DOI:
10.1039/D1RA05760J
(Paper)
RSC Adv., 2021,
11, 29684-29689
Trinorlabdane diterpenoid alkaloids featuring an unprecedented skeleton with anti-inflammatory and anti-viral activities from Forsythia suspensa†
Received
29th July 2021
, Accepted 25th August 2021
First published on 6th September 2021
Abstract
Two unique trinorlabdane diterpenoid alkaloids, forsyqinlingines A (1) and B (2), were isolated from the ripe fruits of Forsythia suspensa. Their structures, including absolute stereochemical configurations, were fully elucidated from extensive spectroscopy experiments, single-crystal X-ray diffraction, and electronic circular dichroism (ECD). In addition, a plausible biosynthetic pathway for the formation of compounds 1 and 2 in Forsythia suspensa was also proposed. In vitro, the two C17-labdane diterpenoid alkaloids exhibited anti-inflammatory activities by inhibiting the release of β-glucuronidase in rat polymorphonuclear leukocytes (PMNs), and antiviral activities against influenza A (H1N1) virus and respiratory syncytial virus (RSV).
Introduction
Forsythia suspensa is widely distributed in China, Japan, Korea, and many European countries as an ornamental plant for its lovely flowers in spring. But, more importantly, the dried fruit of F. suspensa, named Fructus forsythiae, is a well-known TCM, and is documented in every edition of the Chinese Pharmacopoeia and many classical medical books.1 Modern pharmacological studies also indicated its anti-inflammatory, anti-oxidant, anti-bacterial, anti-cancer, anti-virus, anti-allergy, and neuroprotective effects.2–7 Recently, we isolated thirteen labdane diterpenoids from the ripe fruits of F. suspensa that demonstrated a myriad of different anti-inflammatory and antiviral activities.8,9 Using this as an impetus to isolate and identify more biologically active compounds from plant extracts, we further focused our phytochemical investigations on the alkaloid fraction of the EtOH extract of F. suspensa plants grown in the Qinling District, Shanxi, Province, China. From the EtOH extract, two novel C17-labdane diterpenoid alkaloids, named forsyqinlingine A (1) and forsyqinlingine B (2), were isolated, and their chemical structures, including absolute stereochemical configurations, were determined through extensive spectroscopic experiments, single-crystal X-ray diffraction, and electronic circular dichroism (ECD). The two trinorlabdane diterpenoid alkaloids were screened in vitro for their anti-inflammatory activities and their antiviral activities against the influenza A (H1N1) virus and respiratory syncytial virus (RSV).
Results and discussion
The air-dried ripe fruits of Forsythia suspensa were refluxed in 95% EtOH, and the corresponding crude extract was washed with 2% HCl (aq). The pH of the aqueous solution was then adjusted with to ∼10, after which the aqueous phase was extracted with CHCl3 three times. The combined organic fractions were concentrated under reduced pressure to afford the crude alkaloid concentrate, which was successively subjected to column chromatography and preparative high-performance thin layer chromatography (HPTLC) to afford the pure forsyqinlingines A (1) and B (2) (Fig. 1).
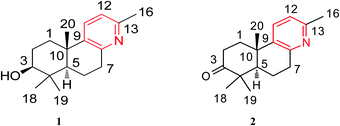 |
| | Fig. 1 Chemical structures of compounds 1 and 2. | |
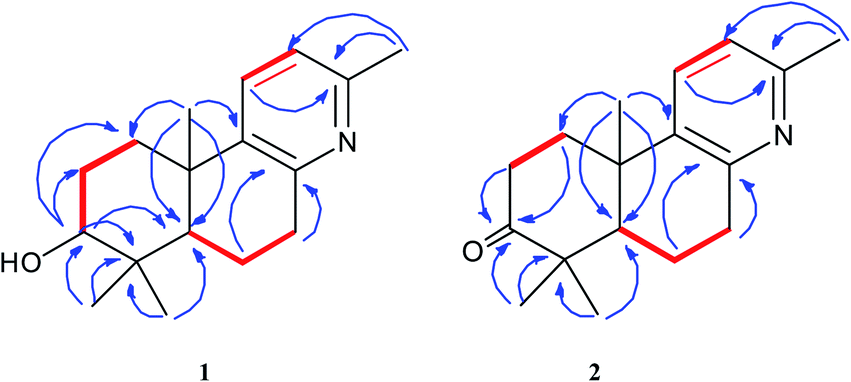
Forsyqinlingine A (1) was obtained as colorless, crystalline needles and showed a positive response to Dragendoff's reagent in MeOH, confirming that it was, indeed, an alkaloid. The molecular formula was determined to be C17H25NO based on the parent quasi-molecular ion peak at m/z 260.2004 ([M + H]+, calcd for C17H26NO, 260.2014) in the HRMS (ESI positive-ion) spectrum. Based on the molecular formula, 1 contained six degrees of unsaturation. The IR spectrum of 1 featured absorption bands at 3425, 1634, 1575, and 1463 cm−1, which corresponded to the hydroxyl and aromatic ring in the molecule. In the 1H NMR spectrum, four tertiary methyl groups at δH 2.33 (s, 3H, H-16), 0.98 (s, 3H, H-18), 0.77 (s, 3H, H-19), and 1.07 (s, 3H, H-20), one oxygenated methine group at δH 3.08 (br t, J = 7.2 Hz, 1H, H-3), one hydroxy group at δH 4.45 (br s, 1H, C3–OH), and two adjacent aromatic protons at δH 7.48 (d, J = 8.1 Hz, 1H, H-11) and 6.94 (d, J = 8.1 Hz, 1H, H-12). The 13C NMR displayed 17 carbon resonances, and the HMQC spectrum indicated the existence of four methyl, four methylene (sp3 hybridized), four methine (two sp2 hybridized and two sp3 hybridized), and five quaternary (three sp2 hybridized and two sp3 hybridized) carbons. After detailed analyses of the 1H and 13C NMR spectra, five sp2 hybridized carbons and two aromatic protons were assigned to a 2,3,6-trisubstituted pyridine ring moiety. Additionally, based not only on the number of carbon resonances in the 13C NMR but also on the spectral information of previously isolated labdane diterpenoid,8,9 it was suspected that compound 1 was a trinorlabdane diterpenoid. As shown in Fig. 2, the 1H–1H COSY spectrum of 1 was consistent with two structural fragments coupled together. The first spin system was traced from a methine group (δH 1.18, dd, J = 2.2, 12.4 Hz, 1H, H-5) and two methylene groups (δH 1.84, m, 1H, H-6a; 1.87, m, 1H, H-6b; 2.77, m, 1H, H-7a; 2.89, dd, J = 5.6, 18.2 Hz, 1H, H-7b), which resulted in the identification of the C5–C6–C7 subunit. The second spin system also included a methine group (δH 3.08, br t, J = 7.2 Hz, 1H, H-3) and two methylene groups (δH 1.70, m, 1H, H-1a; 2.21, m, 1H, H-1b; 1.28, m, 1H, H-2a; 1.62, m, 1H, H-2b), from which the C1–C2–C3 moiety was identified. In the HMBC spectrum of 1, 1H–13C long-range correlations from H2-1 (δH 1.70 and 2.21) to C-10 (δC 37.1) and C-5 (δC 49.7); from H-5 (δH 1.18) to C-10 (δC 37.1), C-1 (δC 36.6), C-4 (δC 39.1), and C-3 (δC 77.0); and from H-3 (δH 3.08) to C-4 (δC 39.1) and C-5 (δC 49.7) confirmed that the two fragments (C1–C2–C3 and C5–C6–C7) were connected at C-4 and C-10. Furthermore, the HMBC cross peaks observed from H2-7 (δH 2.77 and 2.89) to C-8 (δC 154.6) and C-9 (δC 141.5) and from H2-1 (δH 1.70 and 2.21) to C-9 (δC 141.5) indicated that the C5–C6–C7 and C1–C2–C3 structural units were connected to the pyridine ring at C-10 and C-7, respectively; therefore, these observations led to the identification of an octahydrobenzo[f]quinoline skeleton. Moreover, the HMBC correlations between H3-16 (δH 2.33) and C-13 (δC 154.6), H3-18 (δH 0.98) and C-4 (δC 39.1), H3-19 (δH 0.77) and C-4 (δC 39.1), H3-20 (δH 1.07) and C-10 (δC 37.1), and between H-3 (δH 3.08) and C-2 (δC 28.2) and C-4 (δC 39.1) alluded to four methyl groups and one hydroxyl group at C-13, C-4, C-4, C-10, and C-3, respectively. Based on the aforementioned data and comprehensive 2D NMR experiments (1H–1H COSY, HMQC and HMBC), the structure of 1 was ultimately determined to be 3-hydroxyl-4,4,10,13-tetramethyl-1(2),3(4),5(10),6(7)-octahydrobenzo[f]quinolin, belonging to a unique class of planar trinorlabdane diterpenoid alkaloids.
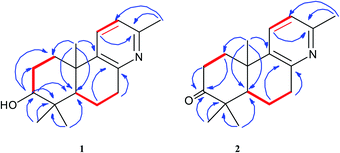 |
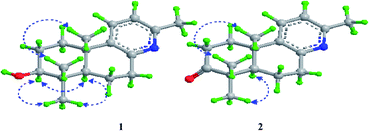
| | Fig. 2 The key 1H–1H COSY ( ) and HMBC ( ) and HMBC ( ) correlations of compounds 1 and 2. ) correlations of compounds 1 and 2. | |
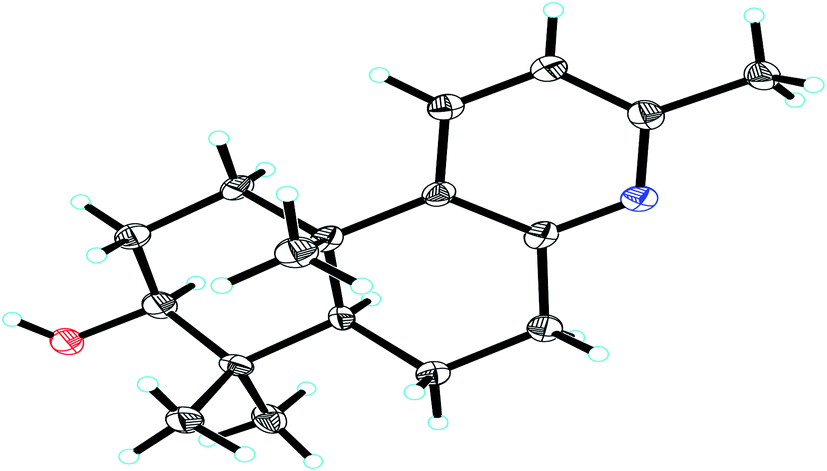
The relative stereochemical configuration of 1 was deduced from the 1H–1H coupling constants (Table 1) and NOESY spectrum (Fig. 3). The coupling constants between H-5 and H-6a/H-6b were calculated to be 2.2 and 12.4 Hz, respectively, which suggested that the cyclohexane and cyclohexene rings were fused together in the trans-configuration, and H-5 was in the axial position. In the NOESY spectrum, H-3/H-5, H-3/H3-18, and H3-19/H3-20 cross peaks indicated that H-3 and H-5 were co-facial and α-oriented, while H3-20 was on the opposite side of the molecular plane and, therefore, β-oriented. Furthermore, the structure of 1 was verified by single-crystal X-ray crystallography using Cu Kα radiation (Fig. 4), which not only confirmed the structural assignment mentioned above but also unambiguously established the absolute configuration of 1 as (3S,5R,10S). Taken together, the complete chemical structure of 1 is shown in Fig. 1.
Table 1 NMR data for compounds 1 and 2 (400 MHz for 1H, and 100 MHz for 13C)a,b
| No. |
1 (DMSO-d6) |
2 (DMSO-d6) |
| δH |
δC |
δH |
δC |
| Chemical shift values were in ppm and J values (in Hz) were presented in parentheses. The assignments were based on HMQC, HMBC, and 1H–1H COSY experiments. |
| 1 |
1.70 (m, H-1a) |
36.6 |
1.76 (m, H-1a) |
36.8 |
| 2.21 (m, H-1b) |
|
2.52 (m, H-1b) |
|
| 2 |
1.28 (m, H-2a) |
28.2 |
2.43 (m, H-2a) |
34.6 |
| 1.62 (m, H-2b) |
|
2.67 (m, H-2b) |
|
| 3 |
3.08 (br t, 7.2) |
77.0 |
|
216.1 |
| 4 |
|
39.1 |
|
47.1 |
| 5 |
1.18 (dd, 2.2, 12.4) |
49.7 |
1.86 (dd, 2.0, 12.4) |
49.7 |
| 6 |
1.84 (m, H-6a) |
18.8 |
1.72 (m, H-6a) |
19.8 |
| 1.87 (m, H-6b) |
|
1.82 (m, H-6b) |
|
| 7 |
2.77 (m, H-7a) |
33.7 |
2.81 (m, H-7a) |
33.5 |
| 2.89 (dd, 5.6, 18.2, H-7b) |
|
2.93 (dd, 5.6, 18.0, H-7b) |
|
| 8 |
|
154.6 |
|
154.1 |
| 9 |
|
141.5 |
|
140.0 |
| 10 |
|
37.1 |
|
36.6 |
| 11 |
7.48 (d, 8.1) |
133.2 |
7.62 (d, 8.2) |
147.0 |
| 12 |
6.94 (d, 8.1) |
121.0 |
7.03 (d, 8.2) |
121.5 |
| 13 |
|
154.6 |
|
154.9 |
| 16 |
2.33 (s, 3H) |
24.0 |
2.37 (s, 3H) |
23.7 |
| 18 |
0.98 (s, 3H) |
28.7 |
1.08 (s, 3H) |
26.8 |
| 19 |
0.77 (s, 3H) |
16.3 |
1.03 (s, 3H) |
21.3 |
| 20 |
1.07 (s, 3H) |
25.1 |
1.19 (s, 3H) |
24.4 |
| 3-OH |
4.45 (br s) |
|
|
|
 |
| | Fig. 3 The key NOESY ( ) correlations of compounds 1 and 2. ) correlations of compounds 1 and 2. | |
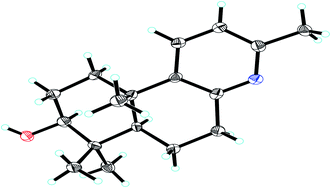 |
| | Fig. 4 X-ray crystal structure of compound 1. | |
Forsyqinlingine B (2), obtained as colorless, crystalline needles after purification of the EtOH extract, also demonstrated a positive response to Dragendoff's reagent in MeOH, confirming that it was, indeed, an alkaloid. Based on the [M + H]+ ion peak at m/z 258.1848 (calcd for C17H24NO, 258.1858) in the HRMS (ESI positive-ion mode) spectrum of 2, a molecular formula of C17H23NO was determined. Detailed comparison of its 1H and 13C NMR spectral data (Table 1) to those of 1 indicated that 2 shared a great deal of structural similarities to 1. The notable differences in the NMR spectra between the two compounds was the absence of the hydroxyl group at C-3 (δH 3.08, t, J = 7.2 Hz, 1H; δC 77.0) in 1 and the presence of one carbonyl group at C-3 (δC 216.1) in 2, which was further supported by the HMBC correlations between H3-19 (δH 1.03, s, 3H) and H3-18 (δH 1.08, s, 3H) and between H-5 (δH 1.86, dd, J = 2.0, 12.4 Hz, 1H) and C-3 (δC 216.1) (Fig. 2). The relative configuration of 2 was elucidated by NOESY experiments in combination with the coupling constants in the 1H–1H COSY spectrum. The coupling constants of JH-5,6a = 12.4 Hz and JH-5,6b = 2.0 Hz implied that H-5 was axially oriented, while the two six-membered rings were fused together in the trans-configuration. Furthermore, the H-5/CH3-18 and CH3-19/CH3-20 NOESY correlations indicated that CH3-20 was in the β-orientation, while H-5 was in the α-orientation (Fig. 3). We attempted to obtain single crystals of 2 that were suitable for determining its absolute configuration by X-ray diffraction, but we were not successful. Therefore, the absolute configuration of 2 was elucidated by comparing the experimental ECD spectrum with the calculated ECD spectra of the enantiomers. As shown in Fig. 5, the measured ECD spectrum of 2 was markedly different from the calculated ECD spectrum of (5S,10R)-2b but was well-fitted to the calculated ECD spectrum of (5R,10S)-2a. Thus, the absolute configurations of the two stereogenic centers in 2 were defined as 5R,10S, providing the complete chemical structure of 2 shown in Fig. 1.
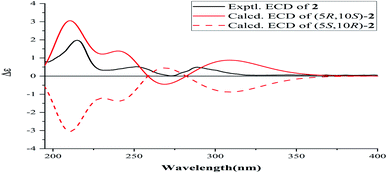 |
| | Fig. 5 ECD spectrum of compound 2. | |
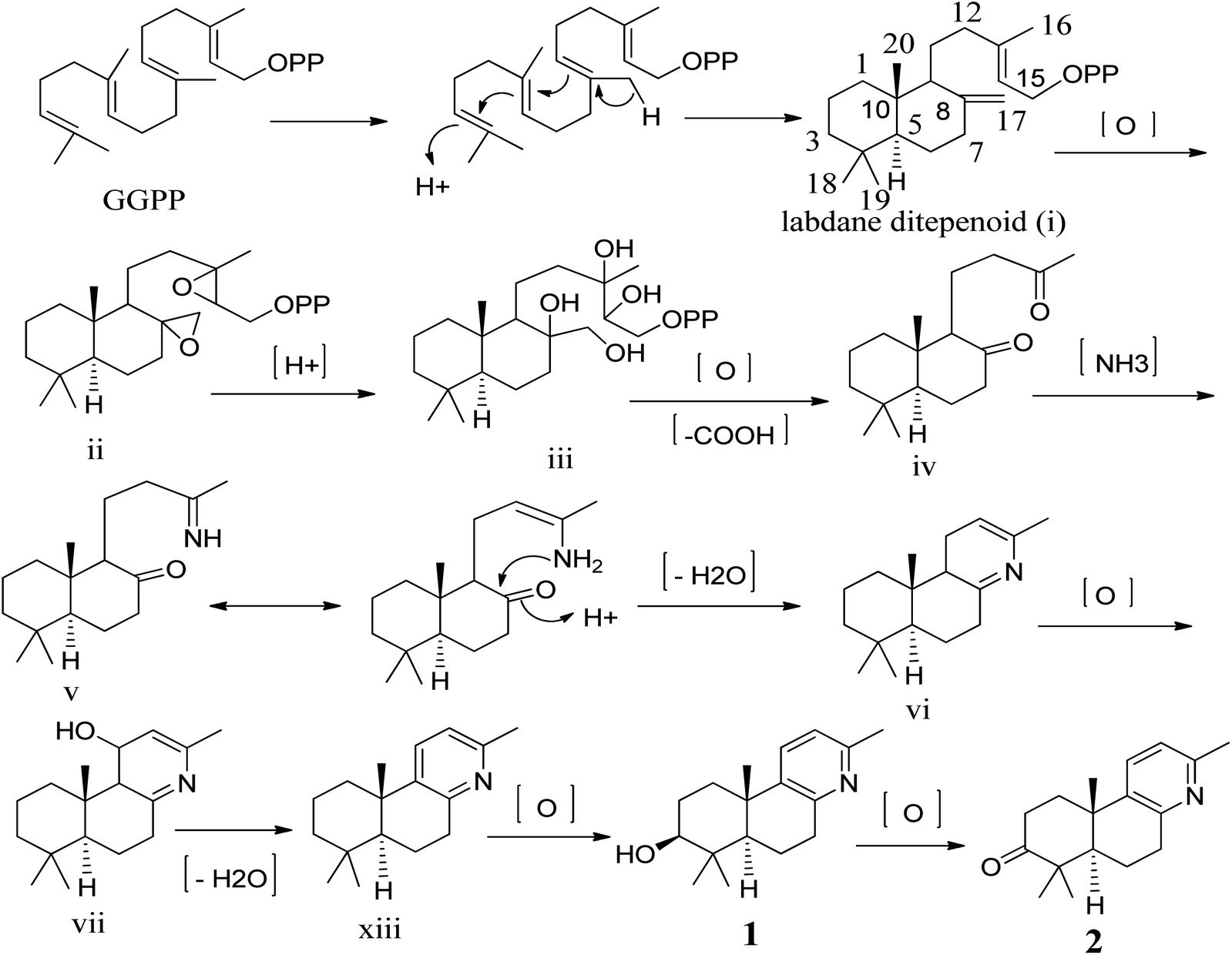
To our knowledge, although norlabdane diterpenoids have previously been isolated as secondary metabolites from various plant sources,10–12 this study was the first to report the isolation of norlabdane diterpene alkaloids. Given the unique and intriguing structures of the two trinorlabdane diterpenoid alkaloids, a plausible biogenetic pathway for biosynthesis of compounds 1 and 2 was proposed (Scheme 1). In this biosynthetic pathway, the precursor to both 1 and 2 was traced back to (E, E, E)-geranylgeranyl diphosphate (GGPP). First, GGPP was converted into the general labdane-type diterpenoid i by an acid–base catalyzed bicyclization reaction,13,14 which was then transformed into ii via the epoxidation of the Δ8(17) and Δ13(14) double bonds. After opening of the epoxide ring, oxidative cleavage (C13–C14 and C8–C17), and decarboxylation at C-14 and C-17, iii was transformed into iv.11,12,15 Through the use of a selective aminotransferase and imine reductase, iv was transformed into v via the condensation of ammonia with the methyl ketone.16,17 Intermediate v further underwent nucleophilic addition onto another ketone, followed by dehydration to yield vi. Through oxidative hydroxylation of C-11 in vi produced vii, which then generated viii by dehydration. Finally, viii was oxidized by hydroxylase into compound 1, which was further oxidized into compound 2.
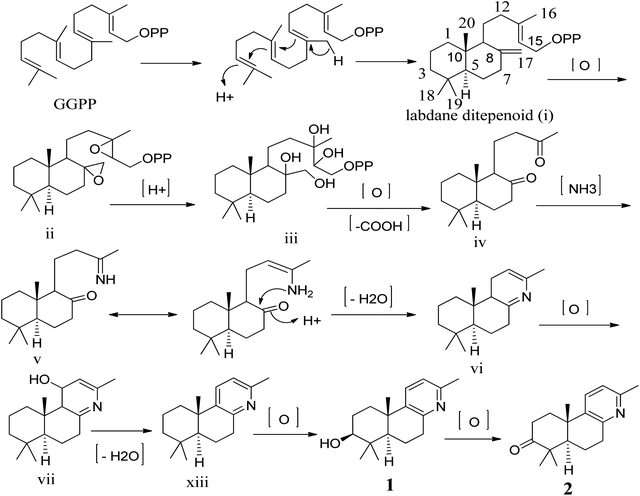 |
| | Scheme 1 Proposed biogenetic pathway of compounds 1 and 2. | |
The effects of compounds 1 and 2 and ginkgolide B (positive control) on the release of β-glucuronidase in rat polymorphonuclear leukocytes (PMNs) induced by platelet activating-factor (PAF) were measured, and the corresponding results are shown in Table 2. Based on the bioassay results, it was concluded that two novel trinorlabdane diterpenoid alkaloids possessed slightly higher anti-inflammatory activities compared to the positive control.
Table 2 Inhibitory activities of compounds 1 and 2 to the release of β-glucuronidasea
| Compounds |
Inhibition rate (%) |
| The inhibitory rates of two novel trinorlabdane diterpenoid alkaloids (1 and 2) and ginkgolide B were tested at a concentration of 10 μM. Positive control substance. |
| Ginkgolide Bb |
54.1 ± 2.35 |
| 1 |
56.7 ± 2.42 |
| 2 |
58.6 ± 3.01 |
In vitro, compounds 1 and 2 were evaluated for their anti-viral activities against the H1N1 virus. Two trinorlabdane diterpenoid alkaloids showed activities; their IC50 values are shown in Table 3. In addition, the anti-viral activities against the RSV of compounds 1 and 2 were also tested. As a result, two trinorlabdane diterpenoid alkaloids showed activities, as shown in Table 3. The first isolated norlabdane diterpene alkaloids showed better anti-viral activities against the H1N1 virus and RSV compared with the previously isolated labdane diterpenoids from this plant.8,9
Table 3 Anti-viral activities of compounds 1 and 2 against influenza A (H1N1) virus and respiratory syncytial virus (RSV)
| Compounds |
IC50 (μM, against H1N1) |
EC50 (μM, against RSV) |
| 1 |
6.9 |
5.0 |
| 2 |
7.7 |
4.8 |
Conclusions
Herein, two trinorlabdane diterpenoid alkaloids (forsyqinlingines A-B, 1 and 2) featuring an unprecedented structural framework were isolated and identified from the ripe fruits of Forsythia suspensa. These compounds enrich the structural diversity of not only labdane diterpenoid but also alkaloids. Furthermore, forsyqinlingines A-B showed significant anti-inflammatory activities by inhibiting the release of β-glucuronidase from polymorphonuclear leukocytes of rats, with inhibition rates in the range 56.7–58.6%, as well as antiviral activities against influenza A (H1N1) virus and RSV, with IC50 values in the range 6.9–7.7 μM and EC50 values in the range 4.8–5.0 μM, respectively. The identification of the chemical structures of these two C17-labdane diterpenoid alkaloids and their potentially important biological activities aim to inspire the further development of research related to phytochemicals, organic synthesis, biosynthesis, and pharmacology.
Experimental section
General experimental procedures
Optical rotations were measured on a Perkin-Elmer 241 polarimeter. CD spectra were obtained on a JASCO J-815 spectropolarimeter. UV spectra were obtained on a Shimadzu UV-160 spectrophotometer. IR spectra were recorded on a Perkin-Elmer 683 infrared spectrometer with KBr disks. HR-ESIMS were recorded on a Bruker Daltonics on a Micromass LCT mass spectrometer. NMR spectra were recorded on an ECZ400S spectrometer, with TMS as the internal standard. Silica gel (200–300 mesh) for column chromatography (CC) and silica gel GF254 for preparative TLC were obtained from Qingdao Marine Chemical Factory, Qingdao, People's Republic of China. Preparative HPTLC (high performance thin layer chromatography) was conducted on Merck precoated silica gel 60 F254 plates, and detected under UV light.
Plant material
The ripe fruits of Forsythia suspensa (Thunb) Vahl (Oleaceae), were collected in Qinling District, Shanxi Province, People's Republic of China, in October 2020, and identified by Professor Yan-Yan Zhao, School of Pharmaceutical Science, Yantai University. Fructus forsythiae was harvested and air-dried at room temperature in the dark. A voucher specimen (YP201165) has been deposited at the Herbarium of School of Pharmaceutical Science, Yantai University.
Extraction and isolation
The air-dried ripe fruits (30.0 kg) of F. suspensa were extracted three times with refluxing EtOH. The solvent was removed under reduced pressure and the resulting extract was subjected to extraction with 2% HCl. Following this, the aqueous solution was adjusted with NH4OH to pH 10 and extracted with CHCl3. The organic fractions were combined, and the solvent was evaporated under vacuum to yield the alkaloid fraction (10.4 g). The crude alkaloid was initially subjected to column chromatography (4 × 100 cm) on silica gel (330 g), eluted with gradient mixture of cyclohexane–acetone [95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 90:10, 85:15, 80:20, 75:25, 70:30, 65:35, 60:40, 55:45, 50:50, v/v] to give ten fractions on the basis of TLC. Fraction 6 (1.1 g) was separated firstly by CC over silica gel (70 g, eluted by petroleum ether–acetone, 95:5-70:30, v/v) and preparative TLC and HPTLC (cyclohexane–ethyl acetate, 60:40, v/v), and subsequently purified on Sephadex LH-20 (150 g, eluting with CHCl3–CH3OH, 50:50, v/v) to afford compound 2 (72 mg). Fraction 7 (1.6 g) was separated by CC over silica gel (90 g, eluted by petroleum ether–acetone, 90:10-65:35, v/v), and preparative TLC and HPTLC (CHCl3–CH3OH, 95:5, v/v), and then purified on Sephadex LH-20 (150 g, eluting with CHCl3–CH3OH, 50:50, v/v) to afford compound 1 (69 mg).
:5, 90:10, 85:15, 80:20, 75:25, 70:30, 65:35, 60:40, 55:45, 50:50, v/v] to give ten fractions on the basis of TLC. Fraction 6 (1.1 g) was separated firstly by CC over silica gel (70 g, eluted by petroleum ether–acetone, 95:5-70:30, v/v) and preparative TLC and HPTLC (cyclohexane–ethyl acetate, 60:40, v/v), and subsequently purified on Sephadex LH-20 (150 g, eluting with CHCl3–CH3OH, 50:50, v/v) to afford compound 2 (72 mg). Fraction 7 (1.6 g) was separated by CC over silica gel (90 g, eluted by petroleum ether–acetone, 90:10-65:35, v/v), and preparative TLC and HPTLC (CHCl3–CH3OH, 95:5, v/v), and then purified on Sephadex LH-20 (150 g, eluting with CHCl3–CH3OH, 50:50, v/v) to afford compound 1 (69 mg).
Forsyqinlingine A (1). Colorless needle crystals (acetone); mp 172–173 °C; [α]25D + 51.4 (c 0.43, MeOH); IR (KBr) νmax: 3425, 3021, 2976, 2875, 1634, 1575, 1463, 1450, 1381, and 1022 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS m/z: 260.2004 [M + H]+ (calcd for C17H26NO, 260.2014).
Forsyqinlingine B (2). Colorless needle crystals (acetone); mp 164–166 °C; [α]25D + 32.7 (c 0.49, MeOH); UV (MeOH) λmax: 196 and 272 nm; CD (MeOH) λmax (Δε): 215 (+5.94) and 251 (+1.56) nm; IR (KBr) νmax: 3019, 2983, 2860, 1739, 1638, 1581, 1456, 1380 and 1024 cm−1; 1H and 13C NMR data, Table 1; HR-ESIMS m/z: 258.1848 [M + H]+ (calcd for C17H24NO, 258.1858).
X-ray crystallographic analysis
Crystal data for compound 1: C17H25NO·2(H2O), M = 295.41, a = 9.7060 (9) Å, b = 24.576 (2) Å, c = 14.6471 (17) Å, α = 90°, β = 109.129 (4)°, γ = 90°, V = 3300.9 (6) Å3, T = 100 (2) K, space group P1211, Z = 8, μ (Cu Kα) = 0.638 mm−1, 47471 reflections measured, 12142 independent reflections (Rint = 0.1023). The final R1 values were 0.1145 (I > 2σ(I)). The final wR (F2) values were 0.3024 (I > 2σ(I)). The final R1 values were 0.1218 (all data). The final wR (F2) values were 0.3132 (all data). The goodness of fit on F2 was 1.371. Flack parameter = −0.24 (14). CCDC: 2095607.
Quantum chemical ECD calculation
The absolute configuration of compound 2 was determined by comparing their experimental ECD spectra with the corresponding quantum chemical calculated ones. One of the two enantiomers for compound (5R,10S)-2 were arbitrary chosen for theoretical studies. Conformational analyses were firstly carried out via Monte Carlo searching using molecular mechanism with MMFF force field in the Spartan 18 program, and (5R,10S)-2 used SYBYL force field. The results showed 3 conformers for (5R,10S)-2 within an energy window of 10 kcal mol−1. The conformers were then reoptimized using DFT at the B3LYP/6-31G (d) level using the Gaussian 09 program. The B3LYP/6-31G (d) harmonic vibrational frequencies were further calculated to confirm its stability.18 Three conformers of (5R,10S)-2 whose relative Gibbs free energies in the range of 0–2 kcal mol−1, were refined and considered for next step. The energies, oscillator strengths, and rotational strengths of the first 60 electronic excitations of (5R,10S)-2 was calculated in methanol. The ECD spectra were simulated by the overlapping Gaussian function (0.35 eV, +10 nm) in horizontal axis for (5R,10S)-2, in which velocity rotatory strengths of the first 5 exited states for (5R,10S)-2 were adopted.19 To get the final ECD spectrum, the simulated spectra of the lowest energy conformers were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy (G). The theoretical ECD curve of (5S,10R)-2 were obtained by directly reverse that of (5R,10S)-2.
Anti-inflammatory assay
The anti-inflammatory activities of two trinorlabdane diterpenoid alkaloids (1 and 2) and ginkgolide B (as positive control) were determined as previously described,8,9 based on the inhibition of the release of β-glucuronidase from rat polymorphonuclear leukocytes (PMNs) induced by platelet activating-factor in vitro.
Anti-viral assay
Neuraminidase inhibition assay. Two trinorlabdane diterpenoid alkaloids (1 and 2) were investigated for their inhibitory effects on NAs from A/PR/8/34 (H1N1). This assay was performed as previously described.8,9 The 50% inhibitory concentration (IC50) was determined by extrapolating the results from various doses tested using a linear equation, with ribavirin as the drug control.
Cytopathic effect reduction assay
Compounds 1 and 2 and ribavirin (as drug control) were tested for their anti-viral activities against RSV using the cytopathic effect (CPE) inhibition assay as described previously.8,9 The concentration reducing CPE by 50% with respect to virus control was estimated from graphic plots and was defined as EC50.
Cytotoxicity assay. Compounds 1 and 2 were evaluated for their cytotoxic activities using MTT assay as described previously.8,9 Each compound was tested in triplicate, and the experiments were repeated three times. The results of MTT assay of compounds 1 and 2 are shown in Fig. 6.
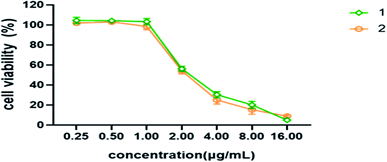 |
| | Fig. 6 Cell viability of different concentrations of compounds 1 and 2 on MDCK cells. | |
Conflicts of interest
There are no conflicts of interest.
Acknowledgements
The authors are grateful to Ms Qianqian Sun (Yantai Branch, Shanghai Institute of Materia Medica, Chinese Academy of Sciences), Zhihua Song (School of Pharmaceutical Science, Yantai University) for the measurements of ESIMS, HR-ESIMS and NMR spectra, respectively. The authors also gratefully acknowledge Mr Yunxue Zhao (School of Basic Medical Sciences, Shandong University) for the bioactivity screenings. This work was financially supported by the Natural Science Foundation of China (21372189) and Natural Science Foundation of Shandong Province (ZR2019HM091).
Notes and references
- F. Zhang, Y. N. Yang, Z. M. Feng, J. S. Jiang and P. C. Zhang, RSC Adv., 2017, 7, 24963–24969 RSC.
- C. C. Chen, H. Y. Chen, M. S. Shiao, Y. L. Lin, Y. H. Kuo and J. C. Ou, Planta Med., 1999, 65, 709–711 CrossRef CAS PubMed.
- H. C. Ko, B. L. Wei and W. F. Chou, J. Ethnopharmacol., 2005, 102, 418–423 CrossRef PubMed.
- W. J. Hu, X. P. Qian, Y. X. Tu, Z. T. Shen, L. X. Yu and B. R. Liu, J. Nanjing Univ. Tradit. Chin. Med., 2007, 6, 379–381 Search PubMed.
- Y. Hao, D. F. Li, X. L. Piao and X. S. Piao, J. Ethnopharmacol., 2010, 128, 412–418 CrossRef PubMed.
- S. Zhang, S. Y. Shao, X. Y. Song, C. Y. Xia, Y. N. Yang, P. C. Zhang and N. H. Chen, NeuroToxicology, 2015, 2, 72–83 Search PubMed.
- M. S. Kim, H. J. Na, S. W. Han, J. S. Jin, U. Y. Song, E. J. Lee, B. K. Song, S. H. Hong and H. M. Kim, Inflammation, 2003, 27, 129–135 CrossRef CAS PubMed.
- K. L. Xiang, R. X. Liu, L. Zhao, Z. P. Xie, S. M. Zhang and S. J. Dai, Phytochemistry, 2020, 173, 112298 CrossRef CAS PubMed.
- L. Zhao, K. L. Xiang, R. X. Liu, L. Zhao, Z. P. Xie, S. M. Zhang and S. J. Dai, Bioorg. Chem., 2020, 173, 112298 Search PubMed.
- J. B. Xu, Y. Y. Fan, L. S. Gan, Y. B. Zhou, J. Li and J. M. Yue, Chem.–Eur. J., 2016, 22, 14648–14654 CrossRef CAS PubMed.
- H. Yin, J. G. Luo, S. M. Shan, X. B. Wang, J. Luo, M. H. Yang and L. Y. Kong, Org. Lett., 2013, 15, 1572–1575 CrossRef CAS PubMed.
- Z. X. Zhang, P. Q. Wu, H. H. Li, F. M. Qi, D. Q. Fei, Q. L. Hu, Y. H. Liu and X. L. Huang, Org. Biomol. Chem., 2018, 16, 1745–1750 RSC.
- M. M. Xu, M. L. Hillwig, A. L. Lane, M. S. Tiernan, B. S. Moore and R. J. Peters, J. Nat. Prod., 2014, 77, 2144–2147 CrossRef CAS PubMed.
- M. M. Xu, M. L. Hillwig, M. S. Tiernan and R. J. Peters, J. Nat. Prod., 2017, 80, 328–333 CrossRef CAS PubMed.
- X. D. Wu, D. Luo, W. C. Tu, Z. T. Deng, X. J. Chen, J. Su, X. Ji and Q. S. Zhao, Org. Lett., 2016, 18, 6484–6487 CrossRef CAS PubMed.
- Y. Luo, X. Z. Li, B. Xiang, Q. Luo, J. W. Liu, Y. M. Yan, Q. Sun and Y. X. Cheng, Fitoterapia, 2018, 125, 135–140 CrossRef CAS PubMed.
- H. D. Peng, E. M. Wei, J. L. Wang, Y. N. Zhang, L. Cheng, H. M. Ma, Z. X. Deng and X. D. Qu, ACS Chem. Biol., 2016, 11, 3278–3283 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. L. X. Caricato, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. C 01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- P. J. Stephens and N. Harada, Chirality, 2010, 22, 229–233 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR (1D and 2D), HR-ESIMS, UV and IR spectra of 1 and 2, X-ray structure of 1, and ECD data of 2. CCDC 2095607. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra05760j |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Lin Zhaoa,
Li-Tong Suna,
Ze-Ping Xie
a,
Lin Zhaoa,
Li-Tong Suna,
Ze-Ping Xie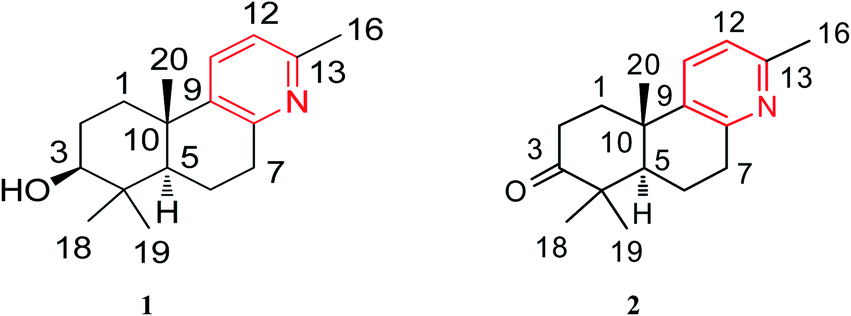
 ) and HMBC (
) and HMBC ( ) correlations of compounds 1 and 2.
) correlations of compounds 1 and 2.
 ) correlations of compounds 1 and 2.
) correlations of compounds 1 and 2.