
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLi-based layered nickel–tin oxide obtained through electrochemically-driven cation exchange†
Anatolii V. Morozov *ab,
Aleksandra A. Savinaa,
Anton O. Boeva,
Evgeny V. Antipovab and
Artem M. Abakumova
*ab,
Aleksandra A. Savinaa,
Anton O. Boeva,
Evgeny V. Antipovab and
Artem M. Abakumova
aSkolkovo Institute of Science and Technology, Bolshoy Boulevard 30, bld. 1, 121205, Moscow, Russia. E-mail: Anatolii.Morozov@skoltech.ru; anatolii.morozov.v@gmail.com; Tel: +7 9254784518
bDepartment of the Material Sciences, Moscow State University, Leninskiye gory 1, 119991, Moscow, Russia
First published on 24th August 2021
Abstract
The Li-based layered nickel-tin oxide Li0.35Na0.07Ni0.5Sn0.5O2 has been synthesized via electrochemically-driven Li+ for Na+ exchange in O3-NaNi0.5Sn0.5O2. The crystal structure of Li0.35Na0.07Ni0.5Sn0.5O2 was Rietveld-refined from powder X-ray diffraction data (a = 3.03431(7) Å, c = 14.7491(8) Å, S. G. R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). It preserves the O3 stacking sequence of the parent compound, but with ∼13% lower unit cell volume. Electron diffraction and atomic-resolution scanning transmission electron microscopy imaging revealed short-range Ni/Sn ordering in both the pristine and Li-exchanged materials that is similar to the “honeycomb” Li/M ordering in Li2MO3 oxides. As supported by bond-valence sum and density functional theory calculations, this ordering is driven by charge difference between Ni2+ and Sn4+ and the necessity to maintain balanced bonding for the oxygen anions. Li0.35Na0.07Ni0.5Sn0.5O2 demonstrates reversible electrochemical (de)intercalation of ∼0.21 Li+ in the 2.8–4.3 V vs. Li/Li+ potential range. Limited electrochemical activity is attributed to a formation of the surface Li/Ni disordered rock-salt barrier layer as the Li+ for Na+ exchange drastically reduces the energy barrier for the Li/Ni antisite disorder.
m). It preserves the O3 stacking sequence of the parent compound, but with ∼13% lower unit cell volume. Electron diffraction and atomic-resolution scanning transmission electron microscopy imaging revealed short-range Ni/Sn ordering in both the pristine and Li-exchanged materials that is similar to the “honeycomb” Li/M ordering in Li2MO3 oxides. As supported by bond-valence sum and density functional theory calculations, this ordering is driven by charge difference between Ni2+ and Sn4+ and the necessity to maintain balanced bonding for the oxygen anions. Li0.35Na0.07Ni0.5Sn0.5O2 demonstrates reversible electrochemical (de)intercalation of ∼0.21 Li+ in the 2.8–4.3 V vs. Li/Li+ potential range. Limited electrochemical activity is attributed to a formation of the surface Li/Ni disordered rock-salt barrier layer as the Li+ for Na+ exchange drastically reduces the energy barrier for the Li/Ni antisite disorder.
1 Introduction
Portable electronics based on Li-ion technology have already become an integral part of human daily life. However, the interest in the new energy storage technologies is driven by the enhancing demands of electric vehicles and power grid markets. Layered LiNiO2 as well as its Mn and Co-substituted analogues LiNixCoyMn1−x−yO2 where x ≥ 0.5 (also called “Ni-rich NMCs”) are considered as promising cathode materials for advanced Li-ion batteries due to their high discharge capacity and cycling rate as well as relatively low cost.1,2 However, their commercial application is limited by low cycling stability and safety problems, which reside in gaseous O2 evolution from the surface of the highly delithiated cathode material causing inflammation risk of organic electrolyte.3 It is commonly known that Ni-rich layered oxides undergo several phase transitions upon charge/discharge,4 among which the high voltage one results in both oxygen release and subsequent structure compression, causing cracking of the cathode particles and subsequent material degradation.5Numerous strategies have been proposed to improve the structural stability and electrochemical performance of Ni-rich layered oxides, including surface coating and chemical doping with a large variety of cationic species.6 Nevertheless, among all doping agents, Sn4+ may be considered as one of the most intriguing from different points of view. On the one hand, Sn4+ was shown to suppress multiple phase transitions in O3 Na-containing layered oxides as well as to raise the average operating voltage, which is beneficial for energy density.7 On the other hand, high Sn–O bonding energy (548 kJ mol−1) in comparison to that of the Ni–O, Co–O, and Mn–O bonds (391.6, 368, and 402 kJ mol−1, respectively) is expected to improve the structural stability of Sn-doped Ni-rich layered cathode materials.8
However, in practice, Li-containing layered oxides turned out to be very insusceptible toward the Sn4+ doping. Although Sn4+ for 3d-metal substitutions have been performed in various systems like Li[Ni0.5Co0.2Mn0.3]1−xSnxO2,8 Li[Ni0.6Mn0.2Co0.2−xSnx]O2,9 LiNi3/8Co2/8Mn3/8−xSnxO2,10 Li[Ni1/3Co1/3Mn1/3]1−xSnxO2,11 Li1.15Ni0.27Mn0.58−xSnxO2,12 Li1.2Ni0.13Co0.13Mn0.54−xSnxO2,13,14 Li[Ni0.82Co0.12Mn0.06]1−xSnxO2,15 Li[Li0.17Ni0.25Mn0.58−xSnx]O2,16 LiNi0.8−xCo0.2SnxO2,17 and finally LiCo1−xSnxO2,18 successful Sn doping has never exceeded a few atomic percents, whereas attempts to further raise the Sn content always resulted in Li2SnO3 admixture. Only for LiCo1−xSnxO2, the doping up to x = 0.10 was reported, but still, the homogeneity of Sn and Co distribution was not directly analyzed.
If the straightforward solid-state synthesis severely limits the homogeneity range of the Sn substitution resulting in exsolution of Li2SnO3, another approach based on electrochemical ion exchange has been proven to be an effective path to obtain compounds, which can't be produced by a direct chemical way. For instance, only electrochemical exchange allowed replacing Li+ for K+ in KVPO4F leading to the (Li,K)VPO4F cathode materials.19 Similar synthetic route was also used to produce mixed Li2−xNaxFePO4F cathode material20 and obtain the olivine-structured NaFePO4 from LiFePO4.21
In contrast to the Li-based layered oxides, up to 50% of transition metal cations can be substituted with Sn4+ in Na-based oxide, as evidenced with the successful direct solid state synthesis of O3-NaNi0.5Sn0.5O2, from which up to 0.9Na can be extracted being charged to 4.2 V in sodium half-cell.7 Therefore, in our study we focused on electrochemical Li+ for Na+ exchange in NaNi0.5Sn0.5O2. The additional argument promoting feasibility of such exchange lies in the fact that NaNi0.5Sn0.5O2 adopts the O3-type structure (denoted after Delmas et al.,22 where O stands for octahedral coordination of alkali cations, with three octahedral layers per repeat period along the c-axis), thus providing the most favorable coordination environment for Li+ in contrast to the P-type phases with trigonal prismatic oxygen coordination of the alkali metals. Herein we report on the synthesis, crystal structure and electrochemical properties of a novel ANi0.5Sn0.5O2 (A = Li, Na) layered oxide and on the evolution of its crystal structure during electrochemical alkali metal substitution investigated by ex situ high-resolution transmission electron microscopy (TEM), providing in-depth study of the exchange process.
2 Materials and methods
Synthesis of Na1−xNi0.5Sn0.5O2
Pristine Na1−xNi0.5Sn0.5O2 was obtained using the synthesis procedure as described by M. Sathiya et al.7 Na2CO3 (Sigma Aldrich), NiO (Sigma Aldrich), and SnO2 (Sigma Aldrich) in the required amounts were mixed and grinded for 10–20 minutes in an agate mortar. Then the resulting powder was ball-milled for 1 h in a SPEX 8000 M mixer/mill under ethanol. Obtained homogeneous mixture was annealed in air for 12 hours at 850 °C (300 °C h−1 heating rate) and then in Ar-flow for 12 hours at 900 °C (60 °C h−1 heating rate, 20 ml min−1 Ar flow dried with P2O5), with intermediate regrinding between the annealing steps. After synthesis, the material was stored in an Ar-filled glove box.Structural characterization
In order to perform phase analysis and crystal structure refinement, powder X-ray diffraction (PXRD) patterns were recorded on a Huber G670 Guinier diffractometer using Co-Kα1 radiation (λ = 1.78892 Å), a curved Ge (111) monochromator, and an image plate detector at room temperature over the 10°–100° 2θ range with the angular step of 0.01°. The unit cell parameters were refined using the Le Bail method and crystal structures were refined using the Rietveld method with the JANA2006 program package.23For the transmission electron microscopy (TEM) studies, a tiny amount of powder sample was grinded with an agate mortar and pestle under ethyl alcohol. The samples after electrochemical cycling were prepared in an Ar-filled glove box by crushing the cathode mass in a mortar with dimethyl carbonate and depositing a drop of suspension onto holey-carbon-coated copper grids. The samples were transported to the microscope column in a Gatan vacuum transfer holder completely excluding contact with air. Electron diffraction (ED) patterns, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images, and EDX compositional maps in the STEM mode (STEM-EDX) were acquired on a probe aberration-corrected Titan Themis Z electron microscope at 200 kV equipped with a Super-X system for energy-dispersive X-ray (EDX) analysis.
Electrochemical measurements and Na/Li exchange
Cathodes for electrochemical measurements and Na/Li exchange were prepared by mixing 80 wt% of the active material, 10 wt% of carbon Super-P and 10 wt% of polyvinylidene fluoride (PVDF). The mixture was dissolved in N-methyl-2-pyrrolidinone (NMP) and stirred to form a homogeneous slurry, which was then coated onto a carbon-coated aluminum current collector using a Zehntner ZAA 2300 applicator and dried at 70 °C until NMP is completely evaporated. The mass loadings of the active material on a round aluminum foil of 16 mm in diameter were ≈5 mg. Two-electrode cells, assembled in Ar-filled glove box (O2, H2O < 1 ppm), were used for both electrochemical measurements and Na/Li exchange. For a preliminary galvanostatic test, the half-cells were assembled consisting of the cathode, metallic Na foil as a counter electrode, and solution of 1 M NaClO4 in a mixture of ethylene carbonate (EC), propylene carbonate (PC) in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with 2 wt% of fluoroethylene carbonate (FEC). For Li for Na exchange and cyclic voltammetry (CV) measurements, metallic Li and 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC)/propylene carbonate (PC)/dimethyl carbonate (DMC) (1:1:3, vol%) was used as a counter electrode and as an electrolyte, respectively. All electrochemical measurements were performed in a 2-electrode setup at 22 °C using Biologic VMP3 potentiostat/galvanostat (EC-Lab software).
:1 with 2 wt% of fluoroethylene carbonate (FEC). For Li for Na exchange and cyclic voltammetry (CV) measurements, metallic Li and 1 M LiPF6 dissolved in a mixture of ethylene carbonate (EC)/propylene carbonate (PC)/dimethyl carbonate (DMC) (1:1:3, vol%) was used as a counter electrode and as an electrolyte, respectively. All electrochemical measurements were performed in a 2-electrode setup at 22 °C using Biologic VMP3 potentiostat/galvanostat (EC-Lab software).
DFT calculation details
Density functional theory (DFT) calculations were performed using Perdew–Burke–Ernzerhof (PBE) flavor24 of the generalized gradient approximation (GGA) to exchange–correlation functional and standard PAW potentials as implemented in the VASP program.25 To take into account the strongly correlated character of d-electrons, a Hubbard-like correction is added within the Dudarev scheme and U value of 6.2 eV for Ni. The energy cutoff of 400 eV and the maximum spacing between k-points of 0.30 Å−1 were chosen for the calculations. The relaxation of atomic positions and cell geometry was performed using the conjugate-gradient method, the maximum force acting on each atom after relaxation was less than 0.025 eV Å−1. To eliminate Pulay errors, the lattice optimization (ISIF = 4) of initial bulk structures was performed at constant volume for several contracted and expanded cells (7 points). All calculations were done with the help of the SIMAN software package used for high-throughput DFT calculations.263 Results and discussion
PXRD pattern of Na1−xNi0.5Sn0.5O2 (Fig. 1a) indicates a rhombohedral O3-layered structure (α-NaFeO2 structure type, S. G. Rm) with the unit cell parameters a = 3.10624(6) Å, c = 16.1653(5) Å that agrees well with the unit cell parameters reported by Sathiya et al.7 The elemental composition and homogeneity were investigated by STEM-EDX (Fig. 2). The material consists of elongated rectangular crystals of 300–500 nm in size and demonstrates the homogeneous distribution of Ni, Sn, and Na. The quantification of the EDX spectra resulted in the Na0.92(5)Ni0.49(4)Sn0.51(4)O2 composition indicating a small Na deficiency.
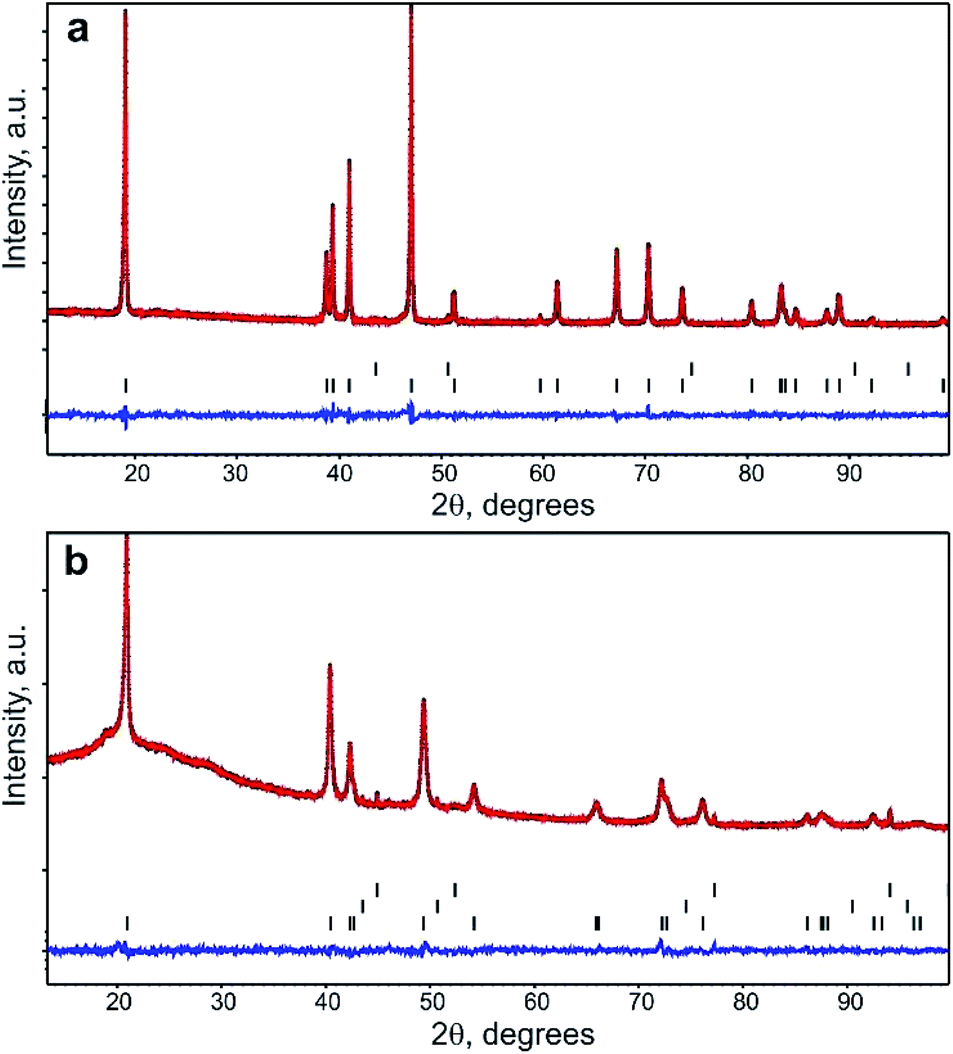 | ||
| Fig. 1 Experimental (black), calculated (red), and difference (blue) powder XRD patterns after the Rietveld refinement for the pristine (a) and Li-exchanged (b) materials. Black vertical lines designate the Bragg reflections positions for NiO (top row) and Na0.95Ni0.5Sn0.5O2 (bottom row) in the pristine sample and Al current collector (top row), NiO (middle row), and Li0.35Na0.07Ni0.5Sn0.5O2 (bottom row) in the Li-exchanged sample. | ||
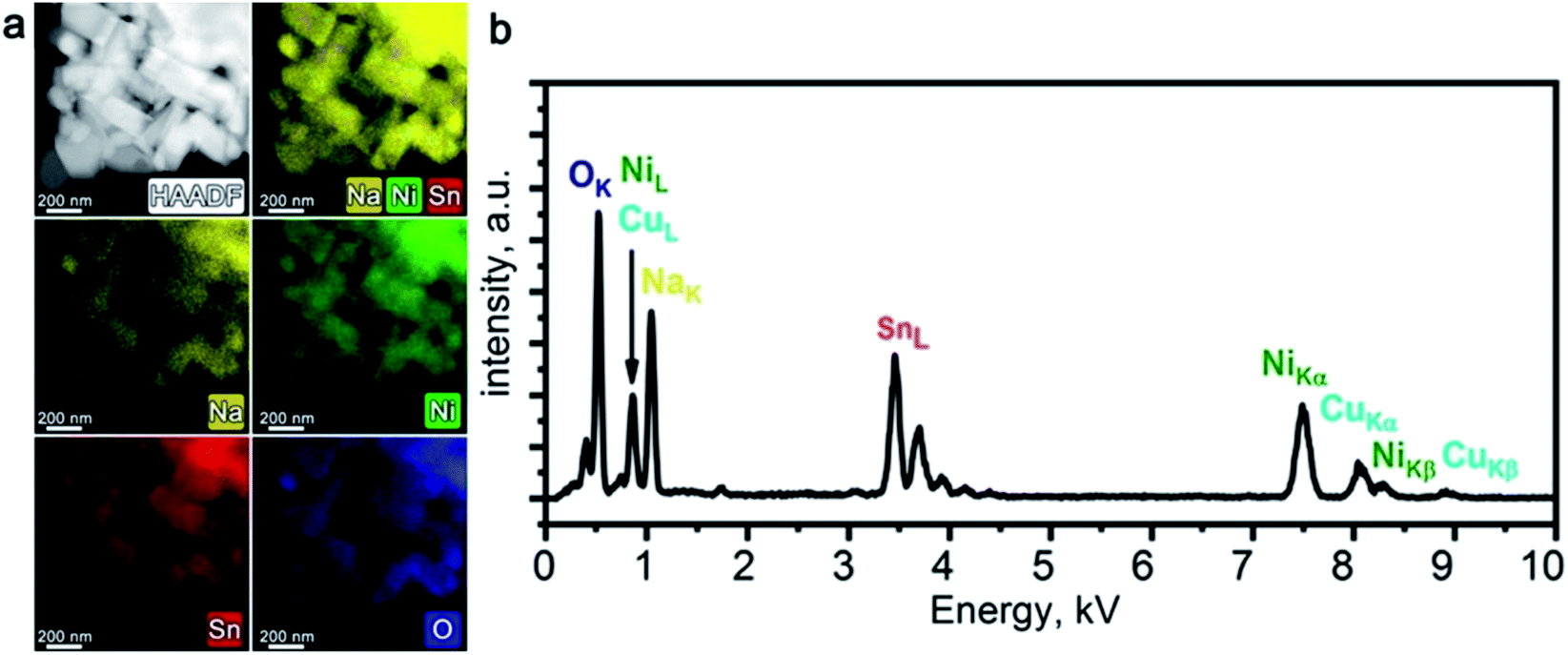 | ||
| Fig. 2 HAADF-STEM image of Na1−xNi0.5Sn0.5O2 along with corresponding colour-coded EDX elemental maps of Na, Ni, Sn, and O (a); typical EDX spectrum of Na1−xNi0.5Sn0.5O2 (b). | ||
For the Rietveld refinement of the Na1−xNi0.5Sn0.5O2 crystal structure, the M1 3a position was jointly populated by Ni and Sn in 50:50 ratio according to the nominal composition and the results of the EDX analysis. The M2 3b position was populated by Na and its occupancy factor was refined resulting in g = 0.95(1), thus confirming the Na deficiency detected with EDX. The final refinement was conducted with a common atomic displacement parameter (ADP) for all positions, also including NiO as an admixture phase (the content amounts to 0.6(2) wt%). Crystallographic parameters from the Rietveld refinement, atomic coordinates, occupancy factors, ADPs, and selected bond distances are presented in Tables S1 and S2 of ESI.† The PXRD profiles after the Rietveld refinement are shown in Fig. 1a. Following the combined EDX and PXRD results, the Na0.95Ni0.5Sn0.5O2 composition is assigned to the pristine material.
Preliminary electrochemical characterization of Na0.95Ni0.5Sn0.5O2 was performed by galvanostatic cycling between 2.7 and 4.0 V vs. Na/Na+. Galvanostatic curves of Na0.95Ni0.5Sn0.5O2 (Fig. 3a) demonstrate the sloped behavior. The initial charge capacity amounts to 110 mA h g−1 that corresponds to the removal of ∼0.6Na, but only ∼0.4Na returns back on subsequent discharge providing the discharge capacity of 74 mA h g−1 and coulombic efficiency of 68% (low first cycle coulombic efficiency was also reported by Sathiya et al.7). The discharge capacity slightly fades to 71 mA h g−1 during the next 10 charge/discharge cycles.
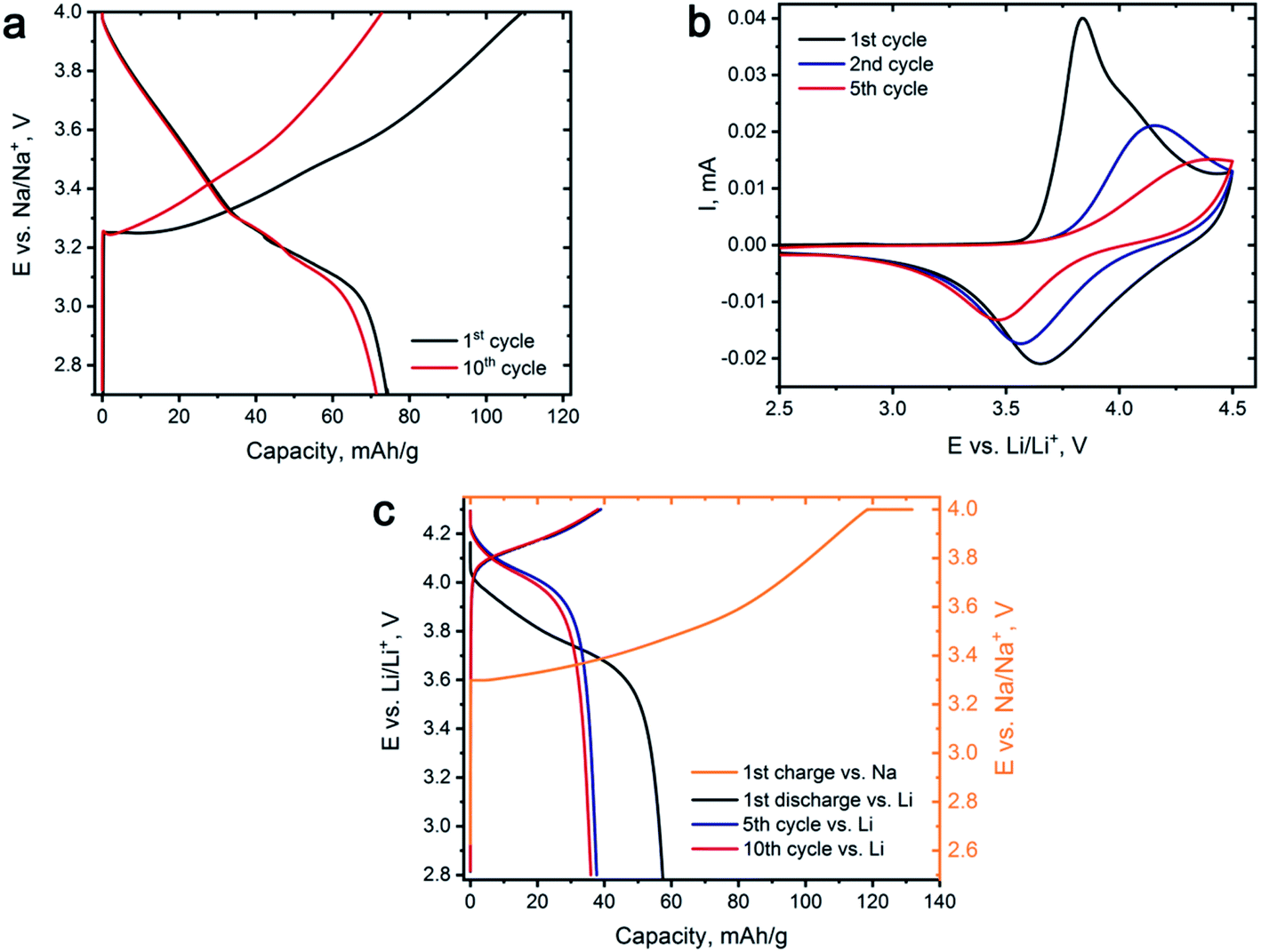 | ||
| Fig. 3 (a) Selected galvanostatic charge–discharge curves for Na0.95Ni0.5Sn0.5O2 cycled vs. Na at 18 mA g−1 current density. (b) Cyclic voltammogram for the 1st, 2nd and 5th cycles of Na0.95Ni0.5Sn0.5O2 vs. Li at 0.1 mV s−1 scanning rate. (c) Galvanostatic charge–discharge curves, representing the conditions of electrochemical ion exchange. | ||
Cyclic voltammetry study of Na0.95Ni0.5Sn0.5O2 was carried out in Li half-cell (Fig. 3b) to identify the voltage regions for Li for Na exchange and select appropriate ion exchange conditions. During the first charge, an intense peak having a shoulder at the high potential side appears on the CV curve at 3.7–4.0 V vs. Li/Li+, which originates from Na removing accompanied by Ni2+ → Ni3+ → Ni4+ oxidation. On the reverse scan, the broad peak near 3.6–3.7 V vs. Li/Li+ is potentially responsible for Li insertion and Ni reduction. The observed current decreases from cycle to cycle on both direct and reverse scans and signals of both anodic and cathodic processes are gradually shifting to higher and lower potentials, respectively, increasing the voltage hysteresis. Such behavior might indicate substantial structural changes in the Li-exchanged material while cycling vs. Li causing increasing kinetic limitations of Li for Na exchange.
The conditions of the electrochemical Li for Na exchange were determined based on CV results. The potential range has been limited to 2.8–4.3 V vs. Li/Li+ as the main electrochemical activity is concentrated in this potential region, and the whole procedure was divided into two steps. Na0.95Ni0.5Sn0.5O2 was first charged in a galvanostatic regime to 4.3 V vs. Na/Na+ at C/18 rate and held at this potential for 10 hours resulting in deintercalation of ∼0.7Na. Then, the charged electrode was removed from the cell, carefully washed with a large amount of DMC, transferred to Li half-cell, and subjected to 10 charge/discharge cycles vs. metallic Li with a current density of 10 mA g−1, corresponding to C/18 rate or 18 hours per charge and discharge (36 hours for the full cycle). The measures, such as short potential range, low cycling rate, and Na+ pre-extraction were aimed at overcoming the kinetic limitations as well as minimizing the risk of joint Na and Li insertion.
Fig. 3c demonstrates the galvanostatic curves taken during the Na+/Li+ ion exchange. After the first charge with the 4.3 V potential plateau, we managed to extract ≈0.73 Na+, which resulted in the Na0.27Ni0.5Sn0.5O2 composition before transferring the electrode to the Li half-cell. After the first discharge, about ∼0.32 Li+ was inserted and then continuous discharge capacity degradation was observed during next five cycles until the steady state is reached corresponding to reversible (de)intercalation of ∼0.21 Li+.
STEM-EDX did not reveal any Ni/Sn segregation in the Li-exchanged material after 10 charge/discharge cycles in Li half-cell (Fig. 4a). Prominent Na depletion is detected on both elemental maps and EDX spectra, but the Na–K peak does not vanish completely (Fig. 4b). The resulting chemical composition corresponds to LixNa0.07(2)Ni0.51(1)Sn0.49(1)O2 (Li cannot be quantified with EDX) indicating that both electrochemical and chemical Li for Na ion exchange occurred, but a tiny amount of Na was not fully removed.
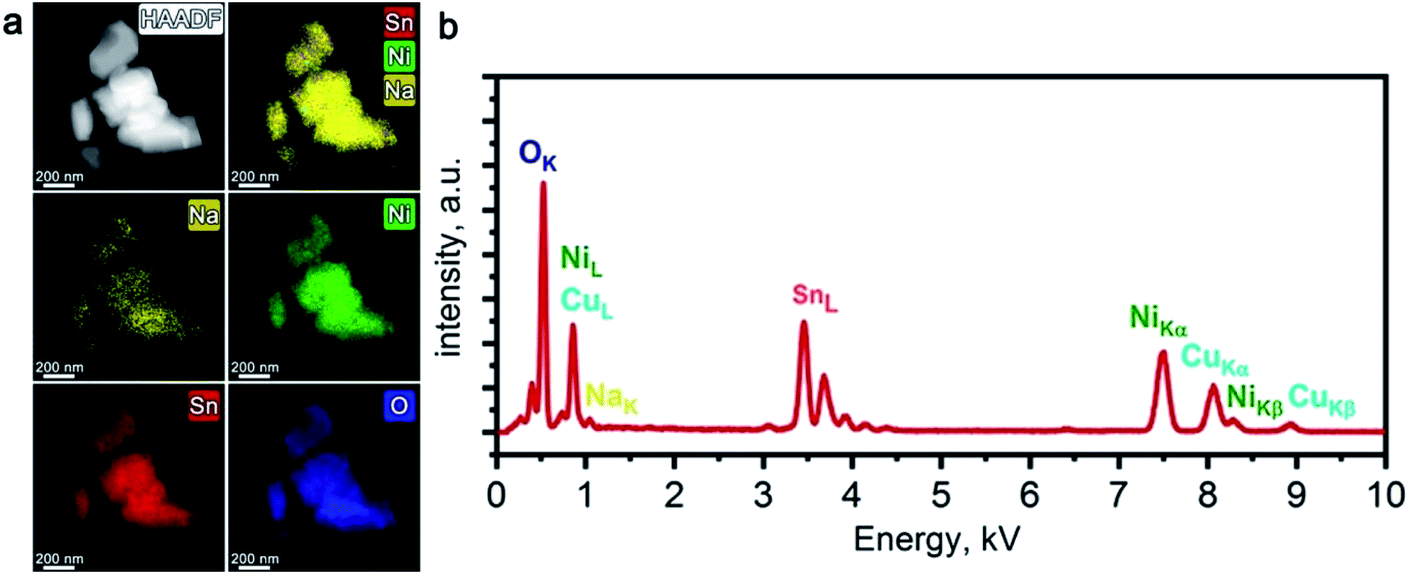 | ||
| Fig. 4 HAADF-STEM image of the Li-exchanged material along with corresponding colour-coded EDX elemental maps of Na, Ni, Sn, and O (a); typical EDX spectrum of the Li-exchanged material (b). | ||
The crystal structure of the Li-exchanged material was refined by the Rietveld method (Fig. 1b, Tables S1 and S2 of ESI†). Similar to pristine Na0.95Ni0.5Sn0.5O2, it crystallizes in the rhombohedral O3-layered structure (α-NaFeO2-type S. G. Rm) with a = 3.03431(7) Å, c = 14.7491(8) Å. Significant shrinking of the unit cell volume from 135.1 Å3 for the pristine material to 117.6 Å3 in the Li-exchanged material evidences successful Na+/Li+ substitution (r(Na+)CN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) VI = 1.02 Å, r(Li+)CNVI = 0.76 Å). The occupation of the M2 position by Ni and Sn was kept intact, whereas the M1 position was first populated by Li only and its occupancy factor was refined, resulting in the electron density corresponding to ∼1.8 e−. This electron density was distributed between 0.07Na (according to the EDX analysis) and Li atoms. The refinement of the occupancy factor for the latter resulted in g = 0.35(2) and the Li0.35Na0.07Ni0.5Sn0.5O2 chemical formula for the Li-exchanged material that corresponds well to ∼0.32 Li+ inserted at the first stage of the cation exchange.
VI = 1.02 Å, r(Li+)CNVI = 0.76 Å). The occupation of the M2 position by Ni and Sn was kept intact, whereas the M1 position was first populated by Li only and its occupancy factor was refined, resulting in the electron density corresponding to ∼1.8 e−. This electron density was distributed between 0.07Na (according to the EDX analysis) and Li atoms. The refinement of the occupancy factor for the latter resulted in g = 0.35(2) and the Li0.35Na0.07Ni0.5Sn0.5O2 chemical formula for the Li-exchanged material that corresponds well to ∼0.32 Li+ inserted at the first stage of the cation exchange.
However, comparative ex situ TEM investigation of pristine Na0.95Ni0.5Sn0.5O2 and Li-exchanged electrodes after 1st and 10th cycles in Li half-cell revealed the structure evolution apparently different from the results of the Rietveld refinement. The [010] and [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10] HAADF-STEM images and corresponding SAED patterns are shown in Fig. 5 and 6, respectively. The main reflections on the SAED patterns of all three materials correspond well with the rhombohedral Rm O3-type structure corroborating the PXRD structure analysis. In the [10] SAED patterns diffuse intensity lines along the h/3,h/3,l, h ≠ 3n reciprocal lattice rows are clearly visible. These diffuse intensity lines are reminiscent of those in the Li-rich NMC layered oxides where they originate from a “honeycomb” ordering of Li and transition metals within the (LixM1−x)O2 (M – transition metal) slabs, in which each LiO6 octahedron tends to be surrounded by six MO6 octahedra.27 As in both Na0.95Ni0.5Sn0.5O2 and Li0.35Na0.07Ni0.5Sn0.5O2 the alkali metals are not present in the Ni/Sn layers in noticeable amount, the diffuse intensity lines must arise from the short-range ordering of the Ni2+ and Sn4+ cations driven by the size and charge difference. The perfect order, however, requires the Ni:Sn = 1:2 atomic ratio (such as in Li2SnO3 = Li[Li1/3Sn2/3]O2), thus only the locally ordered domains can be realized at the Ni:Sn = 1:1 ratio in the studied materials. The tendency to depart from a totally random arrangement of the Ni2+ and Sn4+ cations can be rationalized using bond valence sum (BVS) arguments.28 In the idealized Na0.95Ni0.5Sn0.5O2 structure each oxygen atom is surrounded by 3(Ni/Sn) and 3Na cations resulting in possible 3Sn + 3Na, 2Sn/1Ni + 3Na, 1Sn/2Ni + 3Na and 3Ni + 3Na local configurations which correspond to the BVS values of 2.34, 2.07, 1.81 and 1.54, respectively. For the 3Sn + 3Na and 3Ni + 3Na configurations, the oxygen atoms appear severely overbonded and underbonded (i.e. BVS is significantly larger or smaller than the oxygen formal valency), respectively, and these configurations must be avoided that dictates local cation ordering. In addition to BVS, the bulk energies of ordered and random distribution of Ni/Sn atoms in MO6 layers were compared using DFT calculations. The ordered distribution of Ni and Sn in (Ni,Sn)O6 layers was shown to be more energetically favorable in both LiNi0.5Sn0.5O2 and NaNi0.5Sn0.5O2 (Table S3†).
10] HAADF-STEM images and corresponding SAED patterns are shown in Fig. 5 and 6, respectively. The main reflections on the SAED patterns of all three materials correspond well with the rhombohedral Rm O3-type structure corroborating the PXRD structure analysis. In the [10] SAED patterns diffuse intensity lines along the h/3,h/3,l, h ≠ 3n reciprocal lattice rows are clearly visible. These diffuse intensity lines are reminiscent of those in the Li-rich NMC layered oxides where they originate from a “honeycomb” ordering of Li and transition metals within the (LixM1−x)O2 (M – transition metal) slabs, in which each LiO6 octahedron tends to be surrounded by six MO6 octahedra.27 As in both Na0.95Ni0.5Sn0.5O2 and Li0.35Na0.07Ni0.5Sn0.5O2 the alkali metals are not present in the Ni/Sn layers in noticeable amount, the diffuse intensity lines must arise from the short-range ordering of the Ni2+ and Sn4+ cations driven by the size and charge difference. The perfect order, however, requires the Ni:Sn = 1:2 atomic ratio (such as in Li2SnO3 = Li[Li1/3Sn2/3]O2), thus only the locally ordered domains can be realized at the Ni:Sn = 1:1 ratio in the studied materials. The tendency to depart from a totally random arrangement of the Ni2+ and Sn4+ cations can be rationalized using bond valence sum (BVS) arguments.28 In the idealized Na0.95Ni0.5Sn0.5O2 structure each oxygen atom is surrounded by 3(Ni/Sn) and 3Na cations resulting in possible 3Sn + 3Na, 2Sn/1Ni + 3Na, 1Sn/2Ni + 3Na and 3Ni + 3Na local configurations which correspond to the BVS values of 2.34, 2.07, 1.81 and 1.54, respectively. For the 3Sn + 3Na and 3Ni + 3Na configurations, the oxygen atoms appear severely overbonded and underbonded (i.e. BVS is significantly larger or smaller than the oxygen formal valency), respectively, and these configurations must be avoided that dictates local cation ordering. In addition to BVS, the bulk energies of ordered and random distribution of Ni/Sn atoms in MO6 layers were compared using DFT calculations. The ordered distribution of Ni and Sn in (Ni,Sn)O6 layers was shown to be more energetically favorable in both LiNi0.5Sn0.5O2 and NaNi0.5Sn0.5O2 (Table S3†).
 | ||
| Fig. 5 [010] HAADF-STEM images with corresponding SAED patterns for the pristine Na0.95Ni0.5Sn0.5O2 (a) and Li-exchanged materials after the 1st (b) and 10th charge/discharge cycles (c) in Li half-cell. | ||
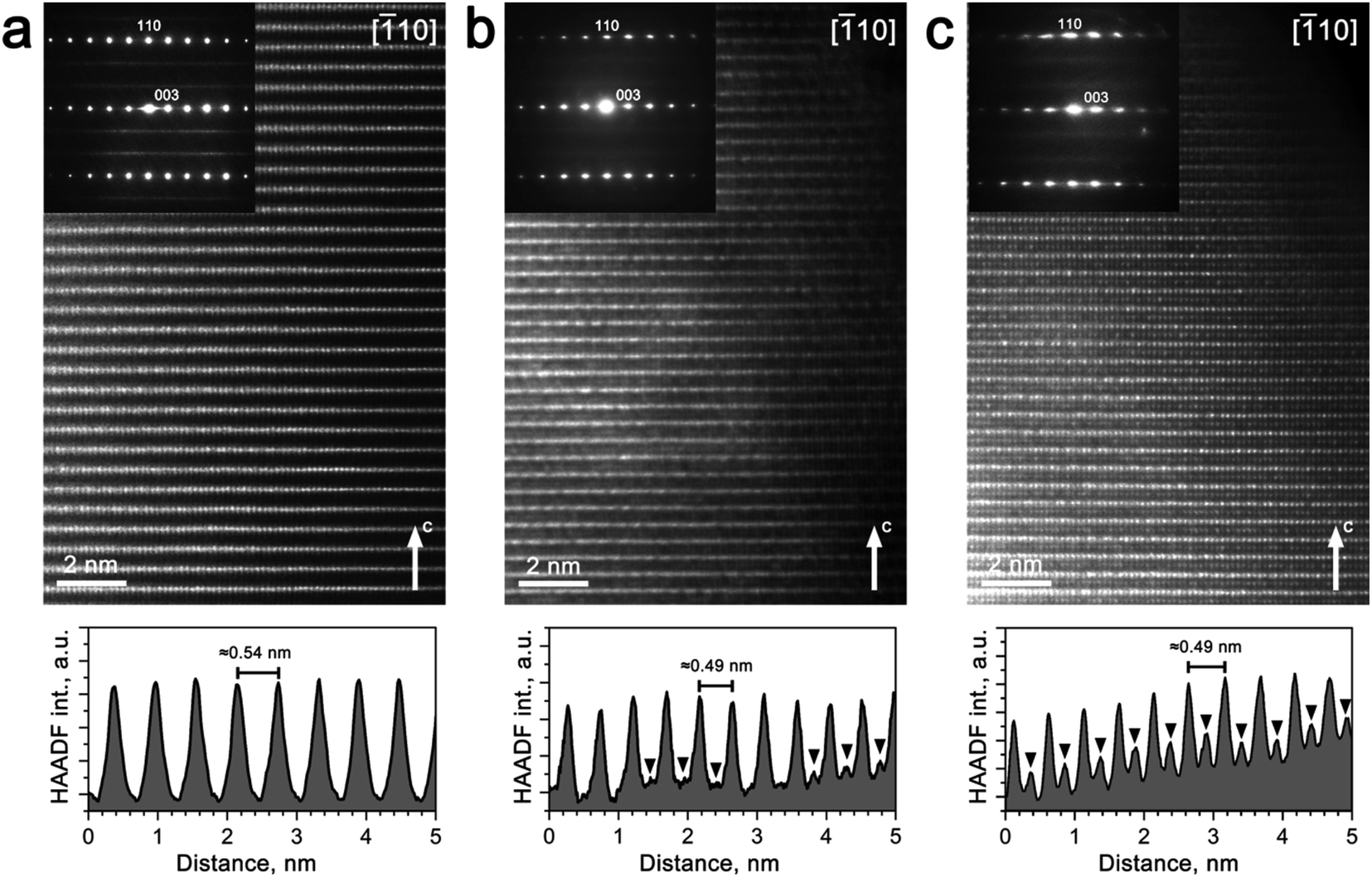 | ||
| Fig. 6 [10] HAADF-STEM images with the SAED patterns for the pristine Na0.95Ni0.5Sn0.5O2(a) and Li-exchanged materials after the 1st (b) and 10th charge/discharge cycles (c) in Li half-cell, along with the corresponding HAADF intensity profiles plotted along the c-axis. Black arrowheads point to the peaks of the HAADF intensity caused by transition metals migrated to the M2 positions in the Na/Li layers. | ||
The virtual absence of HAADF intensity between the rows of bright spots in the [010] and [10] HAADF-STEM images of Na0.95Ni0.5Sn0.5O2 (Fig. 5a and 6a) reflects the well-established layered ordering of the Na and Ni/Sn cations. In the [010] HAADF-STEM images of all three structures, the layers of the bright dots demonstrate systematic lateral shift over 1/3 of the interdot distance that is the signature of the O3-type stacking (Fig. 5). In contrast to the uniform brightness of the dots of Ni/Sn atomic columns expected for the disordered Rm structure, small domains with alternating pairs of bright dots separated by a single fainter dot can be discerned in the [10] HAADF-STEM images (Fig. 6) attributed to local Ni/Sn ordering, in agreement with the SAED patterns.
The Li-exchanged structures differ significantly from the pristine one by shrinking the interlayer separation from ∼0.54 nm to ∼0.49 nm. The cationic migration from the Ni/Sn layers into the Na/Li layers is clearly detected already after the first discharge (Fig. 5b and 6b), while after 10 cycles it becomes even more pronounced (Fig. 5c and 6c). This migration is particularly well seen from the HAADF intensity profiles in Fig. 6 demonstrating a steady increase of scattering density at the M2 positions of the Na/Li layers. Thus, HAADF-STEM imaging points to suppression of the layered Ni/Sn and Na/Li ordering. It should be noted that no extra reflections appear in the SAED patterns of the Li-exchanged materials as could be expected if the layered structure is converted into a distorted spinel one, thus a disordered rock-salt structure is gradually formed with an increasing number of cycles. This might be a reason for the limited amount of Li inserted during the Li for Na exchange, as the metal cations in alkali layers reduce the number of crystallographic positions available for Li and also block the diffusion paths of alkali metal.4
Although this explanation looks plausible at the first glance, it obviously contradicts the Rietveld refinement result for the Li0.35Na0.07Ni0.5Sn0.5O2 structure which demonstrated small scattering density at the M2 position, inconsistent with the massive migration of the Ni/Sn cations. As PXRD probes the whole volume of the crystallites whereas HAADF-STEM imaging is confined to the surface layer, the apparent discrepancy might stem from the difference in cation migration at the surface and in the bulk. Thus, the relative scattering density at the octahedral sites in the M1 and M2 positions was estimated as a function of the distance from the crystal surface (Fig. 7). If at ∼2 nm near-surface layer the HAADF intensity at the M2 positions amounts to ∼50% of the intensity at the M1 positions indicating pronounced cation migration, it gets significantly dumped at ∼4 nm from the surface and almost vanishes at the distance of ∼6 nm. This reflects that the Ni/Sn cation migration is confined to the thin (<10 nm) surface layer and does not affect the interior part of the crystallites much.
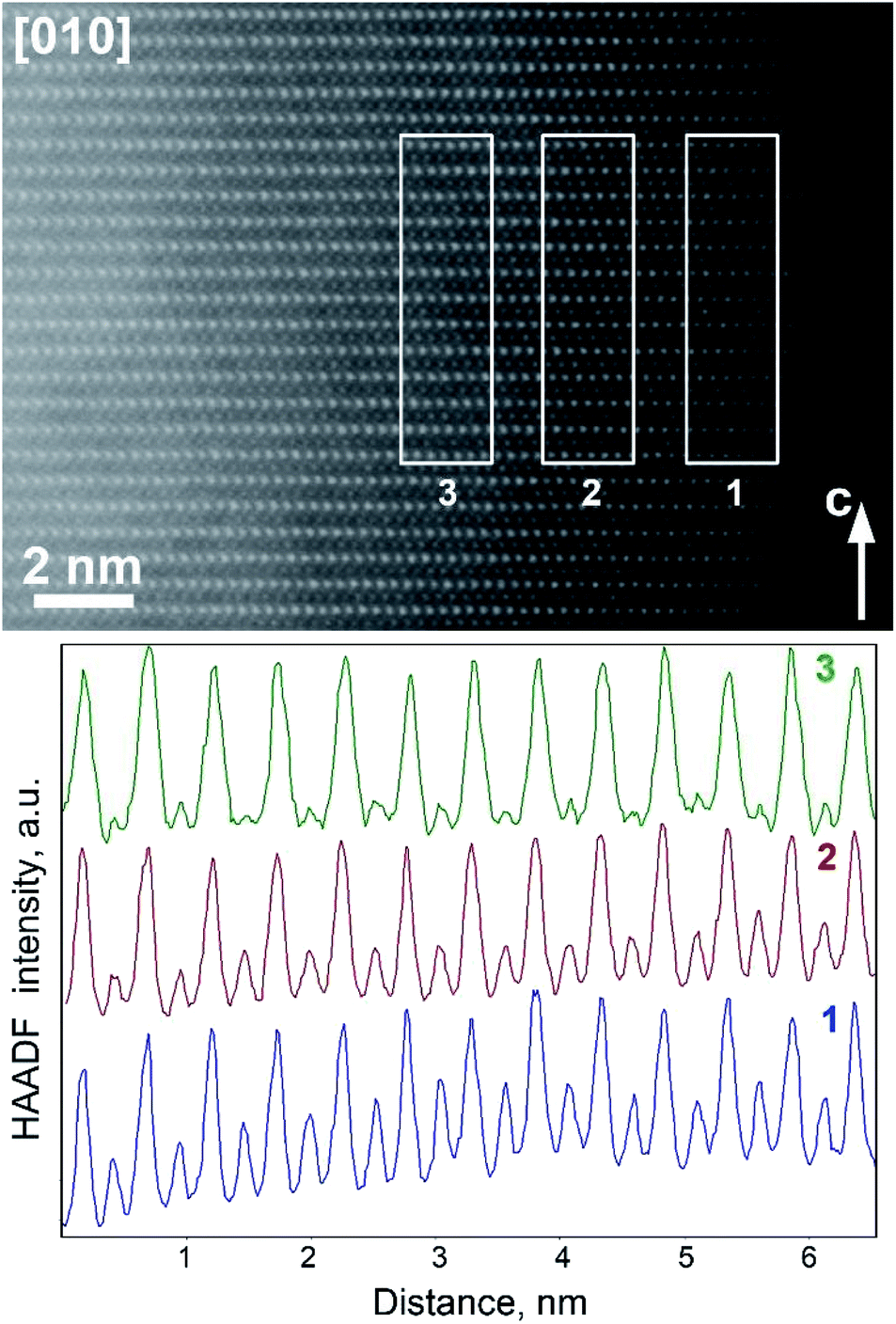 | ||
| Fig. 7 [010] HAADF-STEM image of Li0.35Na0.07Ni0.5Sn0.5O2 and the HAADF intensity profiles integrated over the marked rectangular areas at 2 nm (1), 4 nm (2), and 6 nm (3) from the surface. The profiles are normalized to the height of the strongest peaks corresponding to the M1 atomic columns. Note strong fade of HAADF intensity in the atomic columns of the M2 positions (weaker peaks) upon increasing distance from the surface. | ||
The distinctly different near-surface structure of Li0.35Na0.07Ni0.5Sn0.5O2 becomes evident from the analysis of Fourier transforms (FFTs) (Fig. 8). Sharp reflections from both bulky and near-surface areas belong to the O3-type layered structure, but the near-surface FFT demonstrates structured wave-shaped diffuse intensity which is absent in the FFTs from the interior part of the [010]-oriented crystallites and replaced with the straight diffuse intensity lines due to short-range Ni/Sn ordering in the [10]-oriented crystallites. The wave-shaped diffuse intensity lines are very typical for the f. c. c.-type structures (i.e. for the rock-salt disordered surface layer) and appear due to sectioning of 3D reciprocal space surface corresponding to the cosπh + cosπk + cosπl = 0 equation. This diffuse intensity distribution is characteristic of short-range order with octahedral clusters,29 which can be understood as a violation of random distribution of the cationic species due to the tendency for each O atom to be as homogeneously as possible surrounded by the Li+, Ni2+, Sn4+ cations, and cationic vacancies.
 | ||
| Fig. 8 [010] and [10] HAADF-STEM images of Li0.35Na0.07Ni0.5Sn0.5O2 along with fast Fourier transforms from the encircled areas in the bulky (1) and near-surface (2) areas. Note the difference in diffuse intensity distribution in the Fourier transforms of the areas 1 and 2. | ||
The low electrochemical activity of desodiated Na0.95Ni0.5Sn0.5O2 in Li half-cell looks somewhat surprising as there seemingly no structural and electronic reasons for limiting the reversible electrochemical capacity to just ∼20% of 210 mA h g−1 theoretical capacity for fully lithiated LiNi0.5Sn0.5O2 assuming the realization of the Ni2+ → Ni3+ → Ni4+ redox. One may assume that drastic 13% shrinkage of the unit cell volume could prevent the full occupation of the M2 position by the Li cations because of unfavourable overbonding of the oxygen atoms coordinated by 2Sn/1Ni + 3Li or 1Sn/2Ni + 3Li. However, the BVS values for these coordination environments amount to 2.02 and 1.73 (as calculated with the bond distances in Li0.35Na0.07Ni0.5Sn0.5O2), respectively, indicating that complete Li insertion is needed to properly bound the oxygen atoms. Indeed, the global instability index (GII, 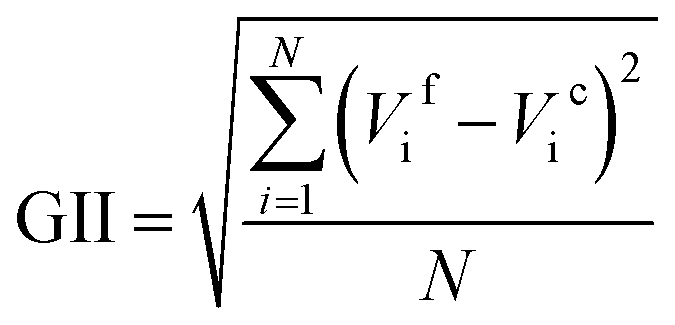 , where Vif and Vic are the formal and BVS-calculated valences of the cation i and N is the number of cations in asymmetric unit30) is equal to 0.326 for idealized NaNi0.5Sn0.5O2 and 0.172 for LiNi0.5Sn0.5O2 demonstrating that the latter looks much better balanced with respect to the chemical bonding. Thus, we tend to attribute the observed low electrochemical activity to a formation of blocking rock-salt-structured disordered layer at the surface of the crystallites, which spans over dozens of unit cells. The driving force behind increased migration of the Ni/Sn cations towards octahedral sites in the alkali layer is the closeness of ionic radii of Ni2+ and Sn4+ (the same ionic radius rVI = 0.69 Å) and Li+ (rVI = 0.74 Å) in contrast to large difference with Na+ (rVI = 1.02 Å). We additionally estimated the formation energies of antisite defect (AS) pairs AM-MA (A = Li, Na; M = Ni, Sn) in the bulk ANi0.5Sn0.5O2 phases using DFT (Table 1). It can be seen that the formation energies of AS pairs in LiNi0.5Sn0.5O2 for both Ni and Sn are twice lower than the same in NaNi0.5Sn0.5O2, while for tin it is twice higher than for nickel in both structures. The obtained data from Table 1 predict more probable Li/Ni exchange in LiNi0.5Sn0.5O2 (Ef = 0.86 eV) when the Sn atoms must remain in their metal layer positions. These results coincide well with previously reported lower AS formation energies in LiNiO2 compared to NaNiO2.31
, where Vif and Vic are the formal and BVS-calculated valences of the cation i and N is the number of cations in asymmetric unit30) is equal to 0.326 for idealized NaNi0.5Sn0.5O2 and 0.172 for LiNi0.5Sn0.5O2 demonstrating that the latter looks much better balanced with respect to the chemical bonding. Thus, we tend to attribute the observed low electrochemical activity to a formation of blocking rock-salt-structured disordered layer at the surface of the crystallites, which spans over dozens of unit cells. The driving force behind increased migration of the Ni/Sn cations towards octahedral sites in the alkali layer is the closeness of ionic radii of Ni2+ and Sn4+ (the same ionic radius rVI = 0.69 Å) and Li+ (rVI = 0.74 Å) in contrast to large difference with Na+ (rVI = 1.02 Å). We additionally estimated the formation energies of antisite defect (AS) pairs AM-MA (A = Li, Na; M = Ni, Sn) in the bulk ANi0.5Sn0.5O2 phases using DFT (Table 1). It can be seen that the formation energies of AS pairs in LiNi0.5Sn0.5O2 for both Ni and Sn are twice lower than the same in NaNi0.5Sn0.5O2, while for tin it is twice higher than for nickel in both structures. The obtained data from Table 1 predict more probable Li/Ni exchange in LiNi0.5Sn0.5O2 (Ef = 0.86 eV) when the Sn atoms must remain in their metal layer positions. These results coincide well with previously reported lower AS formation energies in LiNiO2 compared to NaNiO2.31
| AS defect | Ef, eV | AS defect | Ef, eV |
|---|---|---|---|
| LiNi–NiLi | 0.86 | NaNi–NiNa | 2.11 |
| LiSn–SnLi | 2.03 | NaSn–SnNa | 4.18 |
In fact, the size similarity of Sn4+ and Li+, in contrast to Na+, might be at the origin of thermodynamic instability of LiNi0.5Sn0.5O2 with respect to decomposition to Li2SnO3 and NiO. This instability might be driven by energetically favorable Li+–Sn4+ “honeycomb” ordering in Li2SnO3, in which these cations can occupy the octahedral sites within the same layer without introducing much strain, whereas the Na2SnO3 structure must be more strained because of significant size mismatch between Sn4+ and Na+. Meanwhile, the thermodynamic stability of a compound to be its energy (at zero pressure and T = 0 K) below the ground-state phases, which for phase-separating materials is the linear combination of energies of the stable phase-separated decomposition products.32 Thus, we estimated the energy of ANi0.5Sn0.5O2 phase decomposition into A2SnO3 + NiO (A = Li, Na) products normalized per atom:
| E = [E(A2SnO3) + E(NiO) − E(A2NiSnO4)]/8 |
m) and E(A2NiSnO4) is the doubled energy of ANi0.5Sn0.5O2. (S. G. P2/m). The calculated decomposition energies per atom in ANi0.5Sn0.5O2 are −4.4 meV per atom and 0.3 meV per atom for A = Li and Na, respectively. The negative value for the LiNi0.5Sn0.5O2 phase indicates its instability against decomposition to Li2SnO3 and NiO phases, which inhibits the direct solid-state synthesis of LiNi0.5Sn0.5O2. This conclusion is experimentally supported by our attempt to synthesize ANi0.5Sn0.5O2 (A = Na0.75Li0.25) by a direct solid state reaction which resulted in complete phase separation into NiO and (Na,Li)2SnO3 phase (space group C2/m, a = 5.4159(2) Å, b = 9.3764(3) Å, c = 5.6922(2) Å, β = 108.661(3)°, V = 273.86(2) Å3, Fig. S1†). Detailed investigation of phase stability and cation ordering patterns in this material will be the subject of the forthcoming study.
4 Conclusions
We have demonstrated a topotactic electrochemically-driven Li for Na exchange in the layered O3-Na0.95Ni0.5Sn0.5O2 structure preserving the O3-type stacking and resulting in the Li0.35Na0.07Ni0.5Sn0.5O2 material with ∼13% contracted unit cell volume. However, the capacity of Na0.95Ni0.5Sn0.5O2 during electrochemical cycling in Li half-cell degrades fast, being restricted to a reversible (de)intercalation of only 0.2Li+. A combination of powder X-ray diffraction and aberration-corrected scanning transmission electron microscopy allows relating the limited electrochemical activity to surface passivation with a formation of blocking disordered rock-salt-structured layer. The disorder is likely promoted by the closeness of ionic radii of Ni2+, Sn4+, and Li+, rendering the α-LiFeO2-type rock-salt structure more favorable than the layered α-NaFeO2-type one.Data availability
Experimental data are available by direct request to the corresponding author.Author contributions
Anatolii V. Morozov synthesized the samples, performed Na/Li exchange as well as XRD, electrochemical, EDX and TEM characterization of the materials, and wrote the original draft. Anton O. Boev performed DFT calculations. Artem M. Abakumov, Evgeny V. Antipov, and Aleksandra A. Savina carried out project administration as well as manuscript writing, reviewing, and editing. Additionally, Artem M. Abakumov suggested the general concept and methodology of this research. All authors have read and agreed to the published version of the manuscript.Funding
This research was funded by the Russian Science Foundation (grant no. 20-13-00233).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank Polina A. Morozova and Stanislav S. Fedotov for their advices and help in managing electrochemical experiments. AICF of Skoltech is acknowledged for granting access to the TEM facilities.References
- C. S. Yoon, D.-W. Jun, S.-T. Myung and Y.-K. Sun, ACS Energy Lett., 2017, 2, 1150–1155 CrossRef CAS.
- T. Li, X.-Z. Yuan, L. Zhang, D. Song, K. Shi and C. Bock, Electrochem. Energy Rev., 2020, 3, 43–80 CrossRef.
- S.-M. Bak, E. Hu, Y. Zhou, X. Yu, S. D. Senanayake, S.-J. Cho, K.-B. Kim, K. Y. Chung, X.-Q. Yang and K.-W. Nam, ACS Appl. Mater. Interfaces, 2014, 6, 22594–22601 CrossRef CAS PubMed.
- S. S. Zhang, Energy Storage Mater., 2020, 24, 247–254 CrossRef.
- H.-H. Ryu, K.-J. Park, C. S. Yoon and Y.-K. Sun, Chem. Mater., 2018, 30, 1155–1163 CrossRef CAS.
- T. Weigel, F. Schipper, E. M. Erickson, F. A. Susai, B. Markovsky and D. Aurbach, ACS Energy Lett., 2019, 4, 508–516 CrossRef CAS.
- M. Sathiya, Q. Jacquet, M.-L. Doublet, O. M. Karakulina, J. Hadermann and J.-M. Tarascon, Adv. Energy Mater., 2018, 8, 1702599 CrossRef.
- H. Zhu, R. Shen, Y. Tang, X. Yan, J. Liu, L. Song, Z. Fan, S. Zheng and Z. Chen, Nanomaterials, 2020, 10, 868 CrossRef CAS.
- M. Eilers-Rethwisch, S. Hildebrand, M. Evertz, L. Ibing, T. Dagger, M. Winter and F. M. Schappacher, J. Power Sources, 2018, 397, 68–78 CrossRef CAS.
- J. Li, X. He, R. Zhao, C. Wan, C. Jiang, D. Xia and S. Zhang, J. Power Sources, 2006, 158, 524–528 CrossRef CAS.
- H. Ren, X. Mu, Y. Huang, Z. Li, Y. Wang, P. Cai, Z. Peng and Y. Zhou, Ionics, 2010, 16, 497–502 CrossRef CAS.
- H. Li, Z. Zhu, J. Zhang, H. Qin, Y. Tan, X. Zhang, Z. Yang, G. Pan, W. Wu, M. Yi, S. Hu, K. Qin and M. Chao, Ionics, 2020, 26, 3785–3794 CrossRef CAS.
- A. Paulus, M. Hendrickx, M. Bercx, O. M. Karakulina, M. A. Kirsanova, D. Lamoen, J. Hadermann, A. M. Abakumov, M. K. Van Bael and A. Hardy, Dalton Trans., 2020, 49, 10486–10497 RSC.
- L. Zhou, J. Liu, L. Huang, N. Jiang, Q. Zheng and D. Lin, J. Solid State Electrochem., 2017, 21, 3467–3477 CrossRef CAS.
- G. Kang, K. Lee, K. Kwon and J. Song, Metals, 2017, 7, 395 CrossRef.
- Q.-Q. Qiao, L. Qin, G.-R. Li, Y.-L. Wang and X.-P. Gao, J. Mater. Chem. A, 2015, 3, 17627–17634 RSC.
- X. Ma, C. Wang, J. Cheng and J. Sun, Solid State Ionics, 2007, 178, 125–129 CrossRef CAS.
- S. Valanarasu and R. Chandramohan, Cryst. Res. Technol., 2010, 45, 835–839 CrossRef CAS.
- S. S. Fedotov, N. R. Khasanova, A. S. Samarin, O. A. Drozhzhin, D. Batuk, O. M. Karakulina, J. Hadermann, A. M. Abakumov and E. V Antipov, Chem. Mater., 2016, 28, 411–415 CrossRef CAS.
- H. Ben Yahia, M. Shikano, S. Koike, H. Sakaebe, M. Tabuchi and H. Kobayashi, J. Power Sources, 2013, 244, 87–93 CrossRef CAS.
- S.-M. Oh, S.-T. Myung, J. Hassoun, B. Scrosati and Y.-K. Sun, Electrochem. Commun., 2012, 22, 149–152 CrossRef CAS.
- C. Delmas, C. Fouassier and P. Hagenmuller, Physica B+C, 1980, 99, 81–85 CrossRef CAS.
- V. Petříček, M. Dušek and L. Palatinus, Z. Kristallogr. Cryst. Mater., 2014, 229, 345–352 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- D. A. Aksyonov, S. S. Fedotov, K. J. Stevenson and A. Zhugayevych, Comput. Mater. Sci., 2018, 154, 449–458 CrossRef CAS.
- C. Genevois, H. Koga, L. Croguennec, M. Ménétrier, C. Delmas and F. Weill, J. Phys. Chem. C, 2015, 119, 75–83 CrossRef CAS.
- I. D. Brown, Chem. Rev., 2009, 109, 6858–6919 CrossRef CAS.
- R. De Ridder, G. Van Tendeloo and S. Amelinckx, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 216–224 CrossRef.
- A. Salinas-Sanchez, J. L. Garcia-Muñoz, J. Rodriguez-Carvajal, R. Saez-Puche and J. L. Martinez, J. Solid State Chem., 1992, 100, 201–211 CrossRef CAS.
- A. O. Boev, S. S. Fedotov, A. M. Abakumov, K. J. Stevenson, G. Henkelman and D. A. Aksyonov, Appl. Surf. Sci., 2021, 537, 147750 CrossRef CAS.
- W. Sun, S. T. Dacek, S. P. Ong, G. Hautier, A. Jain, W. D. Richards, A. C. Gamst, K. A. Persson and G. Ceder, Sci. Adv., 2016, 2, e1600225 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2095243 and 2095280. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05246b |
| This journal is © The Royal Society of Chemistry 2021 |