
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trimerization and cyclization of reactive P-functionalities confined within OCO pincers†
Beatrice L. Chinena,
Jakub Hyvl a,
Daniel F. Braytona,
Matthew M. Rieka,
Wesley Y. Yoshidaa,
Timothy W. Chappb,
Arnold L. Rheingoldc and
Matthew F. Cain*a
a,
Daniel F. Braytona,
Matthew M. Rieka,
Wesley Y. Yoshidaa,
Timothy W. Chappb,
Arnold L. Rheingoldc and
Matthew F. Cain*a
aDepartment of Chemistry, University of Hawai'i at Mānoa, 2545 McCarthy Mall, Honolulu, HI 96822, USA. E-mail: mfcain@hawaii.edu
bDepartment of Chemistry, Allegheny College, 520 N. Main Street, Meadville, PA 16335, USA
cDepartment of Chemistry, University of California, 9500 Gilman Drive, La Jolla, San Diego, California 92093, USA
First published on 25th August 2021
Abstract
In order to stabilize a 10–P–3 species with C2v symmetry and two lone pairs on the central phosphorus atom, a specialized ligand is required. Using an NCN pincer, previous efforts to enforce this planarized geometry at P resulted in the formation of a Cs-symmetric, 10π-electron benzazaphosphole that existed as a dynamic “bell-clapper” in solution. Here, OCO pincers 1 and 2 were synthesized, operating under the hypothesis that the more electron-withdrawing oxygen donors would better stabilize the 3-center, 4-electron O–P–O bond of the 10–P–3 target and the sp3-hybridized benzylic carbon atoms would prevent the formation of aromatic P-heterocycles. However, subjecting 1 to a metalation/phosphination/reduction sequence afforded cyclotriphosphane 3, resulting from trimerization of the P(I) center unbound by its oxygen donors. Pincer 2 featuring four benzylic CF3 groups was expected to strengthen the O–P–O bond of the target, but after metal–halogen exchange and quenching with PCl3, unexpected cyclization with loss of CH3Cl was observed to give monochlorinated 5. Treatment of 5 with (p-CH3)C6H4MgBr generated crystalline P-(p-Tol) derivative 6, which was characterized by NMR spectroscopy, elemental analysis, and X-ray crystallography. The complex 19F NMR spectra of 5 and 6 observed experimentally, were reproduced by simulations with MestreNova.
Introduction
Transition metal (TM) catalysis1 has revolutionized the chemical industry, enabling the conversion of cheap feedstocks into valuable products for pharmaceuticals, polymers, and other specialty chemicals.2 However, some of the most commonly employed metals like Ru,3 Rh,4 Ir,5 Pd,6 and Pt7 are scarce, while the supporting ligands are often phosphines,8 N-heterocyclic carbenes (NHCs),9 and/or other elaborate scaffolds including chiral diols,10 functionalized cyclopentadienyl ligands (ansa-metallocenes),11 and rare and/or non-naturally occurring amines.12 These metal–ligand platforms are both expensive and toxic, resulting in a delicate balance between the benefits of chemical synthesis and its harmful effects to the environment and human health. An environmentally friendly and sustainable TM alternative would be to use a more benign and earth-abundant Main Group (MG) element such as phosphorus as the active center. Yet, unlike TMs that have closely spaced HOMO–LUMO gaps, MG compounds feature orbitals that are far apart energetically, which limits their ability to engage in TM-type reactivity like oxidative addition, insertion, and reductive elimination.13 Fortunately, by distorting phosphines away from their classic three-fold symmetry, their frontier orbitals can become energetically accessible.14 For example, Cs-symmetric phosphorus triamide A can promote oxidative addition of alcohols and amines.15–17 Related C2v-symmetric B,18 describable by numerous resonance structures including B′ and B′′ due to extensive conjugation within the ONO ligand19 will oxidatively add H2 from H3N–BH3 and transfer that hydrogen equivalent to azobenzene in a catalytic fashion, producing hydrazines (Fig. 1).20 | ||
| Fig. 1 Structures of geometrically distorted A and B with contributing resonance structures B′ and B′′. | ||
Resonance structure B′′ is a T-shaped 10–P–3 species,21 which contains a frontier orbital environment remarkably similar to a C2v-symmetric, d8 ML3 TM complex like the Ir(PCP) pincer fragment with a low-lying σ acceptor orbital and a higher energy lone pair with π-symmetry,22 capable of engaging in backbonding to an incoming substrate (Fig. 2). In the case of Ir(PCP) pincers,23 this results in the oxidative addition of dihydrogen,24 alkanes,25 and ammonia.26 However, B does not add H2 because the high-energy p-lone pair necessary for backbonding is delocalized into the ligand scaffold.27 Therefore, we speculated that replacing the tridentate ONO ligand with a traditional pincer featuring a central aryl donor would prevent π-delocalization, rendering the p lone pair available for backbonding to small molecules.
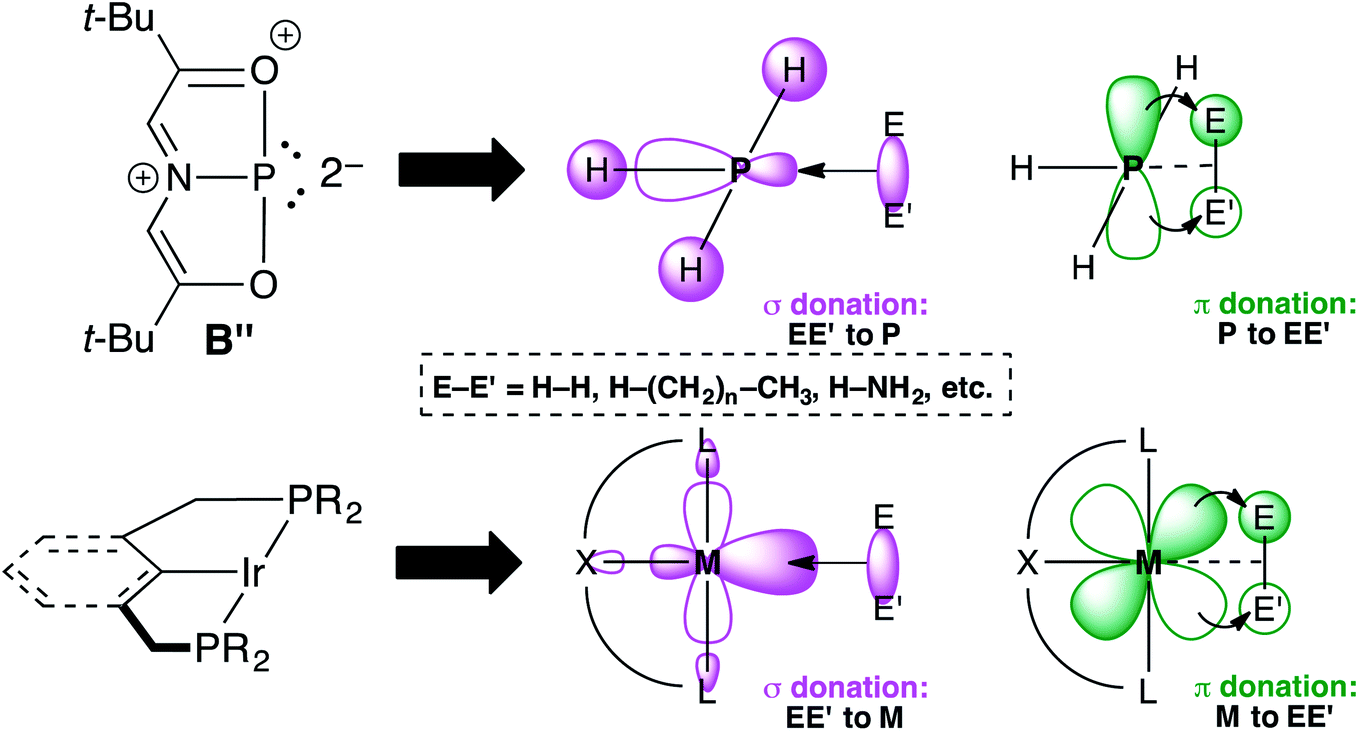 | ||
| Fig. 2 Simplified frontier orbital comparison between B′′ and an Ir(PCP) pincer fragment. | ||
In 2010,28 the Dostál group demonstrated that heavier, C2v-symmetric 10–Bi–3 species C (Scheme 1, inset) could be stabilized within an NCN pincer and similar analogues could perform oxidative addition of the weak bonds of diphenyldichalcogenides29 (ex. PhS–SPh, S–S bond = 55 kcal mol−1).30 This led us to attempt to stabilize a 10–P–3 species within a related NCN scaffold with the hope that substrates with stronger bonds, like those present in dihydrogen (H–H bond = 104 kcal mol−1),31 could be broken at P. Yet, unlike the Bi analogue, reduction of the P(III) intermediate did not afford the desired 10–P–3 species, but rather a Cs-symmetric 10π-electron benzazaphosphole with a tethered imine arm, which existed as a dynamic “bell-clapper” in solution (Scheme 1).32
 | ||
| Scheme 1 Synthesis of a benzazaphosphole “bell-clapper” with 10–Bi–3 species C shown in the inset. | ||
In order to access the targeted 10–P–3 species, we hypothesized that strengthening the 3-center, 4-electron bond between the axial donors and phosphorus (indicated in red, Chart 1, left) could be accomplished using more electron-withdrawing oxygen atoms.19 Additionally, sp3-hybridized benzylic carbons would be employed on the pincer arms to prevent the formation of aromatic P-heterocycles.33 Guided by these ligand design principles, brominated OCO pincers 1 (ref. 34) and 2 were synthesized (Chart 1, right), and we report here on the unexpected trimerization and cyclization chemistry encountered when installing the P-functionality within these scaffolds.
 | ||
| Chart 1 The targeted 10–P–3 species and brominated OCO pincers 1 and 2. | ||
Results and discussion
Synthesis of 1 and the formation of cyclotriphosphane 3 via reduction
Tribromide D 35 was treated with LiOMes (Mes = 2,4,6-trimethylbenzene), generated in situ from BuLi and 2,4,6-trimethylphenol, affording 1 in 64% yield (Scheme 2). | ||
| Scheme 2 Synthesis of 1. | ||
Monobrominated OCO pincer 1 was readily identified by 1H NMR spectroscopy with its downfield shifted benzylic signals (CDCl3: δ 4.93; relative to D, CDCl3: δ 4.55), integrating in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio with the diagnostic doublet/triplet pattern of the central aryl protons. The proligand was further characterized by 13C{1H} NMR spectroscopy and elemental analysis, and its structure was unequivocally confirmed by X-ray crystallography (Fig. 3).
:2:1 ratio with the diagnostic doublet/triplet pattern of the central aryl protons. The proligand was further characterized by 13C{1H} NMR spectroscopy and elemental analysis, and its structure was unequivocally confirmed by X-ray crystallography (Fig. 3).
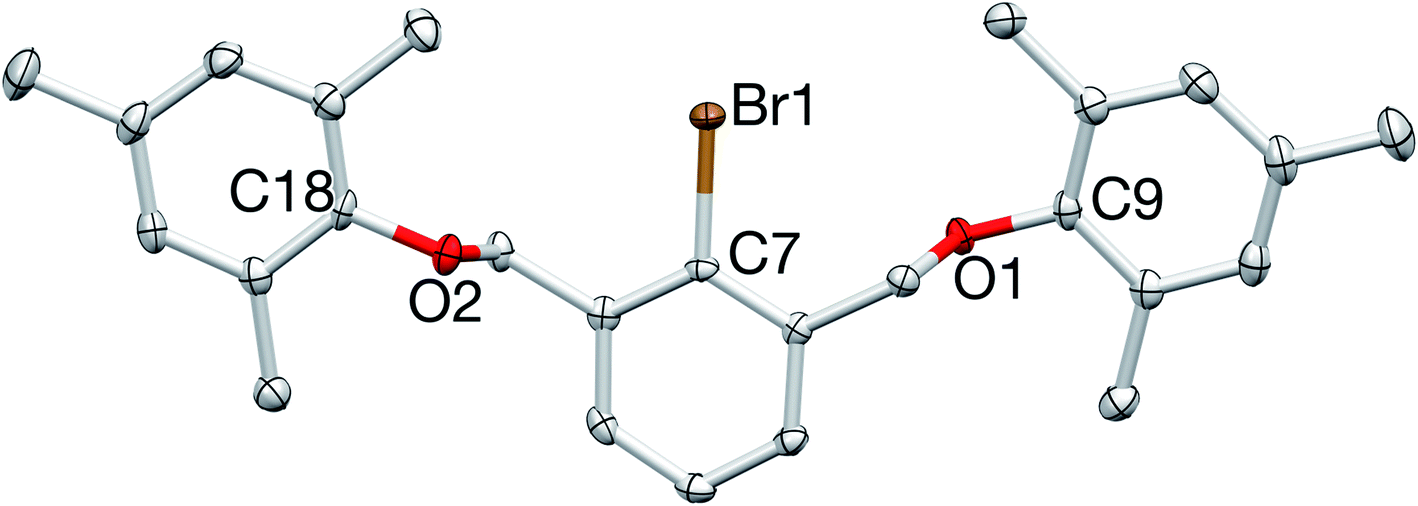 | ||
| Fig. 3 X-ray crystal structure of 1. All bond lengths (Å) and angles (deg) can be found in the ESI.† | ||
Based on literature precedent with heavier pnictogens,28,36 it was anticipated that the bromide in 1 could be substituted by a PCl2 unit via a lithium–halogen exchange/phosphination sequence. Reduction of the intermediate dichlorophosphine would then produce the desired 10–P–3 species. However, treatment of 1 with BuLi, followed by quenching with PCl3 and reduction37 with PMe3 did not afford the target, but rather cyclotriphosphane 3 in 56% yield (Scheme 3). The 31P{1H} NMR spectrum of 3 (inset) contained upfield shifted resonances at −116 and −144 ppm with a JPP = 186 Hz, consistent with a solution structure in which two P atoms are spectroscopically distinct from a third.38 This NMR signature is in line with other (PR)3 species like (PIs)3 (Is = 2,4,6-tri-isopropylbenzene) and (PMes)3,38 but not diphosphenes like Mes*P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PMes* (Mes* = 2,4,6-tri-tert-butylbenzene), which feature bona fide PP double bonds (2.034(2) Å),39 downfield shifted 31P{1H} NMR signals (δ = 493),40 and if unsymmetrical,38,40 large JPP couplings approaching 600 Hz (Mes*PPMes, JPP = 571 Hz). Other higher order monocyclophosphanes like tetrameric [P(t-Bu)]4 are singlets (31P NMR: δ −57.8),41 while (PR)5 pentamers (R = Ph) are complex multiplets (31P NMR: δ −3)42 (Scheme 3, bottom panel).
PMes* (Mes* = 2,4,6-tri-tert-butylbenzene), which feature bona fide PP double bonds (2.034(2) Å),39 downfield shifted 31P{1H} NMR signals (δ = 493),40 and if unsymmetrical,38,40 large JPP couplings approaching 600 Hz (Mes*PPMes, JPP = 571 Hz). Other higher order monocyclophosphanes like tetrameric [P(t-Bu)]4 are singlets (31P NMR: δ −57.8),41 while (PR)5 pentamers (R = Ph) are complex multiplets (31P NMR: δ −3)42 (Scheme 3, bottom panel).
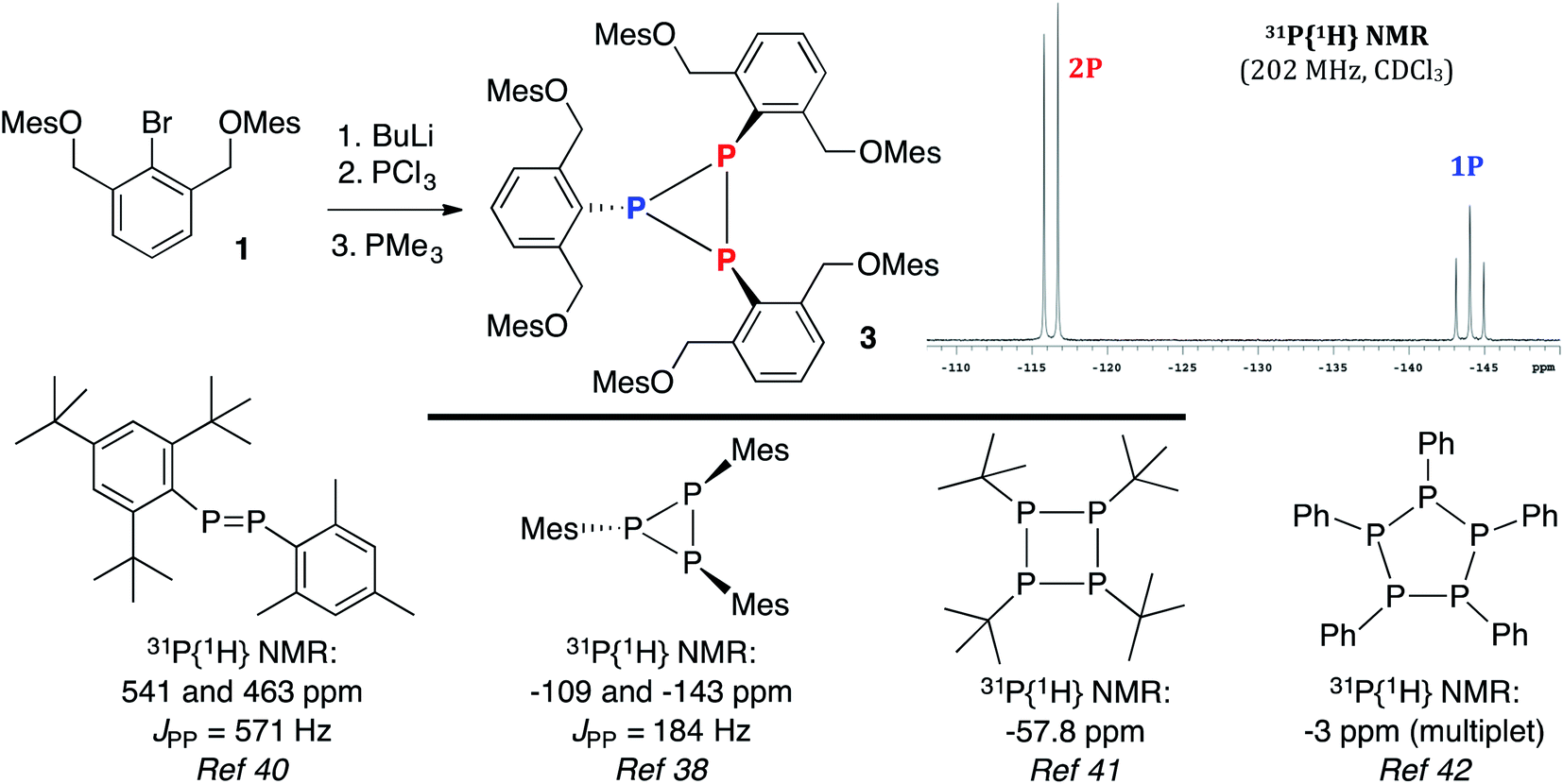 | ||
| Scheme 3 Synthesis of 3 and a selected view of its 31P{1H} NMR spectrum (top) and related (RP)n oligomers (bottom). | ||
The structure of cyclotriphosphane 3 was further corroborated by 1H and 13C{1H} NMR spectroscopy. Specifically, this “2-Down, 1-Up”-type (referring to the organic substituents on P) structure was readily apparent as two distinct aryl O-Mes singlets were observed in the 1H NMR spectrum (CDCl3) at 6.71 and 6.62 ppm in an 8:4 ratio with all the remaining resonances paired (although some broadened) in a similar 2:1 fashion. In addition, the 13C{1H} NMR spectrum displayed two separate benzylic signals and four methyl signals, all consistent with the assignment of 3. Ultimately, the structure of 3, as proposed, was established by X-ray crystallography (Fig. 4). Structural characterization of cyclotriphosphanes is rare,43 but the bond lengths and angles of 3 are quite similar to [P(t-Bu)]3 and [PCH(SiMe3)2]3.44,45 In particular, the P–P bonds measure 2.217(2), 2.194(2), and 2.237(2) Å, respectively with PPP angles of 60.95(7), 59.02(7), and 60.03(7) deg, indicative of P–P single bonds and unhybridized P-centers confined into a small ring system.43 The P–C bonds in 3 (avg = 1.847 Å) are slightly shorter than observed with [P(t-Bu)]3 and [PCH(SiMe3)2]3, which may be due to the presence of an sp2-hybridized46 C-substituent with considerably less bulk and more flexibility than the t-Bu and CH(SiMe3)2 groups. In fact, in comparison with cyclotriphosphanes [P(t-Bu)]3, [PCH(SiMe3)2]3, (PIs)3, and (PMes)3, it is somewhat surprising that 3 adopts a related structure because normally, as steric bulk decreases, the size of the oligomeric fragment increases (note: (PMes)3 versus (PPh)5); however, here, many close contacts (under 4 Å)47 between the aryl rings of the OMes units may impart added stability to the cyclotriphosphane structure.
 | ||
| Fig. 4 X-ray crystal structure of 3. Selected bond lengths (Å) and angles (deg): P1–P2 = 2.194(2), P2–P3 = 2.217(2), P1–P3 = 2.237(2), P1–C1 = 1.839(5), P2–C27 = 1.853(5), P3–C53 = 1.850(5), P3–P2–P1 = 60.95(7), P1–P3–P2 = 59.02(7), P3–P1–P2 = 60.03(7). | ||
Regardless, the structure of 3 suggested that the axial 3-center, 4-electron bond of the targeted 10–P–3 species was still the weak point. In fact, cyclotriphosphane 3 is formally the result of trimerization48 of an OCO-supported P(I) intermediate after the O-donors rotated away from the electron-deficient phosphorus center. In order to elucidate any mechanistic details, the phosphination and reduction steps (shown in Scheme 3) were monitored by 31P{1H} NMR spectroscopy, but did not reveal the generation of RPPMe3 (R = OCO pincer), a proposed precursor to the formation of cyclotriphosphanes such as (PIs)3, nor did a solution of 3, PMe3, and benzaldehyde produce any phosphaalkenes.48
Synthesis of 2 and cyclization of PCl2-functionalized 4
In order to further strengthen the 3-center, 4-electron O–P–O bond of the potential 10–P–3 species, the electronegativity of the O-donors was increased using benzylic CF3 groups. To this end, diol E,49 now accessible in a single step50 from dimethyl 2-bromoisophthalate and TMSCF3 and previously used to stabilize numerous hypervalent MG species51 including 10–Br–3 (ref. 52) and 12–I–5,49 was selected as the building block to brominated OCO pincer 2. After isolation of E in multi-gram quantities,53 dimethylation with MeI/K2CO3 in DMF resulted in the isolation of 2 in 78% yield (Scheme 4). | ||
| Scheme 4 Synthesis of 2. | ||
The 1H NMR spectrum (CDCl3) featured a prominent singlet for the OMe groups at 3.52 ppm, which integrated in a 6:2:1 ratio with the remaining central aryl protons, while the 19F NMR spectrum contained a single resonance at −66.9 ppm, all consistent with the expected C2v symmetry of 2. Additional characterization by 13C{1H} NMR spectroscopy, elemental analysis, and X-ray crystallography confirmed its structure (Fig. 5).
 | ||
| Fig. 5 X-ray crystal structure of 2. All bond lengths (Å) and angles (deg) can be found in the ESI.† | ||
Cognizant that lithium–halogen exchange using BuLi in the presence of fluorine atoms may be complicated by LiF formation54 and that magnesium–halogen exchange is faster with electron-withdrawing substrates and more functional group tolerant,55 2 was exposed to i-PrMgCl·LiCl,56 resulting in smooth in situ conversion to the Grignard reagent, which was subsequently quenched with a precooled solution (−35 °C) of PCl3 in THF (Scheme 5).
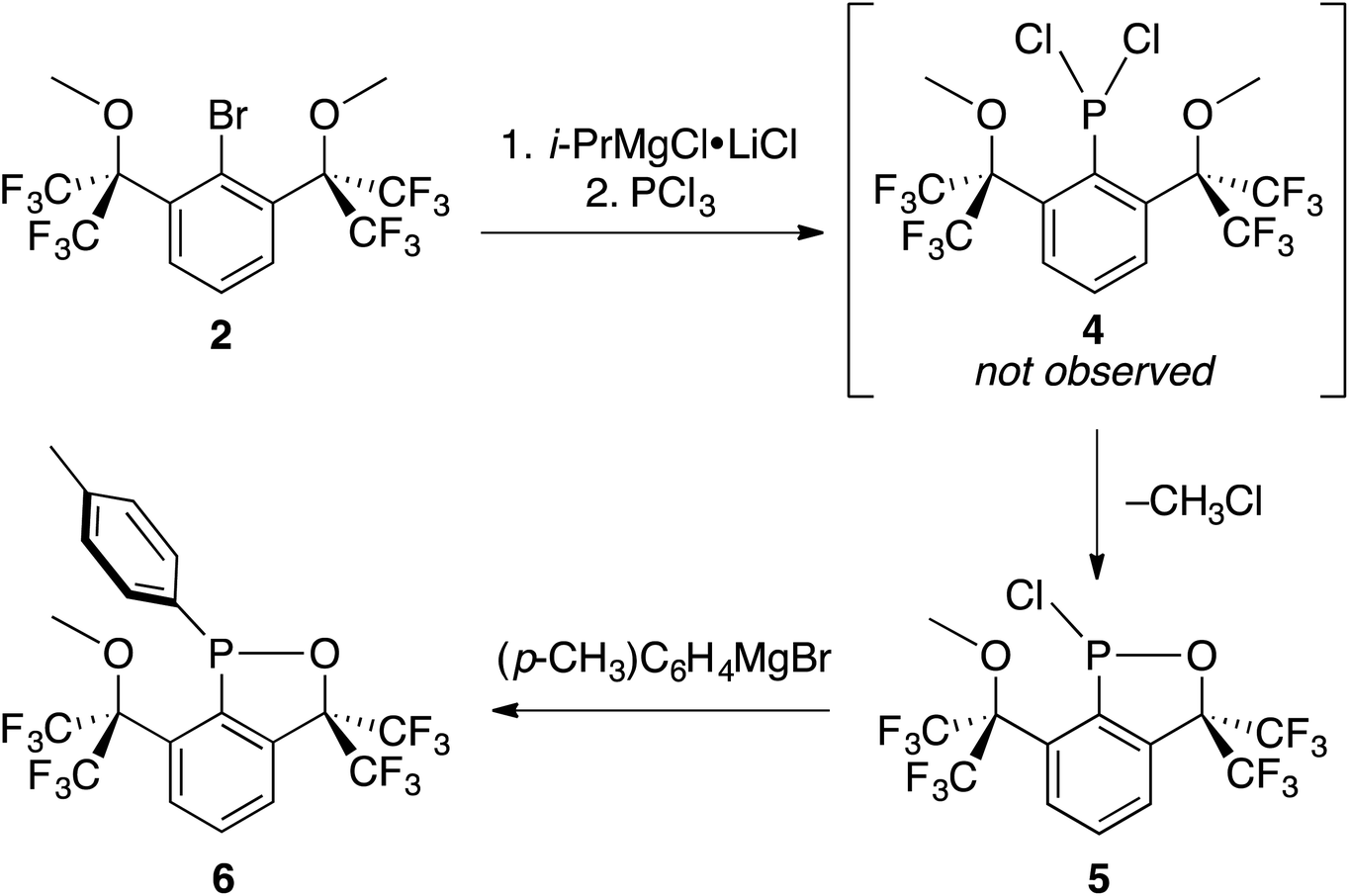 | ||
| Scheme 5 Intramolecular cyclization of 4 to 5 and synthesis of crystalline derivative 6. | ||
The reaction mixture was then analyzed by 31P{1H} NMR spectroscopy (THF), revealing the presence of some unreacted PCl3 (218 ppm), (i-Pr)PCl2 (202 ppm), and an unidentified product (171 ppm). The 19F NMR spectrum (C6D6) displayed four complex multiplet CF3 signals at −68.9, −70.4, −73.4, and −76.3 ppm, demonstrating that the unidentified product no longer contained C2v symmetry and was not PCl2-substituted 4. Instead, we suspected an intramolecular reaction occurred between the highly electrophilic PCl2 functionality and one of the O-donors, resulting in cyclization and the loss of chloride, which subsequently dealkylated57 the O–Me unit affording monochlorinated 5. Using MestreNova,58 simulated 19F NMR signals of 5 that closely matched the experimental spectrum were generated (Fig. 6), revealing the presence of both second order and long range coupling between the diastereotopic CF3 groups, the heterocyclic P atom, and the aryl protons (see ESI† for details).
 | ||
| Fig. 6 Experimental (red) and simulated (blue) 19F NMR signals at −68.9237 (A), −70.4093 (B), −73.3518 (C) and −76.2722 (D) for 5 at 282 MHz. | ||
Experimentally, the cyclization to 5 was confirmed by synthesizing a more crystalline derivative via nucleophilic substitution. Specifically, a solution of 5 in THF at 0 °C was treated with (p-CH3)C6H4MgBr, leading to the isolation of 6 as large off-white crystals in 38% yield (from 2, Scheme 5, above; see Fig. 9 for picture of crystals). The 31P{1H} NMR spectrum (C6D6) of 6 exhibited an apparent septet at 129.7 ppm (4JPF = 6 Hz), while its 19F NMR spectrum, like 5, displayed four distinct resonances for the diastereotopic CF3 groups at −69.5, −69.6, −74.0, and −76.5 ppm. These complex 19F NMR signals could also be reproduced with MestreNova (Fig. 7, see ESI† for details).
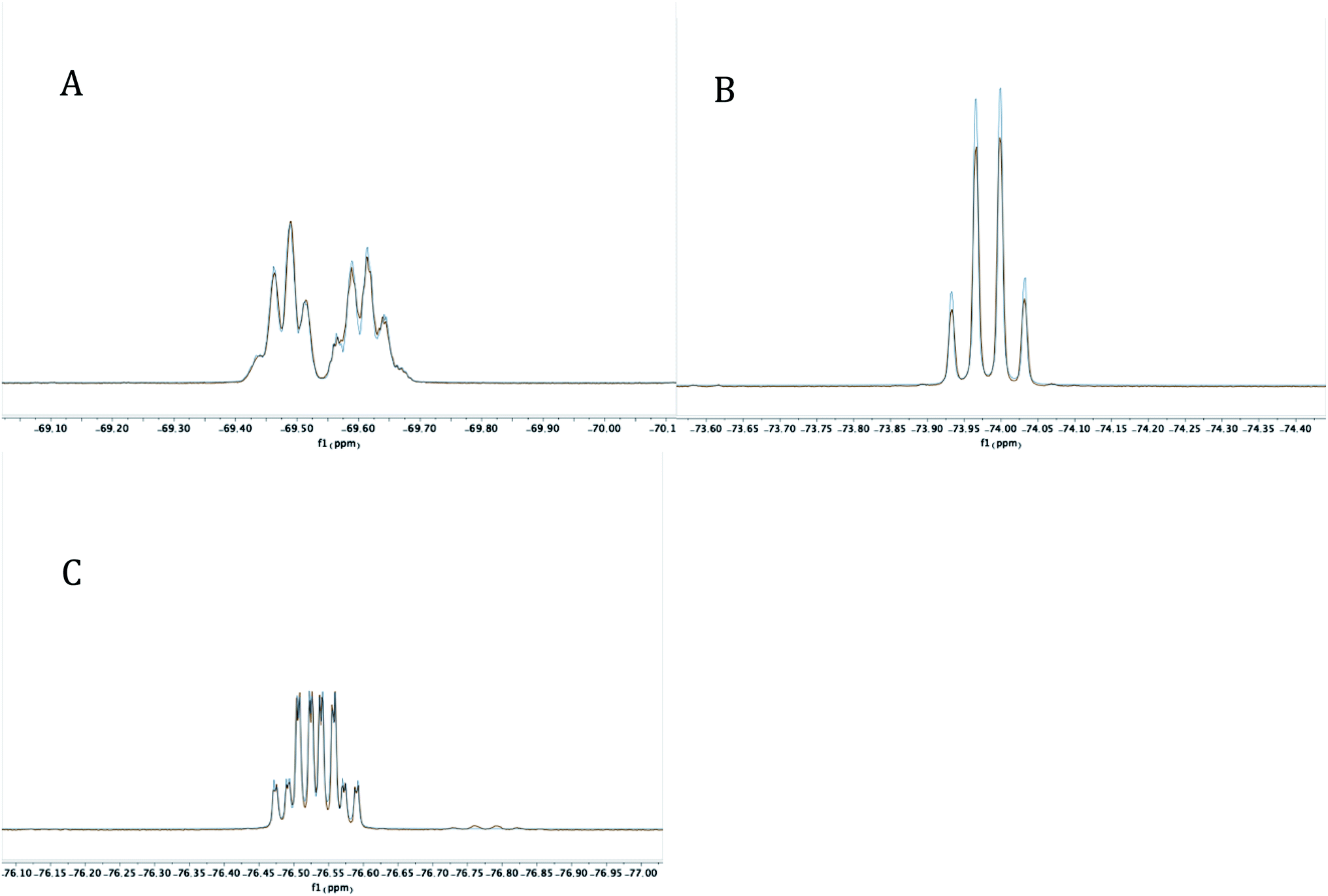 | ||
| Fig. 7 Experimental (red) and simulated (blue) 19F NMR signals at −69.4819 and −69.6176 (A), −73.9876 (B), and −76.5368 (C) for 6 at 282 MHz. | ||
1H NMR spectroscopy (C6D6) further corroborated the structure of 6 as the aromatic signals associated with the p-tolyl group and the three inequivalent protons of the central aryl unit of the OCO pincer integrated in a 2:2:1:1:1 ratio; in addition, two distinct methyl signals (OMe, 3.11 ppm and p-Me, 1.92 ppm) were observed. The 13C{1H} NMR spectrum also highlighted the inequivalency of the CF3 groups with four overlapping signals (see ESI† for zoomed in NMR spectrum): two quartets (JCF ∼ 290 Hz) combined with two quartets of doublets (JCF ∼ 290 Hz and JCP ∼ 9 or 2 Hz, respectively) in the 122–123 ppm range, while the two quaternary carbons C(CF3)2 resonated as a septet (80.8 ppm, JCF = 28 Hz) and a septet of doublets (89.5 ppm, JCF = 31.5 Hz, JCP = 16.5 Hz, Fig. 8).
 | ||
| Fig. 8 A selected view of the 13C{1H} NMR spectrum (C6D6) of 6 displaying the quaternary carbons. | ||
Ultimately, although disordered across a pseudo mirror plane, X-ray crystallography established the structure of 6 with its bulk purity verified by elemental analysis (Fig. 9).
 | ||
| Fig. 9 X-ray crystal structure of 6. All bond lengths (Å) and angles (deg) can be found in the ESI.† A picture of one of the large blocks is shown in the inset. | ||
Unfortunately though, like 1, fluorinated OCO pincer 2 also failed to deliver access to the 10–P–3 target. Here, the increased electron-withdrawing power of four benzylic CF3 groups resulted in, after a metal–halogen exchange/phosphination sequence, highly reactive dichlorophosphine 4, which via an unexpected cyclization57 to 5, partially disassembled the pincer framework before reduction could be attempted.
Conclusions and future directions
Undoubtedly, the hypothesis (vide supra) that the formation of 10–P–3 species over benzazaphosphole “bell-clappers” would be favored by switching from NCN pincers to more electron withdrawing OCO pincers with sp3-hybridized benzylic carbon atoms was overly simplistic. As the phrase goes, “hindsight is 20/20” and here, the biggest oversight was expecting a P-center to adopt a high-energy planarized geometry when lower barrier pathways to phosphorus maintaining its preferred pyramidal geometry exist. For example, 10–P–3 species like B′′ (Fig. 1) and the target (Chart 1, left) can be considered “internally-solvated phosphinidenes”19 that are supported by a 3-center, 4-electron O–P–O bond. However, B′′ features a conjugated ONO ligand that locks the O-donors into place, while CAr–Csp3 or Csp3–O bond rotations within OCO pincer 1 expose the reactive P(I) center, resulting in oligomerization chemistry,43,48,59 a known route by which phosphorus preserves its pyramidal structure and is exemplified by the formation of trimer 3. To prevent this, we aimed to strengthen the 3-center, 4-electron bond using more electron-withdrawing O-donors, but neglected how the enhanced electrophilicity of PCl2-substituted 4 can make the ligand framework susceptible to nucleophilic attack, in this case, cyclization to 5 (and its functionalization to 6). Guided by these lessons, a pincer framework that is rigid featuring electron-withdrawing O-donors, but lacking sp3-hybridized57 carbon atoms prone to nucleophilic attack may provide the stabilization necessary to confine a P-center into a planarized 10–P–3 arrangement and will be investigated.Experimental section
General experimental details
Unless otherwise specified, all reactions were performed under an atmosphere of nitrogen in an MBraun or Vacuum Atmospheres glovebox or using standard Schlenk techniques. All glassware was dried overnight in an oven at 140 °C prior to use. Solvents used in the glove box were purchased directly from chemical suppliers (Aldrich or Acros), pumped directly into the glove box, and stored over oven-activated 4 or 5 Å molecular sieves (Aldrich). Solvents used outside the glove box were purged with N2 for 30 min and stored over molecular sieves. TMSCF3 was dried by cryogenic transfer. 1H, 13C{1H}, 19F, and 31P{1H} NMR spectra were recorded on a Varian Mercury-300 (300/75/282/121 MHz), Varian Unity Inova-500 (500/126/470/202 MHz), or Agilent 600 DD2 (600/151/565/243 MHz) spectrometer at ambient temperature. Chemical shifts are reported in ppm downfield of tetramethylsilane using the solvent as internal standard (1H CDCl3 = 7.27 ppm, 1H C6D6 = 7.16 ppm, 13C CDCl3 = 77.16 ppm, 13C C6D6 = 128.06 ppm). Multiplicities are abbreviated as br (broad), s (singlet), d (doublet), t (triplet), q (quartet), or m (multiplet). Coupling constants (J) are reported in Hertz (Hz). Flash column chromatography was performed on silica gel (40–63 μm, SiliCycle). High-resolution mass spectrometry (HRMS) was recorded on an Agilent 6545 LC-MS Q-ToF spectrometer (NSF-1532310). Dimethyl 2-bromoisophthalate and [Me4N][F] were synthesized according to the published procedures.60,61 All other chemicals were used as received, unless otherwise noted.Synthesis of 1
In the glovebox, 2,4,6-trimethylphenol (3.00 g, 22.03 mmol, 2.5 equiv.) was dissolved in THF (100 mL), and a 1.6 M solution of n-BuLi in hexanes (13.0 mL, 20.8 mmol, 2.3 equiv.) was added dropwise at room temperature. The reaction mixture was stirred for 15 min. Subsequently, tribromide D (3.086 g, 9.0 mmol) was added to the stirred solution, and the reaction mixture was transferred to a Schlenk bomb equipped with a Teflon screw cap and heated at 100 °C for 1 day. The volatiles were then removed under reduced pressure, and the crude residue was extracted with a toluene:hexane mixture (1:1, 3 × 15 mL). The combined extracts were filtered through a Celite plug, and the filtrate was concentrated under vacuum then purified by column chromatography (silica gel, toluene:hexane 1:1, Rf = 0.63). The title product (2.64 g, 5.822 mmol) was obtained as white plates in 65% yield. X-Ray quality crystals were obtained by slow evaporation from a concentrated solution of 1 in n-hexanes.
Anal. Calcd for C26H29BrO2: C, 68.87; H, 6.45. Found: C, 68.77; H, 6.27. 1H NMR (500 MHz, CDCl3): δ 7.78 (d, J = 7.6 Hz, 2H, Ar), 7.51 (t, J = 7.6 Hz, 1H, Ar), 6.89 (s, 4H, Ar), 4.93 (s, 4H, CH2), 2.30 (s, 12H, Me), 2.29 (s, 6H, Me). 13C{1H} NMR (126 MHz, CDCl3): δ 153.5 (Ar), 137.8 (Ar), 133.6 (Ar), 130.9 (Ar), 129.6 (Ar), 127.9 (Ar), 127.7 (Ar), 121.6 (Ar), 73.3 (CH2), 20.9 (CH3), 16.4 (CH3).
Synthesis of 2
Diol E (3.61 g, 7.38 mmol) and DMF (180 mL) were combined in a Schlenk flask with a stir bar. The solution was purged with N2 on the Schlenk line for 30 min, then K2CO3 (5.10 g, 36.9 mmol, 5 equiv.) was added with stirring under positive N2 pressure. Next, MeI (2.30 mL, 36.9 mmol, 5 equiv.) was injected via syringe, and the reaction mixture was stirred overnight, then quenched with NH4Cl (aq) and extracted with toluene (3 × 50 mL). The combined organic layers were washed with water, brine, dried over MgSO4, filtered, and the filtrate concentrated under vacuum, resulting in pale yellow solid 2 (2.97 g, 5.74 mmol, 78%). Recrystallization from acetonitrile (8 mL) at 4 °C afforded crystals suitable for X-ray diffraction.Anal. Calcd for C14H9BrO2F12: C, 32.52; H, 1.75. Found: C, 32.71; H, 1.77. 1H NMR (500 MHz, CDCl3): δ 7.76 (d, J = 10 Hz, 2H, Ar), 7.51 (t, J = 10 Hz, 1H, Ar), 3.52 (s, 6H, Me). 13C{1H} NMR (151 MHz, CDCl3): δ 134.1 (Ar), 129.6 (Ar), 127.0 (Ar), 123.5 (Ar), 122.4 (q, J = 290 Hz, CF3), 86.5 (septet, J = 29 Hz, C(CF3)2), 54.8 (OMe). 19F NMR (471 MHz, CDCl3): δ −66.9 (s).
Synthesis of 3
OCO-supported aryl bromide 1 (1.134 g, 2.50 mmol) was loaded into a Schlenk flask, dissolved in Et2O (60 mL), taken outside of the glovebox, and cooled to −78 °C. A 1.6 M solution of n-BuLi in hexanes (1.7 mL, 2.72 mmol, 1.1 equiv.) was injected (under N2), and the reaction mixture was stirred at −78 °C for 5 min. A second Schlenk flask containing a solution of PCl3 (550 mg, 4.01 mmol, 1.6 equiv.) in Et2O (5 mL) was transferred via cannula to the cooled reaction mixture. The reaction mixture was stirred at −78 °C for 10 min and then at room temperature (RT) for 1 h, leading to the precipitation of a white solid. The volatiles were removed under reduced pressure, the Schlenk flask was brought back into the glovebox, the residue was triturated with Et2O (10 mL), and the volatiles were again removed. The residue was dissolved in THF (20 mL), and PMe3 was added (485 mg, 6.375 mmol, 2.6 equiv.). The reaction mixture was stirred at RT for 1 day. The organic volatiles were removed under reduced pressure, and the residue was extracted with toluene (3 × 20 mL). The combined extracts were filtered through a Celite plug, the filtrate was concentrated to dryness, triturated with n-pentane (10 mL), and again concentrated to dryness under reduced pressure. The resulting residue was triturated with acetonitrile (10 mL) and stirred at RT for 1 h, generating a white precipitate that was collected by filtration and dried (565 mg, 0.466 mmol, 56%). X-Ray quality crystals of 3 were obtained by recrystallization from a solution of hot acetonitrile.Anal. Calcd for C78H87O6P3: C, 77.20; H, 7.23. Found: C, 76.89; H, 7.14. 31P{1H} NMR (202 MHz, CDCl3): δ −116.2 (d, 1JPP = 186 Hz), −144.0 (t, 1JPP = 186 Hz). 1H NMR (500 MHz, CDCl3): δ 7.71 (d, J = 7.7 Hz, 2H, Ar1), 7.40 (t, J = 7.7 Hz, 1H, Ar1), 7.33–7.28 (m, 4H, Ar2), 7.28–7.22 (m, 2H, Ar2), 6.71 (s, 8H, Ar2), 6.62 (s, 4H, Ar1), 5.39 (s, 4H, CH2–Ar1), 4.71 (br, 8H, CH2–Ar2), 2.25 (s, 12H, Ar1), 2.20 (s, 6H, Ar1), 2.02 (s, 12H, Ar2), 1.95 (br, 24H, Ar2). 13C NMR (126 MHz, CDCl3): δ 153.46 (Ar), 153.44 (Ar), 143.5 (Ar), 143.4 (d, JPC = 6.9 Hz, Ar), 133.6 (Ar), 133.0 (Ar), 132.9 (Ar), 130.7 (Ar), 130.5 (Ar), 130.4 (Ar), 129.6 (Ar), 129.4 (Ar), 129.0 (Ar), 128.4 (d, JPC = 10.0 Hz, Ar), 127.9 (Ar), 126.4 (Ar), 73.2 (CH2), 72.8 (d, JPC = 7.6 Hz, CH2), 20.9 (CH3), 16.7 (CH3), 16.4 (CH3), 16.3 (CH3).
Synthesis of 6
Fluorinated aryl bromide 2 (500 mg, 0.967 mmol) was dissolved in 6 mL of THF in a vial with a stir bar inside the glovebox and (i-Pr)MgCl·LiCl was added dropwise via syringe (0.82 mL, 1.064 mmol, 1.1 equiv., 1.3 M in THF), resulting in a homogeneous, yellow reaction mixture. The reaction mixture was stirred for 1 h at room temperature (RT) then directly filtered into 2 mL of a pre-cooled solution (−35 °C, 1 h) of PCl3 (146 mg, 1.064 mmol, 1.1 equiv.) in THF. The solution was warmed to RT for 1 h then analyzed by 31P{1H} NMR spectroscopy, revealing the presence of unreacted PCl3 (218 ppm), i-PrPCl2 (202 ppm) and the chlorophosphine (170 ppm). The “intermediate” product mixture was then concentrated under vacuum to remove the volatile and unwanted phosphorus byproducts (PCl3 and i-PrPCl2), leaving a pale yellow residue that was dissolved in 6 mL of THF and transferred to a Schlenk bomb fitted with a screw-top Teflon cap. The bomb was taken outside of the glovebox, cooled to 0 °C, and (p-CH3)C6H4MgBr (1.06 mL, 1.064 mmol, 1.1 equiv., 1.0 M in THF) was injected via syringe under positive N2 pressure, affording a light orange reaction mixture, which was subsequently warmed to RT. The Schlenk bomb was resealed (under positive N2 pressure), brought back inside the glove box, and an aliquot was analyzed by 31P{1H} NMR spectroscopy displaying a prominent signal at 129 ppm with slight impurities at −2 and −8 ppm. The entire reaction mixture was concentrated under vacuum, extracted with pentane (3 × 50 mL), and filtered through a Kimwipe plug. The filtrate was concentrated under vacuum, dissolved in ether (2 mL) and cooled to −35 °C overnight, resulting in large white/colorless blocks suitable for X-ray diffraction (202 mg, 0.371 mmol, 38% yield).Anal. Calcd for C20H13F12O2P: C, 44.14; H, 2.41. Found: C, 44.09; H, 2.41. 1H NMR (600 MHz, C6D6): δ 7.57 (br d, J = 8 Hz, 1H, Ar), 7.52 (d, J = 8 Hz, 1H, Ar), 7.10 (apparent t, J = 8 Hz, 2H, Ar), 6.88 (t, J = 8 Hz, 1H, Ar), 6.81 (d, J = 8 Hz, 2H, Ar), 3.11 (s, 3H, OMe), 1.92 (s, 3H, p-Me). 13C{1H} NMR (151 MHz, C6D6): δ 143.4 (d, J = 33 Hz, Ar), 141.2 (Ar), 137.8 (d, J = 41 Hz, Ar), 135.7 (d, J = 6 Hz, Ar), 133.0 (d, J = 5 Hz, Ar), 132.2 (d, J = 25 Hz, Ar), 131.1 (Ar), 130.0 (Ar), 129.0 (d, J = 8 Hz, Ar), 126.9 (Ar), four overlapping CF3 signals*: 123.1 (qd, J ∼ 290 and 9 Hz), 123.0 (q, J ∼ 290 Hz), 122.5 (q, J ∼ 290 Hz), and 122.4 (qd, J ∼ 290 and 3 Hz), 89.5 (septet of doublets, J = 31.5 and 16.5 Hz, C(CF3)2 in P-ring), 80.8 (septet, J = 28 Hz, C(CF3)2), 55.3 (OMe), 21.2 (Me). 19F NMR (282 MHz, C6D6):** δ −69.5 (m, JFF ∼ 8 Hz, JPF ∼ 2 Hz, JHF ∼ 0.5–2 Hz), −69.6 (m, JFF ∼ 8 Hz, JPF ∼ 7 Hz, JHF ∼ 0.5–2.5 Hz), −74.0 (m, JFF ∼ 9 Hz, JHF ∼ 0.5–1 Hz), and −76.5 (m, JFF ∼ 9 Hz, JHF ∼ 1–5 Hz). 31P{1H} NMR (121 MHz, C6D6): δ 129.4 (septet, J = 6 Hz). *In CDCl3, the overlapping CF3 signals in the 13C{1H} NMR spectrum are better resolved: δ 122.5 (q, J = 290 Hz), 122.3 (qd, J = 290 and 9 Hz), 122.0 (q, J = 290 Hz), 121.6 (qd, J = 290 and 2 Hz). **Reported coupling constants in the 19F NMR spectrum were determined by simulation with Mestrenova.
Experimental NMR spectra, simulated 19F NMR spectra using MestreNova, and X-ray crystallography
See the ESI† for details.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MFC thanks the University of Hawai'i at Mānoa (UHM) for laboratory space and support and the National Science Foundation (NSF) for a CAREER Award (NSF CHE-1847711). Mass spectroscopic data was obtained at UHM on an Agilent 6545 Accurate-Mass QTOF-LCMS (NSF-1532310). TWC acknowledges support from Allegheny College and the NSF (CHE-1726109). Elemental analyses were conducted by William Brennessel (University of Rochester).Notes and references
- J. F. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010 Search PubMed
.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, Weinheim, 1997 Search PubMed
- Selected examples:
(a) C. Gunanathan and D. Milstein, Bond Activation and Catalysis by Ruthenium Pincer Complexes, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS
- Selected examples:
(a) I. D. Gridnev, M. Yasutake, T. Imamoto and I. P. Beletskaya, Asymmetric Hydrogenation of α,β-Unsaturated Phosphonates with Rh-BisP* and Rh-MiniPHOS Catalysts: Scope and Mechanism of the Reaction, Proc. Natl. Acad. Sci. U.S.A., 2004, 101, 5385–5390 CrossRef CAS PubMed
- Selected examples:
(a) J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS
- Selected Examples:
(a) P. Ruiz-Castillo and S. L. Buchwald, Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS
- Selected examples:
(a) I. E. Marko, S. Sterin, O. Buisine, G. Mignani, P. Branlard, B. Tinant and J.-P. Declercq, Selective and Efficient Platinum(0)-Carbene Complexes as Hydrosilylation Catalysts, Science, 2002, 298, 204–206 CrossRef CAS
- Selected examples:
(a) M. J. Burk, Modular Phospholane Ligands in Asymmetric Catalysis, Acc. Chem. Res., 2000, 33, 363–372 CrossRef CAS
- Selected examples:
(a) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, N-Heterocycles Carbene Complexes in C–H Activation, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed
- As an example: D. Seebach, A. K. Beck and A. Heckel, TADDOLs, Their Derivatives, and TADDOL Analogues: Versatile Chiral Auxiliaries, Angew. Chem., Int. Ed., 2001, 40, 92–138 CrossRef CAS PubMed
- M. A. Bau, S. Wiesler, S. L. Younas and J. Streuff, Strategies for the Synthesis of Chiral Carbon-Bridged Group IV ansa-Metallocenes, Chem.–Eur. J., 2019, 25, 10531–10545 CrossRef CAS PubMed
- J. D. Firth, P. O'Brien and L. Ferris, Revisiting the Sparteine Surrogate: Development of a Resolution Route to the (–)-Sparteine Surrogate, Org. Biomol. Chem., 2014, 12, 9357–9365 RSC
- P. P. Power, Main-Group Elements as Transition Metals, Nature, 2010, 463, 171–177 CrossRef CAS PubMed
- K. Lee, A. V. Blake, A. Tanushi, S. M. McCarthy, D. Kim, S. M. Loria, C. M. Donahue, K. D. Spielvogel, J. M. Keith, S. R. Daly and A. T. Radosevich, Validating the Biphilic Hypothesis of Nontrigonal Phosphorus(III) Compounds, Angew. Chem., Int. Ed., 2019, 58, 6993–6998 CrossRef CAS PubMed
- W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, Reversible Intermolecular E–H Oxidative Addition to a Geometrically Deformed and Structurally Dynamic Phosphorous Triamide, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed
- Phosphorus triamide A can also perform hydroboration by a different mechanism: Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, P–N Cooperative Borane Activation and Catalytic Hydroboration by a Distorted Phosphorous Triamide Platform, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS
- Other strained phosphorus analogues can perform similar additions: T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, E–H Bond Activation of Ammonia and Water by a Geometrically Constrained Phosphorus(III) Compound, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS
- S. A. Culley and A. J. Arduengo, Synthesis and Structure of the First 10–P–3 Species, J. Am. Chem. Soc., 1984, 106, 1164–1165 CrossRef CAS
- A. J. Arduengo and C. A. Stewart, Low-Coordinate Hypervalent Phosphorus, Chem. Rev., 1994, 94, 1215–1237 CrossRef CAS
- N. L. Dunn, M. Ha and A. T. Radosevich, Main Group Redox Catalysis: Reversible PIII/PV Redox Cycling at a Phosphorus Platform, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef CAS
- N.-X.-L. nomenclature, C. W. Perkins, J. C. Martin, A. J. Arduengo, W. Lau, A. Alegria and J. K. Kochi, An Electrically Neutral σ-Sulfuranyl Radical from the Homolysis of a Perester with Neighboring Sulfenyl Sulfur: 9–S–3 Species, J. Am. Chem. Soc., 1980, 102, 7753–7759 CrossRef
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, Orbital Interactions in Chemistry, Wiley, New Jersey, 2013 Search PubMed
- A. Kumar, T. M. Bhatti and A. S. Goldman, Dehydrogenation of Alkanes and Aliphatic Groups by Pincer-Ligated Metal Complexes, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS
- M. Mediati, G. N. Tachibana and C. M. Jensen, Isolation and Characterization of IrH2Cl(η2-H2)[P(i-Pr)3]2: A Neutral Dihydrogen Complex of Iridium, Inorg. Chem., 1990, 29, 5–9 CrossRef
-
(a) M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, A Highly Active Alkane Dehydrogenation Catalyst: Stabilization of Dihydrido Rhodium and Iridium Complexes by a P–C–P Pincer Ligand, Chem. Commun., 1996, 2083–2084 RSC
- J. Zhao, A. S. Goldman and J. F. Hartwig, Oxidative Addition of Ammonia to Form a Stable Monomeric Amido Hydride Complex, Science, 2005, 307, 1080–1082 CrossRef CAS PubMed
- A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, The Synthesis, Structure, and Chemistry of 10–Pn–3 Systems: Tricoordinate Hypervalent Pnictogen Compounds, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef CAS
- P. Simon, F. de Proft, R. Jambor, A. Ruzicka and L. Dostal, Monomeric Organoantimony(I) and Organobismuth(I) Compounds Stabilized by an NCN Chelating Ligand: Syntheses and Structures, Angew. Chem., Int. Ed., 2010, 49, 5468–5471 CrossRef CAS PubMed
- P. Simon, R. Jambor, A. Ruzicka and L. Dostál, Oxidative Addition of Diphenyldichalcogenides PhEEPh (E = S, Se, Te) to Low-Valent CN- and NCN-Chelated Organoantimony and Organobismuth Compounds, Organometallics, 2013, 32, 239–248 CrossRef CAS
- S. W. Benson, Thermochemistry and Kinetics of Sulfur-Containing Molecules and Radicals, Chem. Rev., 1978, 78, 23–35 CrossRef CAS
- S. J. Blanksby and G. B. Ellison, Bond Dissociation Energies of Organic Molecules, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed
- J. Hyvl, W. Y. Yoshida, A. L. Rheingold, R. P. Hughes and M. F. Cain, A Masked Phosphinidene Trapped in a Fluxional NCN Pincer, Chem.–Eur. J., 2016, 22, 17562–17565 CrossRef CAS
- V. Kremlacek, J. Hyvl, W. Y. Yoshida, A. Ruzicka, A. L. Rheingold, J. Turek, R. P. Hughes, L. Dostal and M. F. Cain, Heterocycles Derived from Generating Monovalent Pnictogens within NCN Pincers and Bidentate NC Chelates: Hypervalency versus Bell-Clappers versus Static Aromatics, Organometallics, 2018, 37, 2481–2490 CrossRef CAS
- Related OCO pincers have been prepared: R. Jambor, L. Dostal, A. Ruzicka, I. Cisarova, J. Brus, M. Holcapek and J. Holecek, Organotin(IV) Derivatives of Some O,C,O-Chelating Ligands, Organometallics, 2002, 21, 3996–4004 CrossRef CAS
- L. A. van de Kuil, H. Luitjes, D. M. Grove, J. W. Zwikker, J. G. M. van der Linden, A. M. Roelofsen, L. W. Jenneskens, W. Drenth and G. van Koten, Electronic Tuning of Arylnickel(II) Complexes by Para Substitution of the Terdentate Monoanionic 2,6-Bis[(Dimethylamino)methyl]phenyl Ligand, Organometallics, 1994, 13, 468–477 CrossRef CAS
-
(a) J. Vrana, R. Jambor, A. Ruzicka, A. Lycka, F. De Proft and L. Dostal, N→As Intramolecularly Coordinated Organoarsenic(III) Chalcogenides: Isolation of Terminal As–S and As–Se Bonds, J. Organomet. Chem., 2013, 723, 10–14 CrossRef CAS
- J. Hyvl, W. Y. Yoshida, C. E. Moore, A. L. Rheingold and M. F. Cain, Unexpected Detours and Reactivity Encountered During the Planned Synthesis of Hypervalent 10–Pn–3 Species (Pn = P or As), Polyhedron, 2018, 143, 99–104 CrossRef CAS
- C. N. Smit, Th. A. van der Knapp and F. Bickelhaupt, Stable Unsymmetrical Diphosphenes, Tetrahedron Lett., 1983, 24, 2031–2034 CrossRef CAS
- M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, Synthesis and Structure of Bis(2,4,6-tri-tert-butylphenyl)diphosphene: Isolation of a True “Phosphobenzene”, J. Am. Chem. Soc., 1981, 103, 4587–4589 CrossRef CAS
- A. H. Cowley, J. E. Kilduff, J. G. Lasch, S. K. Mehrotra, N. C. Norman, M. Pakulski, B. R. Whittlesey, J. L. Atwood and W. E. Hunter, Synthesis and Structure of Compounds Containing Double Bonds between the Heavier Group 5A Elements: Diphosphenes, Diarsenes, Phosphaarsenes, and Phosphastibenes, Inorg. Chem., 1984, 23, 2582–2593 CrossRef CAS
- M. H. Holthausen, D. Knackstedt, N. Burford and J. J. Weigand, Phosphenium-Insertion and Chloronium-Addition Reactions Involving the cyclo-Phosphanes (t-BuP)n (n = 3, 4), Aust. J. Chem., 2013, 66, 1155–1162 CrossRef CAS
- P. R. Hoffman and K. G. Caulton, Conformational Stability and Ring-Size Integrity of Cyclic Polyphosphines, Inorg. Chem., 1975, 14, 1997–1999 CrossRef CAS
- M. Baudler and K. Glinka, Monocyclic and Polycyclic Phosphanes, Chem. Rev., 1993, 93, 1623–1667 CrossRef CAS
- For X-ray structure of [P(t-Bu)]3: J. Hahn, M. Baudler, C. Kruger and Y.-H. Tsay, Beitrage zur Chemie des Phosphors, 116, Kernresonanzspektren und Kristallstruktur von Tri-tert-butyl-cycltriphosphan, Z. Naturforsch., B, 1982, 37b, 797–805 CrossRef CAS
- For X-ray structure of [PCH(SiMe3)2]3: P. A. Baldy and J. Estienne, Structure du Tris[bis(trimethylsilyl)methyl]triphosphiranne, Acta Crystallogr. C, 1988, C44, 747–749 CrossRef
- Bonds composed of orbitals that have more s-character are generally shorter: H. A. Bent, An Appraisal of Valence-Bond Structures and Hybridization in Compounds of the First-Row Elements, Chem. Rev., 1961, 61, 275–311 CrossRef CAS
-
(a) L. M. Salonen, M. Ellermann and F. Diederich, Aromatic Rings in Chemical and Biological Recognition: Energetics and Structures, Angew. Chem., Int. Ed., 2011, 50, 4808–4842 CrossRef CAS
- Related trimerizations were observed in phospha-Wittig chemistry:
(a) S. Shah and J. D. Protasiewicz, ‘Phospha-Wittig’ Reactions using Isolable Phosphoranylidenephosphines ArP=PR3 (Ar = 2,6-Mes2C6H3 or 2,4,6-But3C6H2), Chem. Commun., 1998, 1585–1586 RSC
- Original synthesis was an aryl iodide not bromide: T. T. Nguyen, R. L. Amey and J. C. Martin, Tridentate Ligand Useful in Stabilizing Higher Coordination States of Nonmetallic Elements. An Aryldialkoxydifluoroperiodinane, J. Org. Chem., 1982, 47, 1024–1027 CrossRef CAS
- Y. Imada, T. Kukita, H. Nakano and Y. Yamamoto, Easy Access to Martin's Hypervalent Sulfur Anions toward an Electrode Material for Organic Rechargeable Batteries, Bull. Chem. Soc. Jpn., 2016, 89, 546–548 CrossRef CAS
- J. C. Martin, “Frozen” Transition States: Pentavalent Carbon et al, Science, 1983, 221, 509–514 CrossRef CAS
-
(a) T. T. Nguyen, J. C. Martin and A. Dialkoxyarylbrominane, The First Example of an Organic 10–Br–3 Species, J. Am. Chem. Soc., 1980, 102, 7383–7385 CrossRef
- The procedure in ref. 50 was modified slightly: see ESI† for experimental details.
- J. Mohr, M. Durmaz, E. Irran and M. Oestreich, Tris(5,6,7,8-Tetrafluoronaphthalen-2-yl)borane, a Partially Fluorinated Boron Lewis Acid with Fluorination Distal to the Boron Atom, Organometallics, 2014, 33, 1108–1111 CrossRef CAS
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Highly Functionalized Organomagnesium Reagents Prepared through Halogen-Metal Exchange, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed
- A. Krasovskiy and P. Knochel, A LiCl-Mediated Br/Mg Exchange Reaction for the Preparation of Functionalized Aryl- and Heteroarylmagnesium Compounds from Organic Bromides, Angew. Chem., Int. Ed., 2004, 43, 3333–3336 CrossRef CAS
- L. Dostal, R. Jambor, A. Ruzicka, R. Jirasko, J. Holecek and F. De Proft, OCO and NCO Chelated Derivatives of Heavier Group 15 Elements. Study on Possibility of Cyclization Reaction via Intramolecular Ether Bond Cleavage, Dalton Trans., 2011, 40, 8922–8934 RSC
- MestReNova v14.2.0-26256, Mestrelab Research S.L., Santiago de Compostela Spain, 2020 Search PubMed.
- F. Mathey, Developing the Chemistry of Monovalent Phosphorus, Dalton Trans., 2007, 1861–1868 RSC
- F. C. Courchay, J. C. Sworen, I. Ghiviriga, K. A. Abboud and K. B. Wagener, Understanding Structural Isomerization during Ruthenium-Catalyzed Olefin Metathesis: A Deuterium Labeling Study, Organometallics, 2006, 25, 6074–6086 CrossRef CAS
- A. A. Kolomeitsev, F. U. Seifert and G.-V. Roeschenthaler, Simple Preparation of Difluorophosphoranes using Anhydrous Zinc and Tetramethylammonium Fluorides, J. Fluorine Chem., 1995, 71, 47–49 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2095177–2095181. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05926b |
| This journal is © The Royal Society of Chemistry 2021 |