DOI:
10.1039/D1RA06266B
(Paper)
RSC Adv., 2021,
11, 33260-33270
Selective targeting of CD38 hydrolase and cyclase activity as an approach to immunostimulation†
Received
18th August 2021
, Accepted 3rd October 2021
First published on 11th October 2021
Abstract
The ectoenzyme CD38 is highly expressed on the surface of mature immune cells, where they are a marker for cell activation, and also on the surface of multiple tumor cells such as multiple myeloma (MM). CD38-targeted monoclonal antibodies (MABs) such as daratumumab and isatuximab bind to CD38 and promote cancer cell death by stimulating the antitumor immune response. Although MABs are achieving unprecedented success in a percentage of cases, high rates of resistance limit their efficacy. Formation of the immunosuppressive intermediate adenosine is a major route by which this resistance is mediated. Thus there is an urgent need for small molecule agents that boost the immune response in T-cells. Importantly, CD38 is a dual-function enzyme, serving as a hydrolase and a nicotinamide adenine dinucleotide (NAD+) cyclase, and both of these activities promote immunosuppression. We have employed virtual and physical screening to identify novel compounds that are selective for either the hydrolase or the cyclase activity of CD38, and have demonstrated that these compounds activate T cells in vitro. We are currently optimizing these inhibitors for use in immunotherapy. These small molecule inhibitors of the CD38-hydrolase or cyclase activity can serve as chemical probes to determine the mechanism by which CD38 promotes resistance to MAB therapy, and could become novel and effective therapeutic agents that produce immunostimulatory effects. Our studies have identified the first small molecule inhibitors of CD38 specifically for use as immunostimulants.
Introduction
The ectoenzyme known as cluster of differentiation 38 (CD38) has recently re-emerged as a viable target for immunotherapy. CD38 primarily exists as a 34 kDa transmembrane glycoprotein containing a small N-terminal cytosolic tail, a single pass transmembrane domain, and a large C-terminal extracellular domain.1 CD38 is expressed on the surface of mature immune cells, and as adaptive immune cells mature and undergo phenotypic reprogramming, cell surface markers are expressed as part of phenotypic remodelling.2,3 In both B and T cells, CD38 expression is indicative of cellular activation and serves as a receptor for lymphocyte transmigration.3 Recent literature suggests that CD38 expression is highly contextual in that it depends on cellular location and the pathophysiological context in which it is embedded.4
For multiple reasons, CD38 is considered a promising target for small molecule inhibitors. Highly proliferating cells, such as T cells and many tumor cell lines, are highly dependent on nicotinamide adenine dinucleotide (NAD+), suggesting that their activity is likely to be further enhanced by inhibition of the NADase function of CD38. Multiple research groups have targeted CD38 as an NAD boosting therapy in the context of aging, mitochondrial dysfunction, obesity, and diabetes.4–8 Notably, some of these inhibitors increased the levels of NAD+ in vitro and in vivo, but were reportedly toxic in animal models or possessed undesirable pharmacokinetic profiles. In addition, none of these agents were evaluated for antitumor or immunostimulatory effects.4,6,8,9
CD38 is already a validated target for cancer immunotherapy, since it is highly expressed on the surface of a variety of tumor cells, most notably multiple myeloma (MM). The monoclonal antibodies (MABs) daratumumab and isatuximab specifically bind to CD38 on the surface of these tumor cells and promote tumor cell death by Fc-dependent immune effector mechanisms including complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and apoptosis upon secondary cross-linking.10–12 Because CD38 is also highly expressed on the surface of regulatory T-cells (Tregs), regulatory B-cells and myeloid suppressor cells, these MABs also reduce the number of immune suppressors, resulting in an increase in cytotoxic T-cells.13,14
Ironically, resistance to CD38-targeted MABs is mediated by CD38 itself. Importantly, CD38 up regulation appears to be one of the most important factors in mediating resistance to checkpoint blockade in MM and other cancers.15–17 In vitro and in vivo studies demonstrate that CD38 inhibits CD8+ T-cell function via adenosine (ADO) receptor signalling. In addition, resistance to PD-1/PD-L1 blocking antibodies is mediated through upregulation of CD38 and subsequent production of ADO.15 Extracellular ADO binds to ADO receptors on immune cells, including T cells, natural killer (NK) cells, neutrophils, macrophages and dendritic cells, preventing their activation.18,19 ADO produced by CD38 also promotes myeloid-derived suppressor cell (MDSC) expansion, macrophage M2 polarization, and CD4+ Treg generation, all of which support tumor cell progression.20 Taken together, these data indicate that CD38 or ADO receptor blockade are effective strategies to overcome this resistance.21
Role of CD38 in cellular energy metabolism
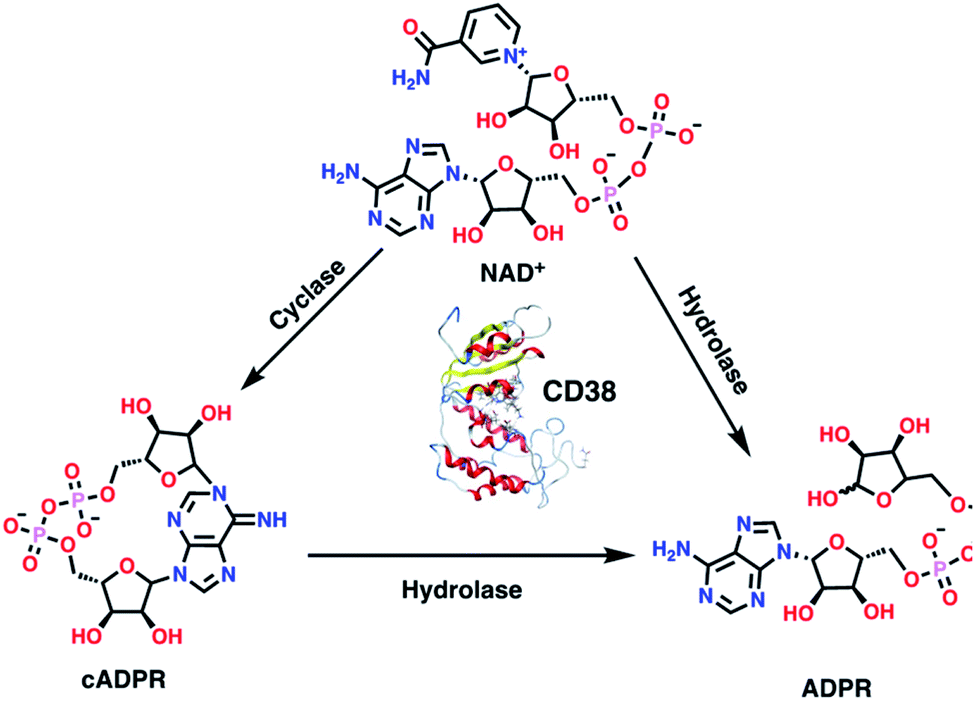
As an ectoenzyme, CD38 plays a critical role in the homeostatic regulation of cellular energetics.4,5 By metabolizing the cofactor NAD+, CD38 removes an essential electron acceptor, thus limiting the energetic capacity of a cell. CD38 is known for its glycohydrolytic activity, though in some circumstances it uses two reactions that may be sequentially coupled (Fig. 1). In the first reaction, CD38 functions as a cyclase, removing nicotinamide from NAD+ and producing cyclic adenosine diphosphate-ribose (cADPR).4,22 CD38 also mediates an energetically more favorable hydrolase reaction that produces adenosine diphosphate-ribose (ADPR) from both NAD+ and cADPR.1,4 CD38 over expression in immune cells and tumor cells within the tumor microenvironment (TME) causes a reduction in NAD+ levels, leading to a down regulation of the immune response against tumor cells.3 CD38-knockout CD4+ T cells more effectively control tumor growth when compared to wild type CD4+ T cells, express twice the level of interferon-gamma (IFNγ), and up regulate Sirt1,23,24 a key NAD-dependent regulator of effector function within T cells.24
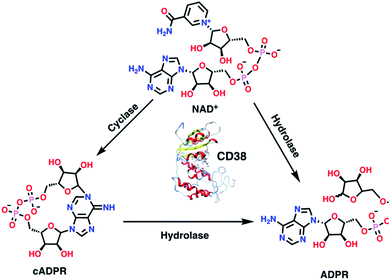 |
| | Fig. 1 Using two distinct catalytic reactions, CD38 produces ADP-ribose (ADPR) from both NAD+ and cADPR. | |
Role of CD38 in ADO generation
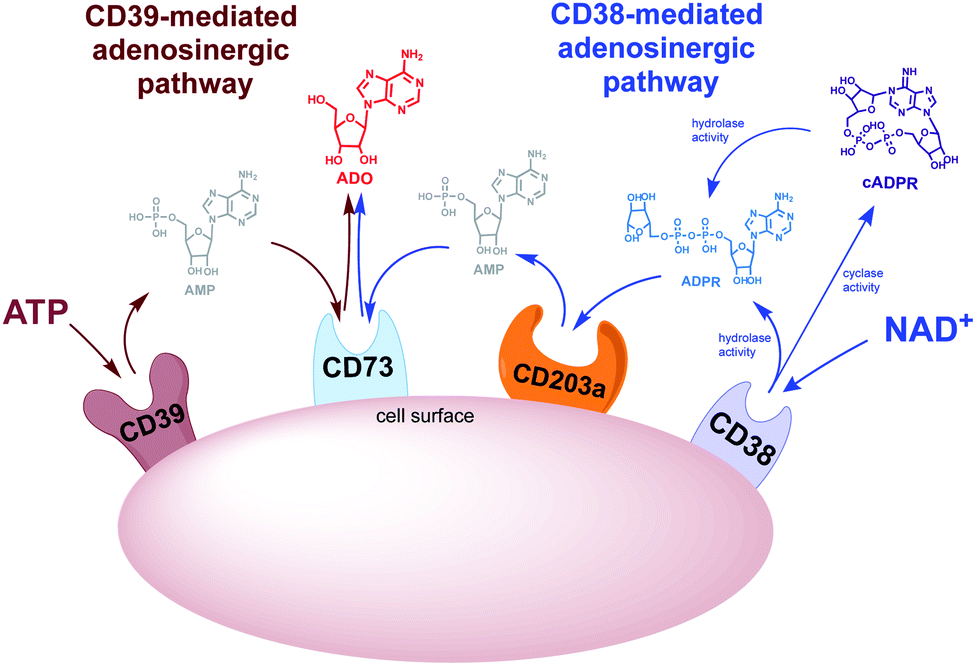
There are two adenosinergic pathways (Fig. 2) associated with exogenous adenosine (ADO) generation. The better-known pathway involves the nucleoside triphosphate diphosphohydrolase known as cluster of differentiation 39 (CD39). CD39 performs two sequential hydrolysis reactions: ADO triphosphate (ATP) is converted to ADO diphosphate (ADP) followed by the conversion of ADP to ADO monophosphate (AMP). Finally, the hydrolysis of AMP to ADO is mediated by the ectoenzyme cluster of differentiation 73 (CD73).25 While the CD39 pathway has been regarded as the predominant source of exogenous ADO, there is skepticism regarding the complete functionality of this pathway in vivo. The optimal pH for CD39-mediated hydrolysis of ATP and ADP is 8.0–8.5, suggesting that ADO production via CD38 ADO may predominate in the acidic TME.26–28 A lesser known adenosinergic pathway is mediated by CD38, which hydrolyzes NAD+ to ADPR. ADPR is in turn converted to ADO by the ectoenzymes cluster of differentiation 203a (CD203a) and CD73 (Fig. 2).29 Extracellular ADO, which is prominent in the TME, stimulates the ADO receptor, A2AR, on the surface of immune effector cells.30,31 Collectively these transformations cause a shift from an ATP-driven proinflammatory environment to an anti-inflammatory milieu induced by ADO-mediated down regulation of immune function.32,33
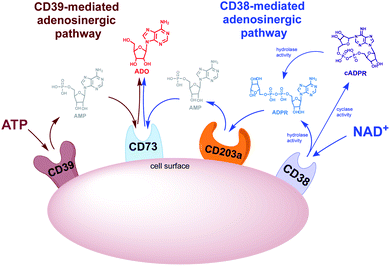 |
| | Fig. 2 CD38- and CD39-mediated adenosinergic pathways. | |
Results and discussion
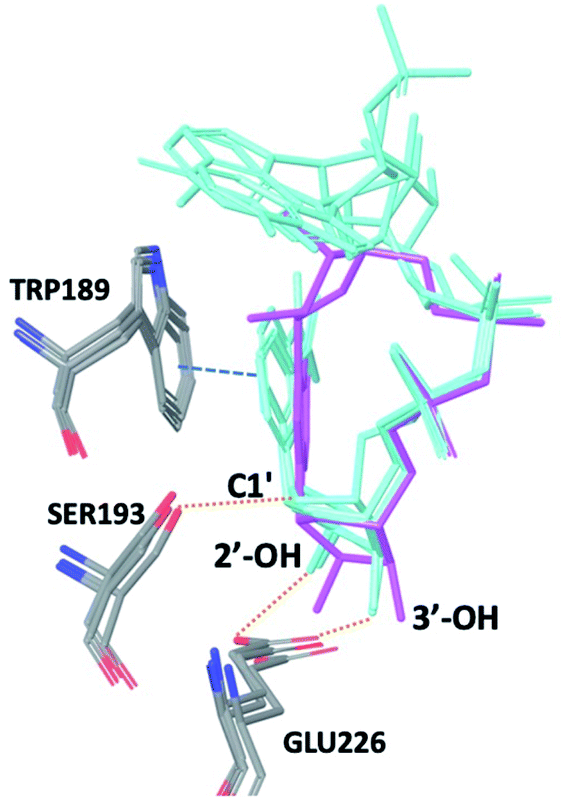
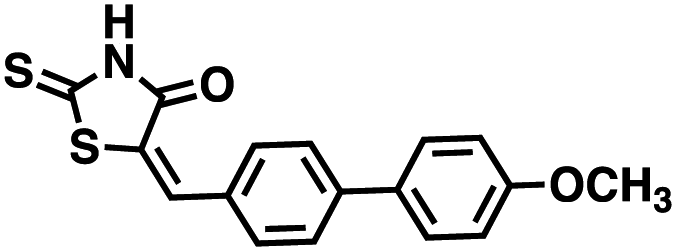
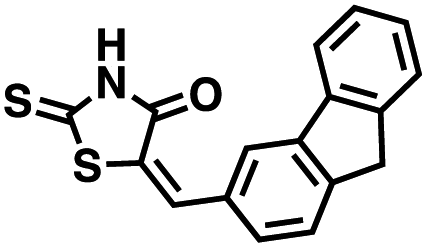
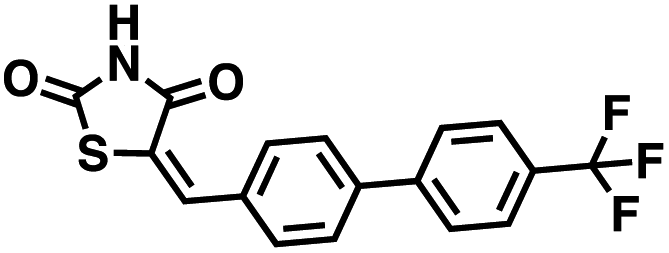
The observations above led us to initiate a screen for small molecule inhibitors of CD38 that are selective for either the cyclase or hydrolase activity of CD38 and to optimize their immunostimulatory activity through structural modification. In order to identify novel non-NAD+ mimetic scaffolds as CD38 inhibitors, we constructed a model consensus structure of CD38 via in silico analysis of published substrate/inhibitor co-crystals of CD38 (PDB 2I65, 2O3U, 4F45, 4XJT and 2O3T) using the Molecular Operating Environment software (Chemical Computing Group, Montreal, CA) (Fig. 3).34–38 After preparing each crystal structure at pH 7.4 and 310 K (see ESI for a complete description†), structures were superimposed to generate a consensus model of the CD38 active site. The model was then used to confirm the importance of GLU226 for catalysis via its ability to H-bond with 2′ and 3′ –OH of the nicotinamide-ribose. Furthermore, SER193 in the model structure of CD38 was observed proximal to the C1′ of the nicotinamide-ribose and has been implicated in the reaction mechanism for NAD+ degradation. The orientation of nicotinamide was mediated through aromatic interactions with TRP189. In both the original X-ray structure 4XJT and the generated model structure, phosphate groups were found stabilizing the complex and active site that were not involved with catalysis. However, despite ligand diversity across substrates and inhibitors, a consistent active site conformation was observed for all ligands screened virtually in the model structure of CD38. This geometric orientation of the active site and the cyclic nature of cADPR suggested the evaluation of heterocycles and macrocycles. Additionally, to mimic substrate interactions with TRP189, SER193, GLU146 and GLU226, nicotinamide and purine bio-isosteres were investigated. Most notably, an emphasis was placed on non-biological bioisosteres with structures not mimicking NAD+, since NAD+ analogues would have a greater potential for off-target effects at other NAD+ binding sites. A curated compound library was then constructed consisting of compounds meeting the above criteria from the commercial Cambridge and Enamine drug screening compound sets, as well as previously synthesized compounds in our laboratory. A total of 100 compounds were selected for the initial in silico screen that included hemi-peptidomimetic macrocycles, as well as mono-, bi-, and tricyclic heterocycles. We then used our consensus active site model to perform virtual screening of the curated library using standard docking experiments (Molecular Operating Environment, Montreal, CA). The 24 compounds with the most favorable docking scores were then selected and purchased for physical screening.
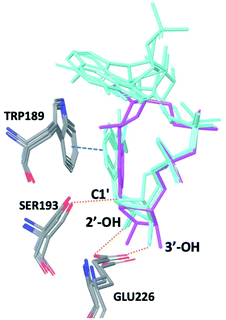 |
| | Fig. 3 Superimposition of PDB codes 2I65, 2O3S, 2O3U, and 4F45 displaying a consensus catalytic site and substrate orientation (NAD+, NGD, cADPr (magenta), and NAADP). The nicotinamide ribose retains a conserved binding pose with substrates: NAD+, NGD+, and NAADP displayed in cyan. Purine location is solvent exposed and tends to lack a consensus binding position. In each structure TRP189, SER193, and GLU226 are responsible for forming critical contracts and mediate catalysis of each substrate. For clarity only the heavy atoms are shown, dotted orange lines represent hydrogen bonds, blue dashed line represent π–π interactions. | |
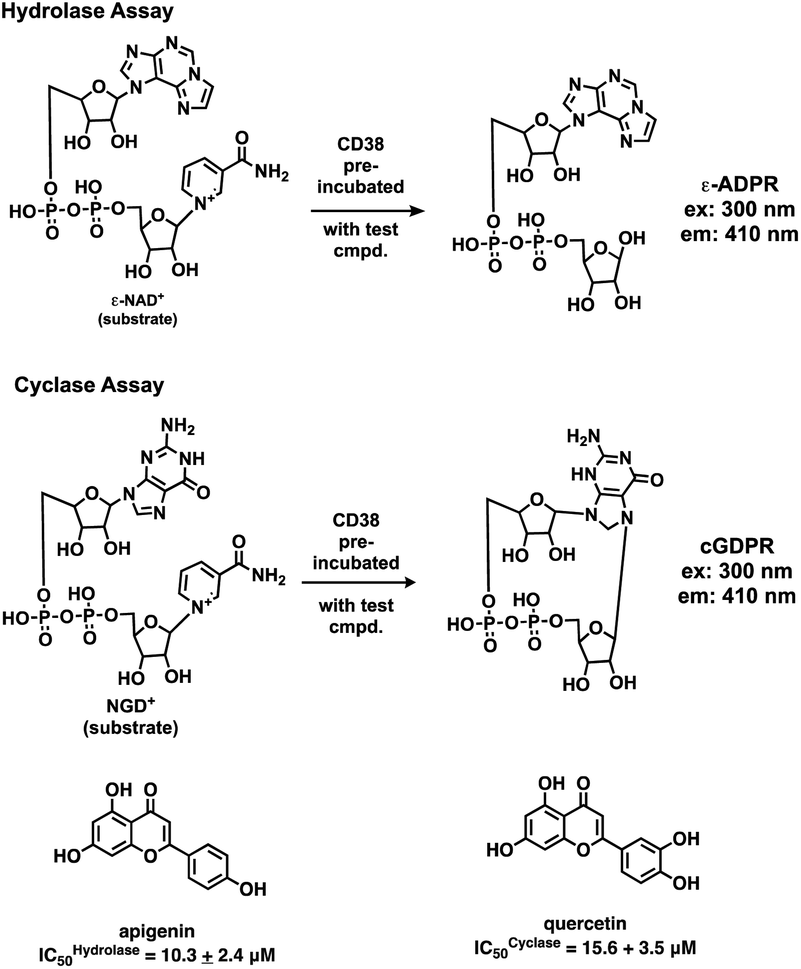
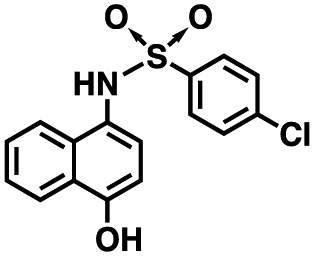
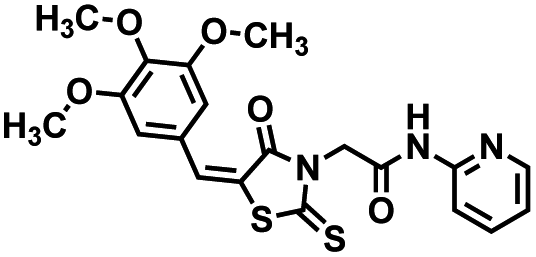
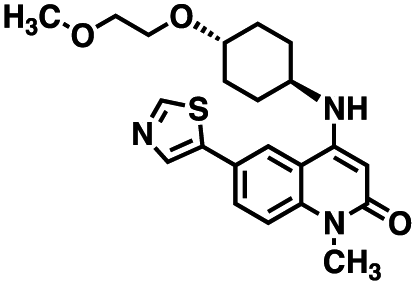
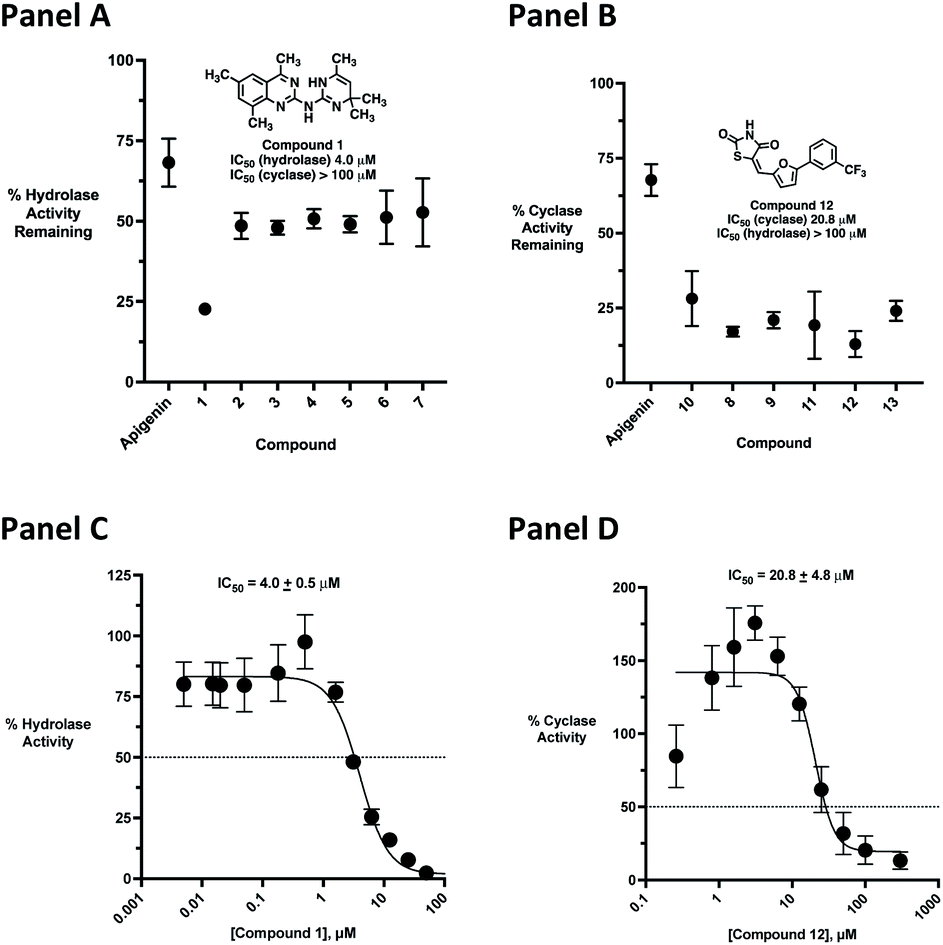
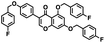
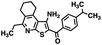
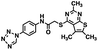
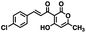
Initial screening was performed at a concentration of 50 μM of each compound in technical and experimental triplicates. Hydrolase or cyclase activities were monitored independently by using N6-ethenonicotinamide adenine dinucleotide (ε-NAD+) or nicotinamide guanine dinucleotide (NGD+), respectively, as substrate, and monitoring fluorescence at 410 nm (Fig. 4).39 The flavanoid apigenin, which has been shown to be an inhibitor of the hydrolase activity of CD38,7 and quercitin, which has been shown to be an inhibitor of the cyclase activity of CD38,7 were used as positive controls for the hydrolase and cyclase reaction, respectively. It should be noted that in our hands, neither compound showed significant selectivity for either activity, and both were significantly less potent than previously reported. Of the 24 selected compounds, 16 produced significant inhibition of CD38 (Table 1) and were then assayed for selectivity between the hydrolase and cyclase activities. Based on these data, compounds 1 (Fig. 5, panel A) and 12 (Fig. 5 panel B) were highly selective for their respective targets and were chosen for further study. Compound 1 exhibited an IC50 value of 4.0 μM against CD38 hydrolase (Fig. 5, panel C) and >100 μM against CD38 cyclase, while 12 had an IC50 of 20.8 μM against CD38 cyclase (Fig. 5 panel D) and >100 μM against CD38 hydrolase. Compound 12 was selected for structural optimization primarily due to its selectivity for the CD38 cyclase activity and because it was the most synthetically feasible for library construction. Other compounds (e.g. 13 and 14) will be elaborated in future studies.
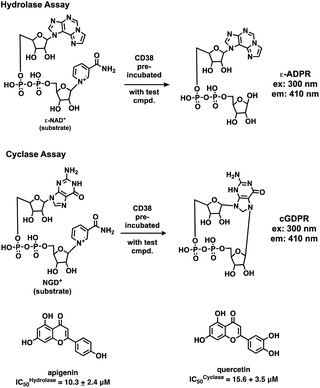 |
| | Fig. 4 Reactions comprising the CD38 hydrolase (top) and cyclase (bottom) assays, and structures of the standard inhibitors apigenin and quercitin. | |
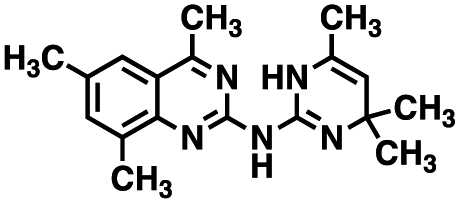
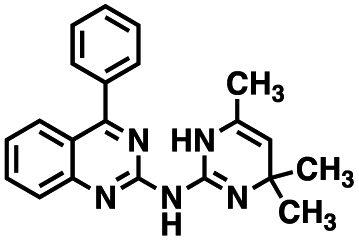
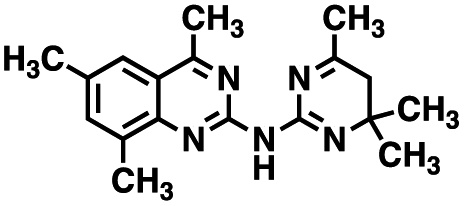
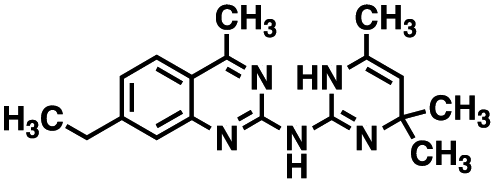
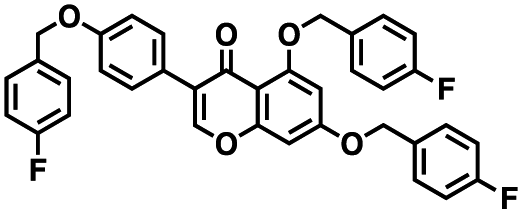
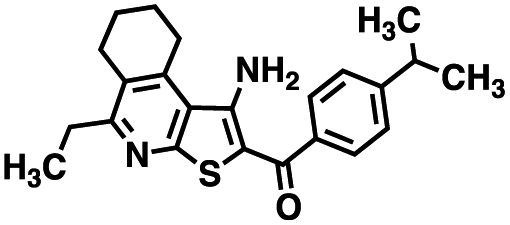
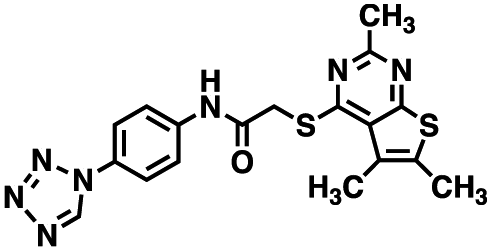
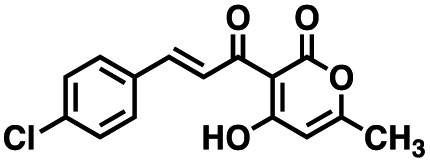
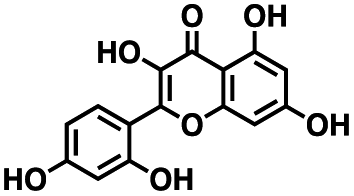
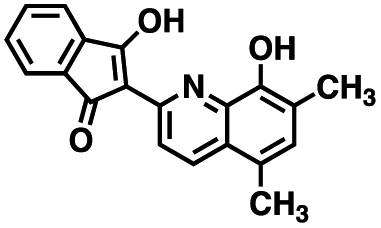
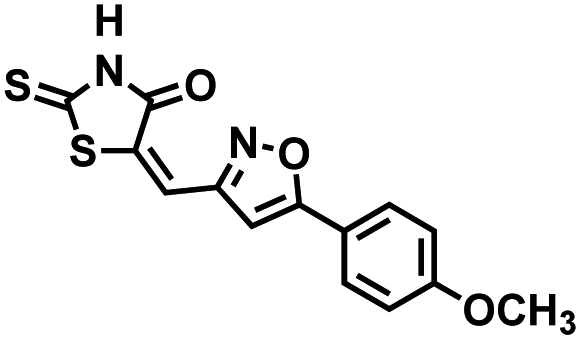
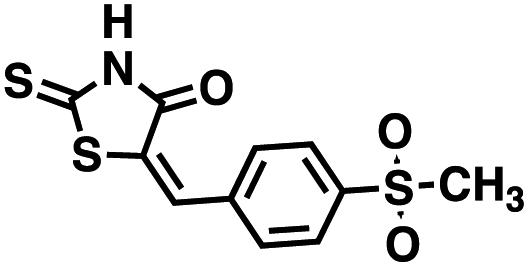
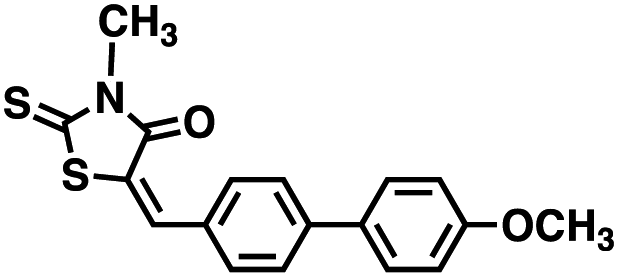
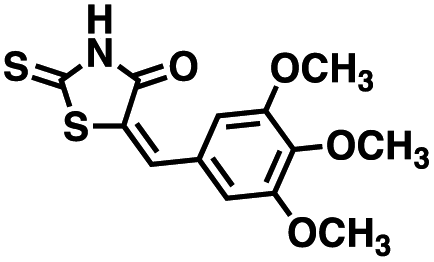
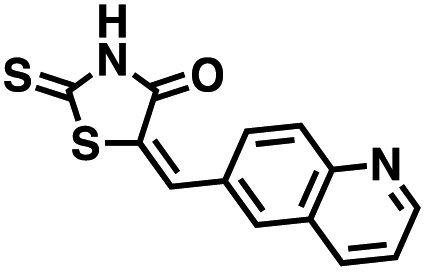
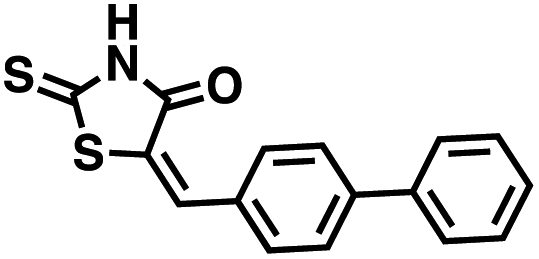
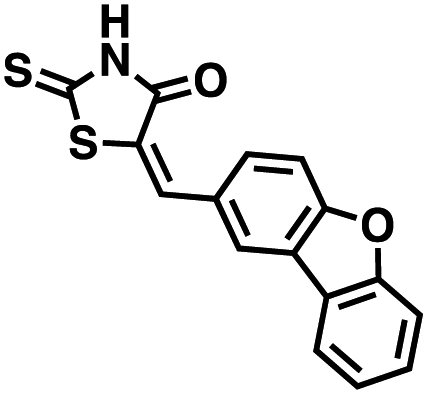
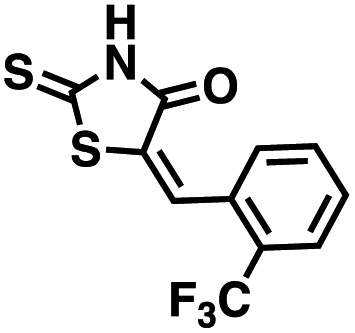
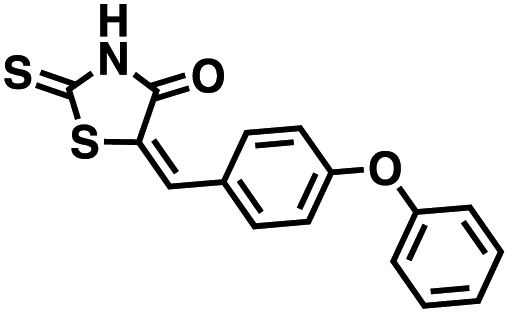
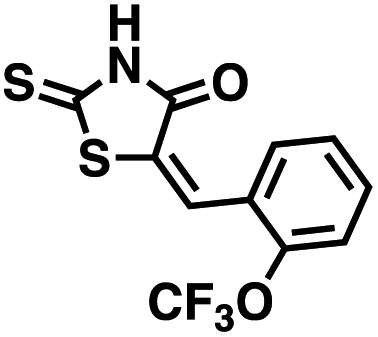
Table 1 Structures of CD38 inhibitors and their activity against CD38 hydrolase and cyclase. *ND = not determined
| Compound |
Structure |
MW |
% hydrolase activity remaining (50 μM) |
% cyclase activity remaining (50 μM) |
IC50 hydrolase (μM) |
IC50 cyclase (μM) |
| 1 |
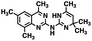 |
309.42 |
22.7 ± 0.40 |
60.9 ± 2.2 |
4.0 ± 0.48 |
>100 μM |
| 2 |
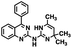 |
343.43 |
48.5 ± 2.3 |
57.0 ± 4.0 |
23.0 ± 1.0 |
ND* |
| 3 |
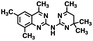 |
309.42 |
47.9 ± 1.2 |
52.1 ± 8.6 |
10.8 ± 0.7 |
ND* |
| 4 |
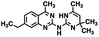 |
309.42 |
50.7 ± 1.7 |
48.0 ± 7.3 |
22.0 ± 1.0 |
ND* |
| 5 |
 |
594.59 |
49.0 ± 1.5 |
40.0 ± 1.2 |
ND* |
ND* |
| 6 |
 |
378.53 |
51.2 ± 4.8 |
28.1 ± 9.2 |
ND* |
36.3 ± 8.6 |
| 7 |
 |
411.50 |
52.7 ± 6.1 |
53.9 ± 49.7 |
ND* |
ND* |
| 8 |
 |
290.70 |
87.3 ± 0.3 |
17.1 ± 1.7 |
ND* |
15.0 ± 3.9 |
| 9 |
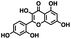 |
302.24 |
87.2 ± 3.4 |
21.0 ± 2.3 |
>100 μM |
35.4 ± 7.6 |
| 10 |
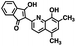 |
317.34 |
ND* |
35.0 ± 4.1 |
ND* |
5.2 ± 1.0 |
| 11 |
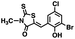 |
364.66 |
103 ± 2.2 |
19.3 ± 11.2 |
ND* |
Insoluble |
| 12 |
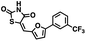 |
333.29 |
80.9 ± 4.4 |
12.9 ± 4.3 |
>100 μM |
20.8 ± 4.0 |
| 13 |
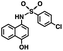 |
333.79 |
95.9 ± 1.4 |
24.0 ± 3.3 |
ND* |
37.5 ± 18.6 |
| 14 |
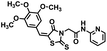 |
445.51 |
80.7 ± 3.7 |
−1.7 ± 10.3 |
ND* |
16.6 ± 0.2 |
| 15 |
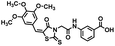 |
488.53 |
98.0 ± 3.9 |
38.4 ± 25 |
ND* |
17.1 ± 4.3 |
| 16 |
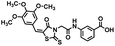 |
392.83 |
0.7 ± 0.4 |
23.1 ± 10.6 |
25.0 ± 3.0 |
23.4 ± 6.9 |
| 17 (50 nM) |
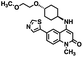 |
413.54 |
60.3 ± 1.8 |
95.7 ± 6.1 |
0.076 + 3.2 |
>1.0 μM |
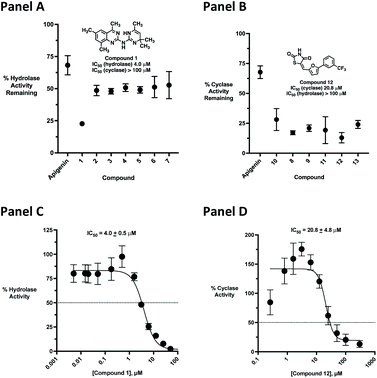 |
| | Fig. 5 Comparison of CD38 hydrolase- and cyclase-selective inhibitors at 50 μM. Panel A: top 7 hydrolase-selective inhibitors; panel B: top 7 cyclase-selective inhibitors; panel C: IC50 determination for compound 1 against CD38 hydrolase; panel D: IC50 determination for compound 12 against CD38 cyclase. All data points are the result of at least 3 determinations + SEM. | |
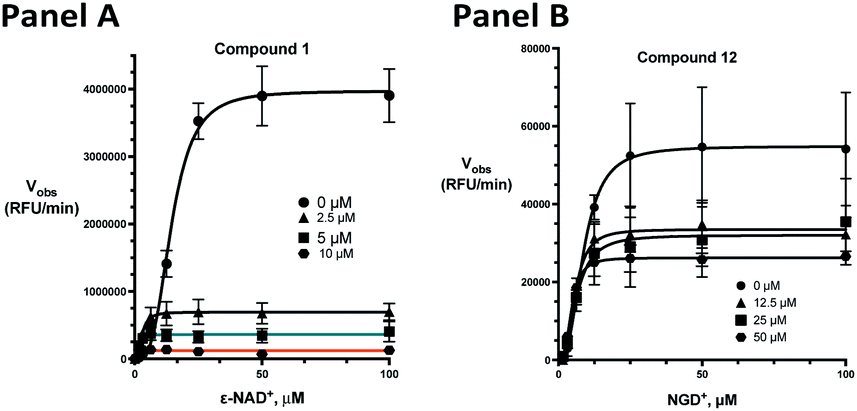
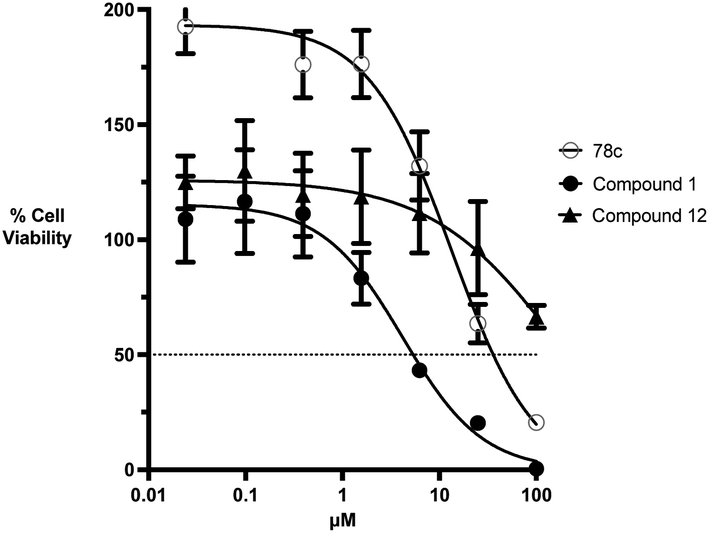
The kinetic mechanism of inhibition for the hydrolase and cyclase activities of CD38 were determined using a standard Michaelis–Menten approach, as shown in Fig. 6. The results suggest that both 1 and 12 produce a mixed/uncompetitive inhibition of the hydrolase and cyclase activities, respectively. In our hands, the known inhibitor 78c (17)8,9 is a potent inhibitor of the hydrolase (IC50 76 nM), but has very little activity against the cyclase. These data are consistent with the reported IC50 value for 17, which exhibited an IC50 of 7 nM against CD38 hydrolase via mixed kinetics.9 The difference in the IC50 value obtained in our laboratory is likely due to differences in assay conditions. The cytotoxicity of 1 was monitored in activated peripheral blood mononuclear cells (PBMCs) over a concentration range of 1 nM to 100 μM, as shown in Fig. 7. Compound 1 began to exhibit cytotoxic effects in PBMC cells at 1.0 μM, which is lower than its IC50 value. By contrast, compound 12 was relatively non-toxic up to 50 μM, which is 2.5-fold higher that its IC50 value.
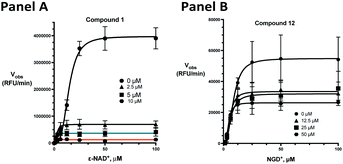 |
| | Fig. 6 Enzyme inhibition kinetics for the hydrolase and cyclase activities of CD38 by compounds 1 and 12. Panel A: inhibition of CD38 hydrolase by compound 1; panel B: inhibition of CD38 cyclase by compound 12. All data points are the result of 3 determinations ± SEM. | |
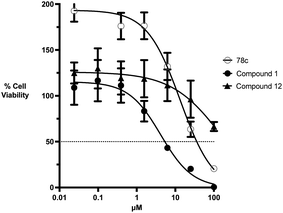 |
| | Fig. 7 Cytotoxicity of 78c, 1 and 12 in naive PBMC cells at concentrations between 0.01 and 100 μM. All data points are the average of 9 determinations (3 donors each run in triplicate in separate experiments) ± SEM. | |
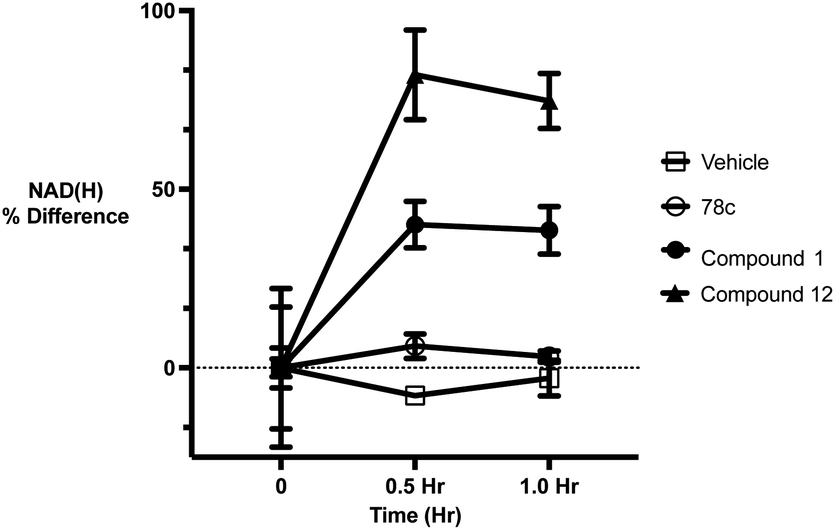
To further demonstrate the effect of 1 and 12 on inhibition of CD38 and to monitor the biological efficacy of our hit molecules, increases in NADH+ in activated PBMCs were monitored at 30 and 60 minutes after addition of the inhibitor. The known CD38 inhibitor 78c9 was used as a control. Compound 78c had a modest effect on NADH+ levels (Fig. 8), while 1 and 12 produced a 40.1 and 82.0% increase at 0.5 h, respectively. In both cases, elevated NADH+ levels remained relatively stable at 1 h.
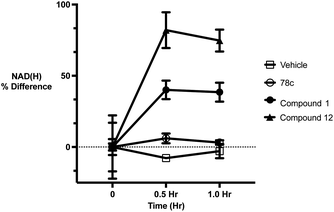 |
| | Fig. 8 Increases in cellular NADH+ levels in activated PBMCs following treatment at 1 μM concentrations of 78c, 1 and 12. All data points are the average of 3 determinations ± SEM. | |
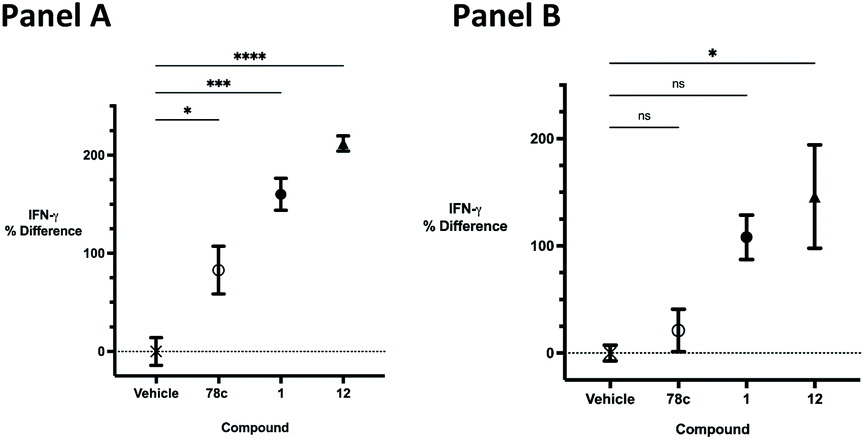
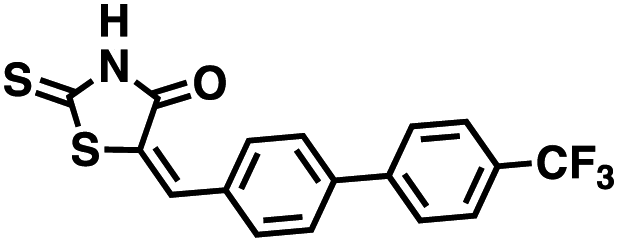
The effects of inhibitors 78c, 1 and 12 on IFNγ levels was next examined after 48 hours and 12 days, as shown in Fig. 9. PBMCs were incubated with the appropriate inhibitor at 10 μM for the indicated time, and IFNγ levels were measured by ELISA. At 48 hours, compounds 1 and 12 produced 160.1 and 212.0% increases in IFNγ, respectively, compared to an 82.9% increase in IFNγ following treatment with 78c. At 12 days, IFNγ levels were 21.1, 108.1 and 145.9% for 78c, 1 and 12, respectively, slightly lower than IFNγ levels observed after 48 hour incubation.
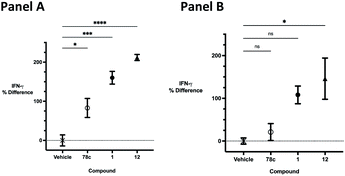 |
| | Fig. 9 Effect of 78c, 1 and 12 at 10 μM on IFNγ levels. Panel A: 48 hours; panel B: 12 days. | |
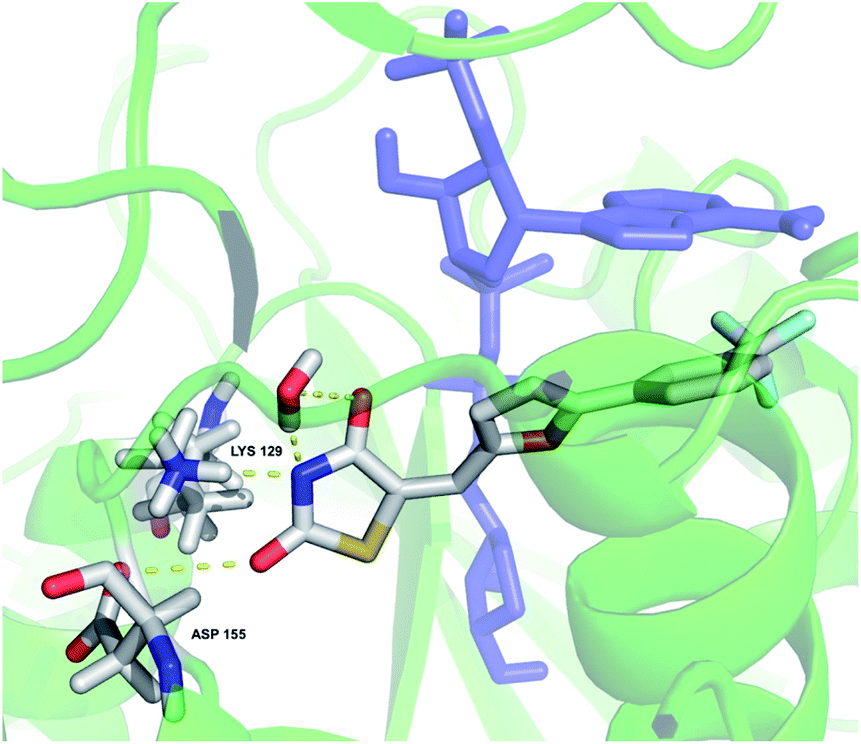
The binding mode of 12 in the CD38 active site was simulated starting with X-ray structure 4XJT, as shown in Fig. 10. The bound inhibitor 4-[(2,6-dimethylbenzyl)amino]-2-methylquinoline-8-carboxamide8 and the covalent bond between ADPR and CD38 were removed, and compound 12 was docked in the active site as described above. In this model, the 2-thioxothiazolidin-4-one ring nitrogen appears to form hydrogen bonds with LYS 129 and ASP 155. In addition, the thiazolidine ring nitrogen and an adjacent carbonyl coordinate a water molecule in the active site. In this orientation, there appears to be a pi stacking arrangement between 12 and ADPR that stabilizes binding. This arrangement is consistent with the observed uncompetitive kinetics of inhibition since optimal binding depends on the presence of the substrate NAD+. As the structure–activity relationships for binding of 12 and its analogues are further elucidated, the in silico model will be refined accordingly.
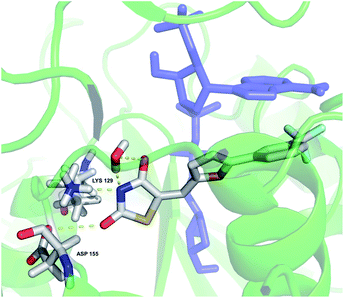 |
| | Fig. 10 In silico representation of compound 12 bound to the CD38 active site. Amino acids involved in binding and 12 are colored by atom type; the all-blue structure is NAD+. | |
Library generation
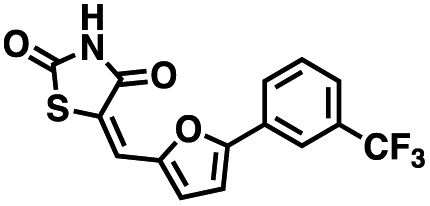
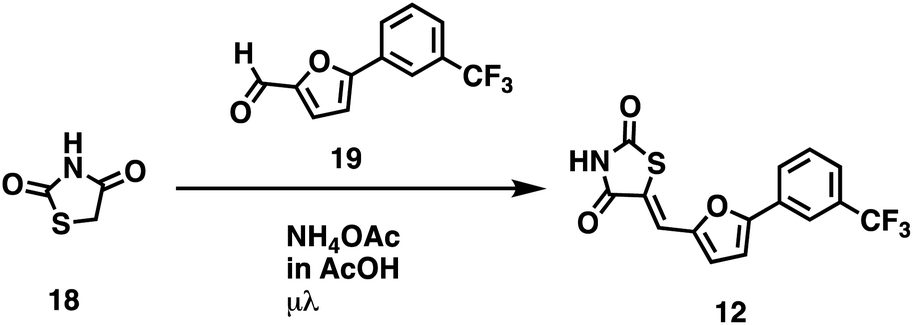
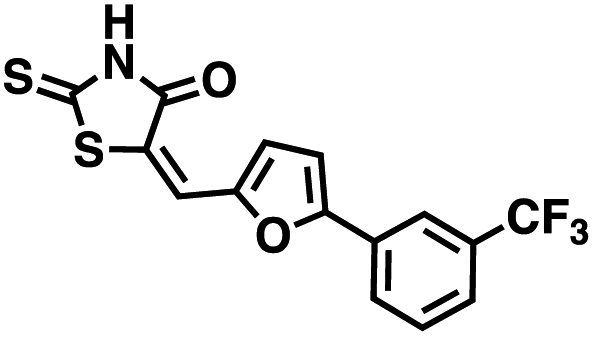
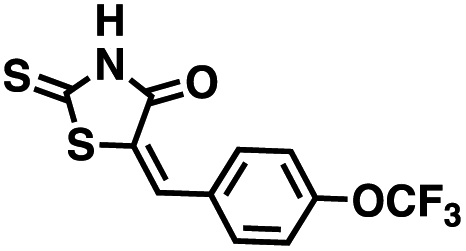
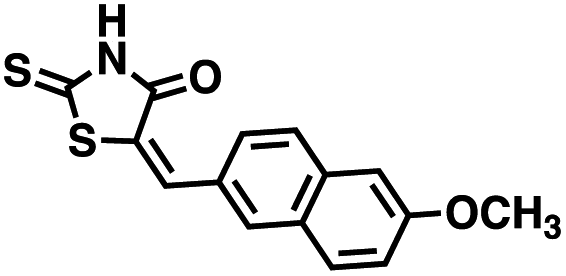
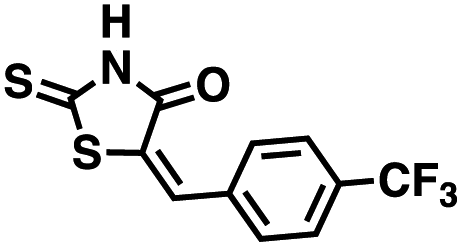
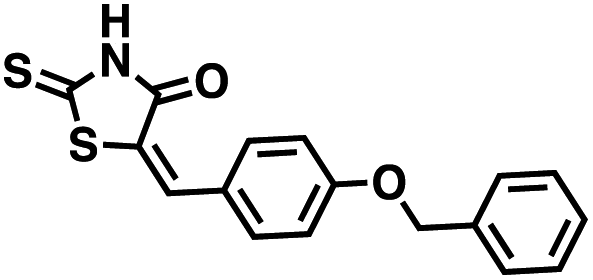
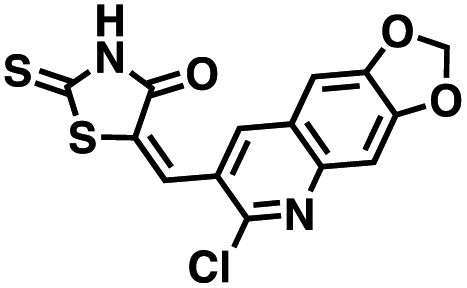
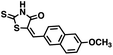
We opted to produce a library of analogues of 12 due to their relative ease of synthesis compared to compound 1. Compound 12 could be synthesized in a single microwave step using a modified Knoevenagel condensation reaction between 2-thioxothiazolidin-4-one 18 and 5-(3-(trifluoromethyl)phenyl)furan-2-carbaldehyde 19 (Scheme 1) predominantly as the Z-isomer.40–42 This synthetic approach was then employed to introduce chemical diversity during structural optimization, resulting in compounds 20–46 (Table 2).
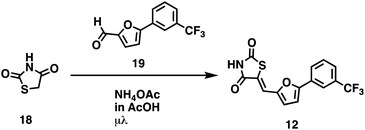 |
| | Scheme 1 | |
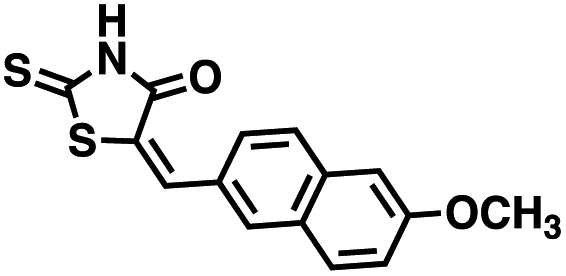
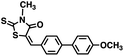
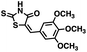
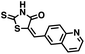
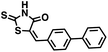
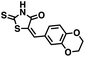
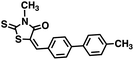
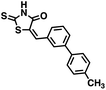
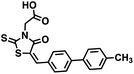
Table 2 Structures of CD38 inhibitors and their activity against CD38 cyclase
| Cmpd |
Structure |
MW |
% cyclase activity remaining (50 μM) |
IC50 cyclase (μM) |
| 20 |
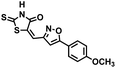 |
318.37 |
17.6 ± 2.1 |
3.71 |
| 21 |
 |
299.38 |
22.1 ± 4.0 |
11.53 |
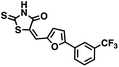
| 22 |
 |
309.25 |
21.7 ± 8.1 |
4.81 |
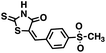
| 23 |
 |
289.29 |
9.3 ± 0.8 |
4.65 |
| 24 |
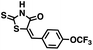 |
341.44 |
90.1 ± 3.9 |
>100 |
| 25 |
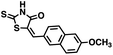 |
272.34 |
68.3 ± 18.2 |
56.0 |
| 26 |
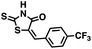 |
311.37 |
77.3 ± 22.4 |
>100 |
| 27 |
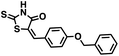 |
313.39 |
88.2 ± 0.6 |
>100 |
| 28 |
 |
279.33 |
93.9 ± 6.9 |
>100 |
| 29 |
 |
311.43 |
98.6 ± 4.7 |
>100 |
| 30 |
 |
350.79 |
70.3 ± 40.7 |
20.9 |
| 31 |
 |
309.40 |
96.3 ± 6.8 |
>100 |
| 32 |
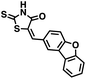 |
365.39 |
64.5 ± 4.2 |
3.15 |
| 33 |
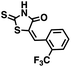 |
311.42 |
39.4 ± 4.9 |
10.0 |
| 34 |
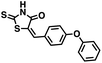 |
298.38 |
66.3 ± 23.4 |
56.0 |
| 35 |
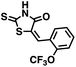 |
355.35 |
81.7 ± 9.0 |
>100 |
| 36 |
 |
301.38 |
90.0 ± 5.4 |
>100 |
| 37 |
 |
295.36 |
84.5 ± 2.2 |
>100 |
| 38 |
 |
327.42 |
84.5 ± 8.5 |
>100 |
| 39 |
 |
311.37 |
64.1 ± 7.2 |
68.6 |
| 40 |
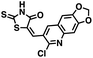 |
297.39 |
22.1 ± 14.3 |
43. 2 |
| 41 |
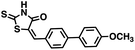 |
289.29 |
81.6 ± 2.4 |
>100 |
| 42 |
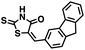 |
305.29 |
87.1 ± 3.4 |
>100 |
| 43 |
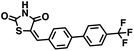 |
325.44 |
29.0 ± 18.8 |
48.2 |
| 44 |
 |
369.45 |
39.1 ± 2.7 |
35.0 |
| 45 |
 |
327.42 |
55.7 ± 2.3 |
53.8 |
| 46 |
 |
349.33 |
49.4 ± 27.1 |
42.7 |
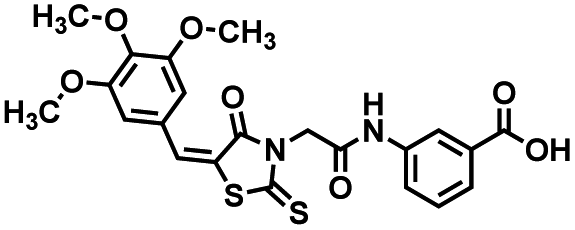
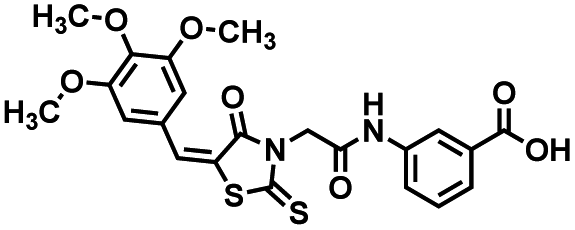
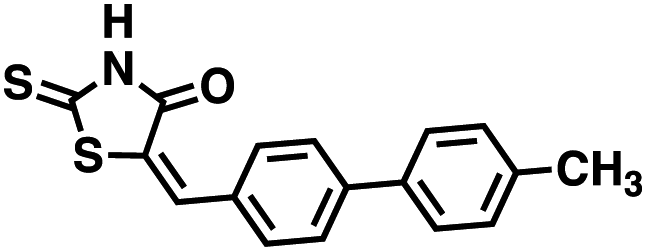
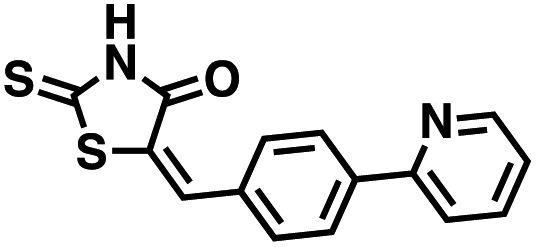
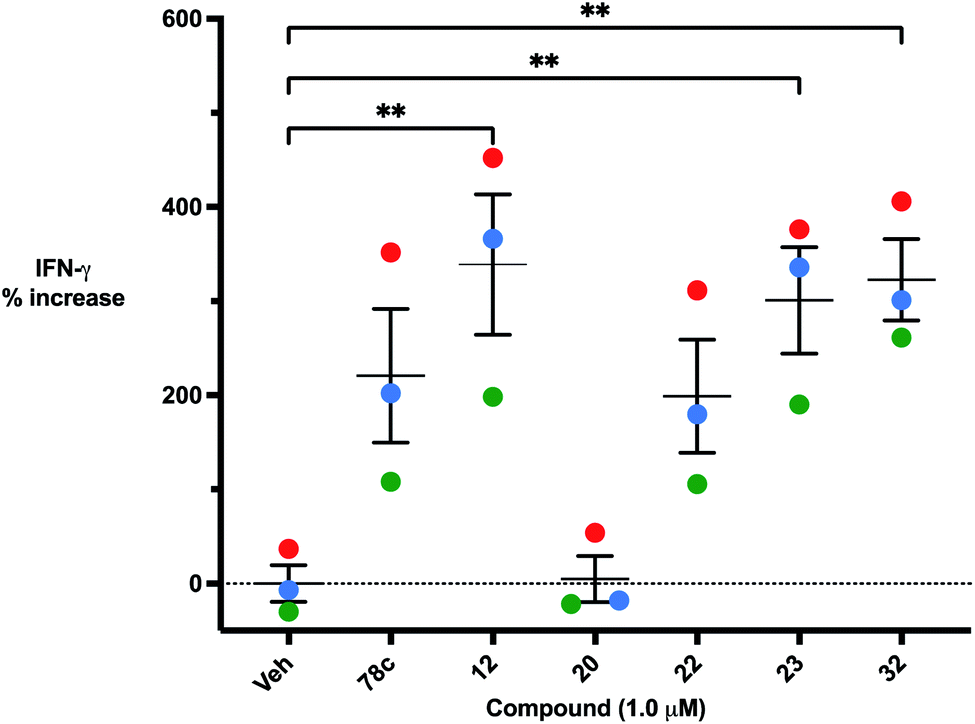
Four of the compounds shown in Table 2 exhibited a lower IC50 against CD38 cyclase activity than 12 (compounds 20, 22, 23 and 32). The ability of these compounds to increase IFNγ content in activated PBMCs from 3 separate donors was determined at 48 hours, as described above, and the results of these studies appear in Fig. 11. Compounds 12, 23 and 32 were more effective than 78c at increasing IFNγ, and the percent increase caused by compounds 22, 23 and 32 was inversely proportional to their observed IC50 values. Among compounds 12, 20, 22, 23 and 32, compound 12 exhibited the greatest effect on IFNγ levels at 1.0 μM, despite having the highest IC50. Because CD38 carries out its enzyme function both inside and outside the cell,43 this finding may be due to an enhanced ability of 12 to penetrate into PBMCs.
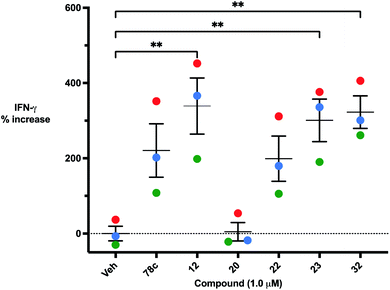 |
| | Fig. 11 48 hour increase in IFNγ in the presence of 1.0 μM compounds 78c, 12, 20, 22, 23 and 24 in activated PBMCs from three separate donors. Each data point is the average of 3 determinations (red = donor 1, blue = donor 2, green = donor 3). These averages were then replotted as % increase in IFNγ ± SEM. | |
Conclusions
A number of small molecule inhibitors of CD38 have been reported, but to date none have been evaluated for stimulatory effects in immune cells. We have identified a series of CD38 inhibitors based on a Z-5-ethylidinethiazolidine-2,4-dione scaffold that are potent inhibitors of CD38, and that in some cases show a marked selectivity for inhibition of cyclase activity. The most potent of these compounds, 32, exhibits an IC50 value of 3.15 μM in the CD38 cyclase assay. We have further shown that these compounds inhibit CD38 by a mixed/uncompetitive mechanism. The parent molecule in this series, 12, produced an 82% increase in NAD+ content in naive PBMCs after 30 minutes, and a 212% increase in IFNγ levels after 48 hours that persisted for 12 days. Importantly, 12 was significantly more potent than the known CD38 inhibitor 78c with regard to promoting increases in IFNγ. Compound 78c was evaluated as an inhibitor of CD38 hydrolase, but not specifically as an inhibitor of CD38 cyclase. Since 12 and its analogues are cyclase-selective, our data suggests that the cyclase plays a greater role in activation of T-cell function than was previously thought.
With regard to structure–activity relationships (SAR), we have not made a sufficient number of analogues to make any meaningful statements concerning 12 and its analogues. However, the most active analogues (12, 20, 22, 23, 30, 32) generally possess large phenyl or biphenyl substituents that are in conjugation with the Z-double bond moiety, and except for 20 are substituted with a halogen-containing substituent, indicating that binding to the cyclase catalytic site depends on π-interactions with nearby residues on the protein. We will refine our SAR model as additional active inhibitors become available. In addition, because these compounds have IC50 values in the micromolar range, the possibility of off-target effects exists. This issue will also be addressed in subsequent studies.
In this manuscript, we have described a number of cyclase-selective inhibitors of CD38 that will be of use in determining the role that the hydrolase and cyclase activities of CD38 play in metabolism, activation of immune cells and in the development of resistance to immune checkpoint inhibitors. It is not currently known whether the CD38 hydrolase or the CD38 cyclase activity makes the greatest contribution to down regulation of the immune response, or which activity contributes to the development of resistance to CD38 immune checkpoint inhibitors. Our data suggests that the CD38 cyclase plays a significant role on down regulation of the immune response and the development of resistance. We are now optimizing CD38 inhibitors that are structurally related to 1 to identify an inhibitor for use as a chemical probe for the hydrolase activity of CD38. With selective hydrolase and cyclase inhibitors as chemical tools, we will be able to determine to what extent selective inhibition of the hydrolase and cyclase activities of CD38 contribute to the activation of immune cells, and which activity plays the greatest role in development of resistance.
The critical role of CD38, which is expressed on B, NK and T-cells as well as macrophages, has been well studied as a target for cancer immunotherapy.44–46 As mentioned above, CD38 metabolism has been shown to be a major factor in the development of resistance to MABs.15–17,47 As such, the CD38 inhibitors described herein may be of great value as adjuncts to existing therapy with checkpoint inhibitors such as daratumumab and isatuximab. However, CD38 inhibition could also be exploited in the treatment of other diseases resulting from depletion of NAD+ and/or over production of ADO. Decreases in NAD+ levels are associated with various metabolic diseases (diabetes, obesity, dyslipidemia and nonalcoholic fatty liver)48,49 and in aging.50–52 It also plays a major role in the immune response to infectious disease53 including COVID-19.54 ADO is a potent immunosuppressant, and plays a similar role in the development and progression of disease.54 Taken together, these observations indicate an increasing medical need for small molecule inhibitors of CD38 for use in diseases where dysregulated NAD+ and ADO metabolism are a factor. For this reason, the optimization of the compounds described in this paper, as well as the discovery of additional scaffolds for CD38 inhibition, is an ongoing concern in our laboratories.
Author contributions
Thomas Z. Benton, conceptualization, data curation, formal analysis, methodology, writing - original draft; Catherine Mills, conceptualization, data curation, formal analysis, methodology, writing – original draft; Jonathan Turner, investigation, validation; Megan J. Francis, investigation; Dalan Soloman, investigation; Pieter B. Burger, formal analysis, methodology, software, visualization; Yuri Peterson, formal analysis, methodology, software, visualization; Nathan Dolloff, formal analysis, methodology; Andre Bachmann, formal analysis, methodology, review and editing; Patrick M. Woster, conceptualization, data curation, formal analysis, funding acquisition, project administration, resources, supervision, writing – original draft, review and editing.
Conflicts of interest
None of the authors of this manuscript have any conflicts to declare.
Acknowledgements
Completion of the experiments delineated in this manuscript were supported in part through participation of the MUSC Drug Discovery Core and the MUSC Nuclear Magnetic Resonance Core.
Notes and references
- H. C. Lee, Mol. Med., 2006, 12, 317–323 CAS.
- L. Fagerberg, B. M. Hallstrom, P. Oksvold, C. Kampf, D. Djureinovic, J. Odeberg, M. Habuka, S. Tahmasebpoor, A. Danielsson, K. Edlund, A. Asplund, E. Sjostedt, E. Lundberg, C. A. Szigyarto, M. Skogs, J. O. Takanen, H. Berling, H. Tegel, J. Mulder, P. Nilsson, J. M. Schwenk, C. Lindskog, F. Danielsson, A. Mardinoglu, A. Sivertsson, K. von Feilitzen, M. Forsberg, M. Zwahlen, I. Olsson, S. Navani, M. Huss, J. Nielsen, F. Ponten and M. Uhlen, Mol. Cell. Proteomics, 2014, 13, 397–406 CrossRef CAS.
- G. Shubinsky and M. Schlesinger, Immunity, 1997, 7, 315–324 CrossRef CAS.
- E. N. Chini, C. C. S. Chini, J. M. Espindola Netto, G. C. de Oliveira and W. van Schooten, Trends Pharmacol. Sci., 2018, 39, 424–436 CrossRef CAS.
- E. N. Chini, Curr. Pharm. Des., 2009, 15, 57–63 CrossRef CAS.
- M. Dong, Y. Q. Si, S. Y. Sun, X. P. Pu, Z. J. Yang, L. R. Zhang, L. H. Zhang, F. P. Leung, C. M. Lam, A. K. Kwong, J. Yue, Y. Zhou, I. A. Kriksunov, Q. Hao and H. C. Lee, Org. Biomol. Chem., 2011, 9, 3246–3257 RSC.
- C. Escande, V. Nin, N. L. Price, V. Capellini, A. P. Gomes, M. T. Barbosa, L. O'Neil, T. A. White, D. A. Sinclair and E. N. Chini, Diabetes, 2013, 62, 1084–1093 CrossRef CAS.
- J. D. Becherer, E. E. Boros, T. Y. Carpenter, D. J. Cowan, D. N. Deaton, C. D. Haffner, M. R. Jeune, I. W. Kaldor, J. C. Poole, F. Preugschat, T. R. Rheault, C. A. Schulte, B. G. Shearer, T. W. Shearer, L. M. Shewchuk, T. L. Smalley Jr, E. L. Stewart, J. D. Stuart and J. C. Ulrich, J. Med. Chem., 2015, 58, 7021–7056 CrossRef CAS.
- C. D. Haffner, J. D. Becherer, E. E. Boros, R. Cadilla, T. Carpenter, D. Cowan, D. N. Deaton, Y. Guo, W. Harrington, B. R. Henke, M. R. Jeune, I. Kaldor, N. Milliken, K. G. Petrov, F. Preugschat, C. Schulte, B. G. Shearer, T. Shearer, T. L. Smalley Jr, E. L. Stewart, J. D. Stuart and J. C. Ulrich, J. Med. Chem., 2015, 58, 3548–3571 CrossRef CAS.
- M. B. Overdijk, J. H. Jansen, M. Nederend, J. J. Lammerts van Bueren, R. W. Groen, P. W. Parren, J. H. Leusen and P. Boross, J. Immunol., 2016, 197, 807–813 CrossRef CAS PubMed.
- N. van de Donk, P. G. Richardson and F. Malavasi, Blood, 2018, 131, 13–29 CrossRef CAS PubMed.
- N. W. van de Donk, M. L. Janmaat, T. Mutis, J. J. Lammerts van Bueren, T. Ahmadi, A. K. Sasser, H. M. Lokhorst and P. W. Parren, Immunol. Rev., 2016, 270, 95–112 CrossRef CAS PubMed.
- J. Krejcik, T. Casneuf, I. S. Nijhof, B. Verbist, J. Bald, T. Plesner, K. Syed, K. Liu, N. W. van de Donk, B. M. Weiss, T. Ahmadi, H. M. Lokhorst, T. Mutis and A. K. Sasser, Blood, 2016, 128, 384–394 CrossRef CAS.
- N. van de Donk, Immunol. Lett., 2018, 199, 16–22 CrossRef CAS.
- L. Chen, L. Diao, Y. Yang, X. Yi, B. L. Rodriguez, Y. Li, P. A. Villalobos, T. Cascone, X. Liu, L. Tan, P. L. Lorenzi, A. Huang, Q. Zhao, D. Peng, J. J. Fradette, D. H. Peng, C. Ungewiss, J. Roybal, P. Tong, J. Oba, F. Skoulidis, W. Peng, B. W. Carter, C. M. Gay, Y. Fan, C. A. Class, J. Zhu, J. Rodriguez-Canales, M. Kawakami, L. A. Byers, S. E. Woodman, V. A. Papadimitrakopoulou, E. Dmitrovsky, J. Wang, S. E. Ullrich, Wistuba II, J. V. Heymach, F. X. Qin and D. L. Gibbons, Cancer Discov., 2018, 8, 1156–1175 CrossRef CAS PubMed.
- S. Koyama, E. A. Akbay, Y. Y. Li, G. S. Herter-Sprie, K. A. Buczkowski, W. G. Richards, L. Gandhi, A. J. Redig, S. J. Rodig, H. Asahina, R. E. Jones, M. M. Kulkarni, M. Kuraguchi, S. Palakurthi, P. E. Fecci, B. E. Johnson, P. A. Janne, J. A. Engelman, S. P. Gangadharan, D. B. Costa, G. J. Freeman, R. Bueno, F. S. Hodi, G. Dranoff, K. K. Wong and P. S. Hammerman, Nat. Commun., 2016, 7, 10501 CrossRef CAS PubMed.
- P. C. Tumeh, C. L. Harview, J. H. Yearley, I. P. Shintaku, E. J. Taylor, L. Robert, B. Chmielowski, M. Spasic, G. Henry, V. Ciobanu, A. N. West, M. Carmona, C. Kivork, E. Seja, G. Cherry, A. J. Gutierrez, T. R. Grogan, C. Mateus, G. Tomasic, J. A. Glaspy, R. O. Emerson, H. Robins, R. H. Pierce, D. A. Elashoff, C. Robert and A. Ribas, Nature, 2014, 515, 568–571 CrossRef CAS PubMed.
- S. Goh, H. H. M. Ng, V. Chew, X. N. Sim, H. Li, S. Lim, J. C. T. Lim, J. J. H. Loh, K. Sabai, C. C. H. Ong, T. Loh, W. Q. Leow, J. L. J. Xin, H. C. Toh, F. Malavasi, D. W. M. Tai, S. Y. Lee, P. Chow, E. Newell, S. P. Choo, J. Yeong and T. K. H. Lim, bioRxiv, 2019, 638981, DOI:10.1101/638981.
- A. Passarelli, M. Tucci, F. Mannavola, C. Felici and F. Silvestris, Tumour Biol., 2019, 42(4), 1–10 Search PubMed.
- B. E. Kennedy, M. Sadek, M.
O. Elnenaei, A. Reiman and S. A. Gujar, Trends Cancer, 2020, 6, 9–12 CrossRef CAS.
- V. Quarona, G. Zaccarello, A. Chillemi, E. Brunetti, V. K. Singh, E. Ferrero, A. Funaro, A. L. Horenstein and F. Malavasi, Cytometry, Part B, 2013, 84, 207–217 CrossRef.
- P. F. Egea, H. Muller-Steffner, I. Kuhn, C. Cakir-Kiefer, N. J. Oppenheimer, R. M. Stroud, E. Kellenberger and F. Schuber, PLoS One, 2012, 7, e34918 CrossRef CAS PubMed.
- S. Chatterjee, A. Daenthanasanmak, P. Chakraborty, M. W. Wyatt, P. Dhar, S. P. Selvam, J. Fu, J. Zhang, H. Nguyen, I. Kang, K. Toth, M. Al-Homrani, M. Husain, G. Beeson, L. Ball, K. Helke, S. Husain, E. Garrett-Mayer, G. Hardiman, M. Mehrotra, M. I. Nishimura, C. C. Beeson, M. G. Bupp, J. Wu, B. Ogretmen, C. M. Paulos, J. Rathmell, X. Z. Yu and S. Mehrotra, Cell Metab., 2018, 27, 85–100 CrossRef CAS PubMed.
- M. R. Fernandez and J. L. Cleveland, Cell Metab., 2018, 27, 3–5 CrossRef CAS.
- G. G. Yegutkin, T. Henttinen, S. S. Samburski, J. Spychala and S. Jalkanen, Biochem. J., 2002, 367, 121–128 CrossRef CAS PubMed.
- D. B. Leal, C. A. Streher, T. N. Neu, F. P. Bittencourt, C. A. Leal, J. E. da Silva, V. M. Morsch and M. R. Schetinger, Biochim. Biophys. Acta, 2005, 1721, 9–15 CrossRef CAS PubMed.
- M. Milosevic, S. Petrovic, N. Velickovic, I. Grkovic, M. Ignjatovic and A. Horvat, Mol. Cell. Biochem., 2012, 371, 199–208 CrossRef CAS.
- E. L. Gordon, J. D. Pearson and L. L. Slakey, J. Biol. Chem., 1986, 261, 15496–15507 CrossRef CAS.
- A. L. Horenstein, A. Chillemi, G. Zaccarello, S. Bruzzone, V. Quarona, A. Zito, S. Serra and F. Malavasi, Oncoimmunology, 2013, 2, e26246 CrossRef PubMed.
- J. Blay, T. D. White and D. W. Hoskin, Cancer Res., 1997, 57, 2602–2605 CAS.
- Tumor-induced immune suppression: mechanisms and therapeutic reversal, ed. D. I. Gabrilovich and A. A. Hurwitz, Springer, 2nd edn, 2014, pp. 411–434 Search PubMed.
- L. Antonioli, P. Pacher, E. S. Vizi and G. Hasko, Trends Mol. Med., 2013, 19, 355–367 CrossRef CAS PubMed.
- A. L. Horenstein, C. Bracci, F. Morandi and F. Malavasi, Front. Immunol., 2019, 10, 760 CrossRef CAS.
- R. Graeff, Q. Liu, I. A. Kriksunov, Q. Hao and H. C. Lee, J. Biol. Chem., 2006, 281, 28951–28957 CrossRef CAS.
- R. Graeff, Q. Liu, I. A. Kriksunov, M. Kotaka, N. Oppenheimer, Q. Hao and H. C. Lee, J. Biol. Chem., 2009, 284, 27629–27636 CrossRef CAS.
- Q. Liu, I. A. Kriksunov, R. Graeff, H. C. Lee and Q. Hao, J. Biol. Chem., 2007, 282, 5853–5861 CrossRef CAS.
- Q. Liu, I. A. Kriksunov, R. Graeff, C. Munshi, H. C. Lee and Q. Hao, Structure, 2005, 13, 1331–1339 CrossRef CAS PubMed.
- Q. Liu, I. A. Kriksunov, R. Graeff, C. Munshi, H. C. Lee and Q. Hao, J. Biol. Chem., 2006, 281, 32861–32869 CrossRef CAS.
- J. Camacho-Perreira, M. G. Tarrago, C. C. S. Chini, C. Escande, G. M. Warner, A. S. Puranik, R. A. Schoon, J. M. Reid, A. Galina and E. N. Chini, Cell Metab., 2016, 23, 1127–1139 CrossRef.
- K. A. Kandeel, Chem. Pap., 2004, 58, 334–340 CAS.
- A. J. Russell, I. M. Westwood, M. H. Crawford, J. Robinson, A. Kawamura, C. Redfield, N. Laurieri, E. D. Lowe, S. G. Davies and E. Sim, Bioorg. Med. Chem., 2009, 17, 905–918 CrossRef CAS PubMed.
- Y. Song, D. T. Connor, R. Doubleday, R. J. Sorenson, A. D. Sercel, P. C. Unangst, B. D. Roth, R. B. Gilbertsen, K. Chan, D. J. Schrier, A. Guglietta, D. A. Bornemeier and R. D. Dyer, J. Med. Chem., 1999, 42, 1151–1160 CrossRef CAS PubMed.
- H. C. Lee and Y. J. Zhao, J. Biol. Chem., 2019, 294, 19831–19843 CrossRef CAS.
- L. Gao, Y. Liu, X. Du, S. Ma, M. Ge, H. Tang, C. Han, X. Zhao, Y. Liu, Y. Shao, Z. Wu, L. Zhang, F. Meng and F. Xiao-Feng Qin, Cell Death Dis., 2021, 12, 680 CrossRef CAS PubMed.
- F. Morandi, A. L. Horenstein, F. Costa, N. Giuliani, V. Pistoia and F. Malavasi, Front. Immunol., 2018, 9, 2722 CrossRef.
- F. Morandi, A. L. Horenstein and F. Malavasi, Front. Immunol., 2021, 12, 658263 CrossRef CAS PubMed.
- F. M. Uckun, Cancers, 2021, 13, 2018–2031 CrossRef.
- D. J. Li, S. J. Sun, J. T. Fu, S. X. Ouyang, Q. J. Zhao, L. Su, Q. X. Ji, D. Y. Sun, J. H. Zhu, G. Y. Zhang, J. W. Ma, X. T. Lan, Y. Zhao, J. Tong, G. Q. Li, F. M. Shen and P. Wang, Theranostics, 2021, 11, 4381–4402 CrossRef CAS PubMed.
- K. Okabe, K. Yaku, K. Tobe and T. Nakagawa, J. Biomed. Sci., 2019, 26, 34 CrossRef.
- R. Zapata-Perez, R. J. A. Wanders, C. D. M. van Karnebeek and R. H. Houtkooper, EMBO Mol. Med., 2021, 13, e13943 CAS.
- H. Massudi, R. Grant, N. Braidy, J. Guest, B. Farnsworth and G. J. Guillemin, PLoS One, 2012, 7, e42357 CrossRef CAS PubMed.
- X. H. Zhu, M. Lu, B. Y. Lee, K. Ugurbil and W. Chen, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 2876–2881 CrossRef CAS.
- B. Groth, P. Venkatakrishnan and S. J. Lin, Front. Mol. Biosci., 2021, 8, 686412 CrossRef.
- N. J. Habeichi, C. Tannous, A. Yabluchanskiy, R. Altara, M. Mericskay, G. W. Booz and F. A. Zouein, Int. Rev. Immunol., 2021, 1–11 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, spectral data for synthesized intermediates. See DOI: 10.1039/d1ra06266b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a