
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTMSOTf-catalyzed synthesis of trisubstituted imidazoles using hexamethyldisilazane as a nitrogen source under neat and microwave irradiation conditions†
Kesatebrhan Haile Asressu ,
Chieh-Kai Chan and
Cheng-Chung Wang*
,
Chieh-Kai Chan and
Cheng-Chung Wang*
Institute of Chemistry, Academia Sinica, Taipei 115, Taiwan. E-mail: wangcc7280@gate.sinica.edu.tw
First published on 19th August 2021
Abstract
In the process of drug discovery and development, an efficient and expedient synthetic method for imidazole-based small molecules from commercially available and cheap starting materials has great significance. Herein, we developed a TMSOTf-catalyzed synthesis of trisubstituted imidazoles through the reaction of 1,2-diketones and aldehydes using hexamethyldisilazane as a nitrogen source under microwave heating and solvent-free conditions. The chemical structures of representative trisubstituted imidazoles were confirmed using X-ray single-crystal diffraction analysis. This synthetic method has several advantages including the involvement of mild Lewis acid, being metal- and additive-free, wide substrate scope with good to excellent yields and short reaction time. Furthermore, we demonstrate the application of the methodology in the synthesis of biologically active imidazole-based drugs.
Introduction
In the drug discovery and development process, obtaining small organic molecules in pure and sufficient amounts is one of the major challenges.1 To access biologically active small molecules, the development of an efficient synthetic methodology from cheap starting materials is highly desirable.2,3 Imidazoles, a class of nitrogen-containing five-membered ring heterocyclic compounds, have a unique chemical behavior and a broad range of biological activities.4–6 The diverse therapeutic properties of these heterocycles include anti-inflammatory, antiproliferative, antimicrobial, anticancer, antihypertensive and inhibition of p38 MAP-kinase, angiotensin II receptor blockers and cytotoxicity.7–13 Imidazole-based compounds have also been used as important precursors in synthesizing pharmaceuticals and natural products like alkaloids.5,9,13–15 Some representative examples of biologically active multisubstituted imidazole drugs are shown in Fig. 1.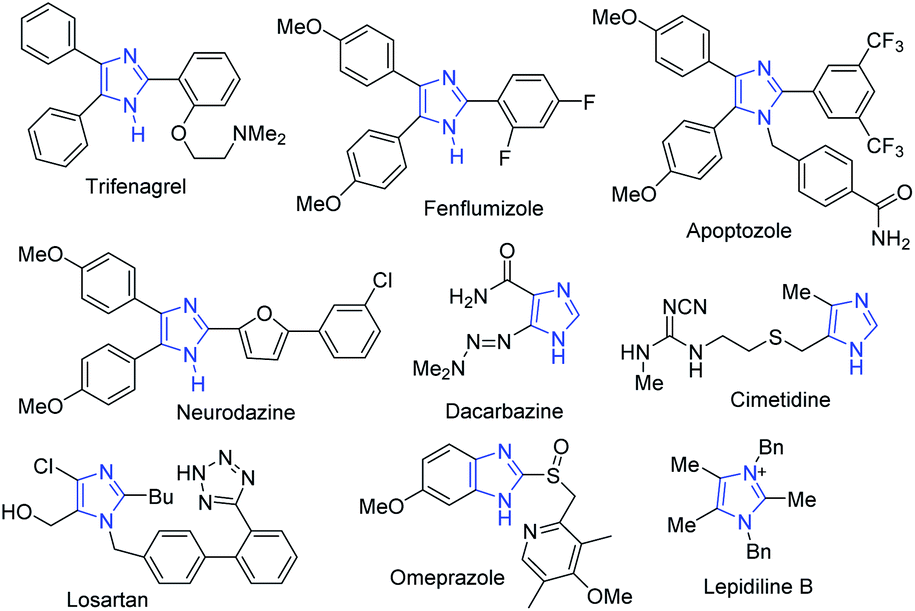 | ||
| Fig. 1 Representative examples of biologically active imidazole-based drugs. | ||
Trifenagrel is a type of imidazole-related drug that inhibits both arachidonate cyclooxygenase and platelet aggregation in many animal species and humans.16,17 Fenflumizol and neurodazine are also bioactive imidazole derivatives having potential neurogenic and anti-inflammatory activities.18,19 The tetraaryl-substituted imidazole apoptozole was reported to induce apoptosis (a programmed cell death) in murine P19 embryonic carcinoma cells and could bind to the heat shock proteins Hsp72 and Hsc70.20,21
The first angiotensin II receptor antagonist, losartan which is mainly used to treat high blood pressure (hypertension) and diabetic kidney disease, whereas the fused imidazole omeprazole is used as proton pump inhibitor.20 The imidazole alkaloid, namely, lepidilines B, was isolated from the root extract of Lepidium meyenii and inhibits potentially cytotoxic reactions against numerous human cancer cell lines,22 while cimetidine is a histamine H-2-receptor antagonist that inhibits gastric acid production.23 Substituted imidazoles also have important applications in the chemistry of materials such as in functional polymers,24 ligands,25 photophysical materials,26,27 ionic liquids,28 semiconductor devices,29 fluorescent probes,30 and N-heterocyclic carbenes.31
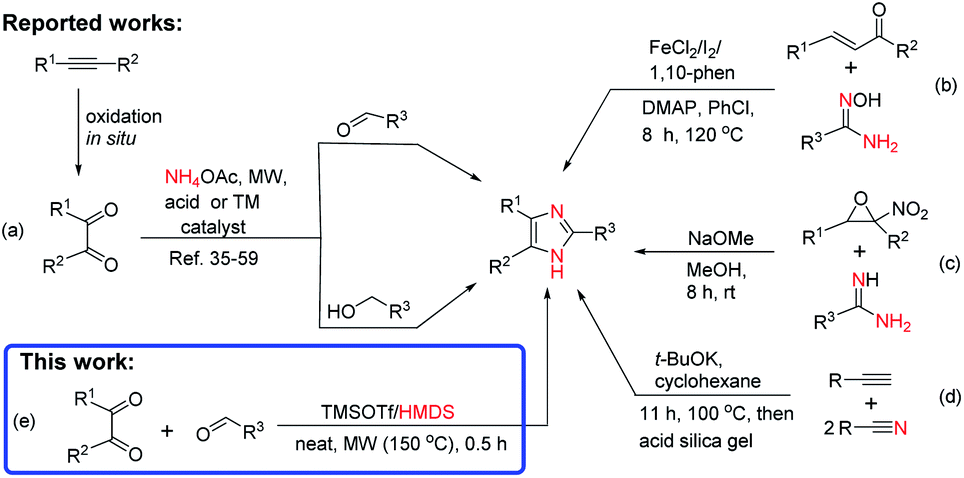
Because of their versatile applications, several approaches have been developed for the chemical synthesis of imidazoles.6,31–39 The most commonly known synthetic methods are (i) the three-component condensation reaction of benzils with -aryl aldehydes, primary alcohols, and ammonium acetate (NH4OAc)/amines using a microwave (MW) irradiation under ionic liquid,13 HOAc,35,36,40,41 Fe,42,43 Ni,44 and Co45 catalysis system, in the presence of acids,6,17,36,39,46,47 urea/hydrogen peroxide (UHP),48 organic bases,49 carbon,50 and transition-metal (TM) catalysts such as TiCl4/SiO2,4 Cu32 and Fe/Ag nanoparticles (NPs),51 Zr NPs,33 La/Mn NPs,52 Au(I)38 and Ru(II)53,54 complexes, urea/ZnCl2,55 MoO3/SiO2,56 I2,57 ZrO2/Al2O3,58 and sulfated SnO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59 (Scheme 1a) (ii) via reaction of α,β-unsaturated ketones/aldehydes and internal alkynes with amidoximes using Fe(II)-catalyzed cross-dehydrogenative coupling60 and Cs2CO3-promoted annulation61 (Scheme 1b) (iii) from α-nitro-epoxides and amidines with an excess base as promoter62 (Scheme 1c), and (iv) from base-promoted nitrile-alkyne domino-type cyclization63 (Scheme 1d).
59 (Scheme 1a) (ii) via reaction of α,β-unsaturated ketones/aldehydes and internal alkynes with amidoximes using Fe(II)-catalyzed cross-dehydrogenative coupling60 and Cs2CO3-promoted annulation61 (Scheme 1b) (iii) from α-nitro-epoxides and amidines with an excess base as promoter62 (Scheme 1c), and (iv) from base-promoted nitrile-alkyne domino-type cyclization63 (Scheme 1d).
 | ||
| Scheme 1 Synthetic methods of 2,4,5-triisubstituted imidazoles. | ||
Although these synthetic strategies are widely established, they are performed in the presence of expensive catalysts, ligands/additives, toxic organic solvents, and under harsh reaction conditions. Owning to the fascinating and wide range of applications of multisubstituted imidazoles in biological, pharmaceutical, and material chemistry, the development of an efficient, environmentally friendly, and economic synthetic method from commercially available starting materials and mild reagents is worth considering.
Hexamethyldisilazane (HMDS), a stable and inexpensive reagent, has been used in silylation64–67 and ring closure68,69 reactions in the presence of protic and Lewis acid66,68 as catalysts. In our previous work on silylation of free sugars, we found that activation of HMDS by trifluoromethanesulfonate (TMSOTf) generated gaseous ammonia (NH3) as a byproduct,65 making it a useful condition for the synthesis of substituted quinazolines.68 Continuing our work on the development of an efficient synthetic methodology for the microwave-assisted synthesis of heterocycles,68,70–72 we herein describe for the first time, TMSOTf-catalyzed solvent-free synthesis of trisubstituted imidazoles using HMDS as nitrogen source under microwave irradiation conditions (Scheme 1e).
Results and discussion
To optimize the reaction condition for the formation of the 2,4,5-triarylated imidazole 3a under microwave irradiation, we selected the commercially available 1,2-diketone (1a, 1 equiv.) and benzaldehyde (2a, 2 equiv.) as model substrates using HMDS (5 equiv.) as the nitrogen source in the absence and presence of various acids as catalysts (0.1 equiv.) (Table 1). To start with, we attempted to synthesize imidazole 3a without any catalyst. Accordingly, 3a was formed in 18% and 16% yields in toluene and water, respectively, which was then improved to 63% when the reaction was carried out in catalyst- and solvent-free systems with microwave heating (150 °C) (Table 1, entries 1–3). This improved yield (63%) indicated that the HMDS can serve both as a reagent and a solvent.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Temperature (°C) | Solvent | Yieldb (%) |
| a Reaction conditions: 1a (0.48 mmol, 1.0 equiv.), 2a (0.95 mmol, 2.0 equiv.), HMDS (2.38 mmol, 5.0 equiv.), catalyst (0.048 mmol, 0.1 equiv.).b Isolated yields. | ||||
| 1 | — | 150 | Toluene | 18 |
| 2 | — | 150 | Water | 16 |
| 3 | — | 150 | — | 63 |
| 4 | AgOTf | 150 | Toluene | 84 |
| 5 | Cu(OTf)2 | 150 | Toluene | 87 |
| 6 | Sc(OTf)3 | 150 | Toluene | 50 |
| 7 | AlCl3 | 150 | Toluene | 49 |
| 8 | TfOH | 150 | Toluene | 90 |
| 9 | TMSOTf | 150 | Toluene | Quant. |
| 10 | TMSOTf | 150 | DCM | 93 |
| 11 | TMSOTf | 150 | MeCN | Quant. |
| 12 | TMSOTf | 150 | — | 96 |
| 13 | TMSOTf | 100 | — | 92 |
| 14 | TMSOTf | 25 | — | 47 |
| 15 | TMSOTf | 200 | — | 75 |
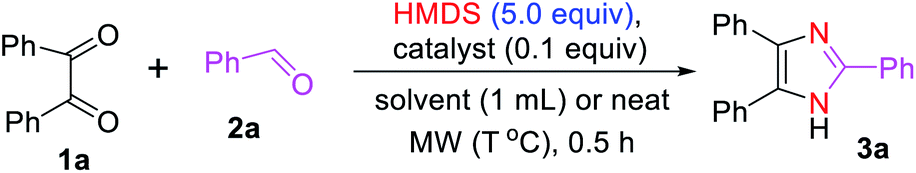
To further prepare compound 3a in a high yield, we conducted the reaction in the presence of Lewis acid as a catalyst. Interestingly, 84% yield of 3a was obtained under the AgOTf catalysis system in toluene (Table 1, entry 4). Based on this promising result, we screened several Lewis acids as catalysts. A slightly higher yield of 3a was obtained with Cu(OTf)2; however, the yield dropped to 50% and 49% when Sc(OTf)3 and AlCl3 were employed, respectively (Table 1, entries 6 and 7) showing their low catalytic activity. Remarkably, a high yield (90%) of imidazole 3a was achieved with protic acid (TfOH) under the same reaction condition (Table 1, entry 8).
We continued to investigate the reaction conditions to obtain a better yield of imidazole 3a. Gratifyingly, the desired product 3a was obtained in a quantitative yield with a catalytic amount of TMSOTf under microwave heating in toluene (Table 1, entry 9). We also examined the role of dichloromethane and acetonitrile on the reaction yields that could achieve good results like that with toluene (Table 1, entries 10 and 11). Importantly, the triaryl substituted imidazole 3a was isolated in 96% yield under solvent-free conditions (Table 1, entry 12). The yield of 3a was not significantly affected by lowering the reaction temperature from 150 to 100 °C, but dropped to 47% when the reaction was performed at 25 °C under a neat condition (Table 1, entries 13 and 14). On the other hand, increasing the reaction temperature to 200 °C decreases the yield of imidazole 3a (Table 1, entry 15). Indeed, the desired product 3a was formed in excellent yields with or without solvents under the TMSOTf-catalyzed system at 150 °C. Therefore, we preferred to use the solvent-free reaction condition (Table 1, entry 12) to investigate the substrate scope based on the principles of green chemistry.73
We also evaluated the preparation of 3a from 1a and 2a under conventional reflux heating condition, and the reaction performed well affording a comparable yield (89%) of 3a as that of microwave-assisted reaction. Muthusubramanian and Esmaeilpour groups have also isolated similar yields of 2,4,5-trisubstituted imidazoles for reactions carried out under thermal and microwave irradiation.43,74 It is reported that microwave-assisted organic synthesis has several advantages such as rapid homogeneous heating, suppression of side product, convenience of handling, simple work-up, short reaction time, and required less energy compared to conventional heating.36,41–43,69,74–77 Bearing of these advantages in mind, we investigated the substrate scope under the microwave irradiation.
Next, we explored the substrate scope and generality of the imidazole formation from various substituted aryl (2b–x) and alkyl (2y, 2z, 2aa, 2ab) aldehydes and benzil (1a) (Table 2) using the optimized reaction conditions. Thus, the aryl aldehydes having electron-withdrawing groups (2b–e, 2i–j, 2l–m, and 2r), electron-donating substituents (2f–h, 2k, and 2n–q), and heterocycles (2u–x) reacted smoothly with the 1,2-diketone 1a to provide the corresponding imidazole derivatives 3b–x in moderate to quantitative yields. The reaction yield of 1a with benzaldehyde derivatives bearing halides (2b–d) and phenyl (2h) groups at para-position was observed to be higher (90–96%) than those using aldehydes 2f–gh and 2k which contained activating groups (methyl and methoxy) either at the para- or meta-positions (58%, 75–79%) and deactivating substituents (2i–j) at meta-positions (73–76%). On other hand, a moderate yield (66%) of 3e was achieved by coupling 1a with the aryl aldehyde 2e having a nitro group, which is a stronger deactivator than halogens.
|
|---|
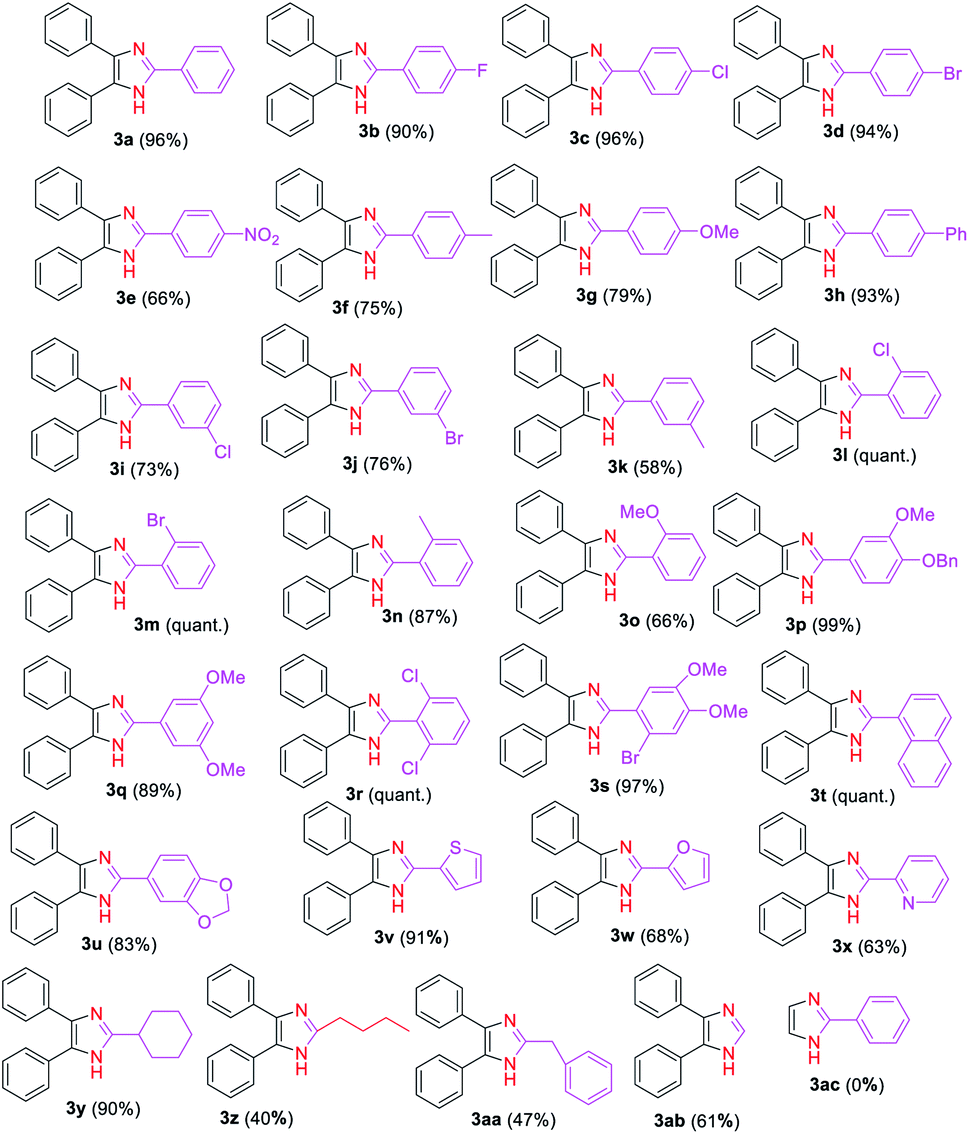 |

The reaction of 1,2-diketone 1a with aryl aldehydes 2l–m which bear weak electron-donating effects, such as chloro, bromo, and methyl groups at ortho-position of the aromatic ring affords in excellent yields of imidazoles 3l–m than those aldehydes with strong electron-rich groups, such as the methoxy group (2o). Similarly, multisubstituted aldehyde substrates 2p–s with electron-donating or electron-withdrawing functionalities performed well to produce high yields of the corresponding imidazole derivatives 3p–s ranging from 89% to quantitative under the established reaction conditions. Interestingly, high yields of trisubstituted imidazoles 3t and 3u were achieved by reacting 1a with bulkier substrates 2t and 2u that contain fused rings, indicating that the steric effect has no influence on the formation of these imidazoles. Additionally, the scope of the reaction on heterocyclic carbaldehydes such as 2-thiophenyl 2v, 2-furanyl 2w, and 2-pyridyl 2x was investigated. Hence, the condensation reaction of 2-thiophenyl 2v with 1a afforded a high yield (91%) of the imidazole 3v, whereas, the reaction of aromatic aldehydes 2w and 2x with 1a gave the desired products 3w and 3x, respectively in moderate yields (63–68%).
After adequately evaluating the scope of substituted aryl aldehydes, we examined the performance of the reaction with alkyl aldehydes such as cyclohexanecarboxaldehyde (2y), valeraldehyde (2z) and phenylacetaldehyde (2aa). Gratifyingly, the alky substituted aldehydes well tolerated to the established reaction conditions, providing the desired 2-alkyl-4,5-diaryl substituted imidazoles 3y–3aa in moderate to excellent yields (40–90%) (Table 2). The yield of imidazole 3y was significantly improved (90%) in this study compared to the literature report yields (18–22%) using NH4OAc as nitrogen source.78,79 Furthermore, the reaction was carried out on formaldehyde (2ab), and the corresponding 4,5-disubstituted derivative 3ab was obtained in 61% yield. Unfortunately, the reaction of glyoxal (2ac) with benzaldehyde (2a) failed to give the desired 2-aryl substituted product 3ac. We believe that the failure of this reaction might be attributed to the low boiling point (51 °C) of glyoxal (2ac) leading to its evaporation before effective reaction with benzaldehyde (2a).
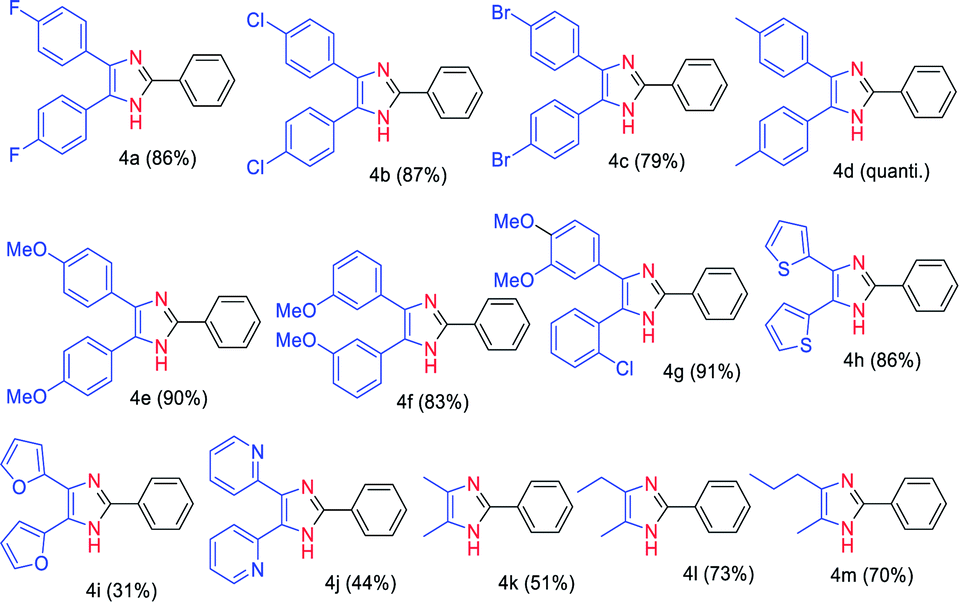
After having screened the substrate scope of the aldehydes, we tested the condensation reaction of benzaldehyde (2a) with various 1,2-diketones containing electron-deficient (1b–d), electron-rich (1e–g), and heteroaromatic (1i–l) functional groups (Table 3). We observed that both electron-deficient (fluoro, chloro, and bromo) and electron-rich (methyl, methoxy) containing 1,2-diketones delivered very good to excellent yields (79% to quantitative) of the corresponding triaryl imidazoles 4a–f under the established reaction condition. These results indicate that the electronic effect of the substrates has no influence on the reaction yields of 4a–f. We also evaluated the reaction condition of unsymmetrical aryl 1,2-diketone 1h having an electron-releasing (R1) and electron-withdrawing group (R2). Gratifyingly, 91% yield of the trisubstituted imidazoles 4g was obtained by coupling of 2-chloro-3′,4′-dimethoxybenzil with 2a. Our methodology also worked on heteroaromatic (1i–l) (R1 = R2 = 2-furyl, 2-thienyl and 2-pyridyl) and aliphatic 1,2-diketones 1m–o (R1 = Me, Et or Pr, R2 = Me). Accordingly, the reaction of 2,2′-thenil (1i) and benzaldehyde (2a) gave a very good yield (86%) of the imidazole 4h. However, in the case of 1,2-diketone substrates bearing O- and N-heteroatoms (1k and 1l), the yield of imidazoles 4i and 4j decreased. The desired products 4k–m were isolated in moderate to good yields (51–73%) through the reaction of an aliphatic 1,2-diketones 1m–o and aldehyde 2a.
|
|---|
 |

The structures of some of the trisubstituted imidazoles 3n, 3q, 3s, 3w, 3z and 4h were confirmed using single X-ray crystallography (Fig. 2).80 X-ray data revealed that 3q and 3w are two independent isomorphous molecules.
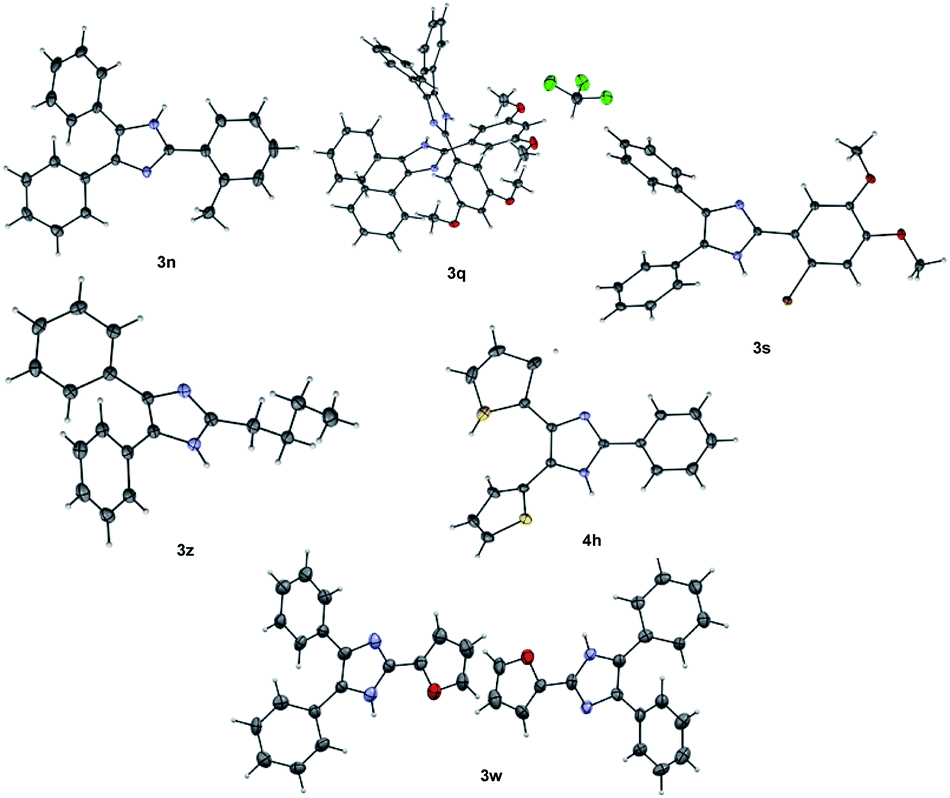 | ||
| Fig. 2 Single X-ray crystal structure of 3n, 3q, 3s, 3w, 3z and 4h. Thermal ellipsoids was drawn at 50% probability. | ||
Compared with the earlier reports, this new synthetic method for trisubstituted imidazoles has several advantages, such as the use of mild TMSOTf as a catalyst, metal- and additive-free condition, wide substrate scope with good to excellent yields and short reaction time. Moreover, the synthesis of an imidazole-based small molecule library is important for the identification of bioactive compounds that can treat several diseases.21,81 To this end, our new synthetic methodology was applied in the synthesis of imidazole-containing drugs. Accordingly, trifenagrel (5a),16,36,55 a novel potent platelet aggregation and arachidonic acid inhibitor in various animal species and humans, was synthesized in a good yield (72%) via the condensation of benzil (1a) with the aldehyde 2ad under the optimized reaction condition (Scheme 2). This drug was previously synthesized in over two steps either from 1a and 2ad in the presence of catalysts and additives such as urea/ZnCl2,55 Co(acac)2/Ag2O,82 and silica/acid83 using NH4OAc as nitrogen source or from the reaction 2-(4,5-diphenyl-1H-imidazol-2-yl)phenol and 2-bromo-N,N-dimethylethanamine under basic condition.82,84
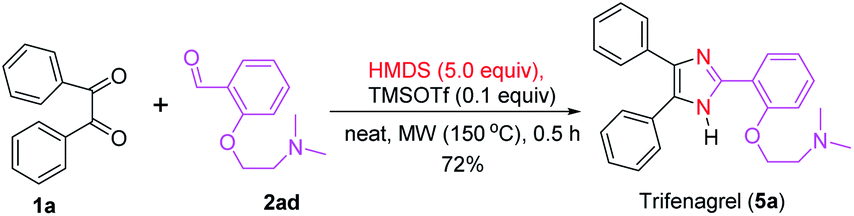 | ||
| Scheme 2 Synthesis of trifenagrel (5a). | ||
This synthesis method described here was also employed in the preparation of fenflumizol (5b),85 a biologically active imidazole-based drug having potential platelet aggregation and anti-inflammatory activities.18,86 The synthesis of drug 5b was achieved by the reaction of the benzil derivative 1f with the aromatic aldehyde 2ae under the established reaction condition. Remarkably, this biologically active compound was obtained in 93% yield (Scheme 3). Previously, this imidazole-based natural product was synthesized from 4,4′-dimethoxybenzil (1f) and 2,4-diflurobenzaldehyde (2ae) for a longer reaction time (3 h) using NH4OAc as the nitrogen source and acetic acid as solvent at 100 °C.85
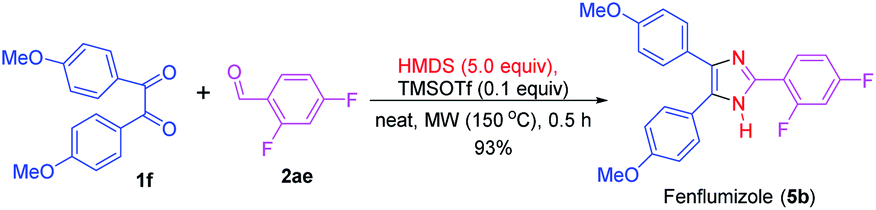 | ||
| Scheme 3 Synthesis of fenflumizol (5b). | ||
Based on the experimental results in Table 1 and literature reports, we propose a plausible reaction mechanism for the formation of 2,4,5-trisubstituted imidazoles (Scheme 4). First, the carbonyl oxygen of aldehyde 2a is activated by TMOTf to form complex A which then leads to a nucleophilic addition with HMDS to afford the tetrahedral intermediate B87 along with the regeneration of TMOTf. Next, nucleophilic addition of the silanamine intermediate B to the carbonyl carbon of the activated benzil C produces complex D. A stepwise elimination of TMOTf and TMSOH from D gave an imine E, which was then followed by increasing the electrophilicity of its carbonyl group through coordination with TMOTf to furnish a complex F. Then the electrophilic carbonyl carbon of F reacts with HMDS to form the tetrahedral intermediate G with a concomitant recovery of TMOTf. Next, elimination of TMSOH87 from G provides 1,2-diimine intermediate H, which in turn undergoes intramolecular cyclization to form the five-membered ring intermediate I upon the expulsion of (TMS)2O. Finally, the desired imidazole 3a is obtained through the aromatization of I under microwave irradiation. Interestingly, the formation of the ethane-1,2-diimine intermediate H was confirmed by mass spectrometry analysis (see ESI†).
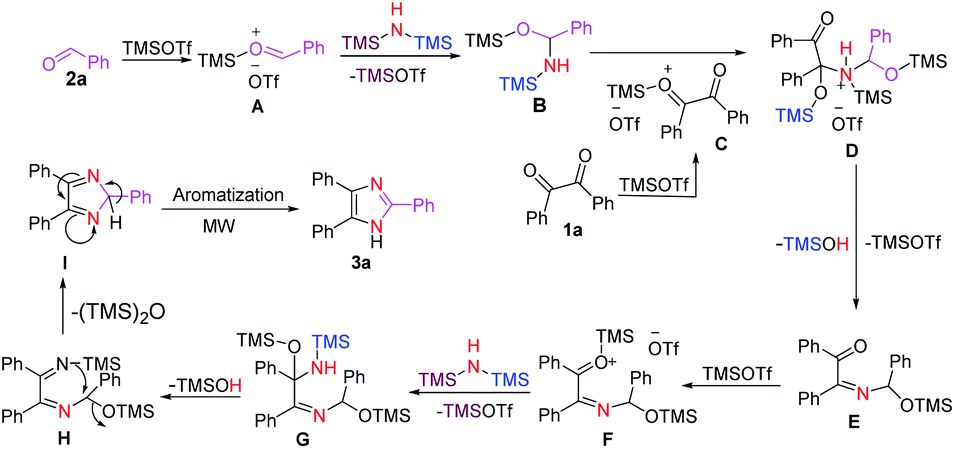 | ||
| Scheme 4 Plausible mechanism for the synthesis of 3a. | ||
Conclusion
We have developed a successfully and highly efficient method for the synthesis of trisubstituted imidazoles by coupling the readily available 1,2-diketones and aldehydes in the presence of TMSOTf as Lewis acid catalyst and HMDS as a nitrogen source under microwave heating and solvent-free conditions. This new synthetic method features wide substrate scope with good to excellent yields, simple starting materials, metal- and additive-free and with a short reaction time. The chemical structures of representative trisubstituted imidazoles were confirmed by X-ray single-crystal diffraction analysis. Furthermore, the potential of the methodology described here was demonstrated through the synthesis of biologically important imidazole-based drugs such as trifenagrel and fenflumizol.Experimental section
General information
All reagents obtained from commercial sources were used without purification, unless otherwise mentioned. Column chromatography was carried out by Silica Gel Geduran® Si 60 (0.040–0.063 mm, E. Merck). TLC was performed on pre-coated glass plates of Silica Gel 60 F254 (0.25 mm, E. Merck); detection was executed by spraying with a solution of Ce(SO4)2 (NH4)2MoO4, and H2SO4 in water and subsequently heating on a hot plate. UV light for TLC analysis was UVGL-25 compact UV lamp (4 watt/254 nm). Melting points were determined with a MP-2D melting apparatus. 1H NMR, 13C NMR, and DEPT spectra were recorded by Bruker DRX500 and AVIII 500. Chemical shifts are in ppm from Me4Si, generated from the DMSO-d6 and CDCl3 lock signal at δ 2.50, 7.24 and 40.00, 77.16 ppm for 1H and 13C NMR, respectively. Multiplicities are reports by using the following abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, s = sextet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets; J = coupling constant values in Hertz. High resolution mass spectrometry (HR-MS) was performed on a Waters Premier XE instrument with ESI source. Structural assignments were made with additional information from selective 1D-TOCSY, 2D-HSQC, 2D-HMBC and X-ray experiments.General procedure for the synthesis of 2,4,5-trisubstituted imidazoles68
1,2-Diketone derivatives 1a–m (0.272 to 1.160 mmol, 1 equiv.), aryl aldehydes 2a–ae (2.32 to 0.543 mmol, 2 equiv.), and trimethylsilyl trifluoromethanesulfonate (0.027 to 0.116 mmol, 0.1 equiv.) in hexamethyldisilazane (1.64 to 5.81 mmol, 5 equiv.) were added into a dried 15 mL microwave vial at 25 °C. The mixture was placed in a microwave irradiation instrument and stirred at 150 °C for 0.5 h. After the reaction completion, the reaction mixture was cooled to 25 °C, dissolved in ethyl acetate and diluted with water. Next, the crude product was extracted from the aqueous phases with ethyl acetate. The organic layer was washed with brine, dried over anhydrous MgSO4, filtered and concentrated in rotary vacuum to afford the crude product. Purification of the crude products by column chromatography using 3 to 15% ethyl acetate in hexane afforded the corresponding pure imidazoles 3, 4 and 5.Author contributions
The manuscript has been prepared with contributions from all the authors.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the Ministry of Science and Technology of Taiwan (MOST 108-2113-M-001-019; MOST 109-2113-M-001-005) and Academia Sinica (MOST 108-3114-Y-001-002; AS-SUMMIT-109). We also thank Dr Ying-Yann Wu and Mr Wei-Tzung Yang for their help with NMR experiments, Dr Yuh-Sheng Wen for the X-ray crystallography experiment, and Ms Ping-Yu Lin for conducting mass spectrometry experiments.Notes and references
- D. C. Blakemore, L. Castro, I. Churcher, D. C. Rees, A. W. Thomas, D. M. Wilson and A. Wood, Nat. Chem., 2018, 10, 383–394 CrossRef CAS.
- S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6699–6702 CrossRef CAS PubMed.
- I. De Toledo, T. A. Grigolo, J. M. Bennett, J. M. Elkins and R. A. Pilli, J. Org. Chem., 2019, 84, 14187–14201 CrossRef CAS.
- Á. Magyar and Z. Hell, Synlett, 2019, 30, 89–93 CrossRef.
- Z. Jin, Nat. Prod. Rep., 2009, 26, 382–445 RSC.
- C. Zhang, S. Sarshar, E. J. Moran, S. Krane, J. C. Rodarte, K. D. Benbatoul, R. Dixon and A. M. M. Mjalli, Bioorg. Med. Chem. Lett., 2000, 10, 2603–2605 CrossRef CAS.
- P. Koch, C. Bäuerlein, H. Jank and S. Laufer, J. Med. Chem., 2008, 51, 5630–5640 CrossRef CAS PubMed.
- A. Siwach and P. K. Verma, BMC Chem., 2021, 15, 1–69 CrossRef.
- N. Kerru, S. V. H. S. Bhaskaruni, L. Gummidi, S. N. Maddila, S. Maddila and S. B. Jonnalagadda, Synth. Commun., 2019, 49, 2437–2459 CrossRef CAS.
- E. M. Perchellet, J. P. Perchellet and P. W. Baures, J. Med. Chem., 2005, 48, 5955–5965 CrossRef CAS.
- A. Mumtaz, A. Saeed, N. Fatima, M. Dawood, H. Rafique and J. Iqbal, Bangladesh J. Pharmacol., 2016, 11, 756–764 CrossRef.
- A. Negi, J. M. Alex, S. M. Amrutkar, A. T. Baviskar, G. Joshi, S. Singh, U. C. Banerjee and R. Kumar, Bioorg. Med. Chem., 2015, 23, 5654–5661 CrossRef CAS PubMed.
- M. Xia and Y.-D. Lu, J. Mol. Catal. A: Chem., 2007, 265, 205–208 CrossRef CAS.
- Z. Jin, Nat. Prod. Rep., 2006, 23, 464–496 RSC.
- D. A. Shabalin and J. E. Camp, Org. Biomol. Chem., 2020, 18, 3950–3964 RSC.
- A. Gail, L. Abrahams, T. Park and N. Carolina, J. Pharmacol. Exp. Ther., 1989, 249, 359–365 Search PubMed.
- L. Nagarapu, A. A. Satyender, G. Chandana and R. Bantu, J. Heterocycl. Chem., 2009, 46, 195–200 CrossRef CAS.
- D. R. Williams, M. R. Lee, Y. A. Song, S. K. Ko, G. H. Kim and I. Shin, J. Am. Chem. Soc., 2007, 129, 9258–9259 CrossRef CAS.
- J. Zielonka, J. Vasquez-Vivar and B. Kalyanaraman, Nat. Protoc., 2008, 3, 8–21 CrossRef CAS PubMed.
- A. Patel, S. Y. Sharp, K. Hall, W. Lewis, M. F. G. Stevens, P. Workman and C. J. Moody, Org. Biomol. Chem., 2016, 14, 3889–3905 RSC.
- D. R. Williams, S. K. Ko, S. Park, M. R. Lee and I. Shin, Angew. Chem. Int. Ed., 2008, 47, 7466–7469 CrossRef CAS PubMed.
- B. Cui, B. L. Zheng, K. He and Q. Y. Zheng, J. Nat. Prod., 2003, 66, 1101–1103 CrossRef CAS PubMed.
- Z. Jin, Nat. Prod. Rep., 2013, 30, 869–915 RSC.
- T. Yamamoto, T. Uemura, A. Tanimoto and S. Sasaki, Macromolecules, 2003, 36, 1047–1053 CrossRef CAS.
- H. Konishi, T. Ueda, T. Muto and K. Manabe, Org. Lett., 2012, 14, 4722–4725 CrossRef CAS PubMed.
- W. Lin, L. Long, L. Yuan, Z. Cao, B. Chen and W. Tan, Org. Lett., 2008, 10, 5577–5580 CrossRef CAS PubMed.
- Y. Dong, J. Qian, Y. Liu, N. Zhu, B. Xu, C. L. Ho, W. Tian and W. Y. Wong, New J. Chem., 2019, 43, 1844–1850 RSC.
- B. A. Omotowa and J. M. Shreeve, Organometallics, 2004, 23, 783–791 CrossRef CAS.
- N. T. Zhang, C. C. Zeng, C. M. Lam, R. K. Gbur and R. D. Little, J. Org. Chem., 2013, 78, 2104–2110 CrossRef CAS.
- A. Uslu, S. O. Tümay, A. Şenocak, F. Yuksel, E. Özcan and S. Yeşilot, Dalton Trans., 2017, 46, 9140–9156 RSC.
- W. A. Herrmann, Angew. Chem. Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- N. V. Gandhare, R. G. Chaudhary, V. P. Meshram, J. A. Tanna, S. Lade, M. P. Gharpure and H. D. Juneja, J. Chinese Adv. Mater. Soc., 2015, 3, 270–279 CrossRef CAS.
- A. Teimouri and A. N. Chermahini, J. Mol. Catal. A: Chem., 2011, 346, 39–45 CrossRef CAS.
- M. Thwin, B. Mahmoudi, O. A. Ivaschuk and Q. A. Yousif, RSC Adv., 2019, 9, 15966–15975 RSC.
- L. Kong, X. Lv, Q. Lin, X. Liu, Y. Zhou and Y. Jia, Org. Process Res. Dev., 2010, 14, 902–904 CrossRef CAS.
- S. E. Wolkenberg, D. D. Wisnoski, W. H. Leister, Y. Wang, Z. Zhao and C. W. Lindsley, Org. Lett., 2004, 6, 1453–1456 CrossRef CAS.
- N. Kerru, L. Gummidi, S. V. H. S. Bhaskaruni, S. N. Maddila and S. B. Jonnalagadda, Mol. Diversity, 2020, 24, 889–901 CrossRef CAS PubMed.
- A. Y. Dubovtsev, D. V. Darin, M. Krasavin and V. Y. Kukushkin, Eur. J. Org. Chem., 2019, 2019, 1856–1864 CrossRef CAS.
- J. Wang, R. Mason, D. VanDerveer, K. Feng and X. R. Bu, J. Org. Chem., 2003, 68, 5415–5418 CrossRef CAS.
- E. Gelens, F. J. J. De Kanter, R. F. Schmitz, L. A. J. M. Sliedregt, B. J. Van Steen, C. G. Kruse, R. Leurs, M. B. Groen and R. V. A. Orru, Mol. Diversity, 2006, 10, 17–22 CrossRef CAS PubMed.
- R. B. Sparks and A. P. Combs, Org. Lett., 2004, 6, 2473–2475 CrossRef CAS PubMed.
- H. Naeimi and D. Aghaseyedkarimi, New J. Chem., 2015, 39, 9415–9421 RSC.
- M. Esmaeilpour, J. Javidi and M. Zandi, New J. Chem., 2015, 39, 3388–3398 RSC.
- T. S. Chundawat, N. Sharma, P. Kumari and S. Bhagat, Synlett, 2016, 27, 404–408 CAS.
- E. Eidi, M. Z. Kassaee and Z. Nasresfahani, Appl. Organomet. Chem., 2016, 30, 561–565 CrossRef CAS.
- A. Hariharasubramanian and Y. D. Ravichandran, RSC Adv., 2014, 4, 54740–54746 RSC.
- A. R. Karimi, Z. Alimohammadi and M. M. Amini, Mol. Diversity, 2010, 14, 635–641 CrossRef CAS PubMed.
- A. Maleki and Z. Alirezvani, J. Chil. Chem. Soc., 2016, 61, 3116–3119 CrossRef CAS.
- S. N. Murthy, B. Madhav and Y. V. D. Nageswar, Tetrahedron Lett., 2010, 51, 5252–5257 CrossRef CAS.
- K. Ramesh, S. N. Murthy, K. Karnakar, Y. V. D. Nageswar, K. Vijayalakhshmi, B. L. A. Prabhavathi Devi and R. B. N. Prasad, Tetrahedron Lett., 2012, 53, 1126–1129 CrossRef CAS.
- A. Maleki, H. Movahed and R. Paydar, RSC Adv., 2016, 6, 13657–13665 RSC.
- H. Sanaeishoar, H. Tavakkoli, M. Asareh and F. Mohave, Iran. J. Catal., 2016, 6, 213–219 Search PubMed.
- G. Vinoth, S. Indira, M. Bharathi, G. Archana, L. G. Alves, A. M. Martins and K. Shanmuga Bharathi, Inorg. Chim. Acta, 2021, 516, 120089 CrossRef CAS.
- S. Sundar and R. Rengan, Org. Biomol. Chem., 2019, 17, 1402–1409 RSC.
- D. Peña-solórzano and C. Ochoa-puentes, Synlett, 2019, 30, 225–229 CrossRef.
- S. V. Bhosale, M. B. Kalyankar, S. V. Nalage, D. S. Bhosale, S. L. Pandhare, T. V. Kotbagi, S. B. Umbarkar and M. K. Dongare, Synth. Commun., 2011, 41, 762–769 CrossRef CAS.
- M. Kidwai, P. Mothsra, V. Bansal, R. K. Somvanshi, A. S. Ethayathulla, S. Dey and T. P. Singh, J. Mol. Catal. A: Chem., 2007, 265, 177–182 CrossRef CAS.
- N. Thimmaraju and S. Z. M. Shamshuddin, RSC Adv., 2016, 6, 60231–60243 RSC.
- S. A. Dake, M. B. Khedkar, G. S. Irmale, S. J. Ukalgaonkar, V. V. Thorat, S. A. Shintre and R. P. Pawar, Synth. Commun., 2012, 42, 1509–1520 CrossRef CAS.
- P. Wu, X. Zhang and B. Chen, Tetrahedron Lett., 2019, 60, 1103–1107 CrossRef CAS.
- H. Mehmood, M. A. Iqbal, L. Lu and R. Hua, Molecules, 2020, 25, 3621 CrossRef CAS PubMed.
- X. Guo, J. Shao, H. Liu, B. Chen, W. Chen and Y. Yu, RSC Adv., 2015, 5, 51559–51562 RSC.
- Q. Wang, X. Chen, X.-G. Wang, H.-C. Liu and Y.-M. Liang, Org. Lett., 2019, 21, 9874–9877 CrossRef CAS PubMed.
- K. H. Asressu and C. C. Wang, Carbohydr. Res., 2017, 453–454, 44–53 CrossRef CAS PubMed.
- A. A. Joseph, V. P. Verma, X. Y. Liu, C. H. Wu, V. M. Dhurandhare and C. C. Wang, Eur. J. Org. Chem., 2012, 744–753 CrossRef CAS.
- D. Zareyee and B. Karimi, Tetrahedron Lett., 2007, 48, 1277–1280 CrossRef CAS.
- B. Karimi and B. Golshani, J. Org. Chem., 2000, 65, 7228–7230 CrossRef CAS PubMed.
- C.-K. Chan, C.-Y. Lai and C.-C. Wang, Org. Biomol. Chem., 2020, 18, 7201–7212 RSC.
- J. C. Burbiel, J. Hockemeyer and C. E. Müller, Beilstein J. Org. Chem., 2006, 2, 1–6 Search PubMed.
- C. K. Chan, C. Y. Lai and C. C. Wang, Synthesis, 2020, 52, 1779–1794 CrossRef CAS.
- K. H. Asressu, C. Chan and C. Wang, ACS Omega, 2021, 6, 7296–7311 CrossRef CAS PubMed.
- C. K. Chan, C. Y. Lai, W. C. Lo, Y. T. Cheng, M. Y. Chang and C. C. Wang, Org. Biomol. Chem., 2020, 18, 305–315 RSC.
- A. Ivanković, Int. J. Sustain. Energy, 2017, 6, 39 Search PubMed.
- M. B. Harisha, P. Dhanalakshmi, R. Suresh, R. R. Kumar, S. Muthusubramanian and N. Bhuvanesh, ChemistrySelect, 2019, 4, 2954–2958 CrossRef CAS.
- E. Chauveau, C. Marestin, F. Schiets and R. Mercier, Green Chem., 2010, 12, 1018–1022 RSC.
- S. Maity, S. Pathak and A. Pramanik, Tetrahedron Lett., 2013, 54, 2528–2532 CrossRef CAS.
- D. Ermolat, K. Van Hecke, L. Van Meervelt and E. Van Der Eycken, Mol. Diversity, 2010, 14, 767–776 CrossRef PubMed.
- S. Naidoo and V. Jeena, Eur. J. Org. Chem., 2019, 1107–1113 CrossRef CAS.
- S. Naidoo and V. Jeena, Tetrahedron, 2020, 76, 131028 CrossRef CAS.
- CCDC 2082023 (3n), 2082029 (3q), 2082025 (3s), 2082032 (3w), 2082036 (4h) and 2098033 (3z) contain the supplementary crystallographic data for this paper†.
- Z. Huang, Chem. Biol., 2002, 9, 1059–1072 CrossRef CAS PubMed.
- J. B. Bharate, S. Abbat, R. Sharma, P. V. Bharatam, R. A. Vishwakarma and S. B. Bharate, Org. Biomol. Chem., 2015, 13, 5235–5242 RSC.
- C. Mukhopadhyay, P. K. Tapaswi and M. G. B. Drew, Tetrahedron Lett., 2010, 51, 3944–3950 CrossRef CAS.
- V. P. Charpe, A. Sagadevan and K. C. Hwang, Green Chem., 2020, 22, 4426–4432 RSC.
- A. H. Bansode and G. Suryavanshi, ACS Omega, 2019, 4, 9636–9644 CrossRef CAS PubMed.
- P. Grande, J. Wadt and T. Corell, Eur. J. Clin. Pharmacol., 1984, 27, 169–172 CrossRef CAS PubMed.
- K. Nishiyama, M. Saito and M. Oba, Bull. Chem. Soc. Jpn., 1988, 61, 609–611 CrossRef CAS.
- J. Jayram and V. Jeena, Green Chem., 2017, 19, 5841–5845 RSC.
- M. Mao, S. Xiao, J. Li, Y. Zou, R. Zhang, J. Pan, F. Dan, K. Zou and T. Yi, Tetrahedron, 2012, 68, 5037–5041 CrossRef CAS.
- S. Das Sharma, P. Hazarika and D. Konwar, Tetrahedron Lett., 2008, 49, 2216–2220 CrossRef CAS.
- S. Samai, G. C. Nandi, P. Singh and M. S. Singh, Tetrahedron, 2009, 65, 10155–10161 CrossRef CAS.
- M. V. Marques, M. M. Ruthner, L. A. M. Fontoura and D. Russowsky, J. Braz. Chem. Soc., 2012, 23, 171–179 CrossRef CAS.
- A. Shaabani, A. Maleki and M. Behnam, Synth. Commun., 2009, 39, 102–110 CrossRef CAS.
- P. Hirapara, D. Riemer, N. Hazra, J. Gajera, M. Finger and S. Das, Green Chem., 2017, 19, 5356–5360 RSC.
- T. B. Gao, Q. Fan, Z. T. Yu and D. K. Cao, Dalton Trans., 2017, 46, 8180–8189 RSC.
- L. Giammanco and F. P. Invidiata, Heterocycles, 1985, 23, 1459–1464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures and spectroscopic data for all compounds and X-ray analysis data for 3n, 3q, 3s, 3w, 4h and 3z. CCDC 2082023, 2082029, 2082025, 2082032, 2082036 and 2098033. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05802a |
| This journal is © The Royal Society of Chemistry 2021 |