
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenchmark calculations for bond dissociation energies and enthalpy of formation of chlorinated and brominated polycyclic aromatic hydrocarbons†
Shenying Xua,
Quan-De Wang *ab,
Mao-Mao Sunb,
Guoliang Yina and
Jinhu Liang*ac
*ab,
Mao-Mao Sunb,
Guoliang Yina and
Jinhu Liang*ac
aFaculty of Materials and Chemical Engineering, Yibin University, Yibin, Sichuan 644000, People's Republic of China. E-mail: quandewang@cumt.edu.cn
bLow Carbon Energy Institute and School of Chemical Engineering, China University of Mining and Technology, Xuzhou 221008, People's Republic of China
cSchool of Environment and Safety Engineering, North University of China, Taiyuan 030051, People's Republic of China. E-mail: jhliang@nuc.edu.cn
First published on 6th September 2021
Abstract
Thermodynamic properties, i.e., bond dissociation energies and enthalpy of formation, of chlorinated and brominated polycyclic aromatic hydrocarbons play a fundamental role in understanding their formation mechanisms and reactivity. Computational electronic structure calculations routinely used to predict thermodynamic properties of various species are limited for these compounds due to large computational cost to obtain accurate results by employing high-level wave function theory methods. In this work, a number of composite model chemistry methods (CBS-QB3, G3MP2, G3, and G4) are used to compute bond dissociation energies and enthalpies of formation of small to medium-size chlorinated and brominated polycyclic aromatic hydrocarbon compounds. The enthalpy of formation is derived via the atomization method and compared against the recommended values. Statistical analysis indicates that G4 is the best method. For comparison, three commonly used density functional theory (DFT) methods (M06-2X, ωB97X-D and B2PLYP-D3) with various basis sets including 6-311++G(d, p), cc-pVTZ, and cc-pVQZ in the prediction of bond dissociation energies and enthalpies of formation have been tested using the optimized geometries at the same M06-2X/6-311++G(d, p) level of theory. It is found that ωB97X-D/6-311++G(d, p) shows the best performance in computing the bond dissociation energies, while ωB97X-D/cc-pVTZ exhibits the best prediction in enthalpy of formation of the studied reaction systems. The structural effect on the bond dissociation energies and enthalpy of formation of chlorinated and brominated polycyclic aromatic hydrocarbons are then systematically analyzed. Based on comparisons of the various methods, reliable DFT methods are recommended for future theoretical studies on large chlorinated and brominated polycyclic aromatic hydrocarbons considering both accuracy and computational cost. This work, to the authors' knowledge, is the first to systematically benchmark theoretical methods for the accurate prediction of thermodynamic properties for chlorinated and brominated polycyclic aromatic hydrocarbons.
1. Introduction
Chlorinated and brominated polycyclic aromatic hydrocarbons (Cl-PAHs and Br-PAHs) are halogenated derivatives of polycyclic aromatic hydrocarbons, which are carcinogenic organic pollutants mainly produced and emitted from combustion processes.1–3 Understanding the sources and formation mechanisms of X-PAHs (X denotes Cl or Br) is important for controlling their emissions and human exposure to these ubiquitous pollutants. Due to the different fuel sources and combustion processes, X-PAHs can be formed in many congeners with different ring numbers, ring structures, substitution position, and number of halogenated atoms.1 These various structures significantly affect their corresponding chemical reactivity.A better understanding of the formation and oxidation mechanisms of these compounds is of central importance to control their formation and emission during combustion process.1,4 For this purpose, it is essential to know their thermodynamic properties including the bond dissociation energies (BDEs) and enthalpy of formation (ΔfH). Specifically, the two parameters are not only important thermochemical properties, but also, they have Evans–Polanyi-type correlations with reaction barrier heights, which can be used to estimate the activation energies of reactions when more complete experiments or theoretical calculations are not available.5,6 Traditionally, experimental determination (primarily calorimetry) is the ideal method to obtain accurate thermochemical data. However, this methodology is very laborious and time-consuming. Further, considering the large number of species involved in detailed combustion chemical kinetic mechanism, it is effectively impossible to obtain the related thermodynamic properties of these species.7 However, with the development of accurate quantum chemistry methods and computational technologies, quantum chemistry calculations now become a major method to obtain thermodynamic properties.8,9
The accuracy of derived thermodynamic properties from quantum chemistry depends largely on the computational methods applied and the size of the molecular system. The current implementation of coupled cluster theory in its CCSD(T) form10 with explicit inclusion of single and double electron excitations and perturbative inclusion of triple electron excitations together with the extrapolation of the computed energies to the basis-set limit (CBS)11 provides computed chemical energies at an accuracy of ∼1 kcal mol−1, i.e., “chemical accuracy”, thus, this method usually denoted as the “gold-standard” of quantum chemistry. However, this method is rarely achievable for large molecular systems due to the large computational cost.
Various efforts have been made to approach chemical accuracy in electronic structure methods focused on approximations to the CCSD(T)/CBS limit based on separability approximations, and the developed composite methods,12 i.e., Gn13,14 and CBS-QB3,15 employs a variety of methods and basis sets to improve computational accuracy. Concretely, the G4 method represents the culmination of the developed composite methods and can achieve a 2σ accuracy of 1.1 kcal mol−1 for a set of 114 combustion relevant heats of formation.13 However, the computational cost remains challenging for larger systems. Thus, the CBS-QB3 scheme of Peterson and coworkers15 has found considerable use in combustion kinetics because it requires less computational resources and can be ready for applications to large systems. The G3MP2 method is selected for comparisons since it has been proven to be efficient for PAH compounds.16–18 These methods have been widely employed in theoretical studies on thermochemistry and kinetic studies of large reaction systems.19–21 Recent years, another different methodology in computational chemistry to obtain accurate chemical energies is the development of various functionals within the density functional theory (DFT) framework.22 These DFT functionals have been developed for various property predictions.23 For thermodynamic and kinetic studies in combustion community, the M06-2X,24 ωB97X-D,25 and B2PLYP-D3 (ref. 26) together with Dunning's correlation consistent basis sets (cc-pVnZ, n = D, T, Q…)11,27,28 have been widely used for geometry optimization and energies calculations of large systems.8,29 In particular, the M06-2X functional has found widespread use in studying main group chemistry as well as in deriving thermodynamic and kinetic parameters of Br-containing systems as flame retardants.30,31 However, accuracy of these DFT functionals and composite methods are usually system-dependent. For the reaction systems considered in this work, fewer studies have been performed to obtain accurate thermodynamic properties.
Based on the above considerations, the major objective of the present study is to determine an appropriate quantum chemical method for studying thermodynamic properties of X-PAHs molecules and then perform a systematical theoretical study of the structural effect on the BDEs and ΔfH for various X-PAHs. The paper is organized as follows: the computational methodology is described in Section 2, and the results and discussion, including benchmark calculations, validations of DFT, and BDEs and ΔfH of X-PAHs, are presented in Section 3. Section 4 summarizes the major conclusions.
2. Computational methodology
Four composite methods, i.e., CBS-QB3,15 G3,14 G4,13 and G3MP2,32 are chosen for benchmarking as part of this work. Detailed procedures of these composite methods can be found in the corresponding references. For DFT calculations, all the geometry optimization of the structures are performed using M06-2X/6-311++G(d, p) method because it achieves a suitable balance between accuracy and computational cost. Analytical harmonic frequency calculations are also carried out at this level to obtain the zero-point vibrational energies (ZPVEs) and to confirm the nature of the stationary points with no imaginary frequency. During the computation of enthalpies, the values of ZPVEs are scaled by 0.97 as recommended for the M06-2X functional.24 Single point energies are then computed using the M06-2X, ωB97X-D, and B2PLYP-D3 functionals with 6-311++G(d, p), cc-pVTZ, and cc-pVQZ, respectively. The three DFT functionals are selected since they exhibit significant better performance for thermochemical and kinetic studies.8,33 Basis set extrapolation are not performed since there is no universal recipe for basis set extrapolation in DFT.34 All electronic structure calculations are performed using the Gaussian 09 suites of programs.35 Finally, compared with the above theoretical methods, the group additivity (GA) method provides a valuable and powerful way to quickly estimate the thermodynamic properties of molecules, which has been widely employed in modeling complex combustion reaction systems because accurate electronic structure computations are prohibitively expensive. Herein, to demonstrate the prediction accuracy of GA method for the studied X-PAHs, the obtained ΔfH298 from GA method is compared with the present theoretical results. The estimation of the ΔfH298 values using GA method are performed via the RMG software.36The BDEs of C–Cl and C–Br bonds in the X-PAHs compounds are computed using the following equation:
| E(BDE) = E0(X-PAH) − E0(R-PAH) − E0(X) | (1) |
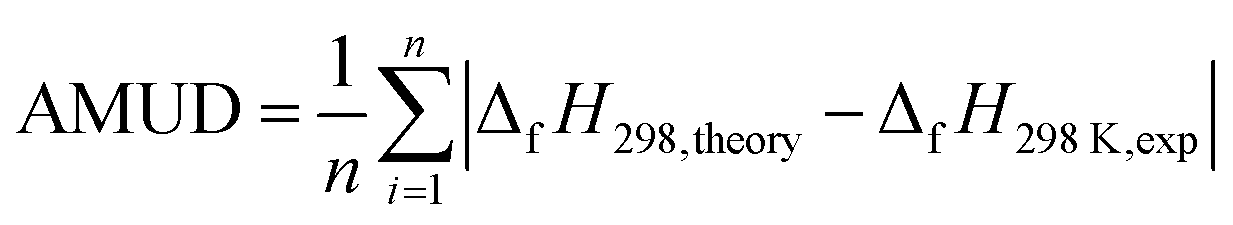 | (2) |
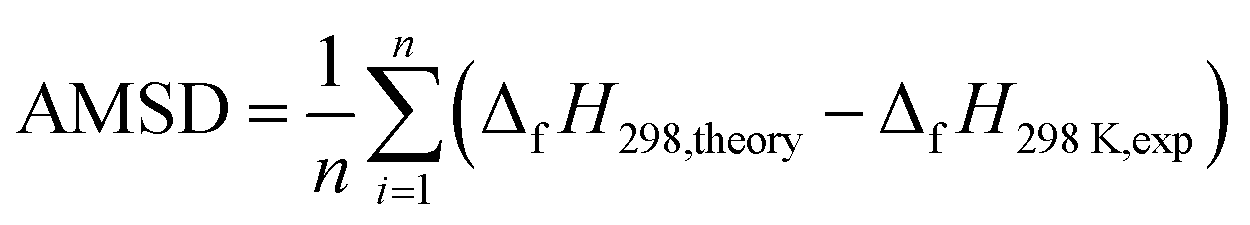 | (3) |
3. Results and discussion
The results and discussion section are organized as follows. First, we assess the ability of the examined composite and DFT methods to accurately reproduce the BDEs and enthalpies of formation for species with experimental values. Then, we proceed to discuss the BDEs and ΔfH of the studied reaction systems. Finally, we discuss the results obtained with group additivity method. The practical recommendations to derive accurate BDEs and ΔfH of the chlorinated and brominated polycyclic aromatic hydrocarbon molecules will be demonstrated.3.1. Benchmark results
Table 1 lists the structures, formula, and the experimental BDE and ΔfH298 values of X-PAHs used as benchmark. AMUD and AMSD with respect to experimental values of BDEs and ΔfH298 K obtained using four composite methods and various DFT functionals combined with 6-311++G(d, p), cc-pVTZ, and cc-pVQZ basis sets are given in Fig. 1. For the prediction accuracy of BDEs, it can be seen that the four composite methods together with the three DFT functionals with cc-pVnZ basis sets tend to overestimate the BDE values, while the three DFT functionals with 6-311++G(d, p) basis set tend to slightly underestimate the BDE values according to AMUD. The selected four composite methods exhibit the following prediction accuracy as G4 > G3 > G3MP2 > CBS-QB3. The most computational expensive G4 method shows the best prediction accuracy with the AMUD and AMSD values of 2.44 and 0.36 kcal mol−1, respectively. The large deviations of the CBS-QB3 method in the prediction of BDEs indicate that this method is not suitable for BDE computations. For the DFT methods, it is interestingly found that the prediction accuracy of BDEs does not increase by increasing the basis set levels. The three DFT functionals with the 6-311++G(d, p) basis set exhibit very good performance in the computation of BDE values, probably contributed by the inclusion of diffusion function in this basis set. It can be seen that the three DFT functionals with 6-311++G(d, p) basis set can provide the AMSD and AMUD values within −0.2 and 1.7 kcal mol−1, respectively, which is even better than the G4 method. Specifically, the ωB97X-D/6-311++G(d, p) method shows the best performance in prediction of BDE values.| Name | Formula | Structure | Expt. C–Cl/C–Br BDEs40 | Expt. ΔfH298 (ref. 41) |
|---|---|---|---|---|
| 1-Chlorobenzene | C6H5Cl |  |
95.5 ± 1.5 | 13.01 |
| 2-Chlorotoluene | C7H7Cl |  |
93.7 | — |
| 1-Chloronaphthalene | C10H7Cl |  |
96.3 ± 2.7 | 27.5 ± 2.3 |
| 2-Chloronaphthalene | C10H7Cl |  |
91.9 ± 2.7 | 32.8 ± 2.4 |
| 1-Bromobenzene | C6H5Br |  |
80.4 ± 1.5 | 25 × 1039 |
| 2-Bromotoluene | C7H7Br |  |
83.9 | — |
| 1-Bromonaphthalene | C10H7Br |  |
79.30 | 41.7 ± 1.3 |
| 2-Bromonaphthalene | C10H7Br |  |
81.7 | 41.97 ± 0.55 |
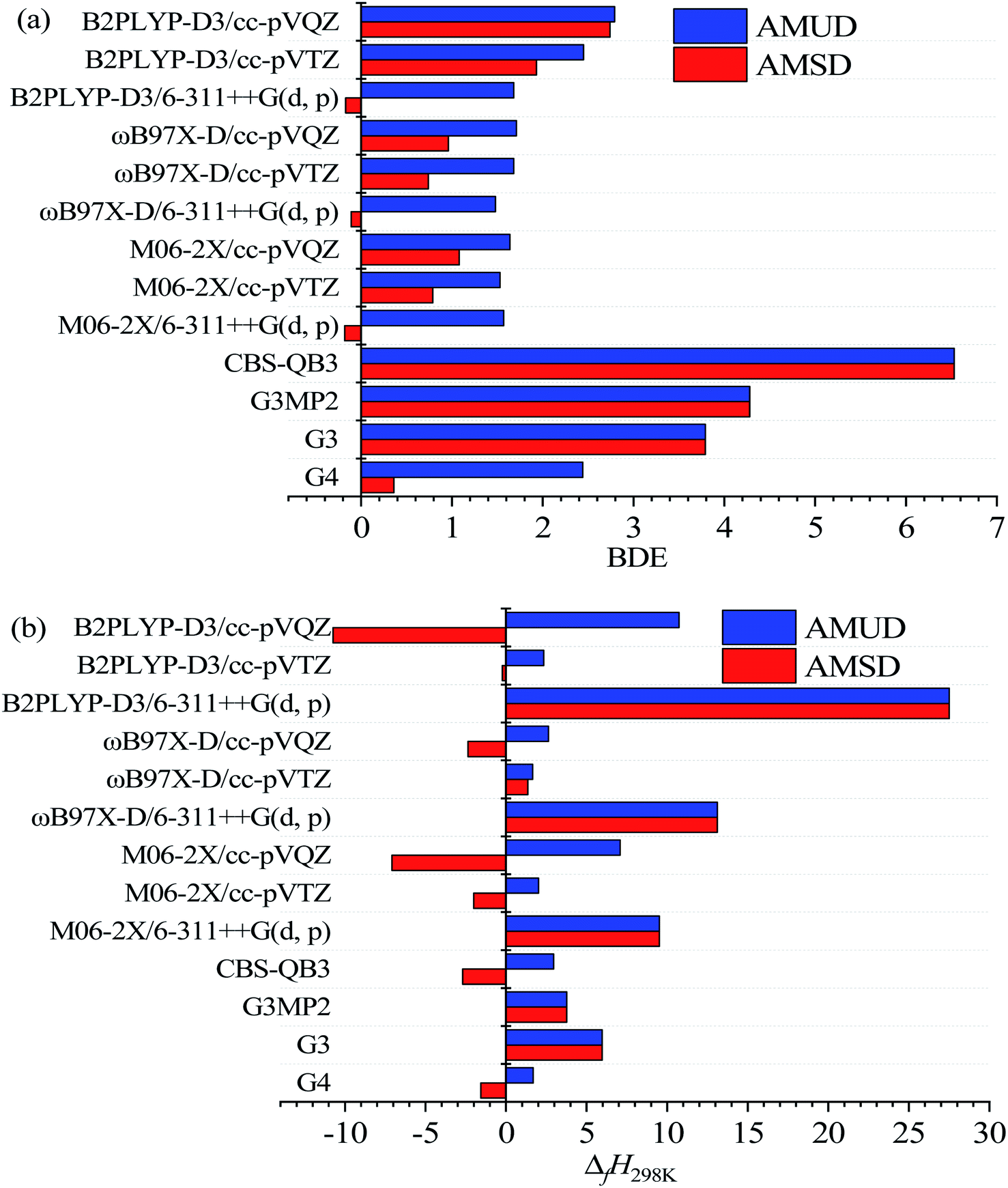 | ||
| Fig. 1 Averaged mean unsigned deviation (AMUD) and averaged mean signed deviation (AMSD) with respect to experimental/recommended values of BDEs (a) and ΔfH298K (b) via single-point energy results using four composite methods and three DFT functionals, i.e., M06-2X, ωB97X-D, and B2PLYP-D3 combined with 6-311++G(d, p), cc-pVTZ, and cc-pVQZ basis sets with the optimized geometry at M06-2X/6-311++G(d, p) level, respectively. | ||
The prediction accuracy of ΔfH298 using the selected methods exhibits very different performance compared with that of BDE results as shown in Fig. 2(b). Although the CBS-QB3 method is not suitable for the prediction of BDEs, the obtained ΔfH298 results can be comparative with the G4 method but with less computational cost. Specifically, the G4 method still is the best composite method for the prediction of ΔfH298, following is the CBS-QB3 method, G3MP2, and G3 methods in order. For the DFT methods, it is obvious that the small 6-311++G(d, p) basis set is not enough to accurately predict the absolute ΔfH298 values. The three DFT functionals with the moderate and widely used cc-pVTZ basis set demonstrate very good performance in the prediction of ΔfH298 values. Specifically, the B2PLYP-D3/cc-pVTZ method shows the smallest AMSD, while the ωB97X-D/cc-pVTZ method shows the smallest AMUD among the all the DFT methods. It is worth noting that the double-hybrid B2PLYP-D3 functional requires more computational cost arising from the MP2 component of the calculation, which limits its application to large X-PAH systems. The M06-2X/cc-pVTZ method is of comparable accuracy to the ωB97X-D/c-pVTZ and B2PLYP-D3/cc-pVTZ methods for the studied reaction systems. Overall, the G4 method demonstrates the best performance in the prediction of both BDEs and ΔfH298, however, the computational cost is also considerably larger compared with other composite methods and DFT methods.
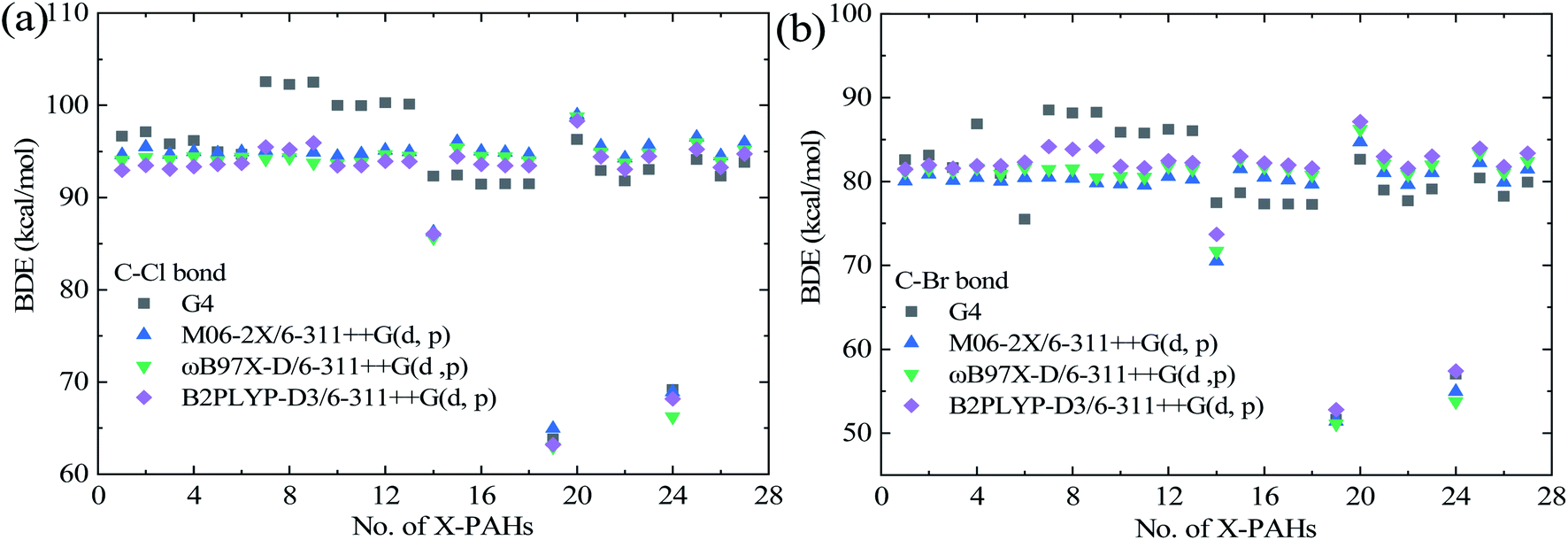 | ||
| Fig. 2 Predicted BDEs of C–Cl (a) and C–Br (b) bond in the studied X-PAH compounds via single-point energy computational results using various theoretical methods. | ||
3.2. BDE results
Although the number of samples used for benchmark is less due to the experimental difficulties to obtain these data, the very large AMSD and AMUD values of some methods as shown in Fig. 1 indicate that they are not reliable for the prediction of BDEs and ΔfH298 results. However, all the methods in the prediction of BDEs and ΔfH298 results for the studied reaction systems are also carried out, and compared with the accurate methods, and details are provided in ESI.† For clarity, Table 2 lists the names and structures of the studied Cl/Br-PAH compounds together with the predicted BDEs and ΔfH298 results using the accurate G4 composite method together with the recommended DFT methods from benchmark analysis. Specifically, the BDEs results from the G4 method and ωB97X-D/6-311++G(d, p) method are explicitly shown, while the predicted ΔfH298 results using the G4 method and ωB97X-D/cc-pVTZ method are listed.| No. | Name (X = chloro/bromo) | Structure (X = Cl/Br) | BDEs | ΔfH298 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G4 | ωB97X-D/6-311++G(d, p) | G4 | ωB97X-D/cc-pVTZ | GA | ||||||||
| Cl | Br | Cl | Br | Cl | Br | Cl | Br | Cl | Br | |||
| 1 | 1-Xbenzene |  |
96.65 | 82.60 | 94.04 | 81.15 | 12.81 | 24.76 | 14.58 | 25.22 | 12.7 | 22.01 |
| 2 | 2-Xtoluene |  |
97.12 | 83.13 | 94.37 | 81.45 | 8.59 | 15.97 | 6.68 | 17.34 | 4.71 | 14.02 |
| 3 | 3-Xtoluene |  |
95.81 | 81.65 | 94.12 | 81.23 | 9.54 | 21.60 | 6.92 | 17.54 | 4.71 | 14.02 |
| 4 | 4-Xtoluene |  |
96.15 | 86.86 | 94.44 | 81.62 | 9.74 | 16.94 | 7.11 | 17.67 | 4.71 | 14.02 |
| 5 | 1-Xnaphthalene | 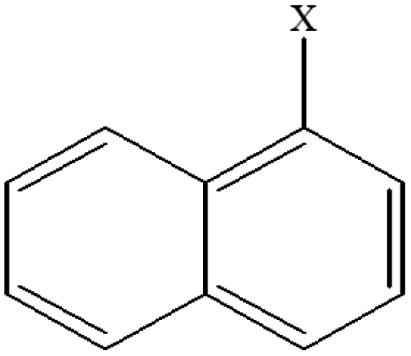 |
94.95 | 80.96 | 93.99 | 80.87 | 27.85 | 39.75 | 32.66 | 43.46 | 27.92 | 37.23 |
| 6 | 2-Xnaphthalene | 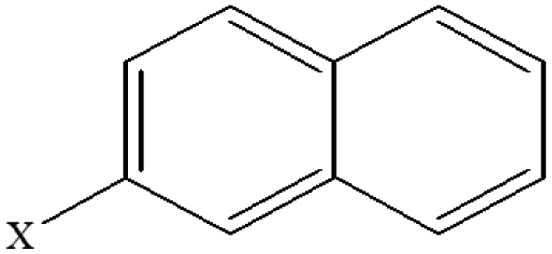 |
94.67 | 75.49 | 94.38 | 81.59 | 27.88 | 39.70 | 31.90 | 42.44 | 27.92 | 37.23 |
| 7 | 1-Xanthracene | 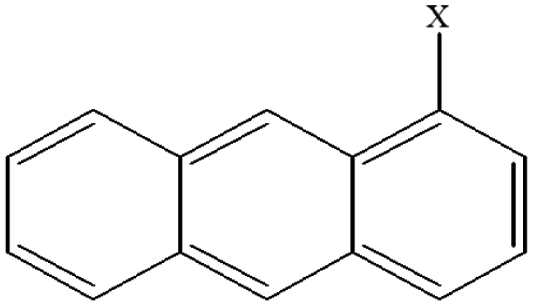 |
102.55 | 88.52 | 94.18 | 81.45 | 35.25 | 65.19 | 54.09 | 64.61 | 43.14 | 52.45 |
| 8 | 2-Xanthracene | 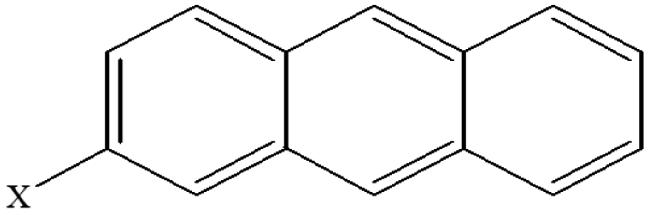 |
102.25 | 88.16 | 94.33 | 81.52 | 35.20 | 65.24 | 53.63 | 64.17 | 43.14 | 52.45 |
| 9 | 9-Xanthracene | 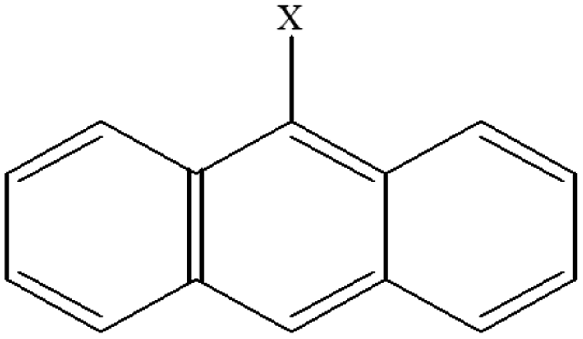 |
102.49 | 88.24 | 93.74 | 80.45 | 35.67 | 65.87 | 55.26 | 66.31 | 43.14 | 52.45 |
| 10 | 9-Xphenanthrene | 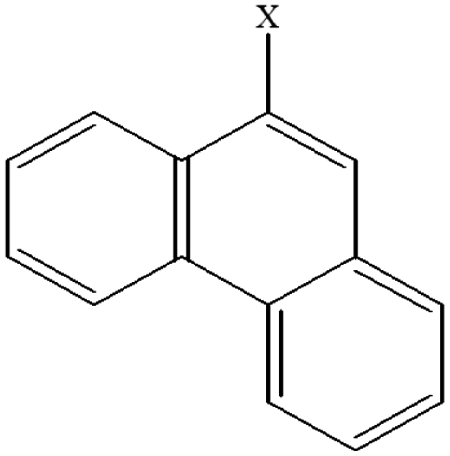 |
99.98 | 85.87 | 93.67 | 80.62 | 30.06 | 60.15 | 48.15 | 58.90 | 40.94 | 50.25 |
| 11 | 1-Xphenanthrene | 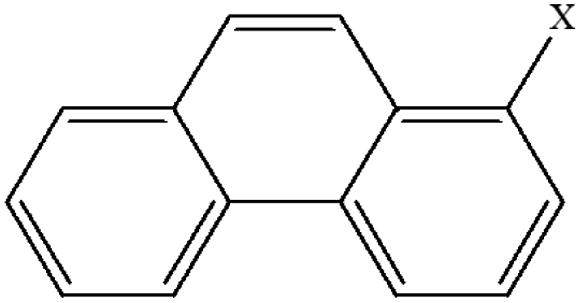 |
99.95 | 85.76 | 93.82 | 80.53 | 30.27 | 60.42 | 48.10 | 59.10 | 40.94 | 50.25 |
| 12 | 2-Xphenanthrene | 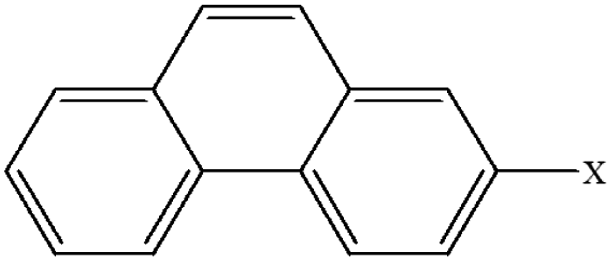 |
100.26 | 86.20 | 94.66 | 81.81 | 29.87 | 59.90 | 47.09 | 57.68 | 40.94 | 50.25 |
| 13 | 3-Xphenanthrene | 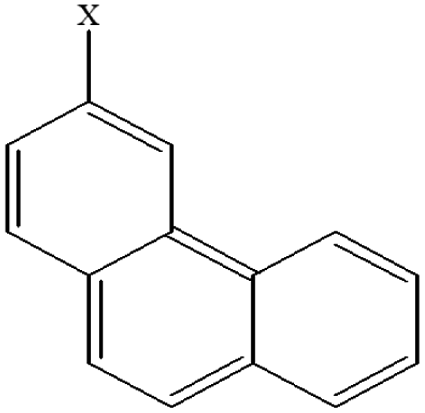 |
100.11 | 86.02 | 94.49 | 81.49 | 29.79 | 59.84 | 47.06 | 57.73 | 40.94 | 50.25 |
| 14 | 4-Xphenanthrene | 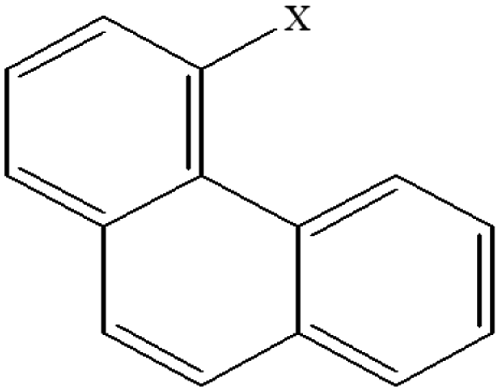 |
92.31 | 77.45 | 85.63 | 71.67 | 36.28 | 67.11 | 54.67 | 66.41 | 40.94 | 50.25 |
| 15 | 1-Xfluorene | 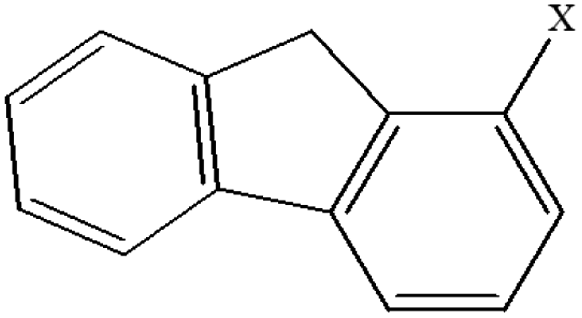 |
92.43 | 78.66 | 95.39 | 82.59 | 24.96 | 53.88 | 40.06 | 50.57 | 35.09 | 44.4 |
| 16 | 2-Xfluorene | 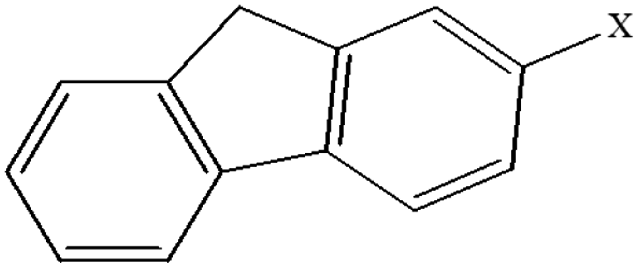 |
91.44 | 77.31 | 94.47 | 81.71 | 26.08 | 55.37 | 40.93 | 51.45 | 35.09 | 44.4 |
| 17 | 3-Xfluorene |  |
91.47 | 77.32 | 94.44 | 81.48 | 26.11 | 55.40 | 40.93 | 51.68 | 35.09 | 44.4 |
| 18 | 4-Xfluorene |  |
91.47 | 77.26 | 93.99 | 80.77 | 26.24 | 55.57 | 41.80 | 52.70 | 35.09 | 44.4 |
| 19 | 9-Xfluorene |  |
63.81 | 51.61 | 62.97 | 51.10 | 21.11 | 48.43 | 43.23 | 52.26 | 33.33 | 44.24 |
| 20 | 1-Xacenaphthylene |  |
96.29 | 82.65 | 98.75 | 86.24 | 44.44 | 71.14 | 61.01 | 71.28 | 44.97 | 56.96 |
| 21 | 3-Xacenaphthylene | 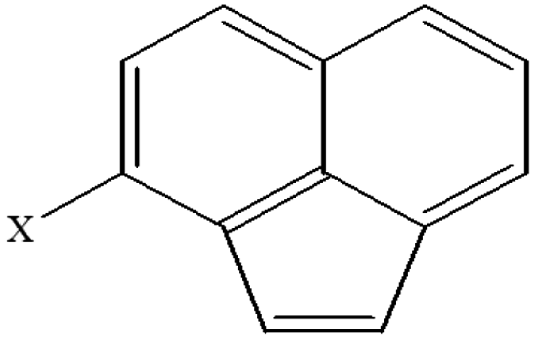 |
92.94 | 78.97 | 94.91 | 82.08 | 43.93 | 70.95 | 60.70 | 71.27 | 44.63 | 53.94 |
| 22 | 4-Xacenaphthylene | 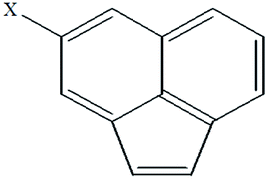 |
91.80 | 77.70 | 93.74 | 80.84 | 45.13 | 72.28 | 61.67 | 72.32 | 44.63 | 53.94 |
| 23 | 5-Xacenaphthylene | 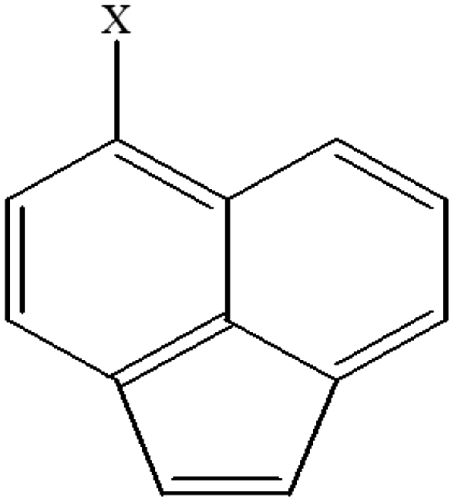 |
93.02 | 79.09 | 94.83 | 81.98 | 44.51 | 71.46 | 61.41 | 71.96 | 44.63 | 53.94 |
| 24 | 1-Xacenaphthene | 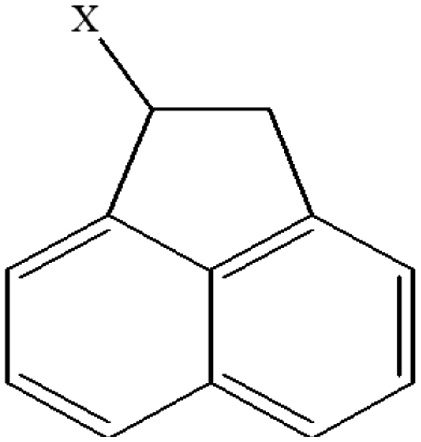 |
69.17 | 57.01 | 66.23 | 53.74 | 13.29 | 39.74 | 33.55 | 43.87 | 20.57 | 31.48 |
| 25 | 3-Xacenaphthene |  |
94.13 | 80.41 | 95.95 | 83.32 | 17.89 | 45.89 | 32.47 | 42.77 | 21.76 | 31.07 |
| 26 | 4-Xacenaphthene |  |
92.32 | 78.23 | 93.94 | 81.01 | 18.97 | 47.35 | 33.31 | 43.96 | 21.76 | 31.07 |
| 27 | 5-Xacenaphthene |  |
93.81 | 79.92 | 95.19 | 82.42 | 18.71 | 46.88 | 33.51 | 43.98 | 21.76 | 31.07 |
Fig. 2 displays the predicted BDE values of C–Cl and C–Br bond in the studied X-PAH molecules using various theoretical methods. From Fig. 2, the predicted BDE values using the three selected DFT methods are close to each other, and the results are also close to that from the G4 methods except for no. 7–14 X-PAHs with anthracene structure. It is noted that no. 7–14 molecules are with three aromatic rings. The DFT functionals may exhibit systematic errors as the system becomes larger.42,43 Considering the accuracy of G4 method in both BDEs and ΔfH298K shown in Fig. 1 and the success of G4 as the culmination of a progressively more accurate set of composite schemes,33,38,44 we also compare the predicted errors using the G4 results as benchmark dataset due to the lack of accurate experimental data for large X-PAHs as shown in Table 3. Compared with the G4 method, it can be seen that the three recommended DFT methods still shows better performance than the other three composite methods from both the AMSD and AMUD analysis. However, the AMUD error analysis results indicate that the DFT methods still exhibit large deviations compared with the G4 method, which mainly induced by the X-PAH molecules with three aromatic rings. Therefore, the structural effects on both BDEs and ΔfH298 are analyzed in detail according to the G4 computational results.
| Method | BDE | |
|---|---|---|
| AMSD | AMUD | |
| G3 | 4.17 | 5.05 |
| G3MP2 | 5.15 | 5.92 |
| CBS-QB3 | 6.28 | 6.67 |
| M06-2X/6-311++G(d, p) | −1.21 | 3.49 |
| ωB97X-D/6-311++G(d, p) | −1.13 | 3.63 |
| B2PLYP-D3/6-311++G(d, p) | −0.69 | 3.34 |
From Fig. 2 and Table 2, the variation trends of BDEs for C–Cl and C–Br bonds for the investigated X-PAH systems are similar, indicating that the structural effect of PAHs on C–Cl and C–Br bonds are identical. However, the absolute BDE value of C–Cl bond is larger than that of the corresponding C–Br bond by an averaged value around 15 kcal mol−1, indicates that the stability of C–Cl bond in X-PAHs is larger that of C–Br bond, which is mainly induced by the larger atomic radius of Cl than Br atom. For both Cl-PAHs and Br-PAHs, the molecules with three aromatic rings tend to enhance the C–Cl and C–Br bonds due to the electron delocalization, which results higher values of BDEs. Besides this, it can be seen that the BDE values among the studied X-PAH systems changes not much except for 4-Xphenanthrene (no. 14), 9-Xfluorene (no. 19), 1-Xacenaphthylene (no. 20), and 1-Xacenaphthene molecules (no. 24). The 9-chloro/bromofluorene shows the weakest C–Cl/C–Br bond among the studies X-PAH molecules, following is the 1-chloro/bromoacenaphthene. The BDE values of 1-chloroacenaphthylene and 1-bromoacenaphthylene tend to slightly larger than the other X-PAH compounds with one or two aromatic rings. Such overall reactivity trends are generally consistent with that of the C–Cl/Br bond in X–CH3, X–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, and X-benzene.40 However, aromatic structures still exhibit large effect on the BDE values.
CH2, and X-benzene.40 However, aromatic structures still exhibit large effect on the BDE values.
Fig. 3 shows the optimized structures of these molecules at the M06-2X/6-311++G(d, p) level of theory. By compared the optimized structures of halogenated phenanthrene, it is found that only 4-Xphenanthrene exhibits a non-planar structure due to the interaction between X and adjacent H atoms, resulting a less stable structure compared with the other planar Xphenanthrene molecules. Thus, the corresponding C–Cl/Br bond is smaller than the other halogenated phenanthrenes. The C–Cl and C–Br bond in 9-chlorofluorene and 9-bromofluorene are generally smaller than that in halogenated methane and halogenated cyclohexane by approximately 20 kcal mol−1, revealing that the effects from the aromatic structure and adjacent H atom are larger to weaken the C–Cl or C–Br bond. The C–Cl and C–Br bonds in 1-chloroacenaphthene and 1-bromoacenaphthene are larger than that in 9-Xfluorene by approximately 6 kcal mol−1, indicating that the synergistic effect from the two aromatic rings reduces the stability of C–Cl and C–Br bonds in 9-Xfluorene. For 1-chloroacenaphthylene and 1-bromoacenaphthylene, the BDEs of C–Cl and C–Br bonds are slightly larger than the others, which can be attributed to the stabilization from the conjugative effect between the CC double bond and the naphthyl ring. The G4 predicted BDEs of C–Cl and C–Br bonds in 1-chloroacenaphthylene and 1-bromoacenaphthylene are also slightly than the corresponding BDE values in Cl–CHCH2 and Br–CHCH2 by 1.5 and 3.3 kcal mol−1, respectively.
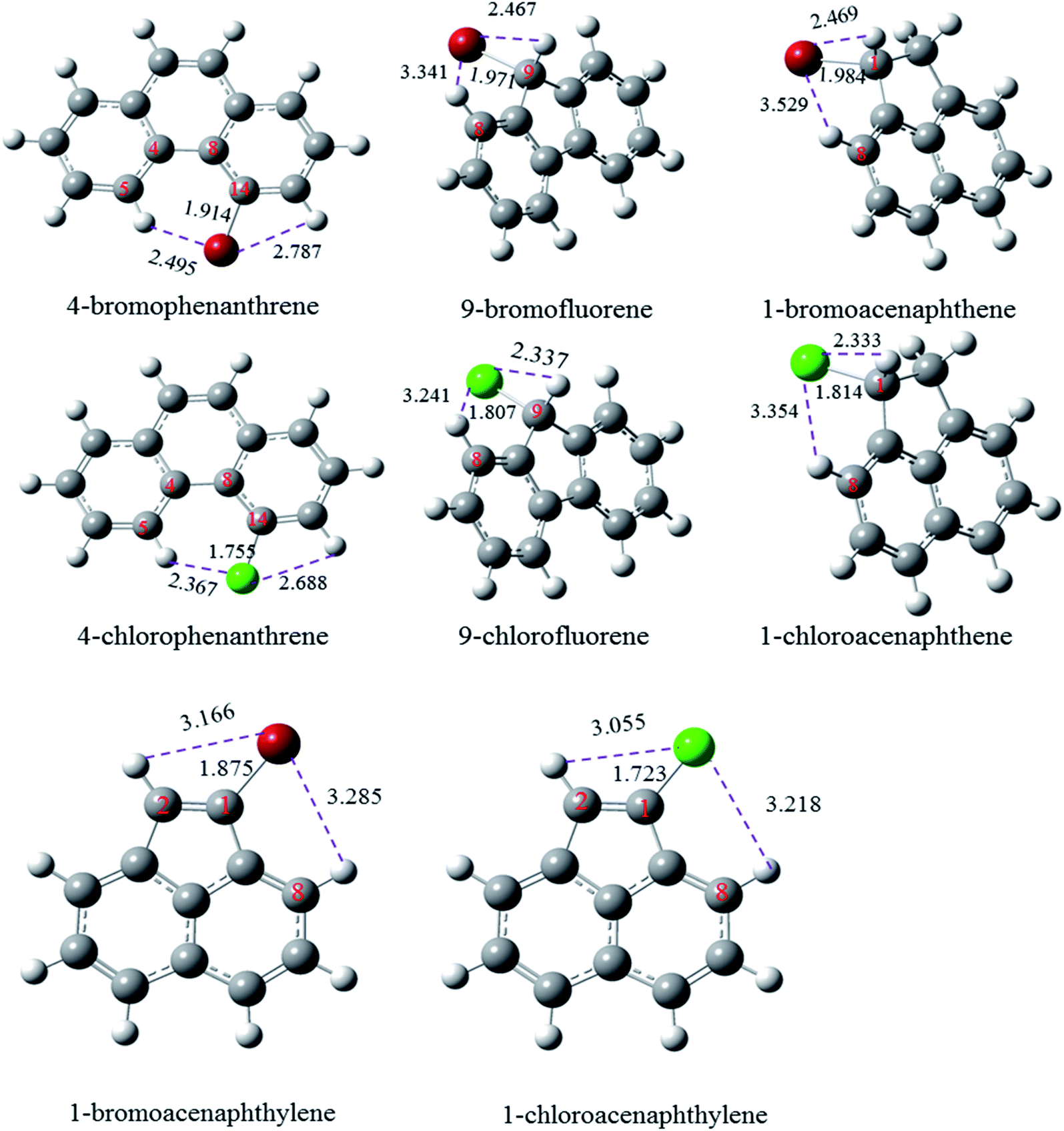 | ||
| Fig. 3 Optimized structures of 4-halogenated phenanthrene, 9-halogenated fluorene, and 1-halogenated acenaphthene at the M06-2X/6-311++G(d, p) level. | ||
3.3. Enthalpies of formation
Table 4 lists the error analysis of the predicted ΔfH298 using different computational methods against the G4 results, while Fig. 4 shows the predicted ΔfH298 values of Cl-PAH compounds (a) and Br-PAH compounds (b) using various theoretical methods. From Table 2, the AMUD values from G3 and G3MP2 methods are significantly larger, indicating that the two composite methods may be not suitable for the prediction of ΔfH298 values of the studied systems. The differences of the AMUD among the three DFT functionals with cc-pVTZ basis set and the CBS-QB3 method are small. The small value of AMSD of the CBS-QB3 methods reveals larger error cancellation effect of this method. Overall, the absolute predicted results using the methods listed in Table 2 still exhibit large deviations compared with the G4 method as explicitly shown in Fig. 4.| Method | ΔfH298 | |
|---|---|---|
| AMSD | AMUD | |
| G3 | 11.92 | 11.92 |
| G3MP2 | 4.59 | 17.94 |
| CBS-QB3 | 0.03 | 7.68 |
| M06-2X/cc-pVTZ | 2.47 | 6.88 |
| ωB97X-D/cc-pVTZ | 6.37 | 7.99 |
| B2PLYP-D3/cc-pVTZ | 4.57 | 8.51 |
| GA | −2.95 | 8.11 |
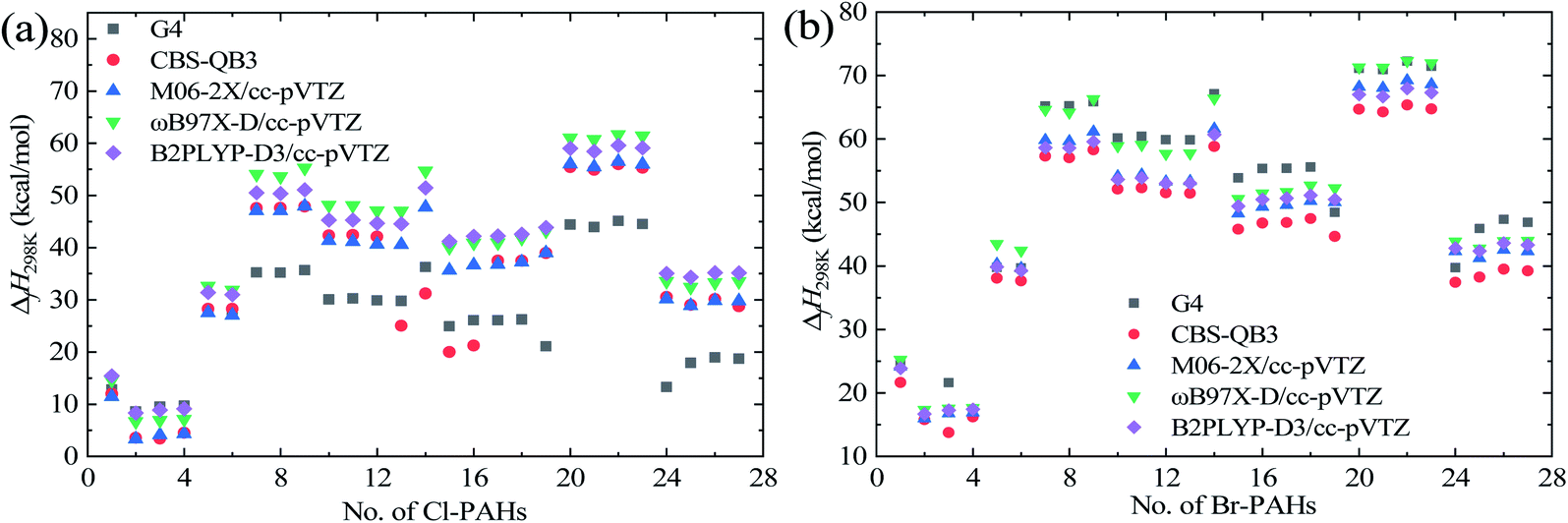 | ||
| Fig. 4 Predicted ΔfH298 values of Cl-PAH compounds (a) and Br-PAH compounds (b) via single-point energy computational results using various theoretical methods. | ||
From Fig. 4, the ΔfH298 values of the corresponding Cl-PAH and Br-PAH molecules show the similar variation trends as the molecular structure changes. However, the absolute ΔfH298 values of Br-PAHs are generally larger than that of the corresponding Cl-PAHs, and the deviations are strongly dependent on the molecular structures. Thus, the thermal stability of Cl-PAHs is generally better than the corresponding Br-PAHs. It can be seen that the halogenated toluene molecules show the smallest ΔfH298 values, while the halogenated acenaphthylene molecules are with the largest ΔfH298 values. The substituted positions of Cl and Br atoms on the PAHs exhibit small effect on the ΔfH298 except for halogenated acenaphthene molecules due to the substitution at the C–C single bond position and 4-Xphenanthrene induced by the specific substituted positions. Further, it can be seen that the ΔfH298 values of halogenated acenaphthylene molecules are larger than that of the halogenated acenaphthene molecules. However, it is hard to found a general relationship between ΔfH298 and the number of aromatic rings for the studied X-PAHs.
The derived enthalpies of the studied X-PAHs and error analysis results from GA method are also shown in Tables 2 and 4. The AMSD and AMUD values for GA method compared with the G4 method are −2.95 and 8.11 kcal mol−1, respectively. This indicates that the overall performance of GA method can be comparable with the DFT methods. However, obvious limitation of the GA method still exists. A major drawback is that the GA method cannot accurately reveal some of the position effect on the ΔfH298 values, i.e., the predicted ΔfH298 of the halogenated naphthalene, and halogenated phenanthrene molecules are identical for different substituted positions. Thus, although the GA method provide an efficient way to quickly estimate the thermodynamic properties, it is not recommended to study the substituted position effect on the X-PAHs. Nonetheless, the number of aromatic rings and structural effect on the ΔfH298 of the studied X-PAHs can be well captures via the GA method, and the obtained absolute results can also be well predicted.
Finally, the computational cost of the employed various theoretical methods is compared as shown in Table 5 for the medium sized molecules, i.e., 1-chloronaphthalene and 1-bromonaphthalene. Considering the smaller difference between 6-311++G(d, p) and cc-pVTZ basis sets in DFT calculations, Table 5 only explicitly shows the relative computational cost from cc-pVTZ basis set in combination with different functionals. The cc-pVQZ basis set does not significantly improves the prediction accuracy for both BDEs and ΔfH298, thus, it also not considered due to the large computational cost compared with the other two basis sets. Assuming the computational time cost for calculations from M06-2X/cc-pVTZ method is 1 by using 20-core Intel Xeon Silver CPU, Table 5 explicitly shows the relative computational cost of typical methods. It is worth noting that the B2PLYP-D3 functional exhibits large computational cost for single-point energy calculations neglecting the optimization process compared with the other functionals due to the inclusion of MP2 component of the calculation, which is not recommended for large similar systems. Although the G3MP2 method shows smaller computational cost compared with the other composite methods, it is not recommended due to large deviations. Further, the relative computational cost of composite methods increases as molecular size increases. Therefore, the composite CBS-QB3 method and ωB97X-D functional are recommended for large reaction systems.
| Method | Relative computational time | |
|---|---|---|
| 1-Chloronaphthalene | 1-Bromonaphthalene | |
| G4 | ∼14 | ∼18 |
| G3 | ∼8.4 | ∼11 |
| G3MP2 | ∼1.8 | ∼3.0 |
| CBS-QB3 | ∼3.4 | ∼5.5 |
| M06-2X/cc-pVTZ | 1 | 1 |
| ωB97X-D/cc-pVTZ | ∼1.05 | ∼1.05 |
| B2PLYP-D3/cc-pVTZ | ∼1.8 | ∼1.8 |
4. Conclusions
In this work, we perform theoretical studies on the BDEs of C–Cl and C–Br bonds of 27 Cl- and Br-PAHs together with their corresponding ΔfH298. Four composite model chemistry methods including CBS-QB3, G3MP2, G3, and G4 together with three widely used DFT functionals with various basis sets in the prediction of the BDEs and ΔfH298 are firstly compared with the experimental results, and statistical analysis indicates that G4 is the only method that can approach the “chemical accuracy” (approximately 1–2 kcal mol−1) for both BDEs and ΔfH298. Due to the lack of accurate data for large X-PAHs, the G4 method is further used as the benchmark for the other methods. It is found that the predicted BDEs of C–Cl and C–Br bonds in X-PAHs using DFT method are dependent on basis set. Specifically, the three M06-2X, ωB97X-D, and B2PLYP-D3 DFT functionals with 6-311++G(d, p) exhibits similar performance, and the ωB97X-D/6-311++G(d, p) method is slightly better than the other two methods. For ΔfH298, the CBS-QB3 method and the three DFT functionals with cc-pVTZ basis set exhibit similar performance, but the deviations remain larger compared with the G4 method, especially for Cl-PAHs. The GA method can provide comparable results with the CBS-QB3 and DFT methods, however, it cannot reveal the position effect of typical halogenated PAHs. Thus, the G4 method is still recommended to obtain accurate ΔfH298 values. However, the GA method provides a quick and efficient way to obtain reliable ΔfH298 results for large X-PAHs when position effect is not the research focus. Using the G4 results, the structural effect and substituted position effect on the BDEs of C–Cl and C–Br bonds in the studied X-PAHs and the ΔfH298 are systematically analyzed. It is found that structural and position effects on the BDEs and ΔfH298 exhibit very similar variation trends. The thermal stability of Cl-PAHs is generally better than the corresponding Br-PAHs from the computed ΔfH298, which results in the C–Br bond is smaller than the corresponding C–Cl bond in X-PAHs. Further, no linear relationship is found between the number of aromatic rings and the BDEs or ΔfH298 values. The present work provides valuable guidance for the future estimation of thermodynamic properties of large X-PAH molecules.Author contributions
Shenying Xu: investigation, data curation, original draft preparation. Quan-De Wang: supervision, project administration, conceptualization, writing-reviewing and editing. Mao-Mao Sun: methodology, formal analysis, data curation. Guoliang Yin: conceptualization, data curation, formal analysis. Jinhu Liang: data curation, visualization, writing-reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank National Supercomputing Center in Shenzhen for providing the computational resources and Gaussian 09 suite of programs (Revision D.01). S. Xu acknowledges financial support from Sailing Project of Yibin University (XJ2020007702). Q.-D. Wang and J. Liang acknowledge financial support from the opening funds of Functional Materials and Resource Utilization Lab of Yibin University.References
- R. Jin, M. Zheng, G. Lammel, B. A. M. Bandowe and G. Liu, Prog. Energy Combust. Sci., 2020, 76, 100803 CrossRef.
- J. L. Sun, H. Zeng and H. G. Ni, Chemosphere, 2013, 90, 1751–1759 CrossRef CAS PubMed.
- C. Ding, H. G. Ni and H. Zeng, Environ. Pollut., 2012, 168, 80–86 CrossRef CAS PubMed.
- Q. Z. Liu, X. Xu, L. Wang and D. H. Wang, Chem. Eng. J., 2020, 400, 125901 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1936, 32, 1333–1360 RSC.
- J. L. Bao, R. Meana-Paneda and D. G. Truhlar, Chem. Sci., 2015, 6, 5866–5881 RSC.
- H. J. Curran, Proc. Combust. Inst., 2019, 37, 57–81 CrossRef CAS.
- S. J. Klippenstein, Proc. Combust. Inst., 2017, 36, 77–111 CrossRef CAS.
- M. Keceli, S. N. Elliott, Y. P. Li, M. S. Johnson, C. Cavallotti, Y. Georgievskii, W. H. Green, M. Pelucchi, J. M. Wozniak, A. W. Jasper and S. J. Klippenstein, Proc. Combust. Inst., 2019, 37, 363–371 CrossRef CAS.
- J. D. Watts, J. Gauss and R. J. Bartlett, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- K. A. Peterson, D. E. Woon and T. H. Dunning Jr, J. Chem. Phys., 1994, 100, 7410–7415 CrossRef CAS.
- K. Raghavachari and A. Saha, Chem. Rev., 2015, 115, 5643–5677 CrossRef CAS PubMed.
- L. A. Curtiss, P. C. Redfern and K. Raghavachari, J. Chem. Phys., 2007, 126, 084108 CrossRef PubMed.
- L. A. Curtiss, P. C. Redfern, V. Rassolov, G. Kedziora and J. A. Pople, J. Chem. Phys., 2001, 114, 9287–9295 CrossRef CAS.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- C. C. Chen, J. W. Bozzelli and J. T. Farrell, J. Phys. Chem. A, 2004, 108, 4632–4652 CrossRef CAS.
- G. Blanquart and H. Pitsch, J. Phys. Chem. A, 2007, 111, 6510–6520 CrossRef CAS PubMed.
- T. C. Allison and D. R. Burgess, J. Phys. Chem. A, 2015, 119, 11329–11365 CrossRef CAS PubMed.
- Q.-D. Wang and Z.-W. Liu, J. Phys. Chem. A, 2018, 122, 5202–5210 CrossRef CAS PubMed.
- Q.-D. Wang, J. Phys. Org. Chem., 2017, 30, e3668 CrossRef.
- L. Lai and W. H. Green, J. Phys. Chem. A, 2019, 123, 3176–3184 CrossRef CAS PubMed.
- N. Mardirossian and M. Head-Gordon, Mol. Phys., 2017, 115, 2315–2372 CrossRef CAS.
- P. Verma and D. G. Truhlar, Trends Chem., 2020, 2, 302–318 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- T. H. Dunning Jr, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning, J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- S. J. Klippenstein, L. B. Harding and B. Ruscic, J. Phys. Chem. A, 2017, 121, 6580–6602 CrossRef CAS PubMed.
- D. Maftei, D.-L. Isac, M. Dumitraş, Ş. Bucur and A.-C. Dîrţu, Struct. Chem., 2018, 29, 921–927 CrossRef CAS.
- M. Altarawneh, A. Saeed, M. Al-Harahsheh and B. Z. Dlugogorski, Prog. Energy Combust. Sci., 2019, 70, 212–259 CrossRef.
- L. A. Curtiss, P. C. Redfern, K. Raghavachari, V. Rassolov and J. A. Pople, J. Chem. Phys., 1999, 110, 4703–4709 CrossRef CAS.
- S. J. Klippenstein and C. Cavallotti, in Mathematical modelling of gas-phase complex reaction systems: pyrolysis and combustion, 2019, pp. 115–167, DOI:10.1016/b978-0-444-64087-1.00002-4.
- P. Kraus, J. Chem. Theory Comput., 2020, 16, 5712–5722 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, A. Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- M. Liu, A. Grinberg Dana, M. S. Johnson, M. J. Goldman, A. Jocher, A. M. Payne, C. A. Grambow, K. Han, N. W. Yee, E. J. Mazeau, K. Blondal, R. H. West, C. F. Goldsmith and W. H. Green, J. Chem. Inf. Model., 2021, 61, 2686–2696 CrossRef CAS PubMed.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, J. Chem. Phys., 1997, 106, 1063–1079 CrossRef CAS.
- B. Ruscic and D. H. Bross, in Mathematical Modelling of Gas-Phase Complex Reaction Systems: Pyrolysis and Combustion, 2019, pp. 3–114, DOI:10.1016/b978-0-444-64087-1.00001-2.
- B. Ruscic, J. Phys. Chem. A, 2015, 119, 7810–7837 CrossRef CAS PubMed.
- Y.-R. Luo, Handbook of bond dissociation energies in organic compounds, CRC press, 2002 Search PubMed.
- P. J. Linstrom and W. G. Mallard, NIST Chemistry WebBook, NIST Standard Reference Database Number 69, retrieved June 6, 2021 Search PubMed.
- R. Sivaramakrishnan, R. S. Tranter and K. Brezinsky, J. Phys. Chem. A, 2005, 109, 1621–1628 CrossRef CAS PubMed.
- P. C. Redfern, P. Zapol, L. A. Curtiss and K. Raghavachari, J. Phys. Chem. A, 2000, 104, 5850–5854 CrossRef CAS.
- J. M. Simmie and K. P. Somers, J. Phys. Chem. A, 2015, 119, 7235–7246 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra05391d |
| This journal is © The Royal Society of Chemistry 2021 |