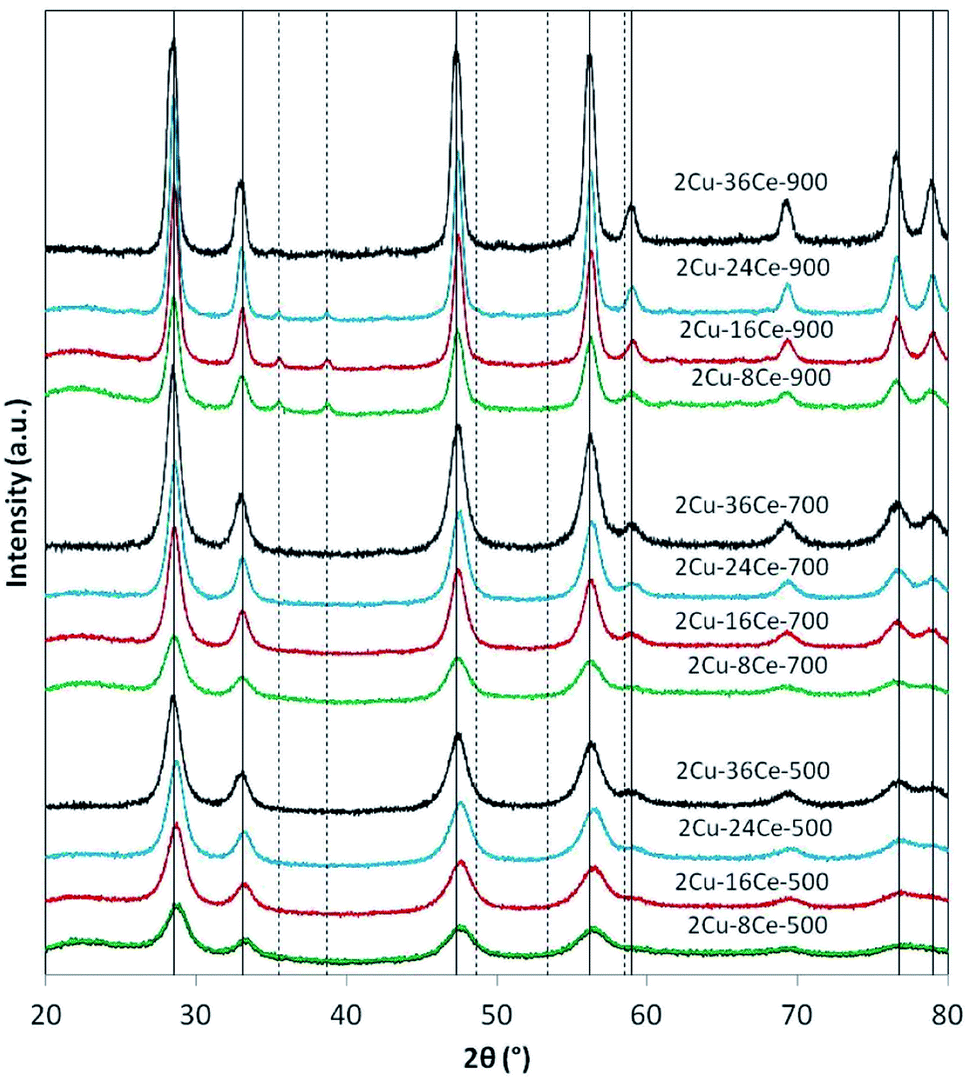 | ||
| Fig. 1 XRD diffractograms of bimetallic catalysts with a constant Cu load (2%) and variable Ce load (8%, 16%, 24% and 36%), calcined at 500, 700 and 900 °C. The nomenclature indicates the Ce loading and the calcination temperature of each catalyst. The characteristics diffraction lines of CuO ( − − ) and CeO2 (—) are indicated as reference. Operation conditions: scan range = 20–80°; scan rate = 0.02 degrees per minute. | ||
The TPR curves are shown in Fig. 2 and the ratios of the experimental consumption of H2 with respect to the theoretical consumption needed to reduce only the CuO to Cu0 are presented in Table 1. As shown in Fig. 2, in the case of 2Cu–8Ce-500 and 2Cu–16Ce-500 catalysts it can be observed two reduction peaks, one with a maximum below 200 °C, attributable to the reduction of highly dispersed Cu species in contact with CeO2, and a second peak with a maximum between 200 and 225 °C that can be attributed to larger cluster-type Cu species also in contact with CeO2.20 As the Ce load increases from 8% to 16%, the first reduction peak moves slightly to lower temperature, and the second peak reduce its intensity, indicating the formation of higher amount of highly dispersed Cu species in contact with Ce. For the 2Cu–24Ce-500 catalyst, a third peak appears as a shoulder at 260 °C. The same peak moves to 280 °C for the 2Cu–36Ce-500 catalyst and it is also more intense. This third peak could be attributed to CuO particles interacting with CeO2 having higher particle size (see Table 1). These CuO species are poorly crystalline or of small size, as they are not detected by XRD. This is an unexpected result, considering that it is well known that CeO2 favors the dispersion of Cu species.19 A more deeply study will be performed to elucidate the nature of this CuO species formed at high Ce loading, but as previously noted, it could be related to the growing of CeO2 particle size, which decreases the contact between Cu species and CeO2. The H2 consumption ratio of catalysts calcined at 500 °C is always superior to 1, indicating an important reduction of CeO2 in contact with Cu species. Calcination at 700 °C produces a different effect in Cu dispersion depending on Ce content. In fact, for the catalysts with 8% and 16% of Ce content (2Cu–8Ce-700 and 2Cu–16Ce-700), the low temperature peaks disappears, and only the reduction peak associated with the reduction of CuO species in contact with CeO2 can be observed in the range between 260–280 °C, thus reflecting the agglomeration of dispersed Cu species. In contrast, the TPR curves of the catalysts with the highest Ce load (2Cu–24Ce-700 and 2Cu–36Ce-700), the high temperature peaks disappears and only peaks with maxima below 200 °C can be observed. In other words, the Cu species migrate and disperse forming highly dispersed CuO species in contact with CeO2. Finally, when calcining at 900 °C, all the catalysts show a main reduction peak close to 300 °C, characteristic of crystalline CuO species or bulk CuO poorly or not interacting with CeO2. Also, the H2 consumption ratio of catalysts calcined at 900 °C is almost 1 in all cases, indicating no reduction of CeO2, i.e., the CuO–CeO2 interface disappears.
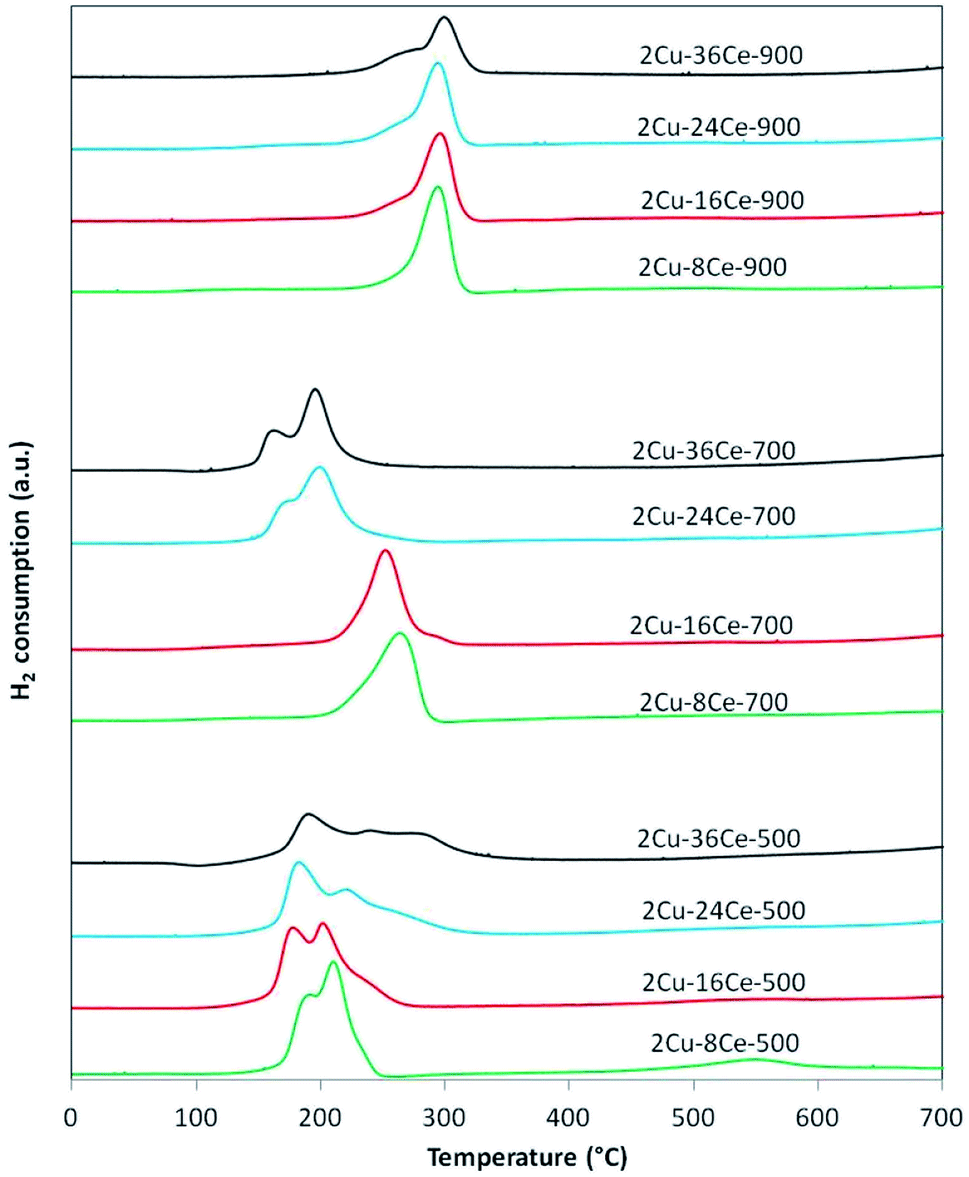 | ||
| Fig. 2 TPR curves of bimetallic catalysts with a constant Cu load (2%) and variable Ce load (8%, 16%, 24% and 36%), calcined at 500, 700 and 900 °C. Experimental conditions: temperature ramp rate: 10 °C min−1, flow rate: 20 cm3 min−1 of 5% H2/Ar. | ||
The results of the conversion of CO with temperature for the different catalysts are shown in Fig. 3. As can be seen in Fig. 3(A), when catalysts are calcined at 500 °C, the activity increases as the Ce content is increased from 8% to 16%, and the activity remains constant when the Ce content is raised to 24%. However, increasing the Ce loading to 36% led to a decrease in activity, which shows the existence of an optimum Ce loading (16% Ce) in catalysts with 2% Cu loading and calcined at 500 °C, and this result is in agreement with a previous study of our group.20 The behavior of these catalysts can be explained by the activity of the different copper species formed on the surface. As discussed in the literature, the highly dispersed Cu species in contact with Ce are the most active species of the CuO–CeO2 system,32 while the bulk CuO species is practically inactive at temperatures below 200 °C.19 Therefore, the increase of activity as Ce loading increases from 8% to 16% is in agreement with the increase in the concentration of highly dispersed Cu species in contact with CeO2 observed in the TPR experiment. The concentration and reducibility of the highly dispersed Cu species in contact with CeO2 (first peak below 200 °C) on the 2Cu–24Ce-500 catalyst are similar to those of the 2Cu–16Ce-500 catalyst, and this explains why their activities are also very similar. When the Ce content increases to 36%, the TPR curve of the 2Cu–36Ce-500 catalyst clearly shows a decrease in the amount of highly dispersed Cu species in contact with CeO2 and an increase of CuO in contact with CeO2, explaining the lower activity of the 2Cu–36Ce-500 catalyst compared to that of the 2Cu–24Ce-500 catalyst.
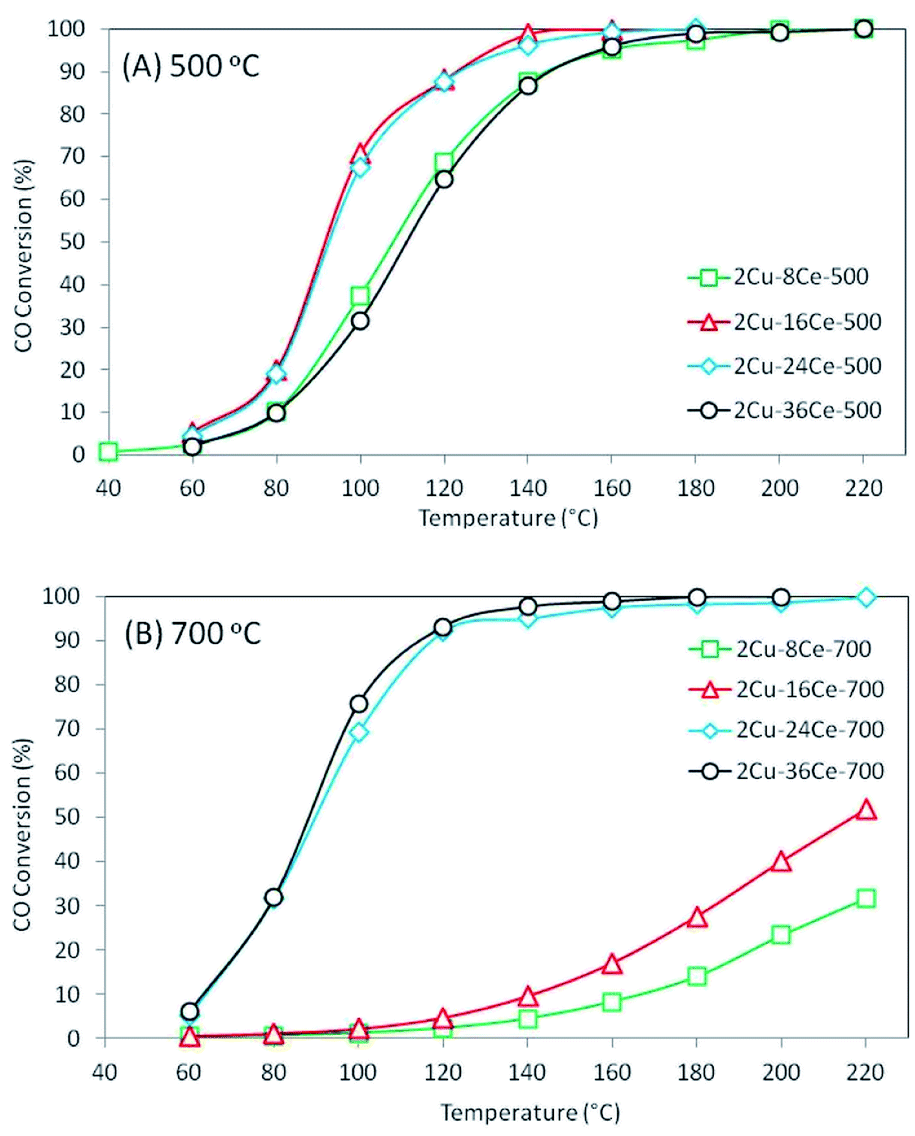 | ||
| Fig. 3 CO conversion curves of bimetallic catalysts with a constant Cu load (2%) and variable Ce load (8%, 16%, 24% and 36%) calcined at 500 °C and 700 °C. (A) Catalysts calcined at 500 °C: 2Cu–8Ce-500 (green); 2Cu–16Ce-500 (red); 2Cu–24Ce-500 (blue); 2Cu–36Ce-500 (black). (B) Catalysts calcined at 700 °C: 2Cu–8Ce-700 (green); 2Cu–16Ce-700 (red); 2Cu–24Ce-700 (blue); 2Cu–36Ce-700 (black). Reaction conditions: temperature ramp rate = 3 °C min−1, CO concentration = 2% v/v, O2 concentration = 3% v/v, mass of catalysts = 0.1 gram, total flow rate = 100 cm3 min−1. | ||
When catalysts were calcined at 700 °C, as shown in Fig. 3(B), the 2Cu–8Ce-700 and 2Cu–16Ce-700 catalysts undergo a substantial loss of activity respect to homologous catalysts calcined at 500 °C (Fig. 3(A)). The TPR curves explain this activity decrease due to the agglomeration of Cu species in contact with CeO2, with the resulting loss of the amount of highly dispersed Cu species. In contrast, the 2Cu–24Ce-700 and 2Cu–36Ce-700 catalysts show very high and similar activities after calcination at 700 °C. In fact, the 2Cu–24Ce-700 catalyst retains almost the same activity to that found when calcined at 500 °C, while the 2Cu–36Ce-700 catalyst substantially increases its activity. The TPR curves of both catalysts, 2Cu–24Ce-700 and 2Cu–36Ce-700, show an increase in the concentration of highly dispersed Cu species in contact with CeO2, while CuO particles interacting with high particle size CeO2 disappears, which explains the very high activity of these catalyst. It is important to note that in these catalysts calcined at 700 °C, the optimum Ce loading is 24%, showing that optimum Ce loads depend on the calcination temperature.
Finally, by calcining at 900 °C, none of the catalysts is active below 200 °C (CO conversion curves were not included) and this is accounted for by the strong sintering and segregation of the CuO and CeO2 species, destroying the active CuO–CeO2 interface and forming mainly CuO crystalline species or bulk CuO, as seen in the TPRs and XRDs of these catalysts. The above is related to that H2 consumption ratios are almost equal to 1 (see Table 1) in these catalysts calcined at 900 °C, indicating that CeO2 reduction does not occur.
Experimental
Catalyst preparation
The catalysts were prepared by coimpregnation of SiO2 support (Aerosil 130, Degussa). This is a non-porous, amorphous, hydrophilic fumed silica of 99.8% of SiO2 content having a specific surface area of 130 m2 g−1, with traces of impurities of metal in the ppb range. This support was coimpregnated with an aqueous solution containing the proper amounts of both Cu and Ce nitrates, to obtain a constant load of 2% Cu and variable loads of 8%, 16%, 24%, and 36% (w/w) of Ce. The catalysts were dried in an oven at 105 °C for 12 hours. Each catalyst was separated into three samples and calcined in a muffle furnace at 500, 700 and 900 °C for 3 hours under air. The catalysts were identified as 2Cu-XCe-Y, where X indicates the Ce load, and Y is the calcination temperature.Catalyst characterization
The samples were characterized by N2 adsorption, X-ray diffraction (XRD), and temperature programmed reduction (TPR) in a hydrogen stream.Determination of the BET specific surface area of the catalysts was made by obtaining the N2 adsorption and desorption isotherms in a Micromeritics equipment, Model ASAP 2010 Sorptometer. The samples were previously degassed at 200 °C.
The crystal structure of the different catalysts was determined on a Siemens D-5000 diffractometer using Cu Kα radiation and a scan rate of 0.02 degrees per minute.
Finally, the temperature programmed reduction analyses were made on a conventional system equipped with a TCD detector, with a flow rate of 20 cm3 min−1 of a gaseous mixture of 5% H2 in Ar, at a heating rate of 10 °C min−1 between ambient temperature and 700 °C.
Measurement of catalyst activity
The kinetics tests were made in a piston flow tubular reactor, with a stream of 2% CO and 3% O2 at a total flow rate of 100 cm3 min−1. After loading the reactor with 0.1 g of catalyst, the sample was pretreated at 300 °C for one hour in O2, and the reactor was cooled down to room temperature. The reactants were then fed, and the temperature was increased at a rate of 3 °C min−1, taking samples every 20 °C to determine the concentration of CO, O2, and CO2, using a PerkinElmer Autosystem Chromatograph with a CTR column (Alltech) and a TCD detector.Conclusions
This work shows that Ce content is a crucial factor to determine the formation of different Cu species with the calcination temperature, in the range 500–700 °C. At Ce content below 24%, the Cu species agglomerates after calcination at 700 °C, decreasing the amount of highly disperse Cu species interacting with CeO2 (CuO–CeO2 interface) leading to low activity in these catalysts. At Ce content of 24% or higher, calcination at 700 °C increases Cu dispersion and the amount of highly disperse CuO–CeO2 species, thus increasing catalysts activity.These new findings open the possibility of synthesizing CuO–CeO2/SiO2 catalysts with a high thermal stability of CuO–CeO2 interface over a wider temperature range, allowing its use in reactions that require high temperatures.
Author contributions
Gonzalo Aguila: conceptualization, methodology, investigation, formal analysis, validation, visualization, writing – original draft. Rafael Calle: initial research, experimental work. Sichem Guerrero: formal analysis, investigation, validation, writing – review & editing. Patricio Baeza: formal analysis, validation, visualization, writing – original draft. Paulo Araya: conceptualization, funding acquisition, project administration, resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge gratefully the financial support of this work under Fondecyt project 1161227 and PUCV project 039.376.Notes and references
- W. Liu and M. Flytzani-Stephanopoulos, J. Catal., 1995, 153, 304–316 CrossRef CAS.
- L. Kundakovic and M. Flytzani-Stephanopoulos, Appl. Catal., A, 1998, 171, 13–29 CrossRef CAS.
- L. Kundakovic and M. Flytzani-Stephanopoulos, J. Catal., 1998, 179, 203–221 CrossRef CAS.
- Y. Li, Q. Fu and M. Flytzani-Stephanopoulos, Appl. Catal., B, 2000, 27, 179–191 CrossRef CAS.
- A. Adamski, W. Zając, F. Zasada and Z. Sojka, Catal. Today, 2012, 191, 129–133 CrossRef CAS.
- H. Zhou, Z. Huang, C. Sun, F. Qin, D. Xiong, W. Shen and H. Xu, Appl. Catal., B, 2012, 125, 492–498 CrossRef CAS.
- M. Zabilskiy, B. Erjavec, P. Djinovic and A. Pintar, Chem. Eng. J., 2014, 254, 153–162 CrossRef CAS.
- M. Zabilskiy, P. Djinovic, B. Erjavec, G. Drazic and A. Pintar, Appl. Catal., B, 2015, 163, 113–122 CrossRef CAS.
- A. Martínez-Arias, M. Fernández-García, O. Gálvez, J. M. Coronado, J. A. Anderson, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207–216 CrossRef.
- W. W. Wang, P. P. Du, S. H. Zou, H. Y. He, R. X. Wang, Z. Jin, S. Shi, Y. Y. Huang, R. Si, Q. S. Song, C. J. Jia and C. H. Yan, ACS Catal., 2015, 5, 2088–2099 CrossRef CAS.
- J. S. Elias, N. Artrith, M. Bugnet, L. Giordano, G. A. Botton, A. M. Kolpak and Y. Shao-Horn, ACS Catal., 2016, 6, 1675–1679 CrossRef CAS.
- X. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2006, 110, 428–434 CrossRef CAS PubMed.
- L. Mo, E. T. Saw, Y. Kathiraser, M. L. Ang and S. Kawi, Int. J. Hydrogen Energy, 2018, 43, 15891–15897 CrossRef CAS.
- J. Wu, X. Zhao, L. Xue, H. Su and S. Zeng, J. Rare Earths, 2020, 38, 46–51 CrossRef CAS.
- S. T. Hossain, Y. Almesned, K. Zhang, E. T. Zell, D. T. Bernard, S. Balaz and R. Wang, Appl. Surf. Sci., 2018, 428, 598–608 CrossRef CAS.
- S. T. Hossain, E. T. Zell, S. Balaz and R. Wang, Appl. Surf. Sci., 2019, 491, 374–382 CrossRef CAS.
- J. Luo, W. Chu, H. Xu, C. Jiang and T. Zhang, J. Nat. Gas Chem., 2010, 19, 355–361 CrossRef CAS.
- J. W. Park, J. H. Jeong, W. L. Yoon, H. Jung, H. T. Lee, D. K. Lee, Y. K. Park and Y. W. Rhee, Appl. Catal., A, 2004, 274, 25–32 CrossRef CAS.
- G. Águila, F. Gracia and P. Araya, Appl. Catal., A, 2008, 343, 16–24 CrossRef.
- J. Astudillo, G. Águila, F. Díaz, S. Guerrero and P. Araya, Appl. Catal., A, 2010, 381, 169–176 CrossRef CAS.
- G. Águila, S. Guerrero and P. Araya, Appl. Catal., A, 2013, 462–463, 56–63 CrossRef.
- Y. Xie, J. Wu, G. Jing, H. Zhang, S. Zeng, X. Tian, X. Zou, J. Wen, H. Su, C. J. Zhong and P. Cui, Appl. Catal., B, 2018, 239, 665–676 CrossRef CAS.
- W. Li, X. Shen, R. Zeng, J. Chen, W. Xiao, S. Ding, C. Chen, R. Zhang and N. Zhang, Appl. Surf. Sci., 2019, 492, 818–825 CrossRef CAS.
- A. F. Zedan, K. Polychronopoulou, A. Asif, S. Y. AlQaradawi and A. S. AlJaber, Surf. Coat. Technol., 2018, 354, 313–323 CrossRef CAS.
- J. Ning, Y. Zhou, A. Chen, Y. Li, S. Miao and W. Shen, Catal. Today, 2020, 357, 460–467 CrossRef CAS.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Appl. Catal., B, 2018, 230, 18–28 CrossRef CAS.
- X. Gong, B. Liu, B. Kang, G. Xu, Q. Wang, C. Jia and J. Zhang, Mol. Catal., 2017, 436, 90–99 CrossRef CAS.
- Z. Guo, L. Song, T. Xu, D. Gao, C. Li, X. Hu and G. Chen, Mater. Chem. Phys., 2019, 226, 338–343 CrossRef CAS.
- S. Yang, F. Zhou, Y. Liu, L. Zhang, Y. Chen, H. Wang, Y. Tian, C. Zhang and D. Liu, Int. J. Hydrogen Energy, 2019, 44, 7252–7261 CrossRef CAS.
- C. Papadopoulos, K. Kappis, J. Papavasiliou, J. Vakros, M. Kusmierz, W. Gac, Y. Georgiou, Y. Deligiannakis and G. Avgouropoulos, Catal. Today, 2020, 355, 647–653 CrossRef CAS.
- M. A. G. Hevia and J. Pérez-Ramírez, Appl. Catal., B, 2008, 77, 248–254 CrossRef CAS.
- S. Yao, K. Mudiyanselage, W. Xu, A. C. Johnston-Peck, J. C. Hanson, T. Wu, D. Stacchiola, J. A. Rodriguez, H. Zhao, K. A. Beyer, K. W. Chapman, P. J. Chupas, A. Martínez-Arias, R. Si, T. B. Bolin, W. Liu and S. D. Senanayake, ACS Catal., 2014, 4, 1650–1661 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |